
Synergistic dual sites of Zn–Mg on hierarchical porous carbon as an advanced oxygen reduction electrocatalyst for Zn–air batteries†
Mincong
Liu
a,
Jing
Zhang
b,
Yan
Peng
a and
Shiyou
Guan
 *a
*a
aDepartment of Chemistry, College of Science, Shanghai University, 99 Shang-Da Road, Shanghai 200444, China. E-mail: syguan@shu.edu.cn
bCollege of Sciences & Institute for Sustainable Energy, Shanghai University, 99 Shang-Da Road, Shanghai 200444, China
First published on 9th May 2024
Abstract
The development of cost-effective and high-performance non-noble metal catalysts for the oxygen reduction reaction (ORR) holds substantial promise for real-world applications. Introducing a secondary metal to design bimetallic sites enables effective modulation of a metal–nitrogen–carbon (M–N–C) catalyst's electronic structure, providing new opportunities for enhancing ORR activity and stability. Here, we successfully synthesized an innovative hierarchical porous carbon material with dual sites of Zn and Mg (Zn/Mg–N–C) using polymeric ionic liquids (PILs) as precursors and SBA-15 as a template through a bottom-up approach. The hierarchical porous structure and optimized Zn–Mg bimetallic catalytic centers enable Zn/Mg–N–C to exhibit a half-wave potential of 0.89 V, excellent stability, and good methanol tolerance in 0.1 M KOH solution. Theoretical calculations indicated that the Zn–Mg bimetallic sites in Zn/Mg–N–C effectively lowered the ORR energy barrier. Furthermore, the Zn–air batteries assembled based on Zn/Mg–N–C demonstrated an outstanding peak power density (298.7 mW cm−2) and superior cycling stability. This work provides a method for designing and synthesizing bimetallic site catalysts for advanced catalysis.
1. Introduction
As resources deplete rapidly and reliance on fossil fuels becomes unsustainable, the imperative lies in the development of renewable energy sources to secure the future well-being of human society.1–4 Energy conversion and storage devices, such as metal–air batteries and fuel cells, have gained significant attention due to their sustainability and efficiency.5–7 However, the slow reaction kinetics of the cathodic oxygen reduction reaction (ORR) significantly hinder the development of related research.8,9 Until now, platinum (Pt)-based catalysts have been considered among the most advanced electrocatalysts in electrolyte systems. However, Pt-based catalysts face challenges such as scarcity, high cost, and poor durability, making their widespread adoption on a large scale difficult.10,11 Therefore, the development of cost-effective, highly active, and durable electrocatalyst alternatives to Pt catalysts is imperative, albeit with significant obstacles.12,13Single metal sites formed by 3d-transition metal and N co-doped carbon materials (M–Nx–C, M = Fe, Co, Ni, etc.) are regarded as highly promising catalytically active sites in the ORR.14–16 Excitingly, recent studies have confirmed that Zn with a fulfilled d-orbital (3d104s2) exhibits catalytic activity when designed as the single metal sites of Zn–Nx.17,18 Nevertheless, the challenge of improving the reaction kinetics of the ORR remains in simply adjusting the coordination environment and local structure of these single metal sites.19,20 In comparison with the case of single metal sites, interactions between bimetallic sites in catalysts can further enhance their intrinsic activity.21 The formation of bimetallic sites is achieved by introducing a second metal near the first metal site, providing a novel strategy for tuning the electronic and geometric structures of M–N–C catalysts.22 However, the focus of coordinating metals has largely been on d-block transition metals.23–25 The main group metals in the S-block exhibit a specific p-orbital electronic state, differing from the electronic structure of the d-orbital in transition metals.26,27 Among them, the utilization of alkaline earth metals as catalytic sites for the ORR has been reported infrequently, and compared with other alkaline earth metals such as Al and Ca, the central position of the p-band increases when Mg coordinates with N, resulting in an optimal adsorption strength for ORR intermediate oxide species.28,29 Also, Mg is the seventh most abundant element on Earth, which translates to a lower cost and a more sustainable option relative to other metals.30,31 Therefore, the exploration of bimetallic sites with both p- and d-orbitals is paramount for synthesizing highly active and stable ORR electrocatalysts.
Poly(ionic liquid)s (PILs) not only possess the characteristics of both nitrogen-rich ILs and polymers,32,33 but also exhibit good coordination ability with metal ions due to the presence of imidazole groups.34–36 Therefore, we selected PILs as precursors and synthesized Zn/Mg bimetallic sites coordinated on hierarchically porous nitrogen-doped carbon materials (Zn/Mg–N–C) through the pyrolysis of a PILs/Zn/Mg complex within SBA-15 channels using a top-down strategy. The introduction of Zn–Mg dual sites optimized the electronic configuration of the Zn/Mg–N–C catalyst, thus enhancing its intrinsic ORR activity. Besides that, the hierarchically porous structure of the Zn/Mg–N–C catalyst can increase the density of active sites and promote mass transport capacity, resulting in improved catalyst performance. The adjusted Zn/Mg–N–C catalyst exhibited an excellent half-wave potential of 0.89 V in alkaline media and a higher power capacity of 298.7 mW cm−2 in Zn–air batteries, which are better than those of the commercial Pt/C catalyst.
