
Fabrication of a Cu(I)-carboxylate metal–organic framework by reduction of metal nodes for an azide–alkyne “click” reaction†
Xiaojiao
Hou
ab,
Wenxiu
He
a,
Xu
Zhai
 a,
Bingbing
Chen
a,
Yuanlin
Fu
a,
Liying
Zhang
a,
Junyi
Chen
*c and
Yu
Fu
*a
a,
Bingbing
Chen
a,
Yuanlin
Fu
a,
Liying
Zhang
a,
Junyi
Chen
*c and
Yu
Fu
*a
aDepartment of Chemistry, College of Sciences, Northeastern University, Shenyang, 110819, P. R. China. E-mail: fuyu@mail.neu.edu.cn
bExperimental Center, LiaoDong University, Dandong, 118003, P. R. China
cEngineering Laboratory of Chemical Resources Utilization in South Xinjiang, College of Chemistry and Chemical Engineering, Tarim University, Xinjiang Uygur Autonomous Region, Alaer, 843300, P. R. China. E-mail: sln5xn@163.com
First published on 7th June 2024
Abstract
Metal–organic frameworks (MOFs) permit significant structural diversity and functional design flexibility due to the various combinations of metal centers and different organic linkers. However, their combinations generally adhere to the classic hard and soft acid and base (HSAB) theory. This makes it impossible to directly synthesize desired MOFs with converse Lewis types of metal ions and ligands. Herein, we present a novel Cu(II) ion cleavage reduction strategy that circumvents the limitations of HSAB theory. We demonstrate this approach by utilizing a metastable CuH2DOBDC MOF, whose inherent structural instability facilitates structural transformations and phase transitions in response to external stimuli. By leveraging the unique structure of ascorbic acid to reduce Cu(II) ions and cleave the H2DOBDC2− linkers, we successfully fabricate a new Cu2H2DOBDC structure composed of a soft acid Cu(I) and a hard base (H4DOBDC). The resultant Cu2H2DOBDC integrates the characteristics of mesoporosity and hierarchical nanostructures, resulting in excellent mass transfer and abundant accessible Cu(I) active sites, which endows the material with outstanding catalytic activity in the azide–alkyne cycloaddition reaction. This is the first example of fabricating stable single-phase Cu(I)-carboxylic acid MOFs with nearly all Cu(I) ions, and this work offers a new perspective on the creation of a new category of MOFs with soft-acid and hard-base type coordination bonds.
Introduction
Metal–organic frameworks (MOFs) are an emerging class of crystalline porous materials composed of metal nodes and organic ligands.1 Due to their tunable structures, accessible open metal sites, and extremely large surface areas, MOFs have shown unprecedented performance in a variety of applications.2–7 To date, millions of distinct metal–organic frameworks have been synthesized by combining metal ions and organic linkers. However, their coupling is not arbitrary and is usually restricted by Pearson's hard and soft acid and base (HSAB) theory. Hard acids prefer to combine with hard bases and soft acids prefer soft bases.8,9 For instance, hard acidic metal ions such as Zr4+, Fe3+, Cr3+, and Al3+ are used to form stable MOFs with carboxylate-based ligands that act as hard bases, such as UiO-66, MIL-101, and MIL-53.10 Soft acidic metal ions such as Cu1+ and Ag1+ are used to form stable MOFs with nitrogen- or sulfur-containing soft base ligands, such as MAF-2 and CityU-7.11,12 Therefore, it is normally difficult to achieve a class of MOFs based on soft-acid and hard-base type coordination bonds via direct reactions of metal ions and organic ligands.Functionalization of metal–organic frameworks (MOFs) with Cu(I) sites holds significant promise for various applications including adsorption, separation, catalysis, and sensing.13–18 However, according to the above HSAB theory, Cu(I)-MOFs are usually fabricated using ligands with pyridyl N- or sulfhydryl S-centers. Although carboxylate-based MOFs constitute the overwhelming majority, Cu(I)-MOFs fabricated with carboxylate-based ligands are expected to exhibit thermal stability and have broad catalytic applications.19 Up to now, the direct synthesis of MOFs with Cu(I) metal ions and carboxylate-based ligands has not been reported. Most commonly, Cu(I)-carboxylate MOFs are synthesized using post-treatment approaches. For example, Chen et al. reported that HKUST-1, in the presence of hydroquinone, was reduced to mixed-valence Cu(I)Cu(II)-BTC and could remarkably boost the D2/H2 isotope separation.14 Sun et al. modulated some of the Cu(II) metal nodes in HKUST-1 to Cu(I) by reducing vapor treatment, thereby significantly enhancing the capture activity of aromatic sulfides.20 To our knowledge, only Qiao et al. have recently reported a “molecular scalpel” to chemically cleave conventional CuBDC with a controlled phase transition and synthesize a Cu2BDC MOF, where the Cu(I)/Cu(II) ratio is 1.21 Although the post-treatment approach is an effective strategy for creating new Cu(I)-carboxylate MOFs, achieving a near equal proportion of Cu(I) and Cu(II) sites in the MOF is far from satisfactory. Hence, it is highly desirable to develop an efficient and controllable approach for creating a new MOF with a high proportion of Cu(I) metal ions and carboxylate-based ligands, but this remained a challenge until now.
Herein, we have successfully fabricated a stable single-phase Cu(I)-carboxylate MOF (denoted as Cu2H2DOBDC), as shown in Fig. 1. We exploited the unique characteristics of metastable MOF structures, which are prone to undergo structural transformation under external stimulation, to achieve full phase transition. Starting with a reported metastable Cu(II)-MOF synthesized from Cu(OAc)2 and 2,5-dihydroxyterephthalic acid (H4DOBDC), we demonstrated that this metastable Cu(II)-MOF could be reduced to construct a novel Cu(I)-carboxylate MOF in the presence of ascorbic acid. A controlled phase transition was achieved by ascorbic acid accurately regulating the chemical state and coordination environment of Cu metal centers. X-ray photoelectron spectrum (XPS) analysis confirmed that nearly all Cu(II) ions were converted to Cu(I) ions. Cu2H2DOBDC, with its high chemical stability, mesoporosity, and uniformly abundant Cu(I) exposure sites, exhibits excellent catalytic capability as a recyclable heterogeneous catalyst for the azide–alkyne cycloaddition (AAC) reaction. To the best of our knowledge, this is the first attempt to fabricate a stable single-phase Cu(I)-carboxylate MOF with near-full Cu(I) ions. It is always fascinating to discover new MOF structures, and this work presents a promising step toward the synthesis of desired MOFs with converse Lewis types of metal ions and ligands, which is impossible to realize by conventional methods.
