
Enantioselective reductive allylic alkylation enabled by dual photoredox/palladium catalysis†
Sheng
Tang
a,
Hong-Hao
Zhang
*ab and
Shouyun
Yu
 *a
*a
aState Key Laboratory of Analytical Chemistry for Life Science, Jiangsu Key Laboratory of Advanced Organic Materials, Chemistry and Biomedicine Innovation Centre (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yushouyun@nju.edu.cn
bSchool of Petrochemical Engineering, Changzhou University, Changzhou 213164, China. E-mail: zhanghonghao@cczu.edu.cn
First published on 3rd January 2023
Abstract
A dual photoredox/palladium catalyzed regio- and enantioselective reductive cross-coupling of allylic acetates with tertiary/secondary alkyl bromides has been achieved, and Hantzsch ester is used as a homogeneous organic reductant. This straightforward protocol enables the stereoselective construction of C(sp3)–C(sp3) bonds under mild reaction conditions. Mechanistic studies suggest that this reaction involves radical pathways and a chiral Pd complex enables the control of the regio- and enantioselectivities.
Over the past decades, transition metal-catalyzed reductive cross-coupling reactions have emerged as one of the most powerful and straightforward protocols to construct carbon–carbon bonds efficiently.1 This procedure eliminates the need for preformed organometallic reagents in conventional transition metal-catalyzed cross-coupling reactions2 by coupling two electrophiles directly in the presence of a terminal reductant. Enantioselectivity remains a significant challenge in this field.3 While considerable efforts have been devoted during the last decade, asymmetric reductive cross-coupling reactions are mainly focused on C(sp2)–C(sp3) couplings (Scheme 1(a), top).3 The development of enantioselective reductive C(sp3)–C(sp3) cross-couplings has been hampered historically by deleterious side-reactions, such as β-H elimination or homocoupling of C(sp3)-electrophiles (Scheme 1(a), bottom).4–6 This is particularly true for the sterically hindered C(sp3) electrophiles, such as tertiary alkyl halides. To meet these challenges, novel reductive and catalytic systems would be highly desirable.7
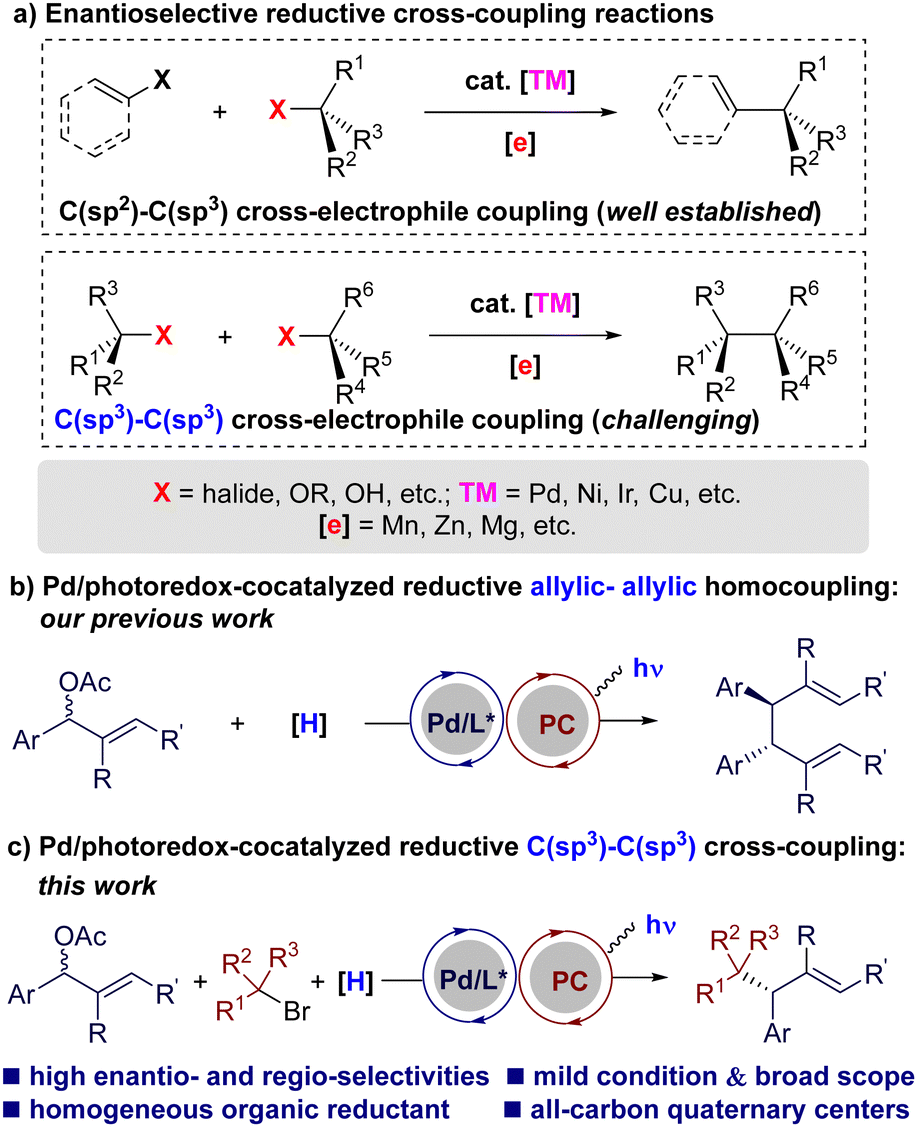 | ||
| Scheme 1 Enantioselective reductive cross-coupling reactions. | ||
