
Direct methylation of benzene with methane over Co/MFI catalysts generated by self-dispersion of Co(OH)2†
Kazu
Okumura
 *a,
Kai
Tanaka
a,
Akimichi
Ohtsuki
a,
Hikaru
Iiyoshi
a and
Naonobu
Katada
b
*a,
Kai
Tanaka
a,
Akimichi
Ohtsuki
a,
Hikaru
Iiyoshi
a and
Naonobu
Katada
b
aDepartment of Applied Chemistry, School of Advanced Engineering, Kogakuin University, 2665-1 Nakano-machi, Hachioji-City, Tokyo 192-0015, Japan. E-mail: okmr@cc.kogakuin.ac.jp
bCenter for Research on Green Sustainable Chemistry, Tottori University, 4-101 Koyama-cho Minami, Tottori 680-8552, Japan
First published on 1st September 2023
Abstract
A methane–benzene reaction was performed over Co/MFI prepared with an aqueous solution of Co(OAc)2 and MFI zeolite. Co(OH)2 was deposited on the MFI zeolite when the Co(OAc)2 solution was stirred at 343 K in the presence of MFI. A decline of the h-peak in NH3 TPD occurred during the loading of Co on MFI. Further thermal treatment at 573–773 K caused the spontaneous formation of the atomically dispersed Co species, which was observed with Co K-edge XAFS. The toluene formation rate of the catalyst prepared with Co(OAc)2 was proportional to the Co loading and was higher than that prepared with Co(NO3)2 due to the large amount of supported Co.
1. Introduction
Methane, the main component of natural gas, is an abundant resource on earth, thus it is expected to be used not only as a fuel but also as a carbon resource. In order to use methane for non-fuel applications, it is usually converted to other molecules by C1 chemistry via synthesis gas (CO + H2) obtained by steam reforming of methane with water vapor using Ni or Ru supported catalysts.1,2 Meanwhile, if methane can be used directly as a carbon source, it is expected that a new synthetic route will be developed. The use of methane as a carbon source is beneficial over conventional ones using petroleum as the carbon source for production of plastics and so on from an economic viewpoint. Various catalysts that directly utilize methane as a carbon resource have been developed including Mo-loaded MFI for the formation of benzene.3 Among them, Co/MFI has been found to be active in the direct methylation of benzene by methane to give toluene and hydrogen.4 The reaction proceeded exclusively when MFI zeolite was employed as the support for Co among various structures of zeolites.5The conventional Co/MFI catalysts have primarily been prepared with an ion-exchange method.6 The Co located at the α sites was active in the methane–benzene reaction7 among three kinds of ion-exchange sites (α, β, γ).8 The adsorption energy generated at the adsorption of benzene on the Lewis acid sites of Co compensated the activation energy required for scission of the C–H bond in methane, so that the reaction between methane and benzene was promoted.7 It could be supposed that one of the ways to improve the catalytic activity of Co/MFI zeolite is to increase the loading of Co with an atomically dispersed form using the ion-exchange sites of MFI. Another possible way to increase the loading of dispersed Co is to make use of the reaction between defect sites of MFI and Co. Such a method has been reported to prepare atomically dispersed species of various elements including Ti and Sb using volatile precursors.9,10 In previous studies on Co/MFI catalysts, Co(NO3)2 has been primarily used as a precursor of Co for the preparation of Co/MFI by the above-mentioned ion-exchange method under non-hydration conditions.11 The hydrolysis of Co2+ ions is inhibited under acidic or low temperature conditions.12 Other possible methods for the preparation of Co/MFI is the impregnation of Co(NO3)2 and the sublimation of volatile Co precursors.13 In the former case, aggregated and dispersed Co oxides formed simultaneously, resulting in a heterogeneous distribution of Co sizes.4 In this research, we employed cobalt acetate tetrahydrate (Co(OAc)2·4H2O) as the precursor of Co/MFI in place of Co(NO3)2 on purpose. The pH of the aqueous solution of Co(OAc)2 was ca. 7.2 at 200 mmol L−1. Under this condition, hydrolysis of Co2+ ions occurs to form Co(OH)2 spontaneously at around 343 K.14 We expected that the deposited Co(OH)2 on the surface of MFI zeolites acted as the possible precursor of active Co species in the methane–benzene reaction, which was generated through the reaction with defect or ion exchange sites in MFI zeolites. This is because Co(OAc)2 was reported to be a promising precursor to obtain well-dispersed Co species in MFI via solid-state ion-exchange reactions.15 MFI treated with an aqueous NaOH solution was also used as the support for Co, taking into account that treatment with an aqueous NaOH solution has been reported to change the porosity of MFI.16
The dispersion process of aggregated to dispersed Co species was monitored with Co K-edge X-ray absorption fine structure (XAFS), and NH3 temperature programmed desorption (NH3-TPD). Particularly, Co K-edge XAFS is powerful technique to obtain information on the structural and electronic state of the Co center, which has been applied for the characterization of Co/zeolites so far.17,18
2. Experimental
2.1. Catalyst preparation