2. Experimental
2.1. Materials
1-Vinyl-3-butylimidazolium bromide ([VBIm]B, sourced from Shanghai Chengjie Chemical Co., Ltd), N,N-dimethylformamide (DMF, 99.8%, obtained from Adamas), zinc chloride (ZnCl2, obtained from Aladdin), anhydrous magnesium chloride (MgCl2, obtained from Adama), 2,2-azobis(2-methylpropionitrile) (AIBN, 98% purity, from Adamas), SBA-15 (pore diameter: 6–7 nm, specific surface area: 550–600 m2 g−1, obtained from XFNANO), hydrofluoric acid (HF, 48%, obtained from Adamas), Pt/C catalyst (20 wt%, manufactured by Johnson Matthey), Nafion polymer (5 wt%, obtained from DuPont), and ethanol (C2H5OH, 99.9% purity, from Adamas) were used. All the materials were used without further purification.2.2. Preparation of electrocatalysts
2.3. Characterization of materials, electrochemical measurements, and DFT calculations
A detailed description of the materials characterization, electrochemical measurements, and DFT calculations can be found in the ESI.†3. Results and discussion
The synthesis process of Zn/Mg–N–C is illustrated in Fig. 1a. First, the prepared mixture of ILs/Zn/Mg was dispersed in DMF solvent, ensuring full contact with the SBA-15 template and complete filling of the channels within SBA-15. Subsequently, the ILs initiated polymerization within the pores of SBA-15 at 80 °C, leading to the formation of a porous structure of the catalyst (PILs/Zn/Mg@SBA-15). Next, PILs/Zn/Mg@SBA-15 was carbonized at high temperature while the excess Zn precursor was evaporated under a protective atmosphere, resulting in the creation of pores. This process yielded a composite of Zn/Mg-based porous carbon materials and SBA-15 (Zn/Mg–N–C@SBA-15). Finally, SBA-15 was further etched with HF to prepare Zn/Mg–N–C.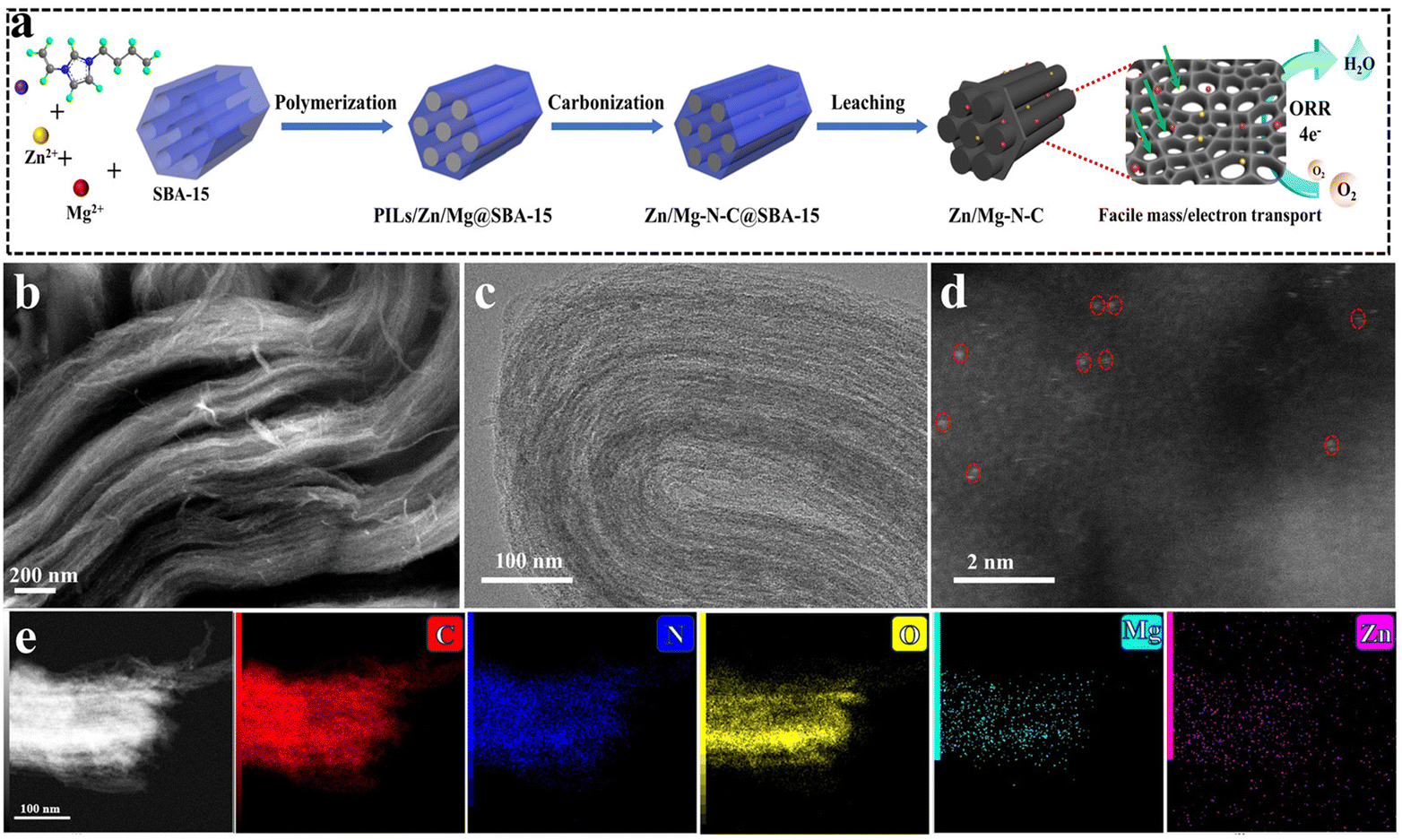 | ||
| Fig. 1 (a) Schematic diagram of the synthesis process of the Zn/Mg–N–C catalyst. (b) SEM and (c) TEM images of Zn/Mg–N–C. (d) HADDF-STEM image of Zn/Mg–N–C. (e) HAADF-STEM image of Zn/Mg–N–C and the corresponding elemental maps. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were utilized to thoroughly examine the catalyst's detailed morphology. According to Fig. 1b, the SEM image of Zn/Mg–N–C showed an abundant pore structure, providing full exposure of active sites and facilitating rapid electron transport.37 Zn–N–C and Mg–N–C prepared using SBA-15 as the template also exhibited similar structures (Fig. S1†). The TEM image of Zn/Mg–N–C clearly showed ordered layered nanoarrays, suggesting that the mesoporous structure primarily originates from the gaps between the nanoarrays caused by the removal of the SBA-15 template (Fig. 1c). The high-resolution transmission electron microscopy (HRTEM) images of Zn/Mg–N–C did not evidence the presence of metal nanoparticles (Fig. S2†). To further confirm the distribution of Mg and Zn in the Zn/Mg–N–C catalyst, it was characterized using high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1d). Bright dots randomly distributed on the carbon support (highlighted with red circles) can be observed, indicating that there are no clusters of Mg and Zn in the catalyst. Additionally, as shown in Fig. 1e, the energy dispersive X-ray spectroscopy (EDS) mapping images confirm that C, N, O, Mg and Zn were uniformly distributed in the Zn/Mg–N–C catalysts. The content of Zn and Mg in Zn/Mg–N–C was found to be 0.57 and 0.21 wt%, respectively, using inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis (Table S1†).