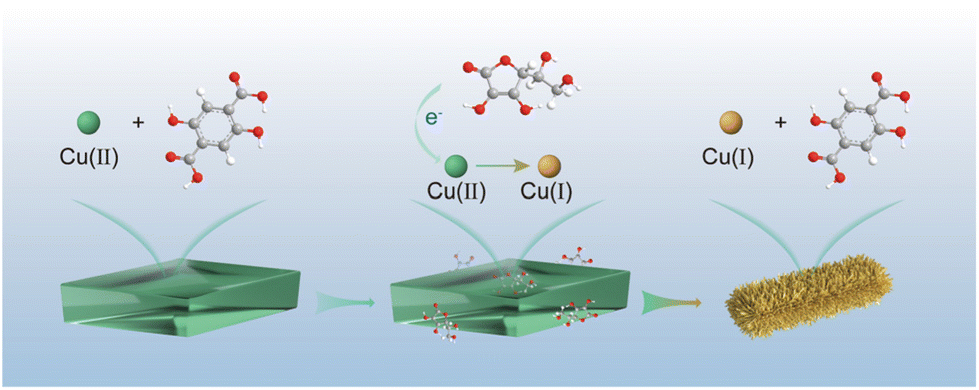 | ||
| Fig. 1 Schematic illustration of the fabrication of a Cu(I)-carboxylate MOF. | ||
Results and discussion
Structural characterization
The metastable Cu(II)-MOF ([Cu(H2DOBDC)(DMF)2]·DMF)22 (denoted as CuH2DOBDC) was synthesized by the reaction of Cu(OAc)2 and H4DOBDC at room temperature, according to our group's previous work with a few modifications. Scanning electron microscopy (SEM) images showed that the as-prepared metastable Cu(II)-MOF exhibited a typical rhombohedron morphology with a smooth surface (Fig. 2a). Single-crystal X-ray diffraction indicated that the metastable Cu(II)-MOF crystallized in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and formed a two-dimensional network layer (Fig. S1†). In addition, the powder X-ray diffraction (XRD) pattern of the crystal was consistent with the pattern simulated based on single-crystal XRD data (Fig. S2†), verifying the crystal phase purity. Following ascorbic acid treatment, it exhibited a well-defined cube structure with a substantial decrease in size (Fig. 2b). Notably, the surface was no longer smooth and had been replaced by hierarchical nanostructures. The crystallinity of the nanostructure was further confirmed by selected area electron diffraction (SAED) data, which indicated that the cube structure is a single crystal, as shown in Fig. 2c and d.
space group and formed a two-dimensional network layer (Fig. S1†). In addition, the powder X-ray diffraction (XRD) pattern of the crystal was consistent with the pattern simulated based on single-crystal XRD data (Fig. S2†), verifying the crystal phase purity. Following ascorbic acid treatment, it exhibited a well-defined cube structure with a substantial decrease in size (Fig. 2b). Notably, the surface was no longer smooth and had been replaced by hierarchical nanostructures. The crystallinity of the nanostructure was further confirmed by selected area electron diffraction (SAED) data, which indicated that the cube structure is a single crystal, as shown in Fig. 2c and d.
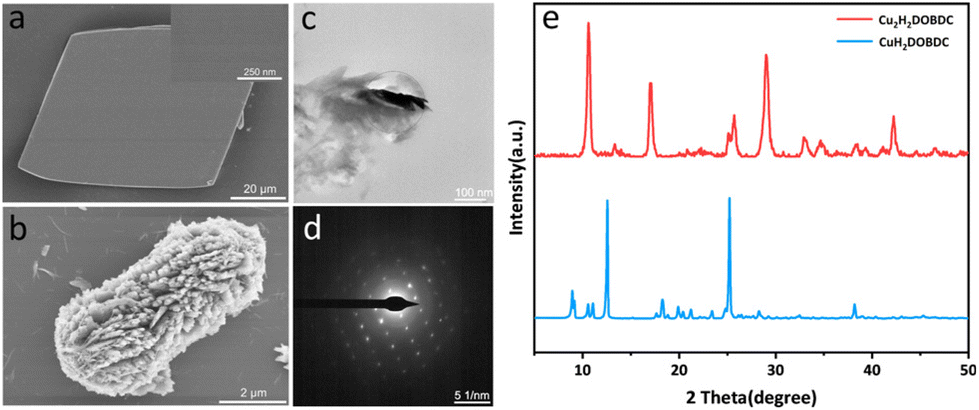 | ||
| Fig. 2 (a) SEM images of CuH2DOBDC. (b) SEM images of Cu2H2DOBDC. (c) TEM images of Cu2H2DOBDC. (d) SAED pattern of the selected area in (c). (e) XRD patterns of CuH2DOBDC and Cu2H2DOBDC. | ||
The XRD pattern showed that the metastable Cu(II)-MOF achieved a full phase transition through the reduction of ascorbic acid (Fig. 2e). As the dosage of the reducing agent increased, the new characteristic peaks at 10.65°, 17.02°, 29.02°, and 42.20° gradually increased in intensity, while the metastable Cu(II)-MOF characteristic peaks gradually faded (Fig. S3†). By adjusting the addition of ascorbic acid, the controlled synthesis of intermediate samples was achieved. The coexistence of two phases is observed in the XRD patterns.