The experiment was carried out primarily using the MFI zeolite supplied by Mizusawa Chemical Co. (52A). In addition to the 52A sample, MFI, FAU, LTL, MOR, and BEA zeolites purchased from Tosoh Co. or obtained from Catalysis Society of Japan and SiO2 (Fuji-Silysia Co.) were employed as the support for Co as well. The composition, abbreviations of the employed zeolites are listed in Table 1. The letters “N” and “H” indicate the NH4 and H forms of zeolites, respectively. The sample treated with an aqueous NaOH solution for zeolite is abbreviated as sample name + AT + treatment time. For example, 822N-AT6h indicated that Co was loaded on the NH4-type 822 treated with an NaOH aqueous solution for 6 h. For the preparation of Co-loaded samples, the zeolite sample (3 g) was ion exchanged with an aqueous solution of NH4NO3 (200 mmol L−1) at 343 K three times for 4 h to obtain the NH4-form prior to the loading of Co. The amount of NH4NO3 used for each ion exchange treatment was ten times as much as the Al present in zeolites. In some sample preparations, Co was loaded on H-type zeolites. Some of the MFI samples were treated with an aqueous solution of NaOH (0.7 mol L−1, 200 mL) at 313 K for 1–16 h. The treated samples were rinsed repeatedly with deionized water and subsequently dried in air. The NaOH-treated MFI samples were transformed to an NH4-type after ion exchange with an NH4NO3 solution (0.5 mol L−1) at 343 K three times, prior to the loading of Co. Cobalt(II) acetate tetrahydrate ((CH3COO)2Co·4H2O) was purchased from Fujifilm Wako Pure Chemical Industries. Zeolites (primarily NH4-form) were added to the aqueous solution of Co(OAc)2 (200 mL), and then they were continuously stirred at 343 K for 4 h to load Co on the zeolites. Co(OAc)2 solutions with different Co concentrations (50–400 mmol L−1) were used for the preparation of Co/MFI. The typical concentration of Co(OAc)2 was 200 mmol L−1. The Co loaded samples were separated by filtration, rinsed repeatedly with deionized water, and dried in an oven kept at 373 K. The loading of Co on silica was carried out in the same manner as for the preparation of Co/zeolites.| Sample name | Structure | Abbreviation | Supplier | Cation type | Si/Al2 |
|---|---|---|---|---|---|
| HSZ-820NHA | MFI | 820 | Tosoh Co. | NH4+ | 23 |
| HSZ-822HOA | MFI | 822 | Tosoh Co. | H+ | 24 |
| HSZ-840HOA | MFI | 840 | Tosoh Co. | H+ | 40 |
| HSZ-860HOA | MFI | 860 | Tosoh Co. | H+ | 72 |
| HSZ-891HOA | MFI | 891 | Tosoh Co. | H+ | 1500 |
| 52A | MFI | 52 | Mizusawa Chemical Co. | Na+, H+ | 52 |
| JRC-Z5-90NA | MFI | 90 | Catalysis Society of Japan | Na+ | 90 |
| HSZ-360UHA | FAU | 360 | Tosoh Co. | H+ | 15 |
| HSZ-500KOA | LTL | 500 | Tosoh Co. | K+ | 6.1 |
| HSZ-640HOA | MOR | 640 | Tosoh Co. | H+ | 380 |
| HSZ-930NHA | BEA | 930 | Tosoh Co. | NH4+ | 27 |
| Q3 | SiO2 | SiO2 | Fuji-Silysia Co. | — | — |
2.2. Sample characterization
Co K-edge XAFS data of the Co(OAc)2-loaded supports were collected using synchrotron radiation. The data were recorded at the BL-9C beamline with the approval of the Photon Factory of the High Energy Accelerator Research Organization (KEK-PF). Proposal numbers were 2020G621 and 2022G581. The samples pressed into a wafer form (one pellet) were subjected to the XAFS measurement under ambient conditions. The data were obtained in a quick scan mode within 5 min using a Si (111) monochromator in the transmission mode. Ion chambers were used for the detection of primary (I0, 100% N2) and transmitted (15% Ar with the balance N2) X-rays. The beam size at the sample position was 0.8 mm (horizontal) × 0.6 mm (vertical). The Co K-edge extended X-ray absorption fine structure (EXAFS) was analyzed by extracting oscillations using a spline smoothing method. The Fourier transform (FT) of the k3-weighted EXAFS oscillations and k3χ(k) from k-space to r-space was conducted in the range of 3–13 Å−1 for curve-fitting analysis. The EXAFS data were analyzed using the REX software (Rigaku Co.). The parameters for the analysis of the Co–O and Co–Co bonds were extracted from Co K-edge EXAFS of CoO and Co foil, respectively. For the analysis of the Co–Si bond, parameters were obtained using the FEFF8.0 code.19 In addition to the ex situ conditions, operando XAFS measurement was carried out at BL-9C of KEK-PF. For this purpose, a wafer sample (40 mg, 10 mm-ϕ) was mounted in a stainless-steel cross-shaped cell. XAFS and IR measurements were performed using crossed X-ray and infrared (IR) beams at the sample position (Fig. S1†). The sample pellet was placed on the optical path at an angle of 45° to the X-ray and IR. Data of the Co K-edge XAFS were obtained under flowing N2 (30 mL min−1) while increasing the temperature (heating rate: 10 K min−1).Co loadings were measured with the atomic absorption (AA) instrument AA-6200 (Shimadzu Co.). The sample was prepared after dissolving Co/MFI with an aqueous solution of HF and subsequently with conc. H2SO4, followed by evaporation. The residue dissolved with hydrochloric acid was subjected to the AA measurements for quantitative analysis of Co. Data of the temperature programmed desorption (TPD) of ammonia were obtained with a BELCAT II equipment (Microtrac Bel Co.). The sample was treated at 773 K in an N2 flow prior to the measurement. Then ammonia (5%) diluted with He was equilibrated with the pretreated sample (0.1 g) at 373 K. The TPD data were collected with a temperature ramping rate of 10 K min−1. The carrier gas (He) flow rate was 30 mL min−1. A mass spectrometer (BELMASS, Microtrac Bel Co.) was used to measure the desorbed NH3. In the measurement, m/z = 16 was monitored to analyze the desorbed NH3. X-ray diffraction (XRD) patterns of the powder samples were obtained using a MiniFlex X-ray diffractometer (Rigaku Co.) with Cu Kα radiation in the 2θ range from 5 to 40° with 10° min−1 scanning speed. N2 adsorption isotherms were recorded on the BELSORP-mini-X (Microtrac Bel Co.) instrument. The samples were dehydrated in a vacuum at 413 K in advance. Transmission electron microscopy (TEM) images were obtained using a JEOL-JEM-2100 microscope. For the preparation of the sample, an ethanol suspension of Co/MFI was dropped onto Cu grids coated with a C-coated porous thin membrane (NEM, Japan) and dried. TEM observations were performed at an operating voltage of 200 kV. Field emission scanning electron microscopy (FE-SEM) images were taken with a JEOL JSM-6701F microscope with an accelerating voltage of 5 kV.
