
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrthopalladated imidazolones and thiazolones: synthesis, photophysical properties and photochemical reactivity†
Darius
Dumitraş
 a,
David
Dalmau
b,
Pilar
García-Orduña
b,
Alexandra
Pop
a,
Anca
Silvestru
a and
Esteban P.
Urriolabeitia
*b
a,
David
Dalmau
b,
Pilar
García-Orduña
b,
Alexandra
Pop
a,
Anca
Silvestru
a and
Esteban P.
Urriolabeitia
*b
aSupramolecular Organic and Organometallic Chemistry Centre, Department of Chemistry, Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, Arany Janos 11, 400028 Cluj-Napoca, Romania
bInstituto de Síntesis Química y Catálisis Homogénea, ISQCH, CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009, Zaragoza, Spain. E-mail: esteban.u.a@csic.es
First published on 2nd May 2024
Abstract
The reaction of Pd(OAc)2 with (Z)-5-arylidene-4-(4H)-imidazolones (2a–e) and (Z)-4-arylidene-5(4H)-thiazolones (3a–e) in trifluoroacetic acid results in the corresponding orthopalladated dinuclear complexes (4a–e, imidazolones; 11a–d, thiazolones) with trifluoroacetate bridges through regioselective C–H activation at the ortho position of the 4-arylidene group. Compound 4e, which contains an imidazolone substituted at 2- and 4-positions of the arylidene ring with methoxide groups and exhibits strong push–pull charge transfer, is an excellent precursor for the synthesis of fluorescent complexes with green yellowish emission and remarkable quantum yields. Breaking the bridging system with pyridine yields the mononuclear complex 5e (ΦF = 5%), while metathesis of trifluoroacetate ligands with chloride leads to the dinuclear complex 6e, also a precursor of fluorescent complexes by breaking the chloride bridging system with pyridine (7e, ΦF = 7%), or by substitution of chloride ligands with pyridine (8e, ΦF = 15%) or acetylacetonate (9e, ΦF = 2%). In addition to notable photophysical properties, dinuclear complexes 4 and 11 also exhibit significant photochemical reactivity. Thus, irradiation of orthopalladates 4a–c and 11a–c in CH2Cl2 with blue light (465 nm) proceeds via [2 + 2] photocycloaddition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of imidazolone and thiazolone ligands, yielding the corresponding cyclobutane-bridging diaminotruxillic derivatives 10a–c and 12a–c, respectively.
C double bonds of imidazolone and thiazolone ligands, yielding the corresponding cyclobutane-bridging diaminotruxillic derivatives 10a–c and 12a–c, respectively.
Introduction
The use of light as a reagent to promote new properties, such as luminescence, and new reactivities, such as photochemistry, has been known for over 100 years.1 Although there has been sustained interest and development since then, it has only been in the last two decades that an unprecedented development has been experienced, and continues so,2–8 due to its numerous and important applications in lighting, imaging, or photoinduced synthetic processes.9–11 In both cases, whether inducing luminescence or promoting chemical processes through light is considered, molecules capable of absorbing light, transitioning to an excited state, and subsequently emitting light or initiating a chemical reaction are required.Among the different photoreactive molecular platforms, azlactone-type substrates, such as oxazolones, have demonstrated excellent properties from both the photophysical and photochemical perspectives. They exhibit intense fluorescence in the solid state,12,13 absorption of two photons,14 and non-linear optical (NLO) properties.15 Additionally, from a photochemical standpoint, they undergo 2 + 2 photocycloadditions and other light-induced processes, resulting in diaminotruxillic and diaminotruxinic-type cyclobutanes that are highly interesting due to their pharmacological properties.16–19
It is evident that the incorporation of a transition metal into the skeleton of a specific organic ligand will have a profound effect on both its photophysical and photochemical properties. This fact is particularly relevant and highly impactful in the case of oxazolones for several reasons. Regarding their photophysical properties, the incorporation of a metal such as palladium as an intramolecular lock between the 4-arylidene ring and the heterocycle20 allows for the amplification of the fluorescence in solution of certain substrates by several orders of magnitude (Fig. 1, past work),21–25 because well-characterized non-radiative deactivation pathways, such as the hula-twist or Z–E isomerizations, are restricted.26–30 Regarding photochemical reactivity, the formation of dinuclear palladacycles is the basis for the building of molecular templates that enable a stereoselective [2 + 2] photocycloaddition in solution, resulting in diaminotruxillic-type cyclobutanes as single diastereoisomers,31–34 in sharp contrast to what occurs with free-state oxazolones.16 Therefore, the significant influence of the incorporation of a metal into the skeleton of these species is evident.
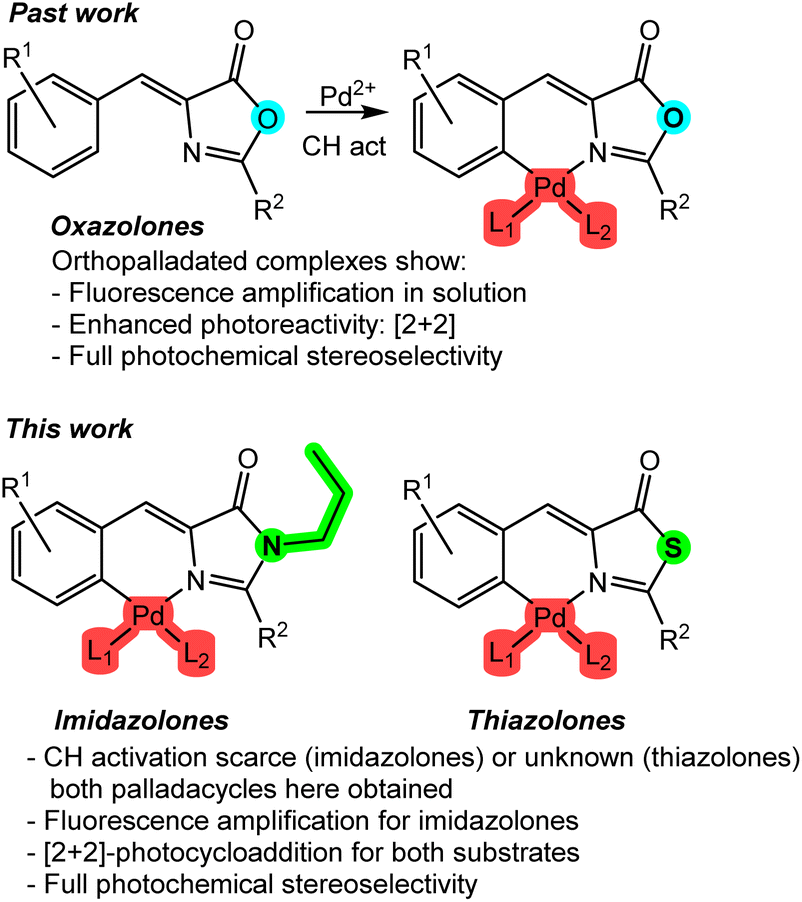 | ||
| Fig. 1 Past work: consequences of the incorporation of Pd into the molecular skeleton of oxazolones on their photophysical properties and photochemical reactivity. This work: results obtained from the palladation of imidazolones and thiazolones concerning their luminescence and photochemical reactivity. | ||