The structure of the catalyst was further investigated through X-ray diffraction (XRD). As depicted in Fig. 2a and Fig. S3,† the XRD pattern of the catalysts shows a broad diffraction peak of carbon at 23° and another prominent peak appeared at 43°, corresponding to the (002) and (101) graphitic planes, respectitively.38 The peak of other metal species was not observed in the XRD spectrum of Zn/Mg–N–C, which is in line with the HRTEM results. The degree of defects and graphitization of the catalyst were characterized by Raman spectroscopy (Fig. S4†). The characteristic peaks of the D band and G band were located at 1350 cm−1 and 1580 cm−1, respectively, representing the degree of carbon defects and the degree of graphitization.39 The intensity ratio (ID/IG) value indicates the degree of defects for the catalysts. The results reveal that the ID/IG ratio of Zn/Mg–N–C is 1.06, exceeding the values of 1.04 for Zn–N–C and 1.05 for Mg–N–C, suggesting that the co-doping of Mg and Zn, along with the use of the SBA-15 template, resulted in a higher presence of disordered graphite defects within the catalyst structure, which is crucial for adsorbing O2 to enhance the electrocatalytic performance.40,41 N2 adsorption–desorption experiments were used to further analyze the specific surface area and pore characteristics of the Mg–N–C, Zn–N–C and Zn/Mg–N–C catalysts (Fig. 2b). The adsorption isotherms of all samples exhibited a combination of type IV hysteresis loops at the relative pressure (P/P0 = 0.4–1.0), indicating the presence of both micropores and mesopores.42 The Mg–N–C, Zn–N–C and Zn/Mg–N–C catalysts showed apparent Brunauer–Emmett–Teller (BET) specific surface areas of 434.8 m2 g−1, 506.3 m2 g−1 and 671.6 m2 g−1 with pore volumes of 0.36 cm3 g−1, 0.51 cm3 g−1 and 0.63 cm3 g−1, respectively. Zn/Mg–N–C demonstrated a higher specific surface area, which may be attributed to the pore-forming effect of the co-doped metal volatilizing during the heat treatment.29,43 Further pore size distribution analysis indicated that Zn/Mg–N–C showed a large number of micropores (<2 nm) and abundant mesopores primarily distributed around 5.2 nm. The excellent stratified porosity, characterized by the coexistence of micropores, mesopores and macropores, is generally considered more conducive to the ORR. Mesopores and macropores are critical for facilitating the mass transfer required during electrochemical reactions, while micropores and mesopores create important three-phase interfaces for reactants and active sites.44
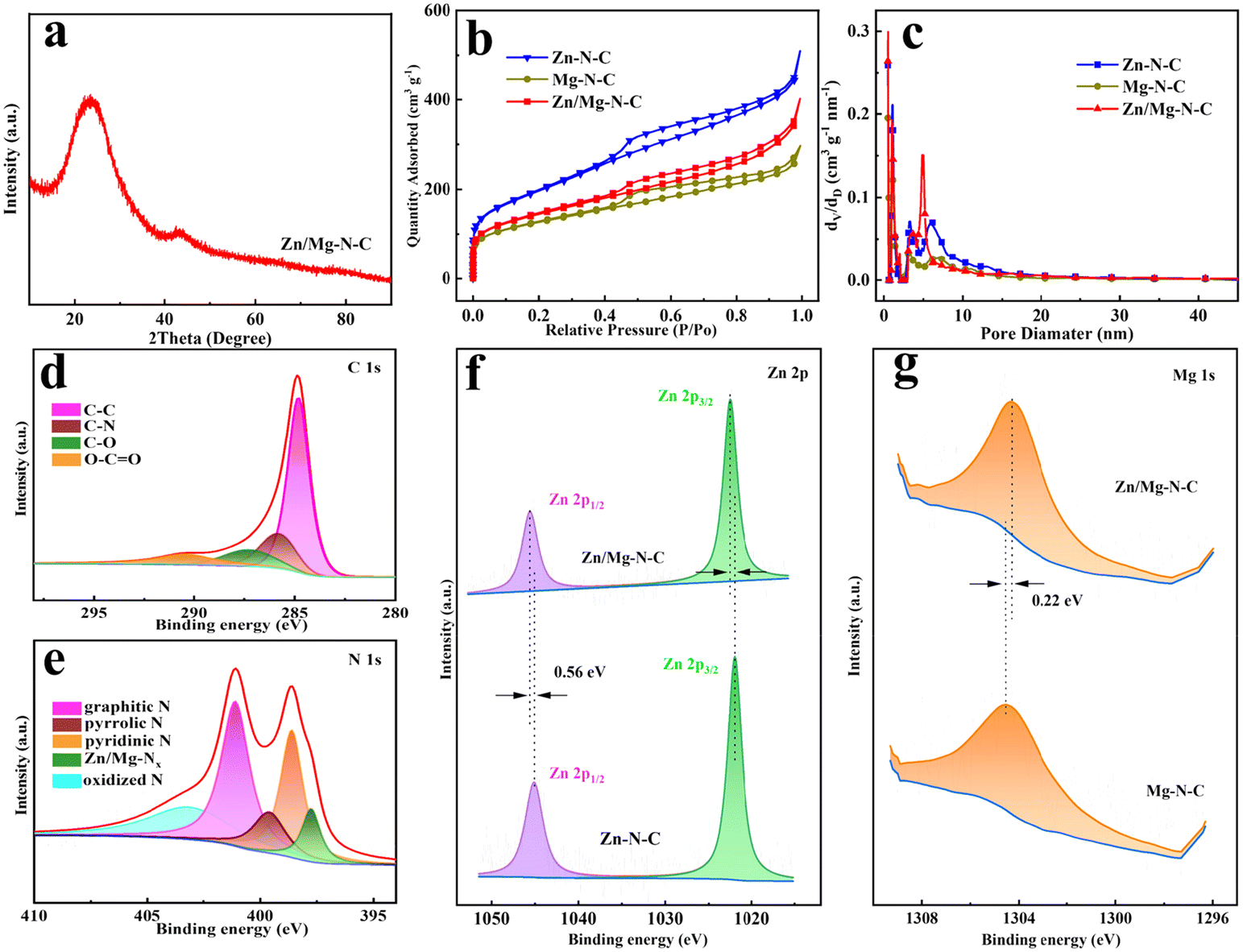 | ||
| Fig. 2 (a) XRD pattern of Zn/Mg–N–C. (b) Nitrogen adsorption/desorption isotherms and (c) pore size distribution curves of Zn/Mg–N–C, Zn–N–C and Mg–N–C. High-resolution XPS spectra of (d) C 1s and (e) N 1s of Zn/Mg–N–C. (f) Zn 2p XPS spectra of Zn/Mg–N–C and Zn–N–C. (g) Mg 1s XPS spectra of Zn/Mg–N–C and Mg–N–C. | ||
X-ray photoelectron spectroscopy (XPS) was employed to gain insights into the surface chemistry and elemental bonding structure of the catalyst. As shown in Fig. 2d, the C 1s spectrum of Zn/Mg–N–C can be deconvoluted into four types, including C–C (284.8 eV), C–N (285.8 eV), C–O (287.3 eV), and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (290.5 eV).45 As depicted in Fig. 2e, the high-resolution N 1s XPS spectrum of Zn/Mg–N–C was fitted into five peaks at binding energies of 397.7 eV (Zn/Mg–Nx), 398.6 eV (pyridinic N), 399.6 eV (pyridinic N), 401.1 eV (graphitic N), and 403.1 eV (oxidized N).17,18 The presence of the Zn–Nx and Mg–Nx bond peaks indicates that stable Zn–N and Mg–N sites had been formed in the structure. Fig. 2f shows the high-resolution Zn 2p spectrum of Zn/Mg–N–C and Zn–N–C, demonstrating two peaks, which are assigned to Zn2+ 2p3/2 and Zn2+ 2p1/2, respectively. In comparison with the Zn–N–C catalyst, the binding energy of Zn in the Zn/Mg–N–C catalyst is positively shifted by 0.56 eV, suggesting that the introduction of Mg has an influence on the electronic structure of Zn sites. Fig. 2g presents the Mg 1s spectra of the Zn/Mg–N–C and Mg–N–C catalysts, which can be deconvoluted into one peak corresponding to the Mg–N bond.28 In contrast to the binding energy of Zn, there is a negative shift of 0.22 eV in the binding energy of Mg, indicating significant electron transfer to the Mg sites. Due to the strong interactions between Zn and Mg as well as with the N ligands, the p-orbital occupancy of the Mg site is altered, adjusting the Mg atom to a reasonable p-band energy level, enabling the Mg site to possess the ability to catalyze the oxidation active species in the ORR.46,47 Therefore, the formation of Mg-based active sites becomes a key factor in enhancing the ORR activity. At the same time, these analyses have validated the structural pathways within Zn/Mg–N–C, including Zn–N/Mg–N and Zn–Mg.