2.3. Catalytic reaction
The catalytic reaction between methane and benzene was performed using a fixed-bed flow reactor. The prepared catalyst was mounted in a reaction tube filled with quartz wool, which was placed at the bottom of the powder catalyst. The catalyst (0.30 g) was pretreated at 823 K in an N2 flow for 1 h prior to the reaction. Neat methane gas (99.995%) was passed through a bubbler containing liquid benzene (Fuji Film Wako Pure Chemical Industries) kept at 273 K with a flow rate of 30 mL min−1. The gas mixture was reacted with the pretreated Co/MFI catalyst at 823 K. The ratio of methane to benzene was 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The effluent gas was analyzed with an on-line FID type gas chromatograph (Shimadzu, GC-2014) equipped with a capillary column (GL Sciences Inc., InertCap 1). The toluene formation rate was calculated based on the benzene feed flow rate (2.7 mmol h−1).
:1. The effluent gas was analyzed with an on-line FID type gas chromatograph (Shimadzu, GC-2014) equipped with a capillary column (GL Sciences Inc., InertCap 1). The toluene formation rate was calculated based on the benzene feed flow rate (2.7 mmol h−1).
3. Results and discussion
3.1. Operando Co K-edge XAFS and IR studies
First, the local structure of Co/MFI(52N) was analyzed with Co K-edge XAFS for the as-prepared samples and the samples pretreated in an N2 flow. Fig. 1(a) shows the Co K-edge X-ray absorption near edge structure (XANES) of the sample in which Co was loaded on MFI using Co(OAc)2 as a precursor (as-prepared) and reference compounds. The XANES of the as-prepared Co/MFI sample was close to that of Co(OH)2, indicating that Co(OAc)2 was hydrated to give Co(OH)2 during heat treatment of a Co(OAc)2 solution in the presence of MFI at 343 K. As shown in Fig. 1(b), the Co K-edge EXAFS of the Co/MFI samples was also close to that of Co(OH)2. The corresponding k3χ(k) data are displayed in Fig. S2.† That is to say, two peaks assignable to the Co–O and Co–(O)–Co bonds appeared at 1.7 and 2.8 Å (phase shift uncorrected) in the EXAFS Fourier transform (EXAFS-FT), respectively. It has been reported that the dehydration of Co(OAc)2 occurred at high pH or during heat treatment under neutral conditions.14 Therefore, it was likely that hydration of Co(OAc)2 proceeded during the preparation of Co/MFI to form Co(OH)2 and acetic acid. The loading of Co in Co/MFI(52N) increased gradually with increasing concentration of Co(OAc)2 measured by AA (Fig. 2(a)). At the same time, the Al content of MFI decreased and the Co loading increased (Fig. 2(b)). However, no shift of the diffraction in the XRD pattern was observed in the two 2θ regions before and after the loading of Co on MFI (Fig. S3†) when the 2θ angle was corrected with MgO as an internal standard. This fact indicated that the dealumination of framework Al hardly occurred. Instead, considering that MFI(52A) contains a large amount of extra framework Al (0.37 mmol g−1), which was larger than that of framework Al (0.25 mmol g−1), the extra framework Al species were probably removed during Co loading. The Co/Al ratio of Co/MFI exceeded 0.5 or 1.0 in many samples, suggesting that Co(OH)2 deposition occurred independently of the Al sites in the MFI (Fig. S4†). | ||
| Fig. 1 Co K-edge (a) XANES and (b) EXAFS-FT of Co/MFI(52N) (Co: 0.5 mmol g−1) and reference compounds measured at 298 K. Fourier transform range: 3–13 Å−1. | ||
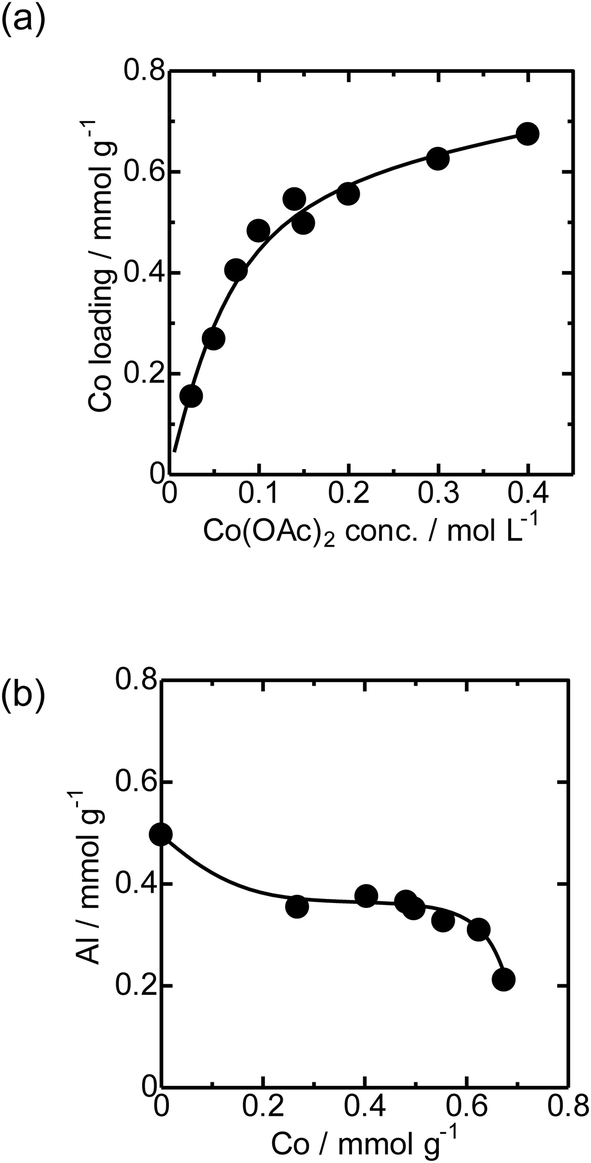 | ||
| Fig. 2 (a) Relationship between the Co loading of Co/MFI(52N) and concentration of Co(OAc)2. (b) Relationship between Co loading and the Al concentration of Co/MFI(52N). | ||
The loading of Co on MFI(52N) was 0.18 mmol g−1 when a 100 mmol L−1 Co(NO3)2 aqueous solution was used as the precursor for Co under acidic (non-hydration) conditions, which has been generally employed as the Co precursor. The loaded amount of Co (0.18 mmol g−1) was comparable to that of ion-exchangeable sites of 52N (0.25 mmol g−1) assuming that the ion-exchange occurred between NH4+ and Co(OAc)2 or Co(OH)2 to form Co(OH)+ species. On the other hand, the Co loading increased to 0.48 mmol g−1 when a 100 mmol L−1 Co(OAc)2 aqueous solution was used for the Co/MFI preparation, which exceeded the ion exchange capacity of 52N (0.25 mmol g−1). The higher Co loading with the use of the Co(OAc)2 precursor suggested that the formation of dispersed Co occurred through not only the ion exchange but also the reaction with defect sites with Co(OH)2.