In this work, the study of the impact of the incorporation of Pd on the photophysical and photochemical properties of two families of compounds structurally related to oxazolones has been addressed. These families include imidazolones and thiazolones (Fig. 1, this work). Imidazolone-type substrates are of particular interest from the fluorescence perspective, as they are chromophores of GFP (green fluorescent protein) and have well-established luminescence properties.35–37 Similar to what has been reported for oxazolones in previous paragraphs, they completely lose fluorescence in solution.26–30 There are very few precedents for fluorescence recovery, and they typically use only boron as an intramolecular lock.38–40 The use of Pd for these substrates is virtually unexplored.21 On the other hand, contrary to what might seem, the structural similarity of imidazolones and thiazolones to oxazolones does not unequivocally imply the same reactivity. For example, there is only one isolated example of [2 + 2] photocycloaddition involving free imidazolones,41 and no photocycloadditions are known under other conditions. Referring to thiazolone-type species, they are even less studied than imidazolones and oxazolones. In this case, there are no fluorescence studies, no incorporation of any metal into their molecular skeleton has been described, and their photochemical reactivity is also very little explored.42
Results and discussion
Synthesis and characterization of orthopalladated complexes from imidazolones 2 and thiazolones 3
The imidazolones 2a–2e and thiazolones 3a–3e shown in Fig. 2 have been synthesized from the corresponding oxazolones 1a–1e,43–46 following reported procedures.21,42,47–50 Representatives with both electron-donating and electron-withdrawing substituents at different positions of the arylidene ring have been chosen to encompass a broader structural variety.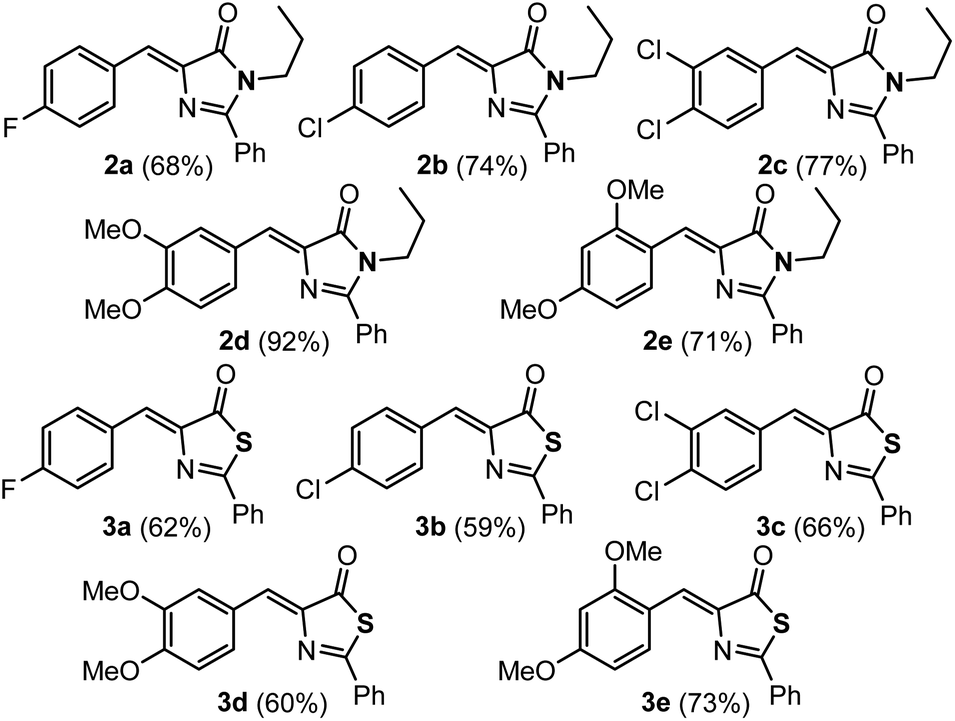 | ||
| Fig. 2 Imidazolones 2a–2e and thiazolones 3a–3e used in this work. | ||
The orthopalladation of the whole set of imidazolones 2 and thiazolones 3 has been attempted through CH bond activation processes, by the reaction of 2 or 3 with Pd(OAc)2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio; OAc = acetate) in trifluoroacetic acid CF3CO2H.20 The incorporation of Pd into the different molecular scaffolds was successful in all attempted cases, except in the case of thiazolone 3e, giving the corresponding dimers with carboxylate bridges 4a–4e or 11a–11d as shown in Fig. 3. The reaction was carried out by heating for 2 h at 75 °C for most of the cases. Only in the case of imidazolone 2e the reaction was performed at room temperature for 17 h; partial conversion was observed at room temperature at shorter reaction times, while decomposition occurred if heat was applied. For thiazolone 3e several different reaction conditions were attempted (temperatures, reaction times, solvents), but the reaction failed in all cases. The yields of isolated compounds 4a–4e and 11a–11d are in most of the cases higher than 78%, showing that the orthopalladation of azlactones through C–H bond activation is a general process.
:1 molar ratio; OAc = acetate) in trifluoroacetic acid CF3CO2H.20 The incorporation of Pd into the different molecular scaffolds was successful in all attempted cases, except in the case of thiazolone 3e, giving the corresponding dimers with carboxylate bridges 4a–4e or 11a–11d as shown in Fig. 3. The reaction was carried out by heating for 2 h at 75 °C for most of the cases. Only in the case of imidazolone 2e the reaction was performed at room temperature for 17 h; partial conversion was observed at room temperature at shorter reaction times, while decomposition occurred if heat was applied. For thiazolone 3e several different reaction conditions were attempted (temperatures, reaction times, solvents), but the reaction failed in all cases. The yields of isolated compounds 4a–4e and 11a–11d are in most of the cases higher than 78%, showing that the orthopalladation of azlactones through C–H bond activation is a general process.
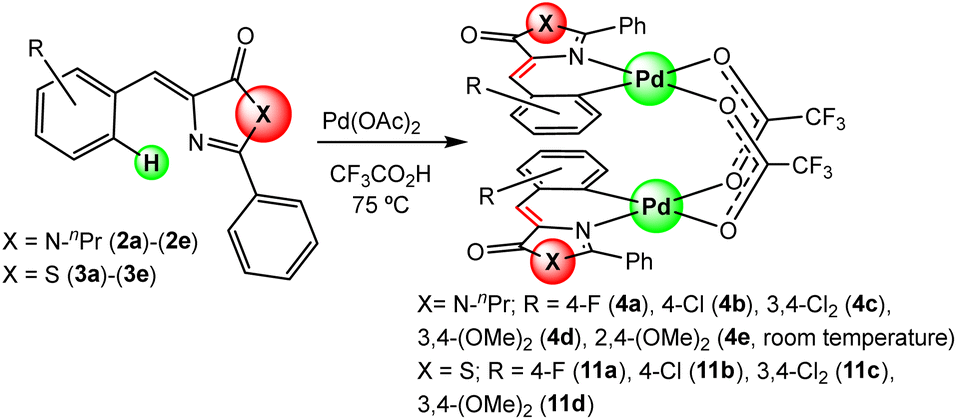 | ||
| Fig. 3 Synthesis of the orthopalladated derivatives 4a–4e (imidazolones) and 11a–11d (thiazolones). | ||
In dimers 4a–4e and 11a–11d, the incorporation of Pd occurred regioselectively at the ortho position of the phenyl ring of the arylidene group, following the trend observed in oxazolones.20 In the cases where two inequivalent accessible positions are present (positions 2 and 6; 2c, 2d, 3c, 3d), palladation took place at the less sterically hindered position (position 6), as deduced from their 1H and 13C NMR spectra. The dinuclear nature of these derivatives was inferred from the observation of peaks in their high-resolution mass spectra (HRMS) with the stoichiometry and isotopic distribution corresponding to the [M + Na]+ cations, where M is the dinuclear complex shown in Fig. 3 (see the ESI†). The relative arrangement of the two orthopalladated fragments is transoid, as deduced from the observation of a single signal in the 19F NMR spectra, because this configuration minimizes intramolecular repulsions.
The determination of the crystal structures of complexes containing orthopalladated imidazolones 4c (Fig. 4), and thiazolones 11a (Fig. 5) and 11b (Fig. 6) provides additional structural information. In all the studied cases, the asymmetric unit is composed of half of the molecule, and then, the dinuclear molecules are formed by two symmetry-related monomers, linked through the carboxylate bridges. The three structures shared common structural features: in all of them the expected open-book structure is observed, placing the two coordination planes containing palladium in close proximity, with an intermetallic distance of 3.0398(2) Å, 3.1569(4) Å and 3.0953(3) Å, in 4c, 11a and 11c, respectively. Although the immediate environments around Pd are square-planar in all three structures, the molecules are highly distorted. Thus, the imidazolone moiety in 4c and thiazolone groups in 11a and 11b deviate significantly from planarity, adopting a U-shaped form, and similarly, they largely deviate from coplanarity with respect to the square-planar metal coordination environments. Both types of distortions are analogous to those recently described in mononuclear complexes derived from oxazolones,22,24 suggesting that they are inherent to this type of structural skeleton and not dependent on the nuclearity of the resulting complexes or the ancillary ligands of palladium. It is noteworthy that the free imidazolone ligands exhibit remarkable coplanarity of the arylidene and heterocycle rings,51–55 which is lost upon metal incorporation.