O (290.5 eV).45 As depicted in Fig. 2e, the high-resolution N 1s XPS spectrum of Zn/Mg–N–C was fitted into five peaks at binding energies of 397.7 eV (Zn/Mg–Nx), 398.6 eV (pyridinic N), 399.6 eV (pyridinic N), 401.1 eV (graphitic N), and 403.1 eV (oxidized N).17,18 The presence of the Zn–Nx and Mg–Nx bond peaks indicates that stable Zn–N and Mg–N sites had been formed in the structure. Fig. 2f shows the high-resolution Zn 2p spectrum of Zn/Mg–N–C and Zn–N–C, demonstrating two peaks, which are assigned to Zn2+ 2p3/2 and Zn2+ 2p1/2, respectively. In comparison with the Zn–N–C catalyst, the binding energy of Zn in the Zn/Mg–N–C catalyst is positively shifted by 0.56 eV, suggesting that the introduction of Mg has an influence on the electronic structure of Zn sites. Fig. 2g presents the Mg 1s spectra of the Zn/Mg–N–C and Mg–N–C catalysts, which can be deconvoluted into one peak corresponding to the Mg–N bond.28 In contrast to the binding energy of Zn, there is a negative shift of 0.22 eV in the binding energy of Mg, indicating significant electron transfer to the Mg sites. Due to the strong interactions between Zn and Mg as well as with the N ligands, the p-orbital occupancy of the Mg site is altered, adjusting the Mg atom to a reasonable p-band energy level, enabling the Mg site to possess the ability to catalyze the oxidation active species in the ORR.46,47 Therefore, the formation of Mg-based active sites becomes a key factor in enhancing the ORR activity. At the same time, these analyses have validated the structural pathways within Zn/Mg–N–C, including Zn–N/Mg–N and Zn–Mg.
The ORR electrocatalytic activities of the obtained catalysts were investigated in N2/O2-saturated 0.1 M KOH using RDE and RRDE techniques. As depicted in Fig. S5,† the cyclic voltammetry curve (CV) of Zn/Mg–N–C was measured at a scan rate of 10 mV s−1, revealing a well-defined peak emerging in the O2-saturated solution. In contrast, all the catalysts did not exhibit distinctly featured peaks under an N2 atmosphere, demonstrating significant catalytic activity towards the ORR in an alkaline electrolyte. Furthermore, linear sweep voltammetry (LSV) curves were recorded to study the catalyst's electrocatalytic behavior in O2-saturated 0.1 M KOH with a rotation speed of 1600 rpm (Fig. 3a and c). The Zn/Mg–N–C catalyst showed an outstanding ORR performance with a half-wave potential (E1/2) of 0.89 V, achieving the highest diffusion kinetic current density (Jk) at 28.86 mA cm−2 among all the catalysts studied in this work (Pt/C, E1/2 = 0.80 V, Jk = 5.58 mA cm−2; Zn–N–C, E1/2 = 0.77 V, Jk = 2.25 mA cm−2; Mg–N–C, E1/2 = 0.73 V, Jk = 1.0 mA cm−2). As can be seen from Fig. 3b, the Tafel slope of Zn/Mg–N–C is 95.1 mV dec−1, which is lower than those of Pt/C (116.4 mV dec−1), Zn–N–C (121.5 mV dec−1), and Mg–N–C (100.9 mV dec−1), proving faster kinetics during the electrocatalytic reaction.48 These results indicate that the rich porous structure and the introduction of Mg into Zn/Mg–N–C contribute to improving the ORR performance. Furthermore, the observed ORR performance of Zn/Mg–N–C surpasses those of the most recently reported ORR catalysts (Table S2†). LSV curves of Zn/Mg–N–C were recorded at different speeds to gain a deeper understanding of its ORR pathway (Fig. 3d). The results demonstrated that the current density increases with an increase in the rotation speed due to the shortening of the diffusion distance of O2 to the catalyst surface. As shown in Fig. 3e, the well parallel straight lines of the Koutecky–Levich (K–L) plot for Zn/Mg–N–C indicate first-order reaction kinetics with the dissolved O2 concentration at different potentials.49 The electron transfer number (n) of Zn/Mg–N–C was calculated and found to be close to 4 according to the K–L plot, suggesting that a four-electron reaction process is a more complete reaction with higher catalytic efficiency. The rotating ring-disk electrode (RRDE) method was used to calculate the electron transfer number (n) and monitor the yield of H2O2 during the ORR process (Fig. 3f). The electron transfer number of Zn/Mg–N–C was also found to be close to 4, confirming that Zn/Mg–N–C mainly operated through the 4e− transfer mechanism during oxygen reduction. The H2O2 yield of Zn/Mg–N–C was below 10%, proving the excellent selectivity for H2O in the ORR process.