Thermogravimetric (TGA) analysis was employed to analyse the thermostability, as variations in coordination mode may present different mass loss curves. As shown in Fig. 3a, following treatment with ascorbic acid, the experimentally determined mass percentage of CuO in the final product was 44.63%. The final product exhibited a weight loss of about 6.28% in the temperature range of 30–220 °C, which can be attributed to the desorption of physically adsorbed water and solvent. Furthermore, a weight loss of about 49.09% was observed in the temperature range of 220–330 °C, which belonged to ligand decomposition. TGA analysis showed that the ratio of Cu to the H4DOBDC ligand significantly changed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1 following ascorbic acid treatment, indicating a potential structural transformation in the material (see detailed analysis in the ESI†). Consequently, the as-synthesized Cu(I)-carboxylate MOF was denoted as Cu2H2DOBDC. Organic elemental analyzer (EA) results further support our conclusions (see detailed analysis in the ESI†). In addition, the dropping H2DOBDC2− ligand was detected by ultraviolet-visible (UV-vis) spectroscopy in a reducing solution (Fig. S4†). There is a gradual increase in the residual Cu mass percentage observed from CuH2DOBDC to Cu2H2DOBDC, a trend that is further supported by ICP-MS analysis (Table S1†). These results further strengthen the conclusion of TGA. The difference thermogravimetry (DTG) analysis suggested that the thermal stability of Cu2H2DOBDC is weakened due to the formation of Cu(I) species and the variation of coordination mode (Fig. S5†).20 Fourier-transform infrared spectroscopy (FTIR) was employed to investigate the coordination mode between Cu and the carboxylate linker.23 Conclusions about the carboxylate coordination mode are often drawn based on the splitting value between the asymmetric and symmetric stretches of the carboxylic vibration, where Δ = νas(COO−) − νs(COO−) (Fig. S6†).21,24 The metastable CuH2DOBDC MOF contains 2,5-dihydroxyterephthalic acid ligands with two coordination modes: carboxylate groups that connect to a single copper atom through monodentate coordination and those that bridge two copper atoms simultaneously. The FTIR spectra of CuH2DOBDC and Cu2H2DOBDC are shown in Fig. 3b. In CuH2DOBDC, the asymmetric stretch of the carboxylic vibration peak appeared at 1595 cm−1, while the symmetric stretch peak presented two distinct bands at 1425 and 1376 cm−1.25 Consequently, the observed splitting values of 170 cm−1 and 219 cm−1 in CuH2DOBDC correspond to bridging and monodentate characteristics, respectively. In addition, the spectra of CuH2DOBDC exhibited a peak at 1670 cm−1 attributed to the carbonyl stretching vibration of DMF. These results are consistent with the CuH2DOBDC crystal structure. In contrast, for Cu2H2DOBDC, the asymmetric stretch of the carboxylic vibration peak also appeared at 1595 cm−1, but the symmetric stretch peak was only observed at 1332 cm−1. Therefore, a splitting value of 263 cm−1 is consistent with the monodentate mode, indicating that in Cu2H2DOBDC, the Cu ions are coordinated to the carboxylate ligands in a monodentate mode. Notably, the absence of a peak corresponding to the carbonyl stretching vibration of DMF in the spectrum suggests that DMF does not participate in coordination within Cu2H2DOBDC. From the N2 adsorption–desorption measurement, Cu2H2DOBDC exhibited a type IV isotherm with a distinct hysteresis loop (Fig. 3c), indicating the existence of mesopores,26–28 which could be attributed to the ligand loss during the reduction process.20 The pore size distributions shown in Fig. S7† reveal that the size of the mesopores is centered at about 17 nm. The SEM images reveal that the surface of Cu2H2DOBDC exhibits a hierarchical nanostructure assembled by nanosheets, where the intersections and stacking of the surface hierarchical nanostructures also contribute to the formation of mesopores.29
:1 to 2:1 following ascorbic acid treatment, indicating a potential structural transformation in the material (see detailed analysis in the ESI†). Consequently, the as-synthesized Cu(I)-carboxylate MOF was denoted as Cu2H2DOBDC. Organic elemental analyzer (EA) results further support our conclusions (see detailed analysis in the ESI†). In addition, the dropping H2DOBDC2− ligand was detected by ultraviolet-visible (UV-vis) spectroscopy in a reducing solution (Fig. S4†). There is a gradual increase in the residual Cu mass percentage observed from CuH2DOBDC to Cu2H2DOBDC, a trend that is further supported by ICP-MS analysis (Table S1†). These results further strengthen the conclusion of TGA. The difference thermogravimetry (DTG) analysis suggested that the thermal stability of Cu2H2DOBDC is weakened due to the formation of Cu(I) species and the variation of coordination mode (Fig. S5†).20 Fourier-transform infrared spectroscopy (FTIR) was employed to investigate the coordination mode between Cu and the carboxylate linker.23 Conclusions about the carboxylate coordination mode are often drawn based on the splitting value between the asymmetric and symmetric stretches of the carboxylic vibration, where Δ = νas(COO−) − νs(COO−) (Fig. S6†).21,24 The metastable CuH2DOBDC MOF contains 2,5-dihydroxyterephthalic acid ligands with two coordination modes: carboxylate groups that connect to a single copper atom through monodentate coordination and those that bridge two copper atoms simultaneously. The FTIR spectra of