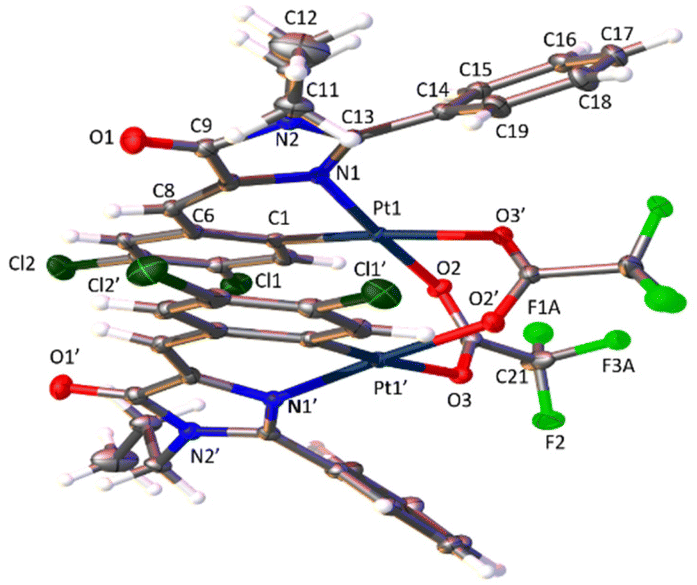 | ||
| Fig. 4 Molecular structure of cyclopalladated 4c. Thermal ellipsoids are drawn at 50% probability. Symmetry code: 1 − x, y, 3/2 − z. | ||
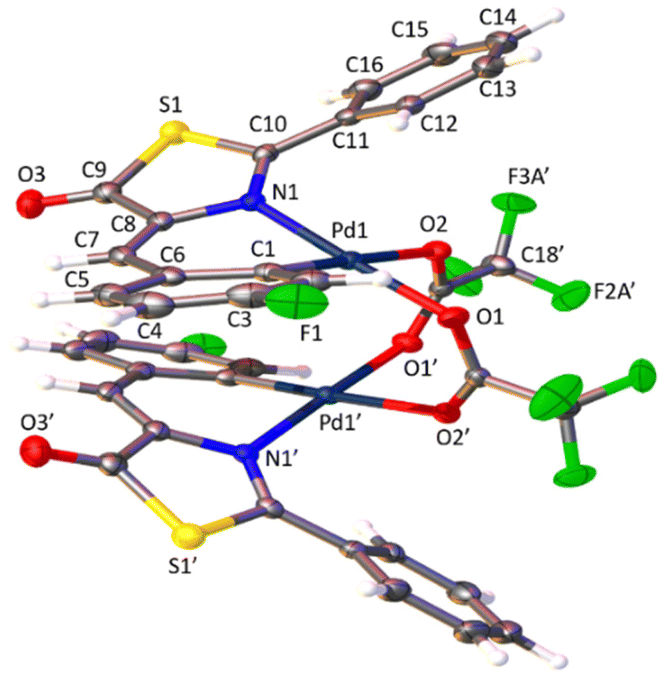 | ||
| Fig. 5 Molecular structure of cyclopalladated 11a. Thermal ellipsoids are drawn at 50% probability. For clarity, only the major component of the disordered fragment has been depicted. Symmetry code: 1 − x, y, 1/2 − z. | ||
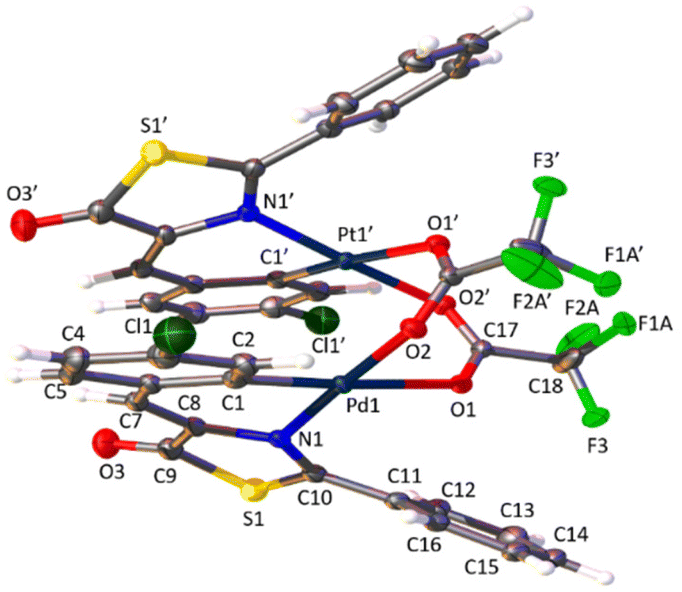 | ||
| Fig. 6 Molecular structure of cyclopalladated 11b. Thermal ellipsoids are drawn at 50% probability. For clarity, only the major component of the disordered fragment have been depicted. Symmetry code: 1 − x, y, 3/2 − z. | ||
Structures involving unsaturated 4-arylidene-5(4H)-thiazolones have not been reported to date by X-ray analysis; therefore no proper comparisons can be made. In each complex, the corresponding imidazolone or thiazolone acts as a C^N-chelating ligand, and the transoid relative arrangement of the two orthopalladated ligands is confirmed, as was deduced from the observation of single peaks in the 19F NMR spectra. Despite the distortion, the internal structural parameters of the orthopalladated arylidene-imidazolone fragment show no significant deviations from the uncomplexed situation, and in general, no relevant variations in bond distances and angles are observed compared to related structural situations found in the literature.56
Synthesis, characterization and photophysical properties of complexes 5e–9e
A preliminary screening with the orthometallated complexes of imidazolones 4a–4e and thiazolones 11a–11d shows that the most promising reactivity from the perspective of preparing fluorescent complexes is realized in the derivative of the dinuclear complex 4e, as shown in Fig. 7. The complexes 5e–9e exhibit fluorescence, albeit with different intensities, as will be discussed later. Attempts to develop reactivity similar to that shown in Fig. 7 with other imidazolone derivatives (e.g., 4d) did not lead to fluorescent complexes (results not shown). Moreover, the reactivity of thiazolone (11d) in reactions analogous to those shown in Fig. 7 resulted in all cases in complex mixtures of products that also did not exhibit fluorescence. Although the results of both reactivities are not shown here, it has been possible to characterize the mononuclear derivatives resulting from the trifluoroacetate bridge-breaking with pyridine in 4d and 11d through X-ray diffraction (CCDC codes 2336747 and 2336748†).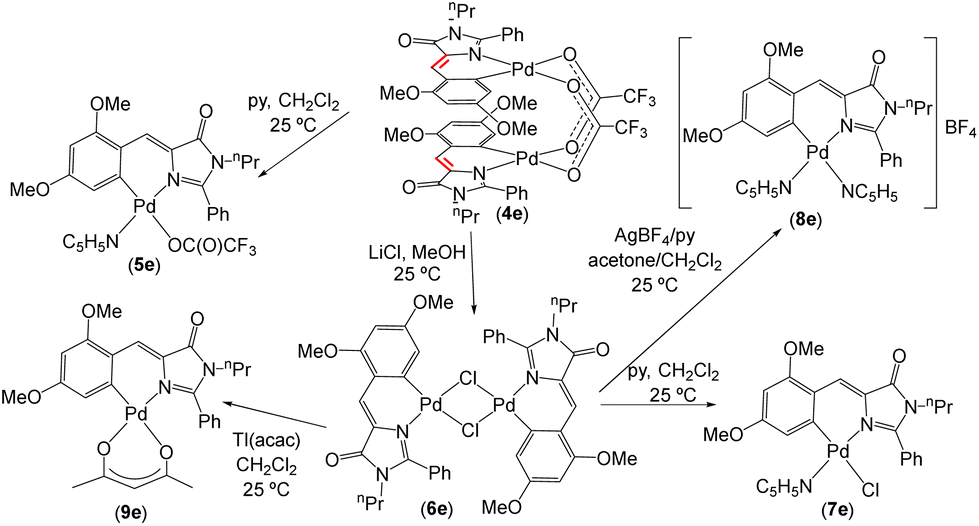 | ||
| Fig. 7 Synthesis of the fluorescent imidazolone derivatives 5e–9e. | ||
Given that in analogous structural situations, both pyridine and trifluoroacetate, chloride, and acetylacetonate ligands have proven to be good ancillary ligands for the development of fluorescence in orthopalladated azlactones, covering an appropriate scope of variation in electronic (trans influence) and steric properties, these same ligands have been selected to complete the coordination sphere of Pd for comparative purposes. As depicted in Fig. 7, the dinuclear derivative 4e reacts with pyridine (1:2 molar ratio) to yield the mononuclear derivative 5e. Treatment of 4e with LiCl (1:4 molar ratio) in MeOH produces the dinuclear chloride-bridging complex 6e, which subsequently reacts with pyridine (1:2 molar ratio) in CH2Cl2 to form mononuclear 7e, with AgClO4 and pyridine (1:2:4 molar ratio) in CH2Cl2/acetone, to give the cationic bis-pyridine derivative 8e, or with Tl(acac) (1:2 molar ratio; acac = acetylacetonate) affording the neutral mononuclear complex 9e. The characterization of all prepared complexes 5e–9e is straightforward from their NMR and HRMS data, as shown in the ESI.† Complexes 5e, 7e, and 8e exhibit dynamic behaviour in solution at room temperature, which is stopped upon lowering the temperature to 233 K. At this temperature, the molecules behave as static during the response time of the NMR. In general, the ligand arrangement follows the antisymbiotic effect shown by the soft Pd(II) center,57 with the pyridine ligand cis to the Pd–C bond.58