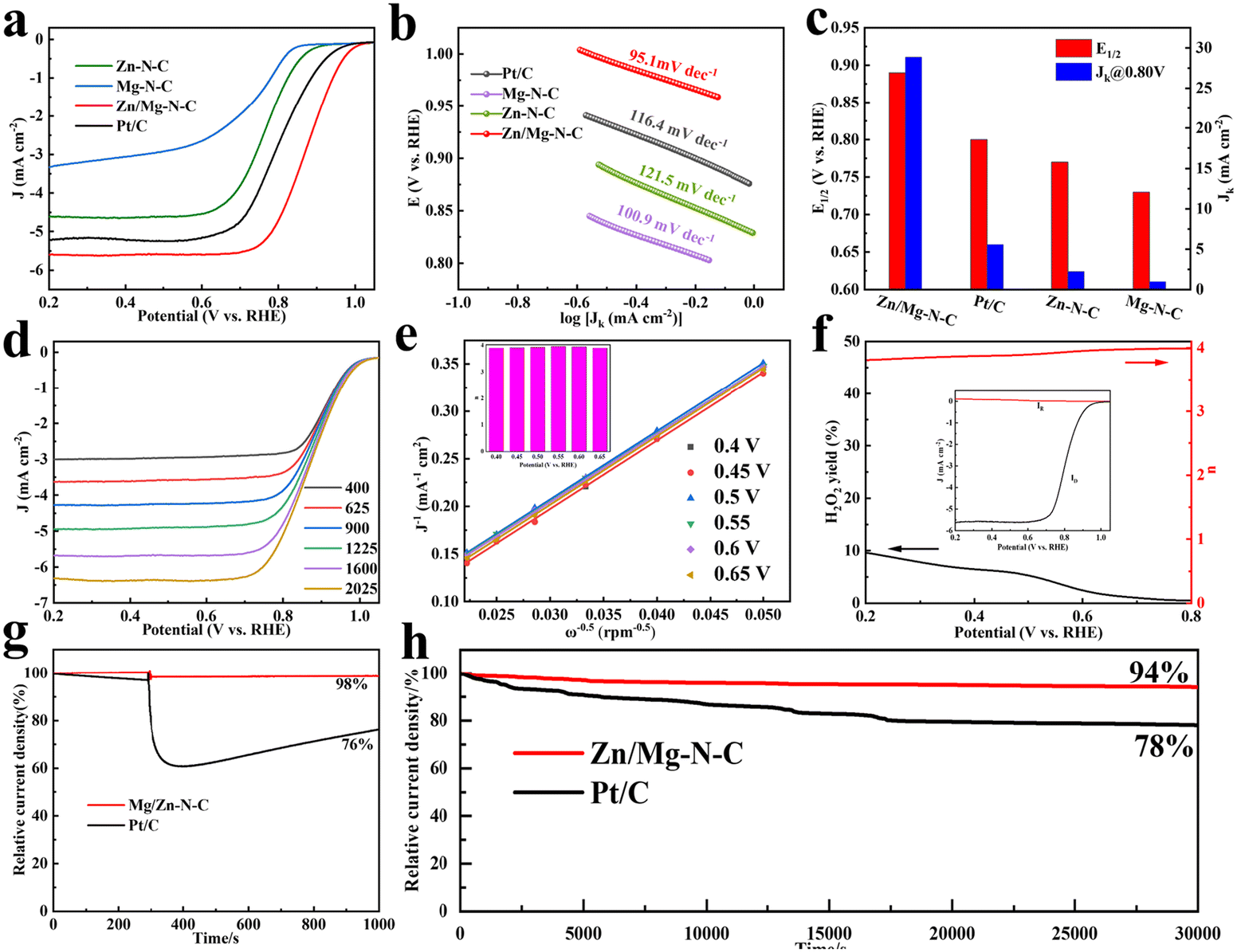 | ||
| Fig. 3 LSV curves of Zn/Mg–N–C, Zn–N–C, Mg–N–C and Pt/C catalysts in O2-saturated 0.1 M KOH (rotation rate: 1600 rpm; sweep rate: 10 mV s−1). (b) Tafel plots of the investigated catalysts. (c) The kinetic current density (Jk) at 0.8 V and E1/2 of the catalysts. (d) LSV curves of Zn/Mg–N–C at various rotation speeds. (e) K–L plots and electron transfer numbers of Zn/Mg–N–C. (f) H2O2 yield and electron transfer number (n) from the RRDE measurement of Zn/Mg–N–C in O2-saturated 0.1 M KOH at 1600 rpm (inset: the corresponding RRDE voltammograms). (g) Methanol tolerance test for Zn/Mg–N–C and Pt/C. (h) Long-term stability of Zn/Mg–N–C and Pt/C. | ||
The cross-toxicity of methanol during the ORR process is an undeniable influencing factor for catalysts. During chronoamperometric measurements, the methanol resistance was studied by introducing 3 M methanol into the electrolyte. In Fig. 3g, it can be observed that the current of Pt/C exhibited drastic fluctuations due to the methanol oxidation reaction on the Pt/C surface when methanol was added to the electrolyte at 300 s during the current measurement process. In contrast, Zn/Mg–N–C showed only slight fluctuations. After 1000 s, the current retention rate of Zn/Mg–N–C reached 98%, while that of Pt/C was only 76%, demonstrating the excellent methanol tolerance capability of the former. Additionally, long-term stability tests were conducted via chronoamperometry to evaluate the stability of Zn/Mg–N–C and commercial Pt/C. As shown in Fig. 3h, Zn/Mg–N–C maintains 94% of its initial current density after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, while under the same conditions, Pt/C shows a loss of 22% in current density, indicating the superior electrochemical stability of Zn/Mg–N–C. Furthermore, the stability of Zn/Mg–N–C was evaluated by an accelerated durability test (ADT) in O2-saturated 0.1 mol L−1 KOH (Fig. S6†). The results demonstrated that the E1/2 of Zn/Mg–N–C only experienced a negative shift of 6 mV, further confirming the excellent stability of the catalyst. The Zn–air batteries (ZABs) were further tested using Zn/Mg–N–C + RuO2 as an oxygen electrocatalyst to investigate the practical application of electrocatalysts in energy storage and conversion equipment (Fig. 4a). For comparison, ZABs based on Pt/C + RuO2 were also assembled. As demonstrated in Fig. 4b, the open-circuit voltage (OVC) of Zn/Mg–N–C + RuO2-based ZABs was 1.48 V, slightly higher than that of Pt/C + RuO2-based ZABs (1.44 V). Notably, Zn/Mg–N–C + RuO2-based ZABs easily illuminated a blue light-emitting diode (LED) (Fig. 4c), indicating the broad prospects of the prepared catalysts for practical applications in energy devices. Furthermore, the Zn/Mg–N–C + RuO2-based ZABs exhibited a polarization voltage equivalent to Pt/C + RuO2-based ZABs during the charging and discharging processes, signifying their excellent reversible oxygen catalytic performance (Fig. S7†).
000 s, while under the same conditions, Pt/C shows a loss of 22% in current density, indicating the superior electrochemical stability of Zn/Mg–N–C. Furthermore, the stability of Zn/Mg–N–C was evaluated by an accelerated durability test (ADT) in O2-saturated 0.1 mol L−1 KOH (Fig. S6†). The results demonstrated that the E1/2 of Zn/Mg–N–C only experienced a negative shift of 6 mV, further confirming the excellent stability of the catalyst. The Zn–air batteries (ZABs) were further tested using Zn/Mg–N–C + RuO2 as an oxygen electrocatalyst to investigate the practical application of electrocatalysts in energy storage and conversion equipment (Fig. 4a). For comparison, ZABs based on Pt/C + RuO2 were also assembled. As demonstrated in Fig. 4b, the open-circuit voltage (OVC) of Zn/Mg–N–C + RuO2-based ZABs was 1.48 V, slightly higher than that of Pt/C + RuO2-based ZABs (1.44 V). Notably, Zn/Mg–N–C + RuO2-based ZABs easily illuminated a blue light-emitting diode (LED) (Fig. 4c), indicating the broad prospects of the prepared catalysts for practical applications in energy devices. Furthermore, the Zn/Mg–N–C + RuO2-based ZABs exhibited a polarization voltage equivalent to Pt/C + RuO2-based ZABs during the charging and discharging processes, signifying their excellent reversible oxygen catalytic performance (Fig. S7†).