CuH2DOBDC and Cu2H2DOBDC are shown in Fig. 3b. In CuH2DOBDC, the asymmetric stretch of the carboxylic vibration peak appeared at 1595 cm−1, while the symmetric stretch peak presented two distinct bands at 1425 and 1376 cm−1.25 Consequently, the observed splitting values of 170 cm−1 and 219 cm−1 in CuH2DOBDC correspond to bridging and monodentate characteristics, respectively. In addition, the spectra of CuH2DOBDC exhibited a peak at 1670 cm−1 attributed to the carbonyl stretching vibration of DMF. These results are consistent with the CuH2DOBDC crystal structure. In contrast, for Cu2H2DOBDC, the asymmetric stretch of the carboxylic vibration peak also appeared at 1595 cm−1, but the symmetric stretch peak was only observed at 1332 cm−1. Therefore, a splitting value of 263 cm−1 is consistent with the monodentate mode, indicating that in Cu2H2DOBDC, the Cu ions are coordinated to the carboxylate ligands in a monodentate mode. Notably, the absence of a peak corresponding to the carbonyl stretching vibration of DMF in the spectrum suggests that DMF does not participate in coordination within Cu2H2DOBDC. From the N2 adsorption–desorption measurement, Cu2H2DOBDC exhibited a type IV isotherm with a distinct hysteresis loop (Fig. 3c), indicating the existence of mesopores,26–28 which could be attributed to the ligand loss during the reduction process.20 The pore size distributions shown in Fig. S7† reveal that the size of the mesopores is centered at about 17 nm. The SEM images reveal that the surface of Cu2H2DOBDC exhibits a hierarchical nanostructure assembled by nanosheets, where the intersections and stacking of the surface hierarchical nanostructures also contribute to the formation of mesopores.29
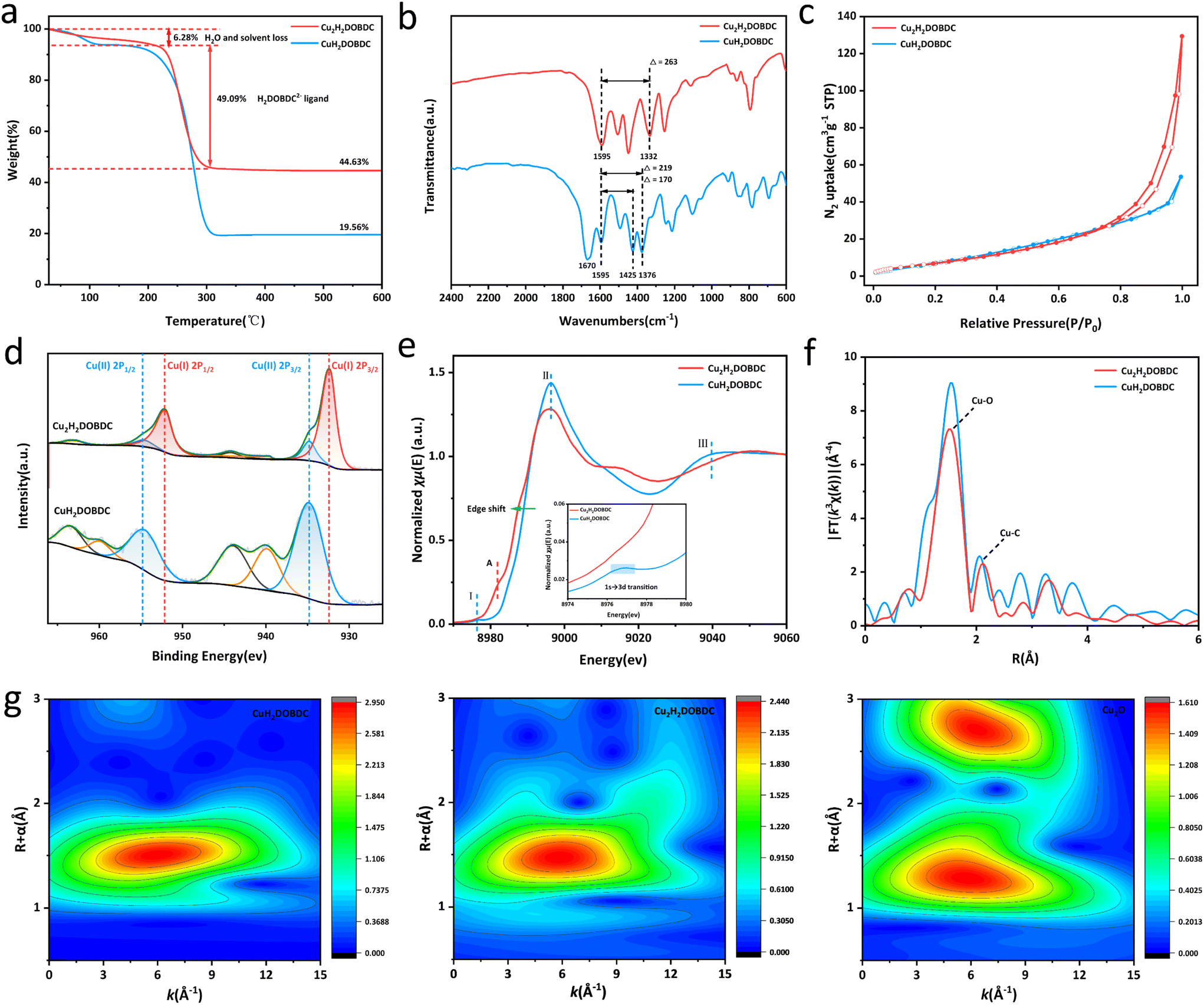 | ||
| Fig. 3 (a) TGA curves for CuH2DOBDC and Cu2H2DOBDC under an air atmosphere. (b) FTIR spectra of CuH2DOBDC and Cu2H2DOBDC. (c) N2 adsorption–desorption isotherms of CuH2DOBDC and Cu2H2DOBDC. (d) XPS spectra for Cu 2p. (e) Cu K-edge XANES spectra. (f) Cu K-edge EXAFS spectra. (g) Wavelet transform plots the Cu K-edge EXAFS signals for CuH2DOBDC, Cu2H2DOBDC and the Cu2O standard. | ||
X-ray photoelectron spectroscopy (XPS) was used for the evaluation of the chemical states and quantitative analysis of Cu species in the samples (Fig. 3d). The Cu 2p spectrum of CuH2DOBDC exclusively demonstrated the presence of Cu species in the form of Cu(II) ions. However, in Cu2H2DOBDC, new characteristic peaks appeared at 932.3 eV and 952.0 eV, which can be attributed to the Cu(I) species. Furthermore, a significant decrease in the intensities of the characteristic peaks associated with Cu(II) was observed, along with the near-absence of satellite peaks. This significant transformation indicates a reduction process, where nearly all Cu(II) ions in CuH2DOBDC were reduced to Cu(I) ions following treatment with ascorbic acid. However, two small residual peaks of Cu(II) at 934.8 eV and 954.59 eV were observed, which could be attributed to the unavoidable partial oxidation of the sample due to its exposure to air during the reaction or sample processing.30,31 The chemical state and coordination environment of the Cu atoms in CuH2DOBDC and Cu2H2DOBDC were further investigated by Cu K-edge X-ray absorption near-edge structure (XANES) spectroscopy and extended X-ray absorption fine structure (EXAFS) spectroscopy. The XANES spectra of the Cu K-edge for both CuH2DOBDC and Cu2H2DOBDC are shown in Fig. 3e. In the spectrum of CuH2DOBDC, distinct characteristic peaks were observed, including a pre-edge peak I at 8977 eV corresponding to the 1s → 3d transition, a white line peak II at 8996 eV, and a resonance peak III at approximately 9042 eV, which indicates that the oxidation state of Cu species is +2.32–34 However, in the XANES spectrum of Cu2H2DOBDC, the aforementioned characteristic peaks associated with Cu(II) are absent. Instead, a new peak at 8983 eV emerged, attributed to the 1s → 4p transition of Cu(I) (peak A), indicating that the predominant oxidation state of Cu species is Cu(I). Furthermore, the significant shift of the edge position towards the lower energy direction provides further evidence for the predominance of the Cu(I) state.33,35 This observation is consistent with the XPS results. In the R-space EXAFS spectra, both samples exhibited a dominant peak at 1.5 Å arising from Cu–O bonding (Fig. 3f). However, for Cu2H2DOBDC, the main peak at 1.5 Å possesses a lower intensity, which suggests a different Cu–O coordination environment. Additionally, when compared to CuH2DOBDC, the Cu–C peak also exhibited a decrease in intensity, which could be attributed to the partial removal of the carboxylate ligand.33 The EXAFS fitting curves showed that the Cu–O configuration predominantly exists in CuH2DOBDC and Cu2H2DOBDC, but with a change in the coordination number of O atoms around the Cu atoms (Fig. S8 and S9 and Table S2†).27 In the wavelet transform (WT) contour plots shown in Fig. 3g, Cu2H2DOBDC exhibited an intensity maximum of Cu–O at approximately 5.85 Å−1, which was very close to that of the reference Cu2O at 5.80 Å−1. Compared with the WT plot of CuH2DOBDC (6.0 Å−1), this provides further evidence that the formation of Cu(I) species leads to a change in the Cu–O coordination environment.36 Therefore, the above results indicate that the valence state and coordination environment of Cu atoms changed significantly after ascorbic acid reduction, resulting in the formation of a novel Cu(I)-carboxylate MOF.