As for the photophysical properties, palladacycles typically exhibit low-efficient emission, only detected at low temperatures, due to the thermal deactivation of the excited state to the low-lying metal-centered orbitals (MC; d–d*) which experience non-radiative decay.25,59–61 On the grounds of previous studies performed in oxazolones where fluorescence amplification was achieved by using Pd as an intramolecular lock,24 we have investigated here the emissive properties of orthopalladated analogs with imidazolones 5e–9e, which result to be fluorescent. Photophysical data of 2a–2e and 5e–9e including their absorption, emission maxima, and emission quantum yields (ΦPL) in solution at room temperature are summarized in Table 1.
Most imidazolones 2 have the strongest absorption maxima in the UV region of the spectrum, due to a π–π* charge transfer from the 4-arylidene ring to the imidazolone heterocyclic moiety.12,62 In previously reported orthopalladated imidazolones the maxima were red-shifted to the blue-green region with respect to free imidazolones.21 We have observed here a similar trend for 5e–9e when compared with 2e. Therefore, the absorption spectra of 5e–9e in CH2Cl2 solution resemble each other and display an intense absorption in the blue region (445–454 nm). The bands with the highest absorbance correspond to intraligand charge transfer transition (ILCT) of the imidazolone fragment, as explained previously. The quantum yield ΦPL values range from 2 to 15% for complexes 5e, 7e, 8e and 9e, which have methoxide substituents at the two meta positions of the arylidene ring with respect to the Pd–C bond (Fig. 7), while it is close to zero for the starting imidazolone 2e (<1%). Therefore, a true recovery of the fluorescence has been achieved. The change of ligands attached to the Pd atom has a significant influence on ΦPL, similar to that determined in oxazolones.24 In the present case, the complex 9e with the acetylacetonate group as the ancillary ligand exhibits the lowest ΦPL (2%), followed by complex 5e with TFA and pyridine (5%), quite similar to that of complex 7e with TFA and Cl (7%). As in related complexes with palladated oxazolones,24 the presence of two pyridines in cis arrangement as ancillary ligands for the Pd (complex 8e) provides the highest performance, reflected in a quantum yield value as high as 15%.
Aiming to provide a detailed explanation about the role of Pd and the ancillary ligands in the recovery and amplification of the fluorescence observed on going from 2e to 5e–9e, the photophysical properties of the most important examples were modelized using computational methods (DFT and TDDFT). The absorption properties of compounds 5e, 7e, and 8e were calculated using the M06-2X functional, as described in the Computational Details section (see Experimental). The obtained results for 5e, 7e and 8e are shown in Fig. 8.
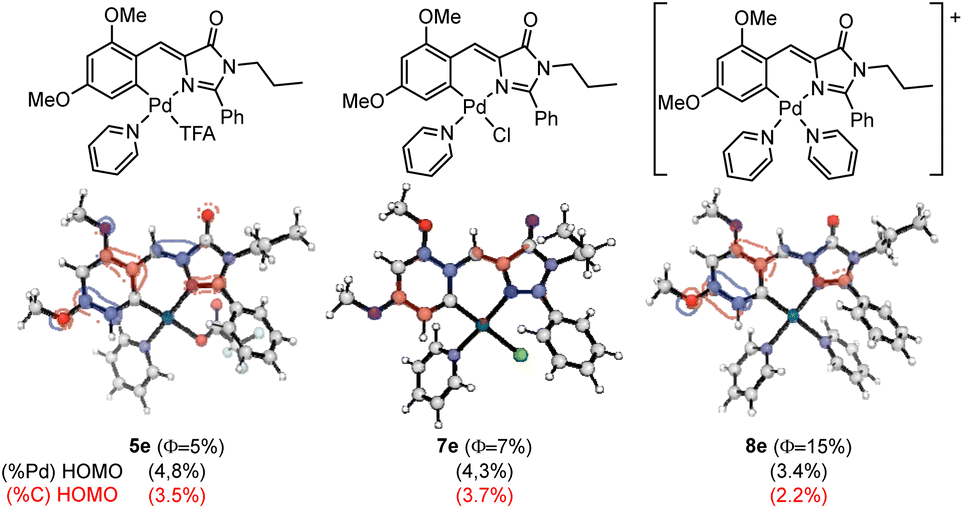 | ||
| Fig. 8 Schematic structures of 5e, 7e and 8e (top), HOMO orbitals (middle) with the corresponding Pd contributions (black) and orthopalladated C atom (red) to HOMO in parentheses (bottom). | ||
The three studied compounds exhibit similar characteristic behavior, for which the absorption properties correspond to the excitation from the ground state (S0) to the first excited singlet state (S1), predominantly involving a HOMO to LUMO transition. In our previous studies in oxazolones,24 we observed a correlation between the participation of Pd and the orthopalladated carbon in the HOMO orbital with the obtained quantum yield ΦPL. We aimed to verify if this pattern extended to complexes with imidazolones. Fig. 8 depicts the HOMO orbitals of the products, showing the percentage of the participation of the Pd orbitals in black parentheses and that for the metallated carbon in red. The comparison of complexes 5e and 7e reveals that the differences in Pd and C participation are negligible, as are the differences in ΦPL. Moreover, the lowest participation of both percentages in Pd and palladated C are observed for 8e, which correlates straightforward with the highest value of ΦPL measured for this compound. Therefore, it seems that once again, data suggest a correlation between the participation of the Pd–C fragment in the HOMO orbital and the value of the quantum yield measured experimentally, this behavior being parallel to that found for complexes with oxazolones.