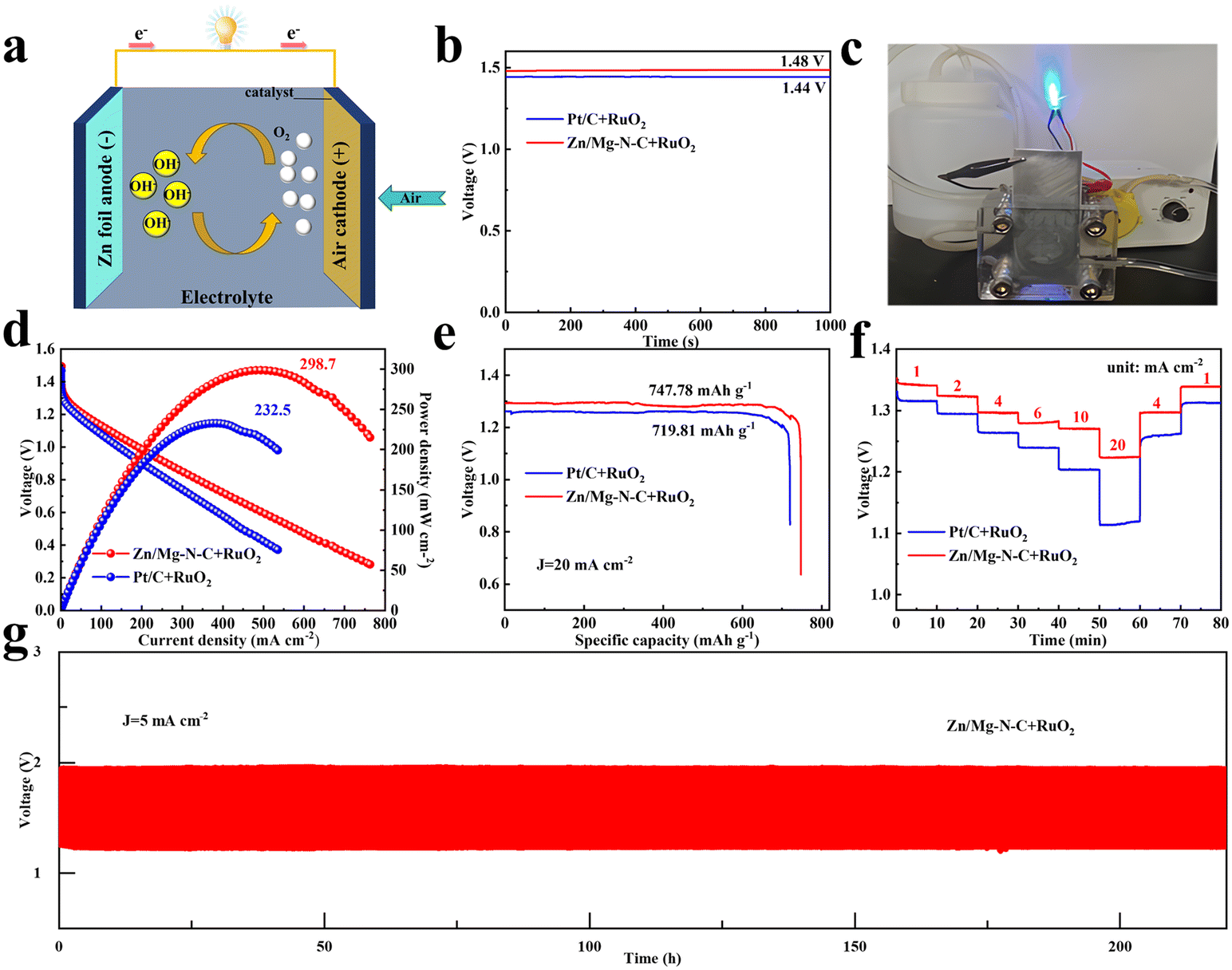 | ||
| Fig. 4 (a) Schematic illustration of the Zn–air battery. (b) Open circuit voltage plots of Zn/Mg–N–C + RuO2 and Pt/C + RuO2. (c) Photograph of the LED lit up using Zn/Mg–N–C + RuO2-based ZABs. (d) The polarization curves of discharging with power density curves of Zn/Mg–N–C + RuO2-based ZABs and Pt/C + RuO2-based ZABs. (e) Specific capacities of Zn–air batteries using Zn/Mg–N–C + RuO2 and Pt/C + RuO2. (f) Discharge voltage and time diagram of catalysts at different current densities. (g) Cycling tests of batteries for Zn/Mg–N–C + RuO2-based ZABs at a current density of 5 mA cm−2. | ||
The Zn/Mg–N–C + RuO2-based ZABs delivered a higher power density of 298.7 mW cm−2 at a current density of 488.8 mA cm−2, surpassing that of Pt/C + RuO2-based ZABs (232.5 mW cm−2 at 369.1 mA cm−2) (Fig. 4d). Additionally, the ZABs assembled with Zn/Mg–N–C + RuO2 exhibited a higher specific capacity at 20 mA cm−2, reaching 747.78 mA h g−1, exceeding that of Pt/C + RuO2-based ZABs (719.81 mA h g−1) (Fig. 4e). Table S2† lists the power densities of other reported electrocatalysts, which demonstrates superiority of Zn/Mg-N-C. Fig. 4f illustrates that the Zn/Mg–N–C + RuO2-based ZABs maintained higher discharge curves and discharge stability than Pt/C + RuO2-based ZABs at various current densities. Moreover, long-cycle tests were conducted at 5 mA cm−2 to verify the durability of Zn/Mg–N–C + RuO2-based ZABs. As demonstrated in Fig. 4g, the Zn/Mg–N–C + RuO2-based ZABs displayed excellent stability after 220 h of cycling, while the voltage gap of Pt/C + RuO2-based ZABs increased during the charging and discharging processes, indicating the poor cycling stability of Pt/C (Fig. S8†). The Zn/Mg–N–C catalyst showed outstanding performance when employed in ZABs, thus highlighting its promising potential for practical applications in energy devices.