Metallaphotoredox catalysis, the merger of photoredox catalysis with transition metal catalysis, has received considerable attention recently.8 It has been applied to the area of reductive reactions, enabling the coupling of electrophiles with homogeneous organic reductants under mild conditions.9 Despite advances, enantioselective versions of metallaphotoredox-catalyzed reductive couplings are still limited.10,11 We recently achieved a photoredox/Pd-cocatalyzed regio-, diastereo-, and enantioselective reductive homocoupling of allylic acetates (Scheme 1(b)).12 Encouraged by the success of the allylic-allylic homocoupling, as well as our lasting research interest in enantioselective palladium metallaphotoredox catalysis,13 we aim to realize the enantioselective reductive C(sp3)–C(sp3) cross-coupling of allylic acetates with tertiary alkyl halides using this state of the art synthetic technology (Scheme 1(c)).
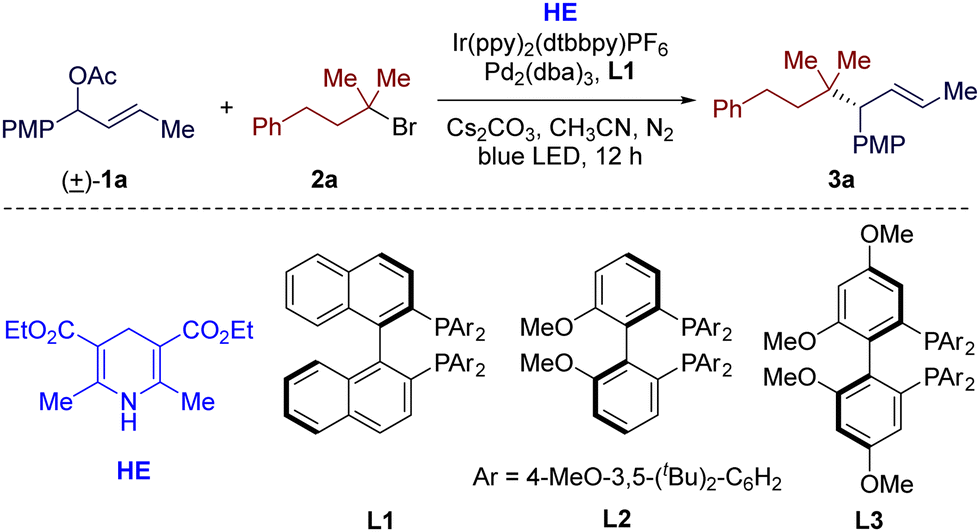
We began our investigations into this reductive cross-coupling protocol using racemic allylic acetate 1a and tertiary alkyl bromide 2a as the model substrates. After a comprehensive evaluation of the reaction parameters (see ESI† for details), the optimal conditions were established. When a solution of 1a (1.0 equiv.), 2a (3.0 equiv.), Hantzsch ester (HE, 2.0 equiv.) and Cs2CO3 (2.0 equiv.) in CH3CN was irradiated by a 45 W blue LED at room temperature for 12 h in the presence of photocatalyst Ir(ppy)2(dtbbpy)PF6 (2 mol%) and Pd2(dba)3 (2.5 mol%)/(R)-DTBM-BINAP (L1, 6 mol%) (Table 1, entry 1), the reductive cross-coupling reaction was achieved and the desired product (3a) was afforded in a good yield (75% GC yield and 70% isolated yield) and excellent regio- and enantioselectivities (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 rr, 96% ee). Replacing the chiral diphosphine ligand L1 with MeO-BIPHEP (L2) or GARPHOS (L3), the optimal ligands of our previous allylic alkylation reactions,12,13 also resulted in excellent regio- and enantioselectivities, but significantly lower yields due to serious homocoupling of allylic acetate 1a (entries 2 and 3). Several other iridium-based photocatalysts, such as fac-Ir(ppy)3 and Ir(dFCF3ppy)2(dtbbpy)PF6, were employed, but were not superior to Ir(ppy)2(dtbbpy)PF6 (entries 4 and 5). No reaction occurred in the absence of reductant HE (entry 5). The choice of homogeneous organic reductants also turned out to be crucial for this transformation, as replacing HE with DIPEA or TEA led to a significant drop in the yield (entries 7 and 8). The increase of concentration had negative effects on yield of this reaction (entry 9). Control experiments verified the necessity of visible light and a photocatalyst (entry 10). The palladium catalyst and the chiral ligand were also essential to this reaction (entry 11).