[2 + 2]-Photocycloaddition of dimers 4 and 11: synthesis of 1,3-diaminotruxillic derivatives 10 and 12
Given the interesting photochemical reactivity previously observed in 4-arylidene-5(4H)-oxazolones to yield bis-amino acids of diaminotruxillic and diaminotruxinic types,16–19 and considering the continued interest in this type of cyclobutanes due to their exceptional pharmacological properties in the treatment of type 2 diabetes,63–66 we have attempted to extend this reactivity to free imidazolone substrates (2). However, irradiation of (2a)–(2e) in a variety of solvents with blue light (465 nm) does not lead to any detectable transformation (Fig. 9a), while irradiation using near-ultraviolet light (365 nm) gradually decomposes the sample, and it is not possible to distinguish cyclobutane-type products in either case. Since the incorporation of Pd into the imidazolone skeleton produces a clear red-shift in absorption, with absorption maxima in the blue region for compounds 4, and dinuclear orthopalladated oxazolones have also shown clear photochemical reactivity in the visible region,31–34 the irradiation of dinuclear complexes (4a)–(4e) with blue light has been attempted. The result of the reaction is shown in Fig. 9b. In the case of thiazolones (3), there is a single report of [2 + 2] photocycloaddition in non-complexed substrates,42 but there are no precedents in the case of orthopalladated thiazolones. For those reasons, irradiation of dimers (11) has been carried out as well.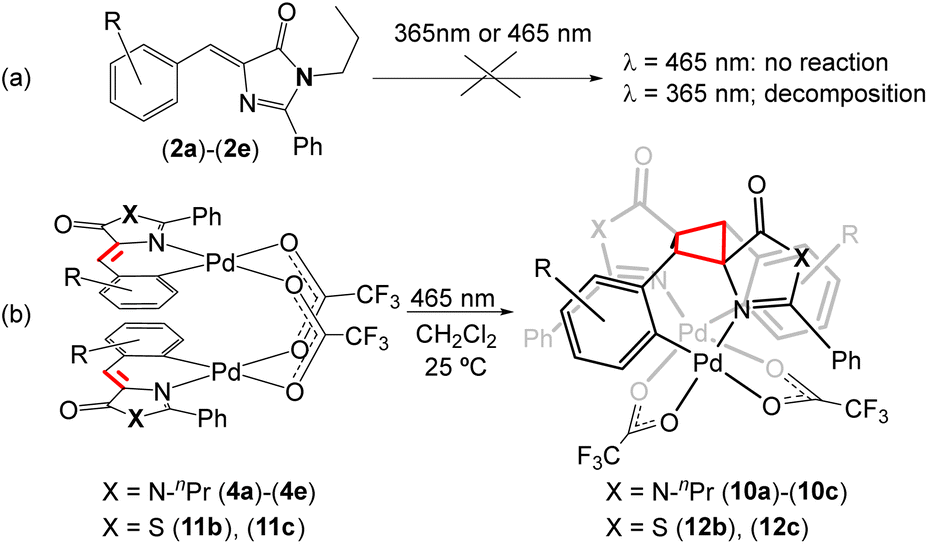 | ||
| Fig. 9 Blue light-promoted [2 + 2]-photocycloaddition of orthopalladated imidazolones (4) and thiazolones (11) to give cyclobutanes (10) and (12), respectively. | ||
The irradiation with blue light (465 nm, Kessil lamps, 40 W) of CH2Cl2 solutions of dinuclear complexes (4a)–(4e), containing orthometallated imidazolones and bridging trifluoroacetate ligands, proceeds through [2 + 2] photocycloaddition of the two CC double bonds of the imidazolones and the selective formation of the corresponding cyclobutanes (10a)–(10c) as the sole diastereoisomers (epsilon ε-isomer). The selectivity of the reaction is dictated by molecular constraints, with the dimer with an open-book structure acting as a molecular template. The optimized reaction time is 24 h, notably shorter than that observed for analogous oxazolones, usually in the range of 48 h–72 h.33,34 The reaction proceeds with complete conversion to a single product in the presence of electron-withdrawing substituents (F 4a; Cl, 4b, 4c) but does not occur (zero conversion) in the case of methoxide substituents (4d, 4e). Despite this limitation, given the complete absence of reactivity in the irradiation of free imidazolones (2) with visible light, the incorporation of Pd into imidazolone provides an advantageous method for accessing new diaminotruxillic-type bis-amino acids. Similarly, the irradiation of CH2Cl2 solutions of dinuclear complexes with orthometallated thiazolones (11b) and (11c) with blue light (465 nm) results in the [2 + 2] photocycloaddition of the exocyclic CCH fragments and the formation of cyclobutanes (12b) and (12c), as shown in Fig. 9b. In the case of complex (11a) a mixture of compounds was obtained – the expected cyclobutane (12a) and another unidentified species which could not be separated from (12a). Due to this fact (12a) was not fully characterized. In all other described cases, the reaction products (10) and (12) are isolated in pure form and with very good yields (even quantitative) by simple evaporation of the solutions and collection of the formed solid. Likewise, all isolated products (10) and (12) are photostable towards further irradiation in the visible region, and also are stable against thermal [2 + 2] retro-cycloaddition.
The observation of a single set of signals in the NMR spectra of (10) and (12) shows that these derivatives are obtained as unique isomers. The NMR signal set indicates that the two orthopalladated ligands, as well as the two carboxylate groups, behave as chemically equivalent, suggesting that (10) and (12) have C2 symmetry. This symmetry, along with the transoid relative configuration of the orthometalated ligands determined in their respective precursors (4) and (11) (Fig. 4–6), implies that the photocycloaddition proceeds with 1,3-head-to-tail C–C coupling, and the regio- and stereoselectively obtained isomer is the epsilon ε-isomer. Key data supporting the formation of the cyclobutane skeleton in (10) and (12) include the disappearance of signals assigned to the vinyl fragment CCH in their 1H and 13C NMR spectra, as well as the appearance of new signals in the 4.9–5.1 ppm (CH) region of the 1H NMR spectra and in the 59 ppm (CH) and 71 ppm (C) regions (62 ppm and 84 ppm, respectively in the case of thiazolones) in the 13C NMR spectra. These changes are in perfect agreement with the Csp2–Csp3 hybridization change occurring in the photocycloaddition.
Experimental
Full details about materials and measurements, crystallography and computational methods are given in the ESI.†Synthetic procedures
CH), 7.10 (dd, 2H, 3JHH = 8.7 Hz, H3/H5, C6H4), 3.74 (t, 2H, 3JHH = 7.5 Hz, NCH2), 1.59 (sext, 2H, 3JHH = 7.4 Hz, CH2), 0.84 (t, 3H, 3JHH = 7.4 Hz, CH3). 13C{1H} NMR (125.77 MHz, CDCl3) δ 171.72 (CO), 163.89 (d, 1JCF = 252.86 Hz, CF, C6H4), 163.00 (CN), 138.67 (C), 134.69 (d, 3JCF = 8.34 Hz, C2/C6, C6H4), 131.46 (Cp, C6H5), 130.78 (d, 4JCF = 3.13 Hz, Ci, C6H4), 129.88 (Ci, C6H5), 128.99 (Cm, C6H5), 128.40 (Co, C6H5), 127.41 (d, 5JCF = 1.34 Hz, CH), 115.99 (d, 2JCF = 21.88 Hz, C3/C5, C6H4), 43.46 (NCH2), 22.68 (CH2), 11.11 (CH3). 19F NMR (376.49 MHz, CDCl3) δ −108.42 (s, CF). HRMS (ESI+) m/z calcd for C19H18FN2O [M + H]+: 309.1403, found: 309.1405.