Based on the aforementioned characteristics, we have formulated a speculation regarding the local structure of Cu2H2DOBDC. Current understanding of its structure reveals that the coordination mode of Cu(I) and the ligand is in the monodentate form, with DMF not participating in the coordination. Furthermore, the molar ratio of Cu(I) to the ligand is 2:1. Consequently, we speculate that the local structure comprises a symmetric unit with two Cu(I) ions (Cu1 and Cu2) linked by an H2DOBDC2− linker (Fig. S10†). Such a possible formation process is shown in Fig. S11.†
Catalytic azide–alkyne “click” reaction
1,2,3-Triazoles are key structural motifs in the preparation of drugs, biochemicals, natural compounds, and functional materials, which are generally obtained by the Cu(I)-catalyzed azide–alkyne cycloaddition reaction (CuAAC).37–39 Cu2H2DOBDC, as a heterogeneous catalyst, has abundant well-dispersed Cu(I) metal sites and a hierarchical structure, which may endow Cu2H2DOBDC with superior catalytic activity. Thus, the catalytic performance of Cu2H2DOBDC is evaluated by the azide–alkyne cycloaddition reaction using benzyl azide and phenylacetylene as the initial exploration substrates (Fig. 4a). Initially, the typical reaction between the benzyl azide and phenylacetylene was investigated in the presence of Cu2H2DOBDC (10 mg) in acetonitrile at room temperature for 2.5 h. However, a yield of only 26% was attained under these reaction conditions (entry 1 in Table S3†). Consequently, the temperature of the catalytic reaction was increased from 25 °C to 60 °C and then to 80 °C. Accordingly, as the temperature increased, the corresponding yields increased to 74% and 99%, respectively (entries 2 and 3 in Table S3†). It is noteworthy that only the single catalytic product 1,4-substituted 1-benzyl-4-phenyl-1H-1,2,3-triazole was obtained by utilizing Cu2H2DOBDC as the catalyst (Fig. S13 and S14†). To further explore the effect of the catalyst amount on the yields, the amount of catalyst was reduced to 5 mg. Nonetheless, a yield of 79% was still achieved (entry 4 in Table S3†). Additionally, experiments were conducted using CuH2DOBDC along with a blank experiment without any catalyst, resulting in yields of only 39% and 19%, respectively (entries 5 and 6 in Table S3†). Under optimal reaction conditions, Cu2H2DOBDC exhibited excellent catalytic performance, with a conversion yield of up to 96% after just 2 h and gradually increasing to 99% after 2.5 h (Fig. 4b). The catalytic performance of Cu2H2DOBDC in the CuAAC reaction is superior to many recently reported related materials (Table S4†).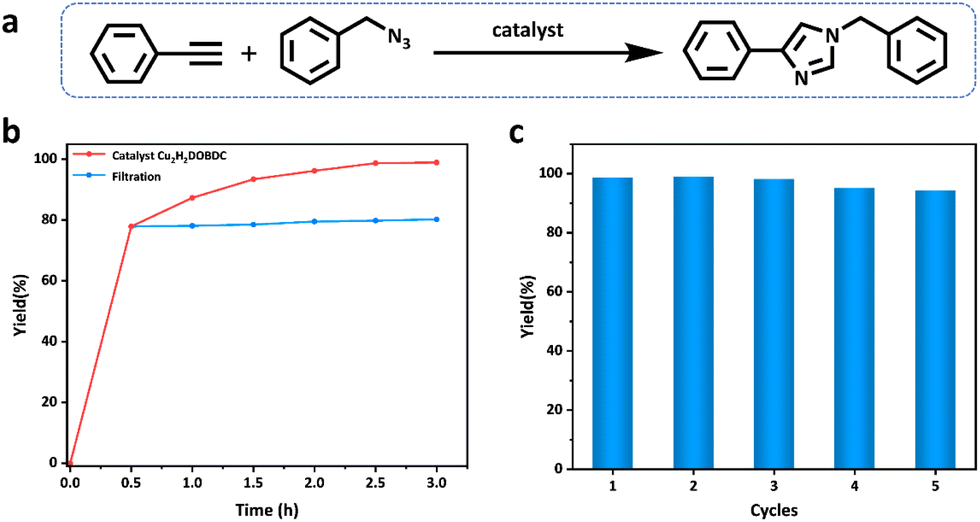 | ||
| Fig. 4 (a) Schematic representation of the “click” reaction between phenylacetylene and benzyl azide. (b) The time profile of the yield and catalyst filtration test for the “click” reaction between phenylacetylene and benzyl azide. (c) Catalytic recyclability of Cu2H2DOBDC for the “click” reaction between phenylacetylene and benzyl azide. | ||
To investigate whether the catalysis was a heterogeneous reaction, the catalysts were separated from the reaction systems by hot filtration after 0.5 h. However, even after continuing the reaction with filtrates, no significant increase in yields was observed (Fig. 4b). The results indicate that Cu2H2DOBDC is an excellent heterogeneous catalyst for the AAC reaction. The recyclability of Cu2H2DOBDC as a heterogeneous catalyst was investigated. Impressively, Cu2H2DOBDC retained satisfactory catalytic activity and could be reused for five consecutive cycles without obvious loss in the catalytic activity, as shown in Fig. 4c. In addition, we further verified the structural stability of the Cu2H2DOBDC catalyst after cycling (Fig. S12†). The SEM results show that the sample morphology remains and the surface still retains hierarchical nanostructures. XRD analysis confirms that the catalyst maintains its crystallinity and structural integrity after cycling. FTIR spectra show no new peaks or shifts in existing peaks, indicating that there is no change in chemical bonds or functional groups. XPS analysis confirms that the predominant oxidation state of Cu species in Cu2H2DOBDC remains as Cu(I) even after undergoing five cycles of reactions. In summary, these results demonstrate the excellent structural and chemical stability of Cu2H2DOBDC, ensuring its potential for long-term and reliable catalytic applications.