:5 rr, 96% ee). Replacing the chiral diphosphine ligand L1 with MeO-BIPHEP (L2) or GARPHOS (L3), the optimal ligands of our previous allylic alkylation reactions,12,13 also resulted in excellent regio- and enantioselectivities, but significantly lower yields due to serious homocoupling of allylic acetate 1a (entries 2 and 3). Several other iridium-based photocatalysts, such as fac-Ir(ppy)3 and Ir(dFCF3ppy)2(dtbbpy)PF6, were employed, but were not superior to Ir(ppy)2(dtbbpy)PF6 (entries 4 and 5). No reaction occurred in the absence of reductant HE (entry 5). The choice of homogeneous organic reductants also turned out to be crucial for this transformation, as replacing HE with DIPEA or TEA led to a significant drop in the yield (entries 7 and 8). The increase of concentration had negative effects on yield of this reaction (entry 9). Control experiments verified the necessity of visible light and a photocatalyst (entry 10). The palladium catalyst and the chiral ligand were also essential to this reaction (entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Variation of reaction conditions | Yieldb/% | eec/% | rrb |
| a Reaction conditions: a solution of 1a (0.1 mmol), 2a (0.3 mmol), HE (0.2 mmol), Cs2CO3 (0.2 mmol), Pd2(dba)3 (2.5 mol%), ligand (6 mol%), and Ir(ppy)2(dtbbpy)PF6 (2 mol%) in CH3CN (4.0 mL) was irradiated by a 45 W blue LED for 12 h. b Yields and regiomeric ratios (rr) were determined by GC analysis. c Enantiomeric excess (ee) values were determined by HPLC on a chiral stationary phase. d Isolated yield. PMP = para-methoxyphenyl. DIPEA = diisopropylethylamine. TEA = triethylamine. | ||||
| 1 | None | 75d (70) | 96 | >95:5 |
| 2 | L2 instead of L1 | 40 | 98 | 94:6 |
| 3 | L3 instead of L1 | 36 | 98 | 95:5 |
| 4 | fac-Ir(ppy)3 | 0 | — | — |
| 5 | Ir(dFCF3ppy)2(dtbbpy)PF6 | 0 | — | — |
| 6 | w/o HE | 0 | — | — |
| 7 | DIPEA in instead of HE | 44 | 92 | 92:8 |
| 8 | TEA in instead of HE | 35 | 94 | 92:8 |
| 9 | CH3CN (2 mL) was used | 67 | 96 | >95:5 |
| 10 | w/o light or Ir(ppy)2(dtbbpy)PF6 | 0 | — | — |
| 11 | w/o Pd2(dba)3 or L1 | 0 | — | — |
With the optimal conditions in hand, we investigated the scope and limitations of this dual photoredox/Pd-catalyzed enantioselective reductive cross-coupling. As shown in Scheme 2(a), the disubstituted allylic acetates 1 were first examined. Under the standard conditions, various allylic acetates 1 were alkylated with tertiary alkyl bromide s(2a or 2b), offering the corresponding