CH), 7.44 (t, 4H, 3JHH = 8.0 Hz, Hm, C6H5), 7.26 (dd, 3JHH = 8.4 Hz, 4JHF = 6.1 Hz, 2H, H2, C6H3), 7.17 (d, broad, 4H, 3JHH = 8.4 Hz, Ho, C6H5), 6.91 (ddd, 2H, 3JHH = 8.0 Hz, 3JHF = 8.0 Hz, 3JHH = 2.4 Hz, H3, C6H3), 6.86 (dd, 2H, 3JHF = 10.5 Hz, 4JHH = 2.5 Hz, H5, C6H3), 3.46 (ddd, 2H, 2JHH = 14.0 Hz, 3JHH = 9.2 Hz, 3JHH = 5.7 Hz, NCH2), 3.19 (ddd, 2H, 2JHH = 14.4 Hz, 3JHH = 9.4 Hz, 3JHH = 6.1 Hz, NCH2), 1.28, 1.14 (m, 4H, CH2), 0.63 (t, 6H, 3JHH = 7.4 Hz, CH3). 13C{1H} NMR (125.77 MHz, CDCl3) δ 165.24 (CO), 164.87 (CN), 164.52 (q, 2JCF = 38.5 Hz, CF3CO2), 161.44 (d, 1JCF = 259.7 Hz, CF), 138.75 (C), 134.90 (CH), 134.15 (d, 2JCF = 8.7 Hz C2, C6H3), 132.67 (Cp, C6H5), 129.14 (Cm, C6H5), 128.69 (Co, C6H5), 128.33 (d, 3JCF = 2.6 Hz, C6, C6H3), 126.51 (d, 4JCF = 1.6 Hz Ci, C6H3), 126.36 (Ci, C6H5), 121.00 (d, 3JCF = 22.0 Hz, C5, C6H3), 114.6 (q, 1JCF = 287.9 Hz, CF3), 112.76 (d, 2JCF = 22.0 Hz, C3, C6H3), 43.67 (NCH2), 21.82 (CH2), 10.94(CH3). 19F NMR (470.55 MHz, CDCl3) δ −74.40 (s, CF3), −106.10 (dt, 3JFH = 10.6 Hz, 4JFH = 7.0 Hz, CF). HRMS (ESI+) m/z calcd for C42H32F8N4NaO6Pd2 [M + Na]+: 1075.0161, found: 1075.0107.
Reactivity of orthopalladated 4e, synthesis of luminescent mononuclear and dinuclear derivatives 5e–9e
:1 molar ratio). Signals at room temperature are broad and preclude a correct characterization. 1H NMR (CD2Cl2, 400.13 MHz, 233K): δ 9.73 (d, 3JHH = 5.6 Hz, 1H, Ho, py), 8.52 (d, 3JHH = 5.2 Hz, 1H, Ho, py), 8.10 (s, 1H, Hv), 8.00 (m, 2H, Ho, Ph), 7.85–7.72 (m, 4H, Ho + Hp, py), 7.67–7.58 (m, 3H, Hv + Ho Ph), 7.55 (m, 2H, Hp, Ph), 7.46–7.37 (m, 2H, Hm, py), 7.36–7.22 (m, 4H, Hm py + Hm Ph), 7.06–6.95 (m, 2H, Hm, Ph), 6.18 (s, br, 1H, C6H2), 6.16 (s, br, 1H, C6H2), 5.66 (s, br, 1H, C6H2), 5.64 (s, br, 1H, C6H2), 3.89, 3.87 (2s, 6H, 2 OMe), 3.66 (t, 3JHH = 7.6 Hz, 2H, NCH2, Pr), 3.59 (t, 3JHH = 7.6 Hz, 2H, NCH2, Pr), 3.51, 3.50 (2s, 6H, 2 OMe), 1.45 (q, 3JHH = 7.7 Hz, 2H, CH2, Pr), 1.33 (q, 3JHH = 7.6 Hz, 2H, CH2, Pr), 0.73 (t, 3JHH = 7.6 Hz, 3H, CH3, Pr), 0.65 (t, 3JHH = 7.7 Hz, 3H, CH3, Pr). 13C{1H} NMR (CD2Cl2, 125.77 MHz, 233K) δ 165.51, 165.12 (CN), 162.47, 161.94 (CO), 161.11, 160.82, 160.17, 159.29 (C–O, C3 + C5, C6H2), 153.13, 152.64 (Co, py), 148.76, 148.25 (C), 139.47, 137.96 (Cp, py), 138.62, 132.10, 131.67, 131.41 (CH + Cp Ph), 129.60, 129.24, 128.93, 128.48 (Co + Cm, Ph), 127.52, 127.46, 127.19, 126.45 (C1 + C2, C6H2), 126.69 (2C, Cm, py), 115.77, 115.56 (Ci, Ph), 114.75, 114.24 (C6, C6H2), 94.48, 94.34 (C4, C6H2), 55.92, 55.76, 55.48, 55.34 (OMe), 43.55 (2NCH2, Pr), 22.68, 22.12 (CH2, Pr), 10.95, 10.85 (CH3, Pr). Peaks assigned to CF3CO2 in the APT spectrum could not be assigned, despite the use of low temperatures and long accumulation trials. 19F NMR (470.55 MHz, CDCl3) δ −74.51, −74.94. HRMS (ESI+) m/z calcd for C28H25F3N3O5Pd [M]+: 646.0781, found: 646.0801.
O, heterocycle), 1570 (νCN, heterocycle), 334 (νPd–Cl).
:1 molar ratio). Only the major isomer could be fully characterized due to extensive overlapping of peaks. Signals at room temperature are broad and preclude a correct characterization. 1H NMR (CD2Cl2, 400.13 MHz, 233K): δ 8.78 (dd, 3JHH = 4.8 Hz, 4JHH = 1.6 Hz, 2H, Ho, py), 8.18 (s, 1H, Hvinyl), 8.00 (m, 2H, Ho, Ph), 7.85 (t, 3JHH = 7.5 Hz, 1H, Hp, py), 7.51 (tt, 3JHH = 8 Hz, 4JHH = 1.2 Hz, 1H, Hp, Ph), 7.41 (m, 2H, Hm, py), 7.20 (d, 4JHH = 2.4 Hz, 1H, H6, C6H2), 6.98 (t, 3JHH = 8 Hz, 2H, Hm, Ph), 6.18 (d, 4JHH = 2.4 Hz, 1H, H4, C6H2), 3.89, 3.87 (2s, 2 OMe), 3.60 (t, 3JHH = 7.2 Hz, 2H, NCH2, Pr), 1.33 (q, 3JHH = 7.5 Hz, CH2, Pr), 0.66 (t, 3JHH = 7.2 Hz, CH3, Pr). 13C{1H} NMR (CD2Cl2, 100.4 MHz, 233 K) δ 165.39 (CN), 161.00 (CO), 160.19, 159.90 (C–O, C3 + C5, C6H2), 153.14 (Co, py), 150.25 (C), 139.10 (Cp, py), 132.03, 131.38 (CH + Cp Ph), 128.98 (Co, Ph), 127.93, 126.51 (C1 + C2, C6H2), 125.34 (Cm, Ph), 124.58 (Cm, py), 117.30 (C6, C6H2), 115.95 (Ci, Ph), 94.78 (C4, C6H2), 55.76 (2C overlapped, OMe), 43.45 (NCH2, Pr), 22.25 (CH2, Pr), 10.89 (CH3, Pr). HRMS (ESI+) m/z calcd for C26H26N3O3Pd [M − Cl + H]+: 534.1004, found: 534.1004.
CH), 8.12 (d, 3JHH = 5.0 Hz, 2H, Ho, py), 7.73 (t, 3JHH = 7.7 Hz, 2H, Hp, py), 7.52–7.35 (m, 7H, Hm py, Ho + Hm + Hp, Ph), 7.04 (dd, 3JHH = 7.8 Hz, 3JHH = 6.0 Hz, 2H, Hm py), 6.15 (d, 3JHH = 2.1 Hz, 1H, H6, C6H2), 5.78 (d, 3JHH = 2.2 Hz, 1H, H4, C6H2), 3.88 (s, 3H, OMe), 3.63 (t, 3JHH = 7.6 Hz, 2H, NCH2), 3.51 (s, 3H, OMe), 1.39 (q, 3JHH = 7.7 Hz, 2H, CH2), 0.69 (t, 3JHH = 7.4 Hz, 3H, CH3). This product was too insoluble at low temperature to obtain reliable 13C NMR data. 19F NMR (470.55 MHz, CDCl3) δ −152.26 (s, BF4). HRMS (ESI+) m/z calcd for C21H21N2O3Pd [M − 2py]+: 455.0587, found: 455.0606.