To further validate the catalytic generality of Cu2H2DOBDC, we employed various terminal alkynes with different functional groups and azides, including methoxy, nitro, heteroaryl, halogen, or methyl moieties, to investigate the catalytic performance under the optimized conditions. As summarized in Table 1, all the selected substrates could efficiently undergo the Cu2H2DOBDC-catalyzed “click” reaction to deliver their corresponding 1,4-disubstituted 1,2,3-triazoles in high yields (Fig. S13–S28†). Cu2H2DOBDC exhibits superior catalytic performance and regioselectivity, which may be attributed to the high density of uniformly distributed Cu(I) active sites and the hierarchical MOF structure, which enhances accessibility to the active sites and facilitates fast diffusion of the reactants.
| Entry | Alkyne | Azide | Product | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: catalyst (10 mg), azide (1 mmol), alkyne (2 mmol), amyl acetate (1 mmol), acetonitrile (4 ml) and 80 °C, 2.5 h. b Isolated yields were calculated by GC, and amyl acetate was employed as the internal standard. | ||||
| 1 |
|
|
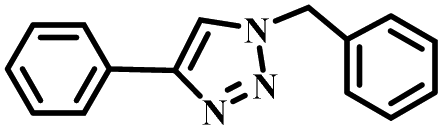
|
99 |
| 2 |
|
|
|
94 |
| 3 |
|
|
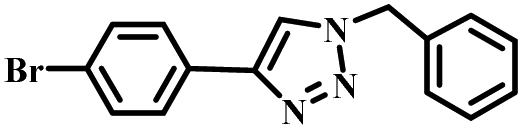
|
99 |
| 4 |
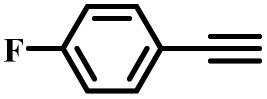
|
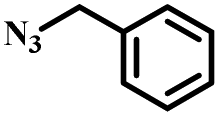
|
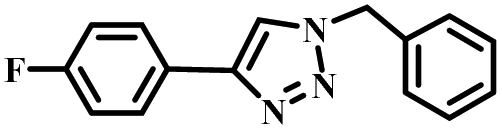
|
99 |
| 5 |
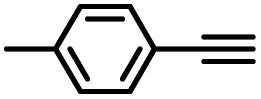
|
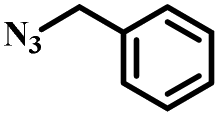
|
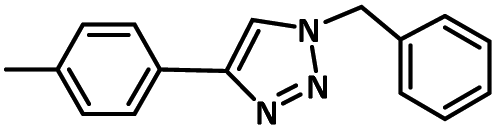
|
95 |
| 6 |
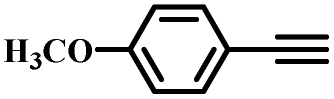
|
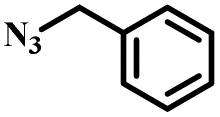
|
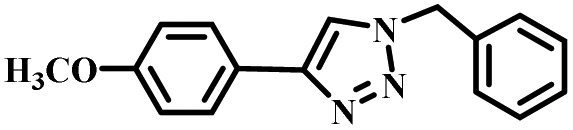
|
99 |
| 7 |
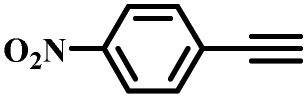
|
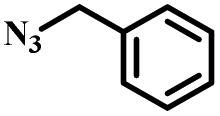
|
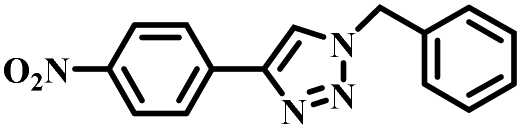
|
96 |
| 8 |
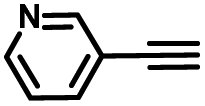
|
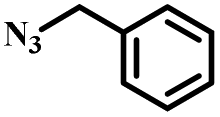
|
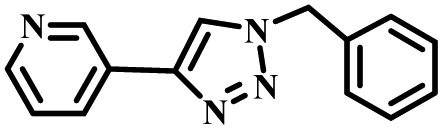
|
97 |
Considering previous reports on the CuAAC reaction,39–42 a possible mechanism, as shown in Fig. S29,† has been proposed. Initially, the Cu(I) sites in Cu2H2DOBDC interact with the alkyne to form the Cu(I)-acetylide intermediate A. Subsequently, azides react with the Cu(I)-acetylide intermediate A to generate intermediate B. Then, the nucleophilic nature of the terminal N3 in intermediate B makes it attack the alkyne to form metallacycle C, which further shrinks to yield the Cu–triazole intermediate D. Finally, the protonation of intermediate D yields the target product 1,4-disubstituted 1,2,3-triazole, accompanied by the recovery of the catalyst.