Then the Co K-edge XAFS data were collected in an N2 flow while the temperature was increased from 303 to 823 K in order to monitor the local structure of Co species accompanied by heat treatments. The XANES data obtained under in situ conditions every 50 K are displayed in Fig. 3(a). A gradual decrease in the white line intensity of the Co K-edge XANES was observed up to 823 K as plotted in Fig. 3(b). The changes up to 573 K observed in XANES may be due to the dehydration of Co(OH)2 to form CoO, consistent with the literature.20 The change occurred progressively in the temperature range between 303 and 823 K, as shown in Fig. S5;† fitting the intermediate-stage XANES with a linear combination of the spectra obtained at room temperature and 823 K showed that the former component continuously decreased and the latter increased instead with increasing temperature. At the same time, the intensity of the pre-edge peak (7708 eV) increased with increasing temperature up to 823 K. This pre-edge peak is attributed to the 1s to 3d transition, which is normally dipole forbidden but allowed when the metal has tetrahedral coordination due to the mixing with p-character.21,22 Therefore, the change may be due to the transformation of the local structure from 6- to 4-coordinated Co centers. This could be caused by the removal of water molecules coordinated to the Co centers during the heat treatment and subsequent reaction of Co with OH groups or acid sites in MFI.
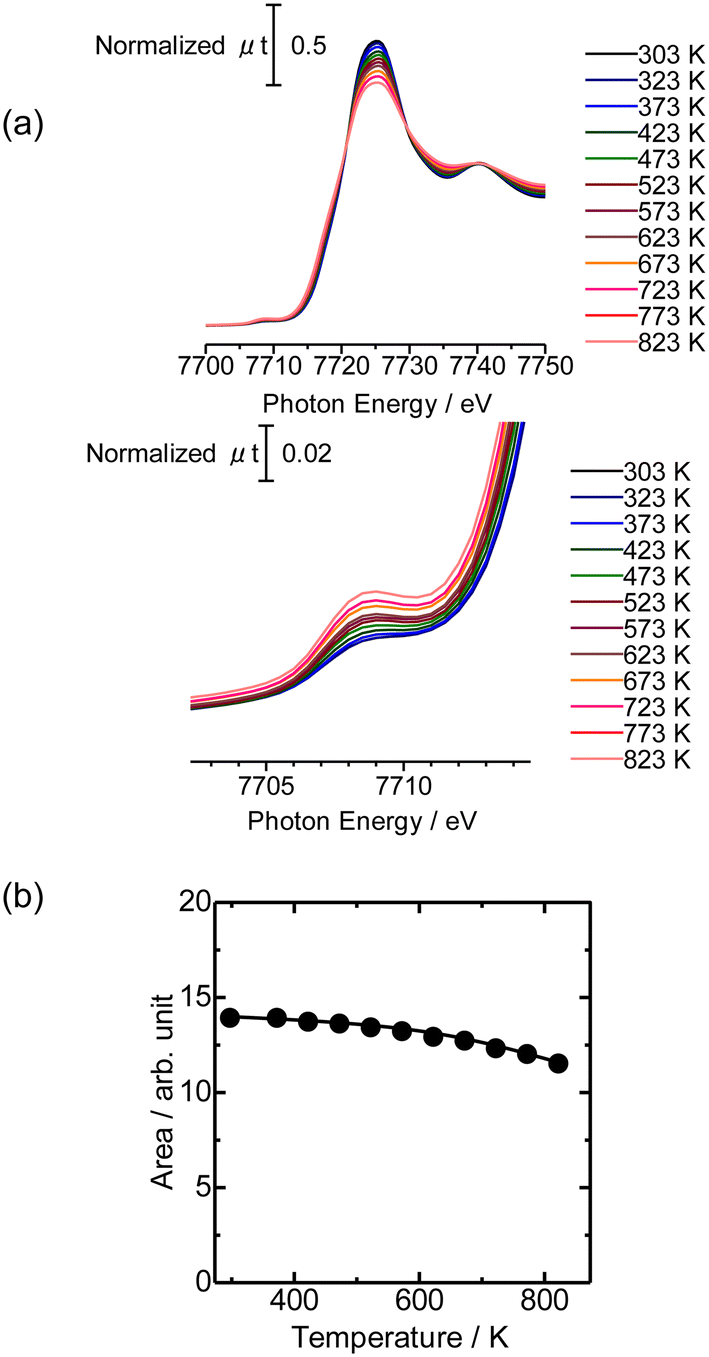 | ||
| Fig. 3 (a) Co K-edge XANES of Co/MFI(52N) (Co: 0.5 mmol g−1) measured under a N2 flow from 303 to 823 K measured under in situ conditions. (b) White line area in Co K-edge XANES of Co/MFI(52N) plotted as a function of the treatment temperature. | ||
Fig. 4(a) shows the Co K-edge EXAFS-FT of the sample in which Co was loaded on MFI(52N) using Co(OAc)2 as a precursor measured during heat treatment. Raw XAFS and k3χ(k) data are displayed in Fig. S6(a) and (b),† respectively. The Co loading of the measured sample was 0.5 mmol g−1. When these samples were heated from 573 to 823 K, the intensity of the Co–O–Co bond (2.8 Å) decreased while the Co–O peak remained almost intact. In the IR spectra measured at the same time, the intensity of isolated hydroxyl groups appearing at 3730 cm−1 remained unchanged, while the NH4+ stretching vibration peak at 3370 cm−1 decreased,23 indicating that the decomposition of NH4+ progressed during heat treatment. The intensity of the acidic hydroxyl group at 3590 cm−1 tended to decrease with increasing temperature, suggesting that ion exchange between H+ or NH4+ occurred with Co2+, but at the same time, the formation of acidic hydroxyl groups progressed as a result of the decomposition of NH4+ (Fig. S7†). The EXAFS oscillation is greatly reduced as the temperature rises because the Debye–Waller factor increases at high temperature. To prevent this temperature effect, XAFS data of the heat-treated samples were collected at 298 K (Fig. 4(b)). The corresponding k3χ(k) data are displayed in Fig. S8(a).† A significant decrease in the intensity of the second peak appearing at 2.8 Å (phase shift uncorrected) was observed in the temperature range of 573–773 K. A similar change was observed in the Co/MFI containing lower Co loading (0.3 mmol g−1, Fig. S8(b) and (c)†). The EXAFS of the as-prepared Co/MFI sample was successfully fitted after assuming Co–O and Co–(O)–Co bonds, while that of the 773 K-treated one was fitted after assuming Co–O and Co–(O)–Si bonds as listed in Table 2 and shown in Fig. S9.† The change observed after treatment at 773 K meant that the aggregated Co(OH)2 underwent self-dispersion through the interaction with MFI zeolite. Such dispersion was also found in Pd on MFI or MOR zeolites.24 Other examples are the formation and segregation of Ir–MgO, and Pt–MgO solid solutions observed in the temperature range between 773 and 1273 K.25,26 A similar change (decline) of the Co–(O)–Co bond appearing at 2.8 Å was observed in Co/SiO2 (Co loading: 0.5 mmol g−1) as displayed in the EXAFS-FT (Fig. S10(b)†); the peak was significantly reduced after the treatment at 773 K. This structural change suggested that the self-dispersion of Co occurred on Co/SiO2, similar to the case of Co/MFI.