CH), 7.87 (dd, 2H, 3JHH = 7.6 Hz, 4JHH = 1.9 Hz, Ho, C6H5), 7.56–7.47 (m, 3H, Hp, C6H5, Hm, C6H5), 7.03 (d, 1H, 4JHH = 2.3 Hz, H3, C6H2), 6.20 (d, 1H, 4JHH = 2.3 Hz, H5, C6H2), 5.05 (s, 1H, CH, acac), 3.90 (s, 3H, OCH3(C4)), 3.84 (s, 3H, OCH3(C2)), 3.69 (t, 2H, 3JHH = 7.7 Hz, NCH2), 1.94 (s, 3H, CH3, acac), 1.51 (sext, 3JHH = 7.6 Hz, 3JHH = 7.7 Hz, 2H, CH2), 1.18 (s, 3H, CH3, acac), 0.76 (t, 3H, 3JHH = 7.4 Hz, CH3). 13C{1H} NMR (125.77 MHz, CDCl3) δ 187.30 (C–O, acac), 185.40 (C–O, acac), 166.16 (CN), 161.58, (C4, C6H2), 160.42 (CO), 159.99 (C2, C6H2), 150.68 (Ci, C6H2), 131.51 (CH), 131.17 (Cp, C6H5), 129.13 (Ci, C6H5), 128.92 (Co, C6H5), 128.68 (C), 126.39 (Cm, C6H5), 116.23 (C6, C6H2), 110.77 (C3, C6H2), 99.05 (CH, acac), 95.88 (C5, C6H2), 55.74 (OCH3(C2)), 55.50 (OCH3(C4)), 43.70 (NCH2), 27.48 (CH3, acac), 26.22 (CH3, acac), 22.34 (CH2), 11.12 (CH3). HRMS (ESI+) m/z calcd for C26H29N2O5Pd [M + H]+: 555.1111, found: 555.1112.
O), 172.91 (CN), 159.45 (d, 1JCF = 252.9 Hz, CF), 138.17 (d, 2JCF = 5.9 Hz, C6, C6H3), 132.65 (Cp, C6H5), 129.11 (Cm, C6H5), 129.04 (C5, C6H3), 128.41 (Co, C6H5), 126.75 (Ci, C6H5), 125.42 (d, 4JCF = 2.6 Hz, Ci, C6H3), 120.40 (d, 3JCF = 19.4 Hz, C2, C6H3), 112.12 (d, 2JCF = 22.3 Hz, C3, C6H3), 71.04 (C cyclobut), 58.87 (CH cyclobut), 44.16 (NCH2), 21.76 (CH2), 10.89 (CH3). 19F NMR (470.55 MHz, CDCl3) δ −75.00 (s, 3F, CF3), −112.82 (tt, 3JFH = 14.9 Hz, 4JFH = 9.1 Hz, 1F, CF). HRMS (ESI+) m/z calcd for C42H32F8N4NaO6Pd2 [M + Na]+: 1075.0156, found: 1075.0154.
Conclusions
The incorporation of Pd into the molecular framework of 4-arylidene-imidazolones (2) and thiazolones (3) through CH bond activation processes generates platforms with remarkable photophysical and photochemical properties. In the case of imidazolones, new systems have been developed where the fluorescence of the free ligands is amplified, achieving quantum yields of orthopalladated complexes of up to 15%. This fluorescence amplification has been explained based on TD-DFT calculations and is related to the involvement of Pd in the HOMO orbitals. However, no apparent amplification occurs in the studied cases of thiazolones. Regarding photochemistry, it has been shown that irradiation in the visible region (blue light), both for orthopalladated imidazolones and thiazolones, proceeds through the [2 + 2] photocycloaddition of exocyclic CC double bonds, leading to the stereoselective generation of the corresponding diaminotruxillics, bis-amino acids with a cyclobutane-type core. In summary, the incorporation of Pd into oxazolone, imidazolone, and thiazolone-type heterocycles causes similar changes in photophysical and photochemical properties, resulting in an amplification of the luminescence of specific substrates and a clear increase in photochemical reactivity due to the templating effect of Pd.
Author contributions
Darius Dumitraş: synthesis, chemical characterization (investigation, validation, data curation, formal analysis, methodology), writing and correction (visualization, writing – original draft, review and editing); David Dalmau: synthesis, chemical characterization, measurement of the photophysical properties (investigation, data curation, validation), computational calculations (formal analysis, methodology), writing and correction (visualization, writing – original draft, review and editing); Pilar García Orduña: measurement of the crystal structures (investigation, validation), writing and correction (visualization, writing – original draft, review and editing); Alexandra Pop: supervision, writing and correction (visualization, review and editing); Anca Silvestru: supervision, funding acquisition, resources, writing and correction (visualization, review & editing); Esteban P. Urriolabeitia: original idea (conceptualization), project administration, supervision, funding acquisition, resources, writing and correction (visualization, writing, original draft, review and editing). All authors analyzed and discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. P. U. and D. Dalmau thank the Spanish Government (Grant PID2019-106394GB-I00, funded by MCIN/AEI/10.13039/501100011033) and Gobierno de Aragón-FSE (Spain, research groups Química Inorgánica y de los Compuestos Organometálicos E17_23R) for funding. D. Dalmau thanks Gobierno de Aragón-FSE for a PhD fellowship. D. Dumitraş is grateful for the financial support from Babes–Bolyai University in the form of a Special Scholarship for Scientific Activity. We acknowledge the support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- N. Hoffmann, Photochem. Photobiol. Sci., 2012, 11, 1613–1641 CrossRef CAS PubMed.
- J. C. Scaiano, Chem. Soc. Rev., 2023, 52, 6330–6343 RSC.
- M. Hertzog, M. Wang, J. Mony and K. Börjesson, Chem. Soc. Rev., 2019, 48, 937–961 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- H. E. Bonfield, T. Knauber, F. Lévesque, E. G. Moschetta, F. Susanne and L. J. Edwards, Nat. Commun., 2020, 11, 804–807 CrossRef CAS PubMed.
- Y. Markushyna and A. Savateev, Eur. J. Org. Chem., 2022, e202200026 CrossRef CAS.
- E. Schroeder and P. Christopher, ACS Energy Lett., 2022, 7, 880–884 CrossRef CAS.
- F. Gomollón-Bel and J. García-Martínez, Angew. Chem., Int. Ed., 2023, 62, e202218975 CrossRef PubMed.
- F. Gomollón-Bel and J. García-Martínez, Nat. Chem., 2022, 14, 113–114 CrossRef PubMed.
- F. Gomollón-Bel, Chem. Int., 2020, 42, 3–9 Search PubMed.
- S. Icli, H. Icli, S. Alp, H. Koc and A. Mckillop, Spectrosc. Lett., 1994, 27, 1115–1128 CrossRef CAS.
- K. Ertekin, S. Alp, C. Karapire, B. Yenigül, E. Hendem and S. Içli, J. Photochem. Photobiol., A, 2000, 137, 155–161 CrossRef CAS.
- C. Rodrigues, I. Mariz, E. Maçôas, C. Afonso and J. Martinho, Dyes Pigm., 2012, 95, 713–722 CrossRef CAS.
- C. Rodrigues, I. Mariz, E. Maçôas, C. Afonso and J. Martinho, Dyes Pigm., 2013, 99, 642–652 CrossRef CAS.
- A. García-Montero, A. M. Rodríguez, A. Juan, A. H. Velders, A. Denisi, G. Jiménez-Osés, E. Gómez-Bengoa, C. Cativiela, M. V. Gómez and E. P. Urriolabeitia, ACS Sustainable Chem. Eng., 2017, 5, 8370–8381 CrossRef.
- S. Sierra, M. V. Gómez, A. I. Jiménez, A. Pop, C. Silvestru, M. L. Marín, F. Boscá, G. Sastre, E. Gómez-Bengoa and E. P. Urriolabeitia, J. Org. Chem., 2022, 87, 3529–3545 CrossRef CAS PubMed.
- S. Sierra, R. López, E. Gómez-Bengoa, L. R. Falvello and E. P. Urriolabeitia, Org. Biomol. Chem., 2023, 21, 3203–3213 RSC.
- S. Sierra, D. Dalmau, J. V. Alegre, A. Pop, C. Silvestru, M. L. Marín, F. Boscá and E. P. Urriolabeitia, Int. J. Mol. Sci., 2023, 24, 7583–7602 CrossRef CAS PubMed.
- D. Roiban, E. Serrano, T. Soler, G. Aullón, I. Grosu, C. Cativiela, M. Martínez and E. P. Urriolabeitia, Inorg. Chem., 2011, 50, 8132–8143 CrossRef PubMed.