Conclusions
In conclusion, we have developed a novel cleavage reduction strategy for fabricating a stable single-phase Cu(I)-carboxylate MOF that circumvents the limitations of HSAB theory. This innovative approach hinges on the distinctive structure of ascorbic acid and the instability characteristics of metastable MOFs. We demonstrated this strategy by chemically cleaving CuH2DOBDC with ascorbic acid, which precisely cleaves the H2DOBDC2− linker and triggers a phase transition. We speculated on the atomic local structure of Cu2H2DOBDC and proposed a possible formation process. The resultant Cu2H2DOBDC acting as a CuAAC catalyst exhibited superior catalytic performance due to the highly dispersed and accessible open Cu(I) metal sites. This new strategy not only circumvents the limitations of HSAB theory but also opens up a new avenue for the creation of unprecedented categories of MOFs. We anticipate that this research will provide new perspectives to advance the development of MOF materials.Author contributions
Xiaojiao Hou, Junyi Chen and Yu Fu designed the project, Xiaojiao Hou performed the experiments and wrote the manuscript, Yu Fu supervised this work, Wenxiu He and Xu Zhai collected the characterization data, and Bingbing Chen, Yuanlin Fu, and Liying Zhang contributed to the data analysis. All the authors contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22175030 and 22365025) and the Open Project of the State Key Laboratory of Supramolecular Structure and Materials (sklssm 202406). Special thanks are due to the instrument from the Analytical and Testing Center, Northeastern University.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341, 1230444 CrossRef.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Secondary building units as the turning point in the development of the reticular chemistry of MOFs, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS.
- E. Martínez-Ahumada, M. L. Díaz-Ramírez, M. d. J. Velásquez-Hernández, V. Jancik and I. A. Ibarra, Capture of toxic gases in MOFs: SO2, H2S, NH3 and NOx, Chem. Sci., 2021, 12, 6772–6799 RSC.
- V. Aggarwal, S. Solanki and B. D. Malhotra, Applications of metal-organic framework-based bioelectrodes, Chem. Sci., 2022, 13, 8727–8743 RSC.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Metal-Organic Frameworks as Platforms for Catalytic Applications, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Metal-organic frameworks for electrocatalysis, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- X.-J. Bai, X. Zhai, L.-Y. Zhang, Y. Fu and W. Qi, Site-directed reduction engineering within bimetal-organic frameworks for efficient size-selective catalysis, Matter, 2021, 4, 2919–2935 CrossRef CAS.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Stable Metal-Organic Frameworks: Design, Synthesis, and Applications, Adv. Mater., 2018, 30, 1704303 CrossRef.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 2002, 85, 3533–3539 CrossRef.
- A. J. Howarth, Y. Y. Liu, P. Li, Z. Y. Li, T. C. Wang, J. Hupp and O. K. Farha, Chemical, thermal and mechanical stabilities of metal-organic frameworks, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu, D. D. Zhou and J. P. Zhang, Flexible Cuprous Triazolate Frameworks as Highly Stable and Efficient Electrocatalysts for CO2 Reduction with Tunable C2H4/CH4 Selectivity, Angew. Chem., Int. Ed., 2022, 61, 202204967 CrossRef.
- J. Liu, R. Xiao, Y. L. Wong, X. P. Zhou, M. Zeller, A. D. Hunter, Q. Fang, L. Liao and Z. Xu, Made in Water: A Stable Microporous Cu(I)-carboxylate Framework (CityU-7) for CO2, Water, and Iodine Uptake, Inorg. Chem., 2018, 57, 4807–4811 CrossRef CAS PubMed.
- J.-X. Qin, P. Tan, Y. Jiang, X.-Q. Liu, Q.-X. He and L.-B. Sun, Functionalization of metal-organic frameworks with cuprous sites using vapor-induced selective reduction: efficient adsorbents for deep desulfurization, Green Chem., 2016, 18, 3210–3215 RSC.
- X. Hu, F. Ding, R. Xiong, Y. An, X. Feng, J. Song, L. Zhou, P. Li and C. Chen, Highly Effective H2/D2 Separation within the Stable Cu(I)Cu(II)-BTC: The Effect of Cu(I) Structure on Quantum Sieving, ACS Appl. Mater. Interfaces, 2023, 15, 3941–3952 CrossRef CAS PubMed.
- L. Zhang, K. Jiang, L. Yang, L. Li, E. Hu, L. Yang, K. Shao, H. Xing, Y. Cui, Y. Yang, B. Li, B. Chen and G. Qian, Benchmark C2H2/CO2 Separation in an Ultra-Microporous Metal-Organic Framework via Copper(I)-Alkynyl Chemistry, Angew. Chem., Int. Ed., 2021, 60, 15995–16002 CrossRef CAS PubMed.
- B.-B. Lu, J. Yang, G.-B. Che, W.-Y. Pei and J.-F. Ma, Highly Stable Copper(I)-Based Metal-Organic Framework Assembled with Resorcin[4]arene and Polyoxometalate for Efficient Heterogeneous Catalysis of Azide-Alkyne “Click” Reaction, ACS Appl. Mater. Interfaces, 2018, 10, 2628–2636 CrossRef CAS.
- S. Zhou, Y. Chen, K. Li, X. Liu, T. Zhang, W. Shen, M. Li, L. Zhou and R. He, Photophysical studies for Cu(I)-based halides: broad excitation bands and highly efficient single-component warm white-light-emitting diodes, Chem. Sci., 2023, 14, 5415–5424 RSC.
- A.-G. Liu, Y. Chen, P.-D. Liu, W. Qi and B. Li, Conversion of CO2 to epoxides or oxazolidinones enabled by a CuI/CuII-organic framework bearing a tri-functional linker, Inorg. Chem. Front., 2022, 9, 4425–4432 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular synthesis and the design of new materials, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. C. Qi, X. Y. Qian, Q. X. He, K. J. Miao, Y. Jiang, P. Tan, X. Q. Liu and L. B. Sun, Generation of Hierarchical Porosity in Metal-Organic Frameworks by the Modulation of Cation Valence, Angew. Chem., Int. Ed., 2019, 58, 10104–10109 CrossRef CAS PubMed.