Furthermore, density functional theory (DFT) calculations were performed to theoretically elucidate the origin of the enhanced oxygen reduction reaction (ORR) performance and excellent ORR kinetic activity resulting from the co-doping of Zn–Mg into the Zn/Mg–N–C catalyst. The structural XPS analysis of Zn/Mg–N–C led to the establishment of computational models for Zn/Mg–N–C (Fig. 5a). Additionally, models for Zn–N–C and Mg–N–C were also constructed (Fig. S9 and S10†). Fig. 5a also illustrates the optimized structures of *O2, *OOH, *O, and *OH intermediates adsorbed on the active site of Zn/Mg–N–C in the ORR pathway (the models of Zn–N–C and Mg–N–C are shown in Fig. S11 and S12†). At the electrode position of 0 V, the Gibbs free-energies (ΔG) of Zn/Mg–N–C, Zn–N–C, and Mg–N–C decrease during the ORR process, indicating that the ORR can spontaneously occur at these active sites (Fig. S13†). As can be seen from Fig. 5b, the rate-determining step (RDS) for Zn–N–C and Mg–N–C during the ORR process was the transition from *OOH to *O, whereas for Zn/Mg–N–C, the RDS is the final step. The ΔG of the RDS for Zn/Mg–N–C (0.665 eV) is smaller than those for Zn–N–C (0.831 eV) and Mg–N–C (1.136 eV) at the equilibrium potential (U = 1.23 V). This manifests that Zn/Mg–N–C is more effective than Zn–N–C and Mg–N–C for the ORR, suggesting that the catalyst co-doped with Zn and Mg exhibits superior ORR performance.
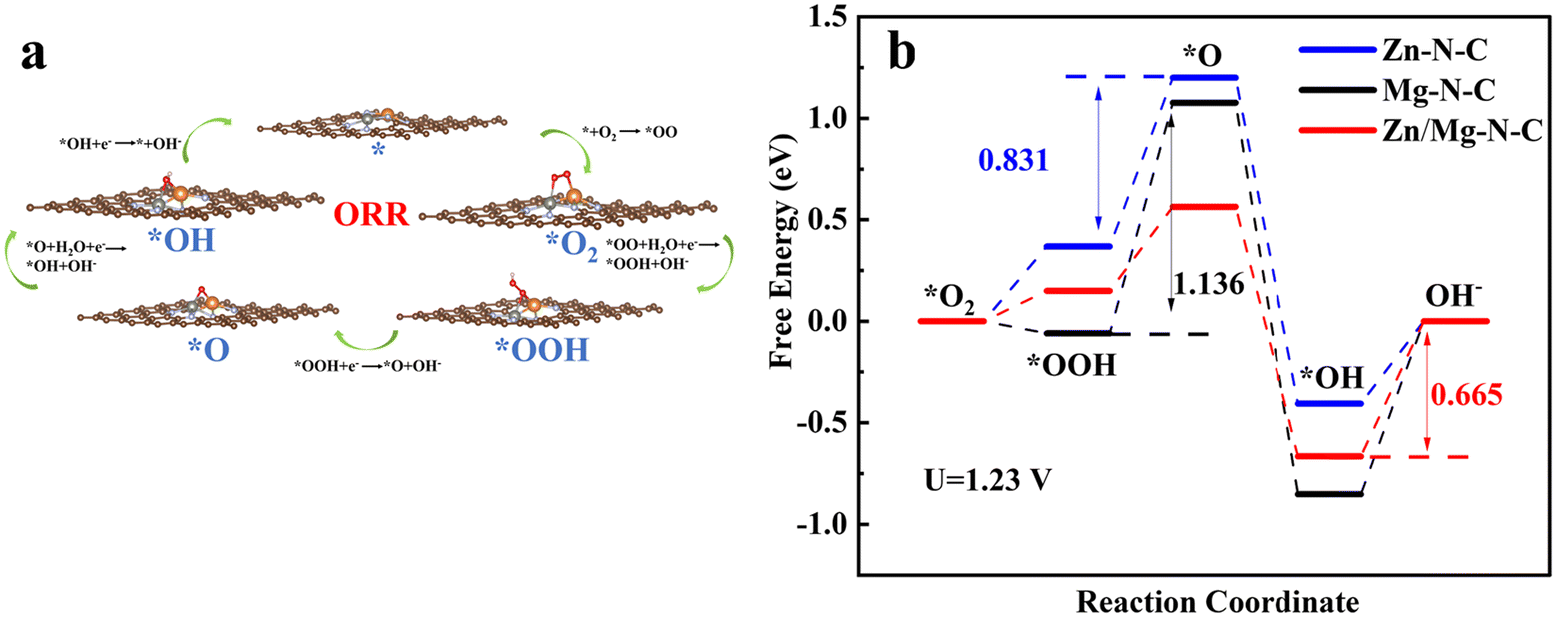 | ||
| Fig. 5 (a) The reaction pathway of the ORR by using Zn/Mg–N–C as an example. (b) The Gibbs free energy diagram of the ORR on Zn/Mg–N–C, Zn–N–C and Mg–N–C obtained from DFT calculations (U = 1.23 V). | ||
4. Conclusions
In summary, this work reports a bottom-up strategy for preparing hierarchical porous carbon materials containing Zn and Mg dual sites (Zn/Mg–N–C). The hierarchical porous structures promote the number of active sites and enhance mass transfer. Additionally, the introduction of metallic Mg sites can optimize the free energy of ORR intermediates and adjust the rate-determining steps of the reaction, thereby reducing the overpotential of the direct four-electron ORR. Impressively, the Zn/Mg–N–C catalyst exhibits an excellent E1/2 of 0.89 V and exceptional durability under alkaline conditions, surpassing commercial Pt/C. Furthermore, the practical application of Zn/Mg–N–C in ZABs reveals an impressive peak power density (298.7 mW cm−2). This study demonstrates that Zn/Mg–N–C holds promising prospects as a non-noble metal ORR catalyst for application in energy devices.Author contributions
M. L.: conceptualization, methodology, investigation, resources, and writing–original draft. J. Z.: validation, formal analysis, visualization, software, data curation, and formal analysis. Y. P.: data curation and formal analysis. S. G.: resources, writing–review and editing, supervision, and data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial aid from the High-level Talent Foundation of Shanghai University (no. 13-G210-21-239).References
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- L. Yang, X. Yao, C. Du, Z. Han, M. Jin, S. Peng, X. Ma, Y. Zhu, M. Zou and C. Cao, Chem. Eng. J., 2024, 481, 148598 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Z. Song, J. Li, Q. Zhang, Y. Li, X. Ren, L. Zhang and X. Sun, Carbon Energy, 2023, 5, e342 CrossRef CAS.
- K. Srinivas, Z. Chen, F. Ma, A. Chen, Z. Zhang, Y. Wu, M.-q. Zhu and Y. Chen, Appl. Catal., B, 2023, 335, 122887 CrossRef CAS.
- L. Yang, Y. Zhu, X. Yao, C. Du, Z. Han, J. Tian, X. Liu, X. Ma and C. Cao, Energy Storage Mater., 2023, 63, 102972 CrossRef.
- M. Liu, J. Zhang, G. Ye, Y. Peng and S. Guan, Dalton Trans., 2023, 52, 16773–16779 RSC.
- J. Balamurugan, P. M. Austeria, J. B. Kim, E.-S. Jeong, H.-H. Huang, D. H. Kim, N. Koratkar and S. O. Kim, Adv. Mater., 2023, 35, 2302625 CrossRef CAS PubMed.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- M. Luo and M. T. M. Koper, Nat. Catal., 2022, 5, 615–623 CrossRef CAS.