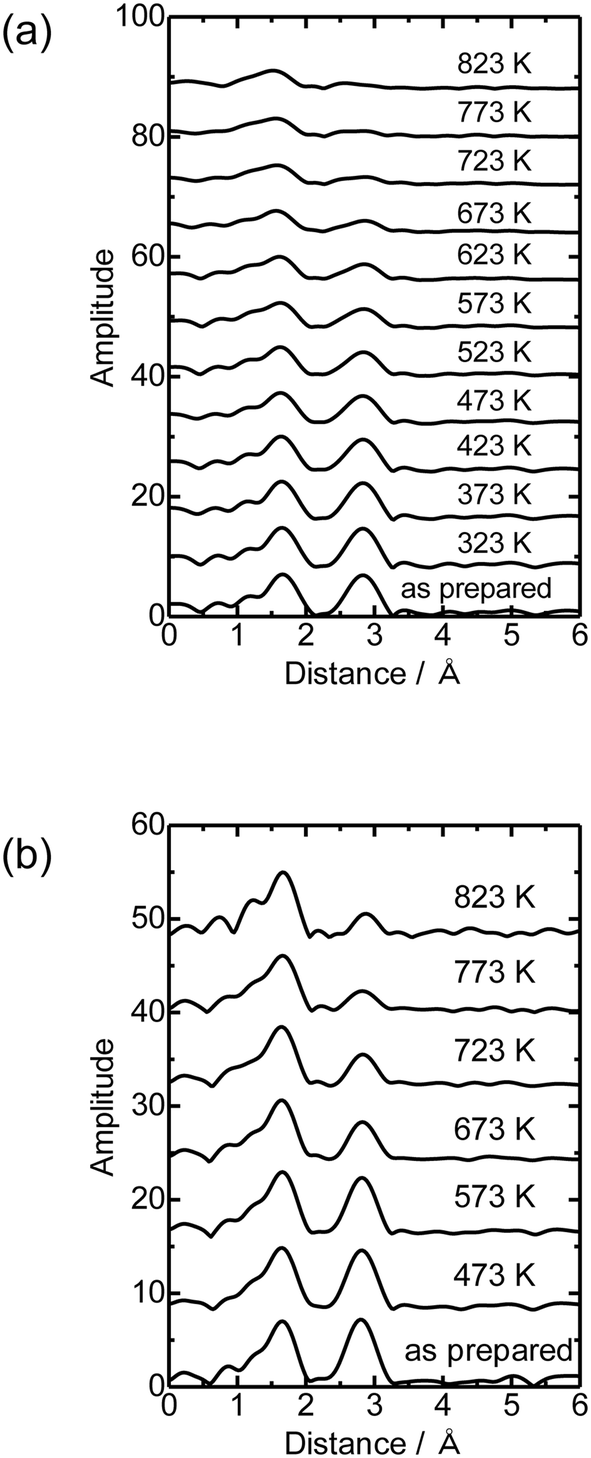 | ||
| Fig. 4 Co K-edge EXAFS-FT of Co/MFI(52N) (Co: 0.5 mmol g−1) (a) measured under a N2 flow from 323 to 823 K under in situ conditions, the as-prepared sample and (b) measured under ambient conditions. | ||
| Sample | Scatterer | CNa | R/Åb | ΔE0c/eV | DWd/Å | R f /% |
|---|---|---|---|---|---|---|
| a Coordination number. b Bond distance. c Difference in the origin of photoelectron energy between the reference and the sample. d Debye–Waller factor. e Residual factor. f Data of X-ray crystallography. g Fourier transform range: 3–13 Å−1. Fourier filtering range: 1.0–3.2 Å. | ||||||
| As-prepared Co/MFI(52N) | O | 5.2 ± 1.1 | 2.07 ± 0.02 | 1 ± 3 | 0.084 ± 0.027 | 0.6 |
| Co | 5.1 ± 1.3 | 3.14 ± 0.02 | 4 ± 2 | 0.086 ± 0.023 | ||
| Co/MFI(52N) treated at 773 K | O | 4.9 ± 0.8 | 2.08 ± 0.01 | 4 ± 2 | 0.087 ± 0.020 | 2.3 |
| Si | 2.0 ± 1.0 | 3.27 ± 0.03 | −4 ± 5 | 0.062 ± 0.067 | ||
| Co(OH)2f | O | (6) | (2.13) | |||
| Co | (6) | (3.20) | ||||
3.2. NH3 TPD and XRD patterns
Fig. 5 shows the NH3 TPD profile of Co/MFI(52N) with different Co loadings. The intensity of the h-peak appearing around 730 K was much lower than that of unloaded MFI(52N), while the change in the l-peak at ca. 500 K was small. The NH3 TPD profile did not change depending on the Co loading. The decline of the h-peak (730 K) observed after the loading of Co might be caused by the ion exchange of H+ with Co2+. | ||
| Fig. 5 NH3 TPD profile of unloaded MFI(52N) and Co/MFI(52N). The samples were treated in an N2 flow at 773 K prior to the TPD measurement. The numbers indicate the loading of Co (mmol g−1). | ||
NH3 TPD profiles of the employed MFI zeolites (NH4+ form) and those of untreated 822N and those treated with an aqueous solution of NaOH, followed by treatment with an NH4NO3 solution at 343 K are displayed in Fig. S11(a) and (b),† respectively. In the latter case, the intensity of the h-peak decreased in the MFI treated with an NaOH solution for 16 h. This means that the number of Al atoms in the MFI framework decreased after the NaOH treatment. In agreement with this, the XRD patterns revealed that partial collapse of the MFI crystals occurred after treatment with NaOH for 16 h as will be shown later. In general, the treatment of zeolite with NaOH solution resulted in the preferential dissolution of Si.27 Consistent with this, the total Al concentration in the MFI increased with treatment time, as shown in Fig. S12.† As for Al distribution, the amount of framework Al decreased, while that of extra-framework Al increased with increasing treatment time with an aqueous solution of NaOH (Table S1†).
