
A copper complex covalently grafted on carbon nanotubes and reduced graphene oxide promotes oxygen reduction reaction activity and catalyst stability†
Abstract
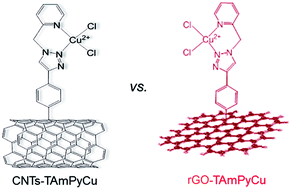
Developing new non-precious-metal catalysts for the oxygen reduction reaction (ORR) is very important to the final substitution of a platinum-based noble metal catalyst for large-scale commercialization of fuel cells. Herein, we report two new composites CNTs-TAmPyCu and rGO-TAmPyCu as ORR electrocatalysts, where a triazole-pyridine coordinated copper complex TAmPyCu was covalently grafted onto multi-walled carbon nanotubes (CNTs) and reduced graphene oxide (rGO), respectively. Covalent immobilization of a copper complex on CNTs or rGO remarkably improves the catalyst ORR activity and stability compared with that of the physisorbed counterparts. Furthermore, the rGO-TAmPyCu composite significantly enhanced the selectivity of 4e− versus 2e− reduction of O2 relative to the CNTs-TAmPyCu catalyst, suggesting the beneficial effect of the flat rGO as a supporting material for copper complexes. The ORR activity of the rGO-TAmPyCu catalyst was also compared with a reported multi-nuclear copper assembly. The results from this study suggest that creating more proximal dinuclear Cu sites on a support is an effective approach to promote the oxygen 4e− reduction process, and a multinuclear Cu assembly is crucial for efficient O2 reduction with impressively lowered overpotential.
 Please wait while we load your content...
Please wait while we load your content...