- J. Zhao, C. Fu, K. Ye, Z. Liang, F. Jiang, S. Shen, X. Zhao, L. Ma, Z. Shadike, X. Wang, J. Zhang and K. Jiang, Nat. Commun., 2022, 13, 685 CrossRef CAS PubMed.
- D. Zhang, Z. Wang, F. Liu, P. Yi, L. Peng, Y. Chen, L. Wei and H. Li, J. Am. Chem. Soc., 2024, 146, 3210–3219 CrossRef CAS PubMed.
- R. Z. Snitkoff-Sol, A. Friedman, H. C. Honig, Y. Yurko, A. Kozhushner, M. J. Zachman, P. Zelenay, A. M. Bond and L. Elbaz, Nat. Catal., 2022, 5, 163–170 CrossRef CAS.
- L. Peng, J. Yang, Y. Yang, F. Qian, Q. Wang, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS PubMed.
- Z. Chen, S. Zhang, J. Zhao, H. Zhang, J. Wang, Q. Dong, W. Zhang, X. Han and W. Hu, Chem. Eng. J., 2023, 462, 142030 CrossRef CAS.
- Y. Wang, J. Liu, T. Lu, R. He, N. Xu and J. Qiao, Appl. Catal., B, 2023, 321, 122041 CrossRef CAS.
- P. Song, M. Luo, X. Liu, W. Xing, W. Xu, Z. Jiang and L. Gu, Adv. Funct. Mater., 2017, 27, 1700802 CrossRef.
- J. Li, S. Chen, N. Yang, M. Deng, S. Ibraheem, J. Deng, J. Li, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2019, 58, 7035–7039 CrossRef CAS PubMed.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS.
- J. Lian, J. Zhao, X. Wang and Q. Bai, Carbon, 2023, 213, 118257 CrossRef CAS.
- Y.-X. Zhao, J.-H. Wen, P. Li, P.-F. Zhang, S.-N. Wang, D.-C. Li, J.-M. Dou, Y.-W. Li, H.-Y. Ma and L. Xu, Angew. Chem., Int. Ed., 2023, 62, e202216950 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, W. Zhao, J. Wang, Y. Hu, C. Huang, X. Zou, Y. Liu, D. Zhang, X. Lu, H. Fan and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202314303 CrossRef CAS PubMed.
- S. Liu, M. Liu, X. Li, S. Yang, Q. Miao, Q. Xu and G. Zeng, Carbon Energy, 2023, 5, e303 CrossRef CAS.
- H. Shang and D. Liu, Nano Res., 2023, 16, 6477–6506 CrossRef CAS.
- G. Yasin, S. Ali, S. Ibraheem, A. Kumar, M. Tabish, M. A. Mushtaq, S. Ajmal, M. Arif, M. A. Khan, A. Saad, L. Qiao and W. Zhao, ACS Catal., 2023, 13, 2313–2325 CrossRef CAS.
- M. Mazzucato, L. Gavioli, V. Balzano, E. Berretti, G. A. Rizzi, D. Badocco, P. Pastore, A. Zitolo and C. Durante, ACS Appl. Mater. Interfaces, 2022, 14, 54635–54648 CrossRef CAS PubMed.
- Y. Gu, B. J. Xi, H. Zhang, Y. C. Ma and S. L. Xiong, Angew. Chem., Int. Ed., 2022, 134, e202202200 CrossRef.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- O. Y. Bisen and K. K. Nanda, Appl. Mater. Today, 2020, 21, 100846 CrossRef.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- C.-H. Shin, H.-Y. Lee, C. Gyan-Barimah, J.-H. Yu and J.-S. Yu, Chem. Soc. Rev., 2023, 52, 2145–2192 RSC.
- L. Zhang, J. Yuan, Q. Xu, F. Zhang, Q. Sun and H. Xie, Int. J. Biol. Macromol., 2023, 242, 125110 CrossRef CAS PubMed.
- L. Liu, B. Farhadi, J. Li, S. Liu, L. Lu, H. Wang, M. Du, L. Yang, S. Bao, X. Jiang, X. Dong, Q. Miao, D. Li, K. Wang and S. Liu, Angew. Chem., Int. Ed., 2023, 33, e202317972 Search PubMed.
- J.-K. Sun, Z. Kochovski, W.-Y. Zhang, H. Kirmse, Y. Lu, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2017, 139, 8971–8976 CrossRef CAS PubMed.
- E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- X.-W. Zhu, D. Luo, X.-P. Zhou and D. Li, Coord. Chem. Rev., 2022, 455, 214354 CrossRef CAS.
- X. Song, Y. Jiang, F. Cheng, J. Earnshaw, J. Na, X. Li and Y. Yamauchi, Small, 2021, 17, 2004142 CrossRef CAS PubMed.
- R. Liu, C. von Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2011, 133, 10372–10375 CrossRef CAS PubMed.
- M. Shen, J. Qi, K. Gao, C. Duan, J. Liu, Q. Liu, H. Yang and Y. Ni, Chem. Eng. J., 2023, 464, 142719 CrossRef CAS.
- H. Yu, J. Hou, R. B. Namin, Y. Ni, S. Liu, S. Yu, Y. Liu, Q. Wu and S. Nie, Carbon, 2021, 173, 800–808 CrossRef CAS.
- J. Rong, E. Gao, N. Liu, W. Chen, X. Rong, Y. Zhang, X. Zheng, H. Ao, S. Xue, B. Huang, Z. Li, F. Qiu and Y. Qian, Energy Storage Mater., 2023, 56, 165–173 CrossRef.
- J. Ding, H.-F. Wang, X. Yang, W. Ju, K. Shen, L. Chen and Y. Li, Natl. Sci. Rev., 2023, 10, nwac231 CrossRef CAS PubMed.
- J. Li, S. Chen, W. Li, R. Wu, S. Ibraheem, J. Li, W. Ding, L. Li and Z. Wei, J. Mater. Chem. A, 2018, 6, 15504–15509 RSC.
- J. Xu, Z. Li, D. Chen, S. Yang, K. Zheng, J. Ruan, Y. Wu, H. Zhang, J. Chen, F. Xie, Y. Jin, N. Wang and H. Meng, ACS Appl. Mater. Interfaces, 2021, 13, 48774–48783 CrossRef CAS PubMed.
- W.-X. Hong, W.-H. Wang, Y.-H. Chang, H. Pourzolfaghar, I. H. Tseng and Y.-Y. Li, Nano Energy, 2024, 121, 109236 CrossRef CAS.
- A. Rosa and E. J. Baerends, Inorg. Chem., 1994, 33, 584–595 CrossRef CAS.
- C.-Y. Lin, L. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2017, 29, 1606635 CrossRef PubMed.
- Y. Liu, Z. Li, Y. Shen, D. Li and Z. Chen, Chem. Eng. J., 2022, 430, 132315 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00152d |
| This journal is © The Royal Society of Chemistry 2024 |