XRD diffraction patterns of the as-prepared Co/MFI(52N) and those treated at 773 K as well as unloaded MFI(52N) are displayed in Fig. 6(a) and (b), respectively. For Co-loaded MFI(52N), no diffraction other than those ascribed to the MFI zeolite was observed after the loading of Co(OAc)2 (Fig. 6(a)), while the Co–(O)–Co bond appeared at 2.8 Å in the Co K-edge EXAFS as already shown in Fig. 4(a and b), indicating that Co existed on the MFI in an aggregated form. The contradictory data suggested that the crystallite size of Co(OH)2 was less than several nanometers. In agreement with the observation, no clear aggregate of Co(OH)2 was observed in the TEM image as will be noted later. Meanwhile, the overall intensity of the diffraction peaks decreased after the loading of Co. The extent was enhanced by increasing the Co loadings. This feature did not change after the thermal treatment in N2 at 773 K (Fig. 6(b)).
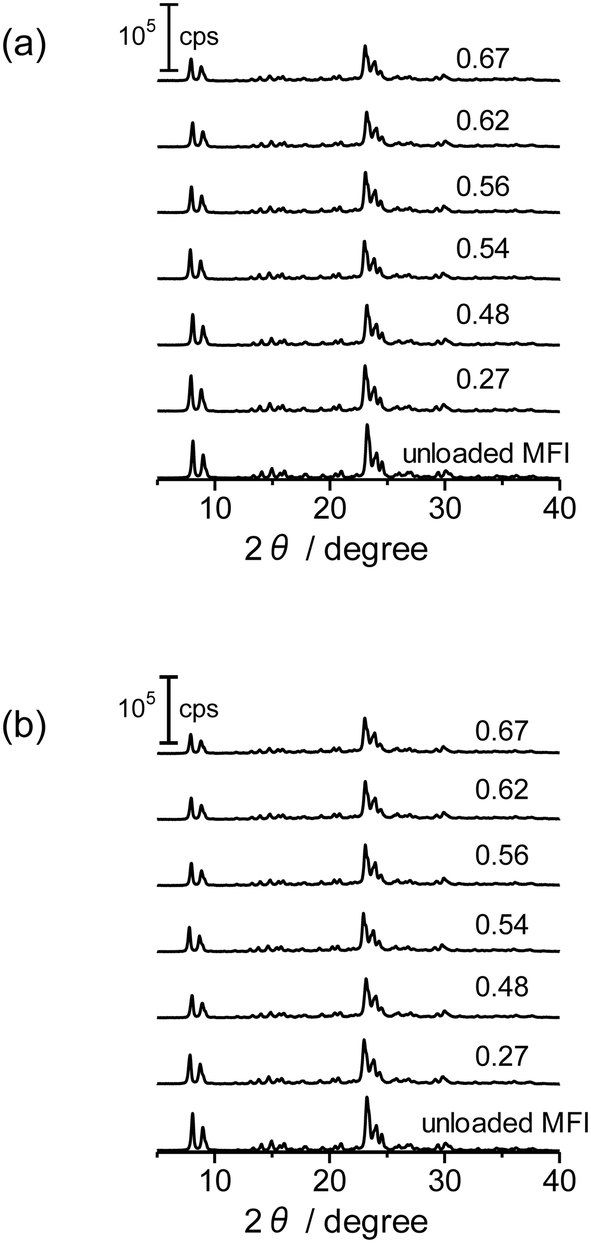 | ||
| Fig. 6 XRD patterns of Co-unloaded MFI(52N) and Co/MFI(52N). (a) The as-prepared and (b) 773 K-treated samples. The numbers indicate the Co loading (mmol g−1). | ||
XRD patterns of the as-received MFI(822N) and those treated with NaOH are displayed in Fig. S13.† The overall intensity of the diffraction declined accompanied by an increase in the concentration of the NaOH solutions, suggesting that the partial collapse of the MFI framework took place during the treatment with the NaOH solution and the extent of which was enhanced as the concentration of the NaOH solution increased.
3.3. N2 adsorption isotherms, SEM and TEM images
The N2 adsorption isotherms of the unloaded MFI(52N) and Co-loaded ones are displayed in Fig. S14(a).† The isotherms of the Co/MFI (0.4 mmol g−1) was identical to that of the Co-unloaded one. The specific surface areas derived from the isotherm are plotted as a function of Co loadings in Fig. 7. A gradual decrease in surface area was observed with increasing Co loading between 0.4 and 0.7 mmol g−1. The tendency was consistent with the XRD pattern; a decrease in diffraction intensity was observed with increasing Co loading (Fig. 6).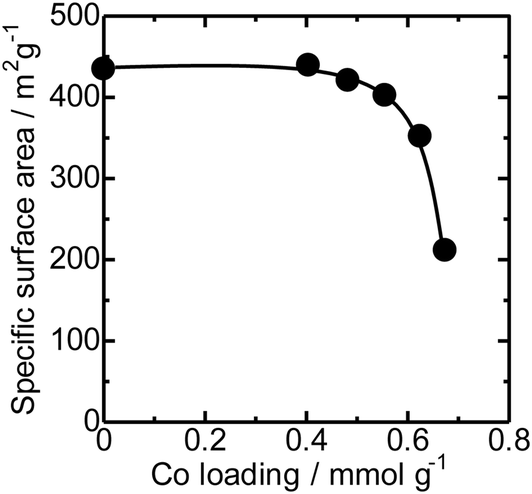 | ||
| Fig. 7 Specific surface area plotted as a function of Co loadings of Co/MFI(52N). | ||
FE-SEM images of the representative MFI(52N, 840N, 822N, and 822N-16h) employed as the support for Co are displayed in Fig. 8. The average crystal size of 840N was approximately several micrometers (Fig. 8(b)). The large crystal size was associated with the appearance of the N2 adsorption isotherm in that the isotherm of the samples was assignable to type I as shown in Fig. S14(b).† The size of the primary particle of 52N and 822N was much smaller than that of 840N; in the former case, the size was ca. 50 nm. Consistent with this SEM figure, a progressive increase in the nitrogen adsorption isotherm curve was observed at pressures above p/p0 = 0.01 (Fig. S14(b)†).