cross-coupling products 3a–3l in moderate to high yields (34–70%), good enantioselectivities (64–96% ee), and excellent regioselectivities (>95:5 rr). Electron-donating or electron-withdrawing substituents at different positions of the phenyl moiety were all tolerated in this reaction. In comparison, substrates with electron-donating groups could give higher yields (3avs.3d and 3e). Polycyclic aromatic allylic acetate 1g was also applicable for this reaction, but with poor yield. Besides methyl, the alkyl group of the allylic acetates could be ethyl (3h), propyl (3i), and even longer pentyl (3j) groups. Cyclic allyl acetates could also be applicable to this transformation and provided the corresponding products 3k and 3l in moderate yields (54% and 45% yield, respectively) and enantioselectivities (68% and 64% ee, respectively). When monosubstituted allyl acetate 1m was alkylated with 2a under the established conditions, the desired product 3m was obtained (32% yield and 88% ee), together with significant homocoupling and over-reduction side products, which could be responsible for the poor yield. Allylic gem-alkyl, aryl-disubstituted acetate 1n was also applicable, affording the desired product 3n with all-carbon quaternary stereogenic centres with good enantioselectivity (78% ee) and 30% yield. We next investigated the scope of tertiary alkyl bromides. As indicated in Scheme 2(b), this protocol was amenable to tertiary alkyl bromides with phenyl groups bearing either electron-donating or electron-withdrawing substituents at different positions, affording the desired cross-coupling products (3o–3v) in 48–68% yields and with excellent enantio- and regioselectivities (90–96% ee, >95:5 rr). Non-aryl substituted tertiary alkyl bromides were compatible with the coupling conditions to give the desired products 3w–3y with moderate yields (50–64%) and good enantioselectivities (90–96% ee). The allylic acetate could be alkylated with the bulkier tertiary alkyl bromide 2m to give 3z. Cyclic tertiary bromides also undertook this transformation, but the yield of 3aa was only 25%. The reductive cross-coupling products with secondary alkyl bromides were also feasible, but at a lower temperature (0 °C). The desired reductive cross-coupling products 3ab and 3ac were obtained in moderate yields (40% and 42%, respectively), and excellent regio- and enantioselectivities (>95:5 rr, 94% and 88% ee, respectively). All attempts to couple with primary bromides failed (for more details, see Fig. S7 in ESI†).