- S. Collado, A. Pueyo, C. Baudequin, L. Bischoff, A. I. Jiménez, C. Cativiela, C. Hoarau and E. P. Urriolabeitia, Eur. J. Org. Chem., 2018, 6158–6166 CrossRef CAS.
- E. Laga, D. Dalmau, S. Arregui, O. Crespo, A. I. Jiménez, A. Pop, C. Silvestru and E. P. Urriolabeitia, Molecules, 2021, 26, 1238–1257 CrossRef CAS PubMed.
- C. Garcia-Sanz, A. Andreu, B. de las Rivas, A. I. Jiménez, A. Pop, C. Silvestru, E. P. Urriolabeitia and J. M. Palomo, Org. Biomol. Chem., 2021, 19, 2773–2783 RSC.
- D. Dalmau, O. Crespo, J. M. Matxain and E. P. Urriolabeitia, Inorg. Chem., 2023, 62, 9792–9806 CrossRef CAS PubMed.
- D. Dalmau and E. P. Urriolabeitia, Molecules, 2023, 28, 2663 CrossRef CAS PubMed.
- J.-S. Yang, G.-J. Huang, Y.-H. Liu and S.-M. Peng, Chem. Commun., 2008, 1344–1346 RSC.
- S. R. Meech, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC.
- Q. Zhang, X. Chen, G. Cui, W. H. Fang and W. Thiel, Angew. Chem., Int. Ed., 2014, 53, 8649–8653 CrossRef CAS PubMed.
- A. Usman, O. F. Mohammed, E. T. J. Nibbering, J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2005, 127, 11214–11215 CrossRef CAS PubMed.
- X. He, A. F. Bell and P. J. Tonge, FEBS Lett., 2003, 549, 35–38 CrossRef CAS PubMed.
- D. Roiban, E. Serrano, T. Soler, C. Cativiela, I. Grosu and E. P. Urriolabeitia, Chem. Commun., 2009, 4681–4683 RSC.
- E. Serrano, A. Juan, A. García-Montero, T. Soler, F. Jiménez-Márquez, C. Cativiela, M. V. Gomez and E. P. Urriolabeitia, Chem. – Eur. J., 2016, 22, 144–152 CrossRef CAS PubMed.
- C. Carrera, A. Denisi, C. Cativiela and E. P. Urriolabeitia, Eur. J. Inorg. Chem., 2019, 3481–3489 CrossRef CAS.
- P. Sánchez, A. Pop, C. Silvestru, E. Laga, A. I. Jiménez, C. Cativiela and E. P. Urriolabeitia, Beilstein J. Org. Chem., 2020, 16, 1111–1123 CrossRef PubMed.
- O. Shimomura, Angew. Chem., Int. Ed., 2009, 48, 5590–5602 CrossRef CAS PubMed.
- M. Chalfie, Angew. Chem., Int. Ed., 2009, 48, 5603–5611 CrossRef CAS PubMed.
- R. Y. Tsien, Angew. Chem., Int. Ed., 2009, 48, 5612–5626 CrossRef CAS PubMed.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089–4096 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Splichenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- M. S. Baranov, K. M. Solntsev, N. S. Baleeva, A. S. Mishin, S. A. Lukyanov, K. A. Lukyanov and I. V. Yampolsky, Chem. – Eur. J., 2014, 20, 13234–13241 CrossRef CAS PubMed.
- P. Naumov, J. Kowalik, K. M. Solntsev, A. Baldridge, J.-S. Moon, C. Kranz and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 5845–5857 CrossRef CAS PubMed.
- S. Sierra, D. Dalmau, S. Higuera, D. Cortés, O. Crespo, A. I. Jimenez, A. Pop, C. Silvestru and E. P. Urriolabeitia, J. Org. Chem., 2021, 86, 12119–12140 CrossRef CAS PubMed.
- J. Plöchl, Chem. Ber., 1883, 16, 2815 CrossRef.
- E. Erlenmeyer, Justus Liebigs Ann. Chem., 1893, 275, 1 CrossRef.
- H. E. Carter, Ch. 5 of the book series Organic Reactions, Azlactones, 1946, 3, 198 Search PubMed.
- Y. S. Rao and R. Filler, Oxazoles, in The Chemistry of Heterocyclic Compounds, ed. I. J. Turchi, John Wiley & Sons, Inc., New York, 1986, ch. 3, vol. 45, pp. 363–691 Search PubMed.
- M. Muselli, L. Colombeau, J. Hédouin, C. Hoarau and L. Bischoff, Synlett, 2016, 2819–2825 CAS.
- R. Filler and Y. S. Rao, J. Org. Chem., 1962, 27, 3730–3731 CrossRef CAS.
- H. Behringer and H. W. Stein, Chem. Ber., 1949, 82, 209–212 CrossRef CAS.
- H. Behringer and J. B. Jepson, Chem. Ber., 1952, 85, 138 CrossRef CAS.
- G. Bhattacharjya, G. Savitha and G. Ramanathan, CrystEngComm, 2004, 6, 233 RSC.
- B. K. Rajbongshi and G. Ramanathan, J. Chem. Sci., 2009, 121, 973 CrossRef CAS.
- G. Bhattacharjya, G. Savitha and G. Ramanathan, J. Mol. Struct., 2005, 752, 98–103 CrossRef CAS.
- C. I. C. Esteves, I. da S. Fonseca, J. Rocha, A. S. Silva and S. Guieu, Dyes Pigm., 2020, 177, 108267 CrossRef CAS.
- W.-T. Chuang, C.-C. Hsieh, C.-H. Lai, C.-H. Lai, C.-W. Shih, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Org. Chem., 2011, 76, 8189–8202 CrossRef CAS PubMed.
- A. Guy-Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83 RSC.
- R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS.
- A. J. Deeming, I. P. Rothwell, M. B. Hursthouse and L. New, J. Chem. Soc., Dalton Trans., 1978, 1490–1496 RSC.
- M. Ghedini, I. Aiello, M. La Deda and A. Grisolia, Chem. Commun., 2003, 2198 RSC.
- P.-K. Chow, W.-P. To, K.-H. Low and C.-M. Che, Chem. – Asian J., 2014, 9, 534 CrossRef CAS PubMed.
- F. Neve, A. Crispini, C. Di Pietro and S. Campagna, Organometallics, 2002, 21, 3511 CrossRef CAS.
- S. Icli, A. O. Doroshenko, S. Alp, N. A. Abmanova, S. I. Egorova and S. T. Astley, Spectrosc. Lett., 1999, 32, 553 CrossRef CAS.
- Z. Cong, Q. Zhou, Y. Li, L.-N. Chen, Z.-C. Zhang, A. Liang, Q. Liu, X. Wu, A. Dai, T. Xia, W. Wu, Y. Zhang, D. Yang and M.-W. Wang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200155119 CrossRef CAS PubMed.
- C. de Graaf, D. Donnelly, D. Wootten, J. Lau, P. M. Sexton, L. J. Miller, J.-M. Ahn, J. Liao, M. M. Fletcher, D. Yang, A. J. H. Brown, C. Zhou, J. Deng and M.-W. Wang, Pharmacol. Rev., 2016, 68, 954–1013 CrossRef CAS PubMed.
- M. Méndez, H. Matter, E. Defossa, M. Kurz, S. Lebreton, Z. Li, M. Lohmann, M. Löhn, H. Mors, M. Podeschwa, N. Rackelmann, J. Riedel, P. Safar, D. S. Thorpe, M. Schäfer, D. Weitz and K. Breitschopf, J. Med. Chem., 2020, 63, 2292–2307 CrossRef PubMed.
- B. Manandhar and J.-M. Ahn, J. Med. Chem., 2015, 58, 1020–1037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental section with references, copies of the NMR spectra of all new prepared species, absorption and excitation–emission spectra of imidazolones 2 and complexes 5e–9e, computational details and Cartesian coordinates of all calculated species. CCDC 2332942–2332944, 2336747 and 2336748. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00730a |
| This journal is © The Royal Society of Chemistry 2024 |