 | ||
| Fig. 8 FE-SEM images of (a) 52N, (b) 840N, (c) 822N, and (d) 822N-AT16h. | ||
No difference was found between TEM images of Co/MFI(52N) (as-prepared sample and those thermally treated at 823 K) and Co-unloaded MFI(52N) as shown in Fig. S15,† suggesting that the degree of the aggregation of the deposited Co(OH)2 was limited in the preparation step. This fact was consistent with the XRD patterns in that no diffraction assignable to the crystalline Co(OH)2 was observed (Fig. 6(a)).
3.4. Catalytic reaction
Fig. 9 shows the time-course change in the rate of toluene formation over Co loaded on different kinds of zeolites. A slight decrease in formation rate was observed in the highly active samples of Co/MFI, but the activity of the catalyst was relatively stable. It can be seen from Fig. 9(a) and (b) that the formation rate was dependent on the kinds of employed MFI zeolites. Co loaded on zeolites other than MFI was almost inactive in the reaction (Fig. 9(c)). The specific high activity of Co supported on MFI was also found with the catalyst using Co(NO3)2 as the precursor.4 The H+- and Na+-MFI have strong Brønsted and Lewis acid character, respectively.28,29 Therefore, we speculate that the strong adsorption of benzene on the Lewis sites of Co2+ promoted the reaction with methane, the presence of which was confirmed with IR spectra of adsorbed NH3.7 The rate of toluene formation at 3 h of time on stream from the beginning of the reaction in Co/MFI is plotted as a function of Co loading in Fig. 10(a). The toluene formation rate was almost proportional to the loading of Co up to 1.0 mmol g−1 except for the Co/MFI(822N-AT16h), in which excessive Co was loaded on the MFI support (Co loading: 1.5 mmol g−1). Since the formation rate of H-type MFI and Co-loaded samples of NH4-type MFI follow the same line, cationic species of MFI during catalyst preparation had no influence on the catalytic activity of Co. The linear relationship indicated that the catalytic activity of Co/MFI depended only on Co loading. The highest toluene formation rate (11.3 μmol gcat−1 min−1) was higher than that (6.2 μmol gcat−1 min−1) of Co/MFI(52N) (Co loading: 0.34 mmol g−1) prepared using Co(NO3)2 as the Co precursor with an impregnation method. The turnover frequency (TOF) value of Co/MFI prepared with Co(OAc)2 was distributed in the range of 0.58–0.85 h−1 with the exception of several samples (Fig. 10(b)). | ||
| Fig. 9 Time course change of the toluene formation rate over Co loaded on different kinds of zeolite supports: (a) NH4-form MFI, (b) H-form MFI, and (c) zeolites other than MFI. The Co/MFI catalysts were prepared using a 200 mmol L−1 Co(OAc)2 solution at 343 K for 4 h. The numbers in the parentheses indicate the loading of Co (mmol g−1). | ||
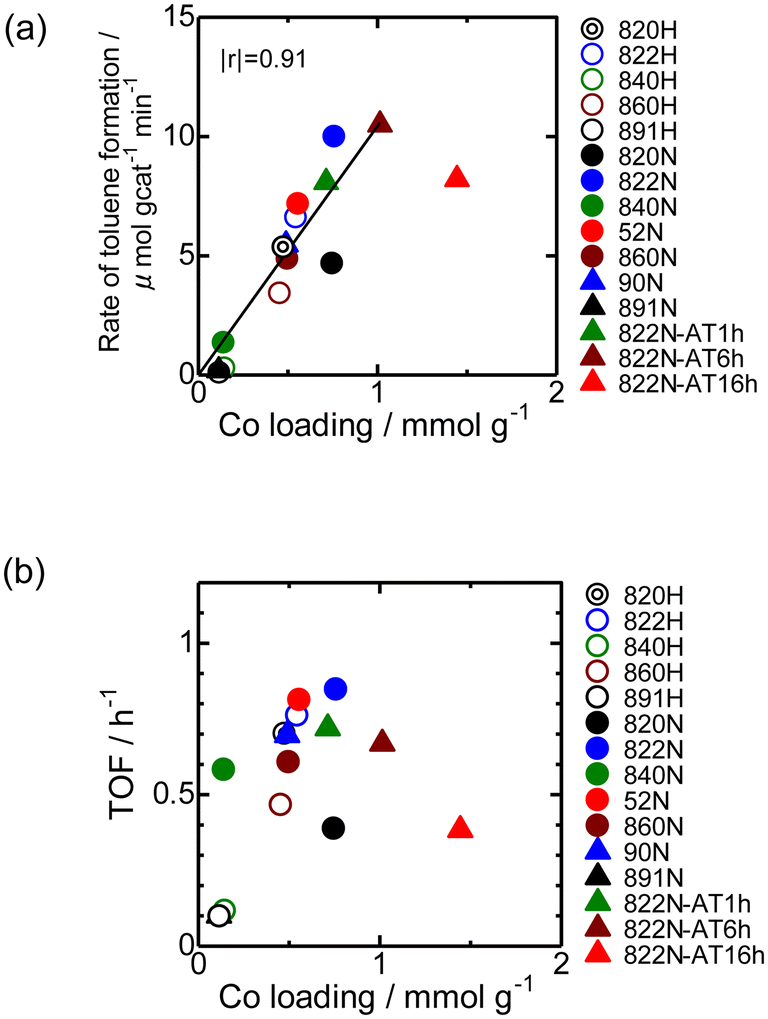 | ||
| Fig. 10 (a) Toluene formation rate at 3 h of time on stream plotted as a function of Co loading for Co/MFI catalysts. The Co/MFI catalysts were prepared using a Co(OAc)2 solution (200 mmol L−1) at 343 K for 4 h. The linear plot was made without the addition of the data of 822N-AT16h. (b) TOF (h−1) plotted as a function of Co loading. The TOF values were calculated based on the toluene formation rate at 3 h of time on stream from the beginning of the reaction and the Co loading. | ||
4. Conclusions
In this study, we found that Co(OH)2 precipitated on MFI when Co(OAc)2 was used as a precursor for Co/MFI. Further heat treatment led to self-dispersion of Co2+ species between 573 and 773 K. The toluene formation rate in the Co/MFI-catalyzed methane–benzene reaction was proportional to Co loading up to 1.0 mmol g−1, suggesting that the catalytic activity was simply determined by the Co loading. Traditionally, atomically dispersed active species using zeolite defect sites have been prepared using volatile precursors. However, as found here, more readily available metal salt (Co(OAc)2·4H2O) can be another precursor by making use of the self-dispersion of active species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JST CREST Grant Number JPMJCR17P1, Japan and JSPS KAKENHI Grant Number 22K04833, Japan. Technical support for operando IR-XAFS measurements was provided by Dr. K. K. Bando (AIST).References
- J. P. Van Hook, Catal. Rev.: Sci. Eng., 1980, 21, 1–51 CrossRef CAS.