- X. Zhou, J. Dong, Y. Zhu, L. Liu, Y. Jiao, H. Li, Y. Han, K. Davey, Q. Xu, Y. Zheng and S. Z. Qiao, Molecular Scalpel to Chemically Cleave Metal-Organic Frameworks for Induced Phase Transition, J. Am. Chem. Soc., 2021, 143, 6681–6690 CrossRef CAS PubMed.
- L. Shao, F. Q. Fan, X. Y. Dai, H. X. Fu, W. Z. Li, W. Qi, F. B. Meng and Y. Fu, Pseudomorphic Replacement in the Transformation between Metal-Organic Frameworks toward Three-Dimensional Hierarchical Nanostructures, Chem. Mater., 2022, 34, 5356–5365 CrossRef CAS.
- V. Zeleňák, Z. Vargová and K. Györyová, Correlation of infrared spectra of zinc(II) carboxylates with their structures, Spectrochim. Acta, Part A, 2007, 66, 262–272 CrossRef PubMed.
- F. Verpoort, T. Haemers, P. Roose and J. P. Maes, Characterization of a Surface Coating Formed from Carboxylic Acid-Based Coolants, Appl. Spectrosc., 2016, 53, 1528–1534 CrossRef.
- N. Zou, J. Chen, T. Qiu and Y. Zheng, Direct hydrogenation of CO2 to ethanol at ambient conditions using Cu(I)-MOF in a dielectric barrier discharge plasma reactor, J. Mater. Chem. A, 2023, 11, 10766–10775 RSC.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou, X. Dong, J. Zhai, J. Li, J. He, D. Jiang, Y. Yamauchi, M. He, H. Wu and B. Han, Adjacent Copper Single Atoms Promote C-C Coupling in Electrochemical CO2 Reduction for the Efficient Conversion of Ethanol, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS PubMed.
- C.-H. Zhang, B.-X. Zhou, X. Lin, J.-X. Wu, L.-H. Wu, S. Cai, J. Fan, W.-G. Zhang, Y. Yan and S.-R. Zheng, High-Capacity Iodine Adsorption and Nonporous to Porous Structural Transformation in an Originally Nonporous Coordination Polymer, Inorg. Chem. Front., 2024, 11, 769–778 RSC.
- X. Shang, G. Li, R. Wang, T. Xie, J. Ding and Q. Zhong, Precision loading of Pd on Cu species for highly selective CO2 photoreduction to methanol, Chem. Eng. J., 2023, 456, 140805 CrossRef CAS.
- W. Hao, S. Chen, Y. Cai, L. Zhang, Z. Li and S. Zhang, Three-dimensional hierarchical pompon-like Co3O4 porous spheres for high-performance lithium-ion batteries, J. Mater. Chem. A, 2014, 2, 13801–13804 RSC.
- J. Wen, K. Wu, D. Yang, J. Tian, Z. Huang, A. S. Filatov, A. Lei and X. M. Lin, Low-Pressure Flow Chemistry of CuAAC Click Reaction Catalyzed by Nanoporous AuCu Membrane, ACS Appl. Mater. Interfaces, 2018, 10, 25930–25935 CrossRef CAS.
- C.-H. Zhang, T.-D. Hu, Y.-T. Zhai, Y.-X. Zhang and Z.-L. Wu, Stepwise engineering of the pore environment within metal-organic frameworks for green conversion of CO2 and propargylic amines, Green Chem., 2023, 25, 1938–1947 RSC.
- V. A. Streltsov and J. N. Varghese, Substrate mediated reduction of copper-amyloid-β complex in Alzheimer's disease, Chem. Commun., 2008, 3169–3171 RSC.
- C. Huang, J. Dong, W. Sun, Z. Xue, J. Ma, L. Zheng, C. Liu, X. Li, K. Zhou, X. Qiao, Q. Song, W. Ma, L. Zhang, Z. Lin and T. Wang, Coordination mode engineering in stacked-nanosheet metal-organic frameworks to enhance catalytic reactivity and structural robustness, Nat. Commun., 2019, 10, 2779 CrossRef PubMed.
- J. L. Fulton, M. M. Hoffmann, J. G. Darab, B. J. Palmer and E. A. Stern, Copper(I) and Copper(II) Coordination Structure under Hydrothermal Conditions at 325 °C: An X-ray Absorption Fine Structure and Molecular Dynamics Study, J. Phys. Chem. A, 2000, 104, 11651–11663 CrossRef CAS.
- Z. Zhang, H. Yoshikawa and K. Awaga, Monitoring the Solid-State Electrochemistry of Cu(2,7-AQDC) (AQDC = Anthraquinone Dicarboxylate) in a Lithium Battery: Coexistence of Metal and Ligand Redox Activities in a Metal-Organic Framework, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS.
- R. Yang, J. Duan, P. Dong, Q. Wen, M. Wu, Y. Liu, Y. Liu, H. Li and T. Zhai, In Situ Halogen-Ion Leaching Regulates Multiple Sites on Tandem Catalysts for Efficient CO2 Electroreduction to C2+ Products, Angew. Chem., Int. Ed., 2022, 61, 202116706 CrossRef.
- A. K. Agrahari, P. Bose, M. K. Jaiswal, S. Rajkhowa, A. S. Singh, S. Hotha, N. Mishra and V. K. Tiwari, Cu(I)-Catalyzed Click Chemistry in Glycoscience and Their Diverse Applications, Chem. Rev., 2021, 121, 7638–7956 CrossRef CAS PubMed.
- L. Liang and D. Astruc, The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- G. Vilé, G. Di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou, A. Savateev, M. Antonietti and G. Pacchioni, Azide-Alkyne Click Chemistry over a Heterogeneous Copper-Based Single-Atom Catalyst, ACS Catal., 2022, 12, 2947–2958 CrossRef.
- M. Y. Yu, T. T. Guo, X. C. Shi, J. Yang, X. Xu, J. F. Ma and Z. T. Yu, Polyoxometalate-Bridged Cu(I)- and Ag(I)-Thiacalix[4]arene Dimers for Heterogeneous Catalytic Oxidative Desulfurization and Azide-Alkyne “Click” Reaction, Inorg. Chem., 2019, 58, 11010–11019 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi00922c |
| This journal is © the Partner Organisations 2024 |