- Y. Zhao, H. Sohn, B. Hu, J. Niklas, O. G. Poluektov, J. Tian, M. Delferro and A. S. Hock, ACS Omega, 2018, 3, 11117–11127 CrossRef CAS PubMed.
- T. Armaroli, M. Bevilacqua, M. Trombetta, F. Milella, A. d. G. Alejandre, J. Ramírez, B. Notari, R. J. Willey and G. Busca, Appl. Catal., A, 2001, 216, 59–71 CrossRef CAS.
- K. Nakamura, A. Okuda, K. Ohta, H. Matsubara, K. Okumura, K. Yamamoto, R. Itagaki, S. Suganuma, E. Tsuji and N. Katada, ChemCatChem, 2018, 10, 3806–3812 CrossRef CAS PubMed.
- K. Nakamura, K. Okumura, E. Tsuji, S. Suganuma and N. Katada, ChemCatChem, 2020, 12, 2333–2340 CrossRef CAS.
- Y. Yan, X. Zhang, J. Wei, M. Chen, J. Bi and Y. Bao, ACS Omega, 2022, 7, 17811–17821 CrossRef CAS PubMed.
- H. Matsubara, K. Yamamoto, E. Tsuji, K. Okumura, K. Nakamura, S. Suganuma and N. Katada, Microporous Mesoporous Mater., 2021, 310, 110649 CrossRef CAS.
- J. Dědeček, D. Kaucký and B. Wichterlová, Microporous Mesoporous Mater., 2000, 35, 483–494 CrossRef.
- P. Wu, T. Komatsu and T. Yashima, J. Phys. Chem., 1996, 100, 10316–10322 CrossRef CAS.
- K. Yamagishi, S. Namba and T. Yashima, Stud. Surf. Sci. Catal., 1989, 49, 459–467 CrossRef.
- R. Da Cruz, A. Mascarenhas and H. M. C. Andrade, Appl. Catal., B, 1998, 18, 223–231 CrossRef CAS.
- Y. Kishi, S. Shigemi, S. Doihara, M. Mostafa and K. Wase, Hydrometallurgy, 1998, 47, 325–338 CrossRef CAS.
- X. Wang, H. Chen and W. Sachtler, Appl. Catal., B, 2001, 29, 47–60 CrossRef CAS.
- M. Eguchi and A. Yazawa, Nippon Kogyo Kaishi, 1975, 91, 39–44 CAS , (in Japanese).
- M. Mhamdi, S. Khaddar-Zine and A. Ghorbel, Appl. Catal., A, 2009, 357, 42–50 CrossRef CAS.
- M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23–30 CrossRef CAS.
- L. Drozdová, R. Prins, J. Dědeček, Z. Sobalík and B. Wichterlová, J. Phys. Chem. B, 2002, 106, 2240–2248 CrossRef.
- H. Matsubara, E. Tsuji, Y. Moriwaki, K. Okumura, K. Yamamoto, K. Nakamura, S. Suganuma and N. Katada, Catal. Lett., 2019, 149, 2627–2635 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. Rehr and S. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- T. Jiang and D. Ellis, J. Mater. Res., 1996, 11, 2242–2256 CrossRef CAS.
- S. DeBeer George, P. Brant and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 667–674 CrossRef CAS PubMed.
- M. D. Wirt, I. Sagi, E. Chen, S. M. Frisbie, R. Lee and M. R. Chance, J. Am. Chem. Soc., 1991, 113, 5299–5304 CrossRef CAS.
- A. Zecchina, L. Marchese, S. Bordiga, C. Paze and E. Gianotti, J. Phys. Chem. B, 1997, 101, 10128–10135 CrossRef CAS.
- K. Okumura, J. Amano, N. Yasunobu and M. Niwa, J. Phys. Chem. B, 2000, 104, 1050–1057 CrossRef CAS.
- K. Okumura, H. Hoshi, H. Iiyoshi and H. Takaba, ACS Omega, 2022, 7, 27458–27468 CrossRef CAS PubMed.
- K. Okumura, H. Hoshi and H. Iiyoshi, Catal. Surv. Asia, 2023, 27, 95–106 CrossRef CAS.
- S. Abello, A. Bonilla and J. Perez-Ramirez, Appl. Catal., A, 2009, 364, 191–198 CrossRef CAS.
- N. Katada, H. Igi, J.-H. Kim and M. Niwa, J. Phys. Chem. B, 1997, 101, 5969–5977 CrossRef CAS.
- R. Yoshimoto, K. Hara, K. Okumura, N. Katada and M. Niwa, J. Phys. Chem. C, 2007, 111, 1474–1479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00305a |
| This journal is © The Royal Society of Chemistry 2023 |