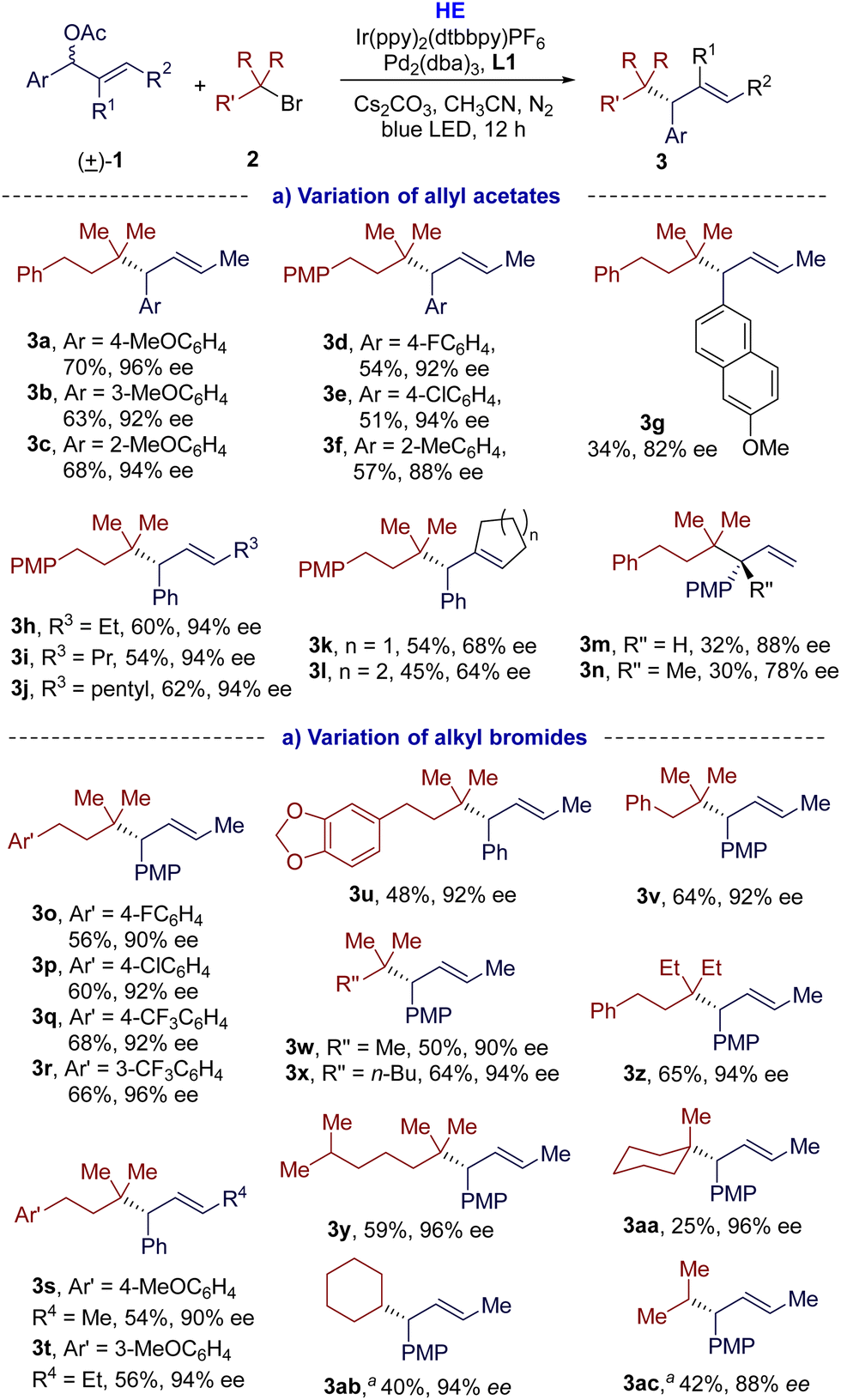 | ||
| Scheme 2 Substrate scope and limitation. aThe reaction was run at 0 °C. | ||
To gain additional insight into the reaction, we performed several control experiments. Under the standard conditions, allyl acetates 1o, 1o′ (isomer of 1o), and an equimolar mixture of them gave similar results, respectively (Scheme 3(a), eqn (1)). When optically pure allyl acetate (S)-1o′ was used as a substrate, the stereochemistry of the product is controlled by the chiral ligand (Scheme 3(a), eqn (2)). These results demonstrate that this reaction goes through a π-allylpalladium intermediate. When the radical trapping reagent TEMPO was introduced to the reaction mixture, the desired reaction could be terminated completely and the TEMPO-trapped product 4 could be observed by high-resolution mass spectrometric analysis (Scheme 3(a), eqn (3)). These phenomena, together with the results of the control experiments in Table 1 (entry 10), suggest that this reaction undergoes a photoredox catalysis process and an alkyl radical might be its key intermediate. Stern–Volmer quenching experiments indicate that the excited photocatalyst is quenched by HE (Fig. S5 in ESI†).
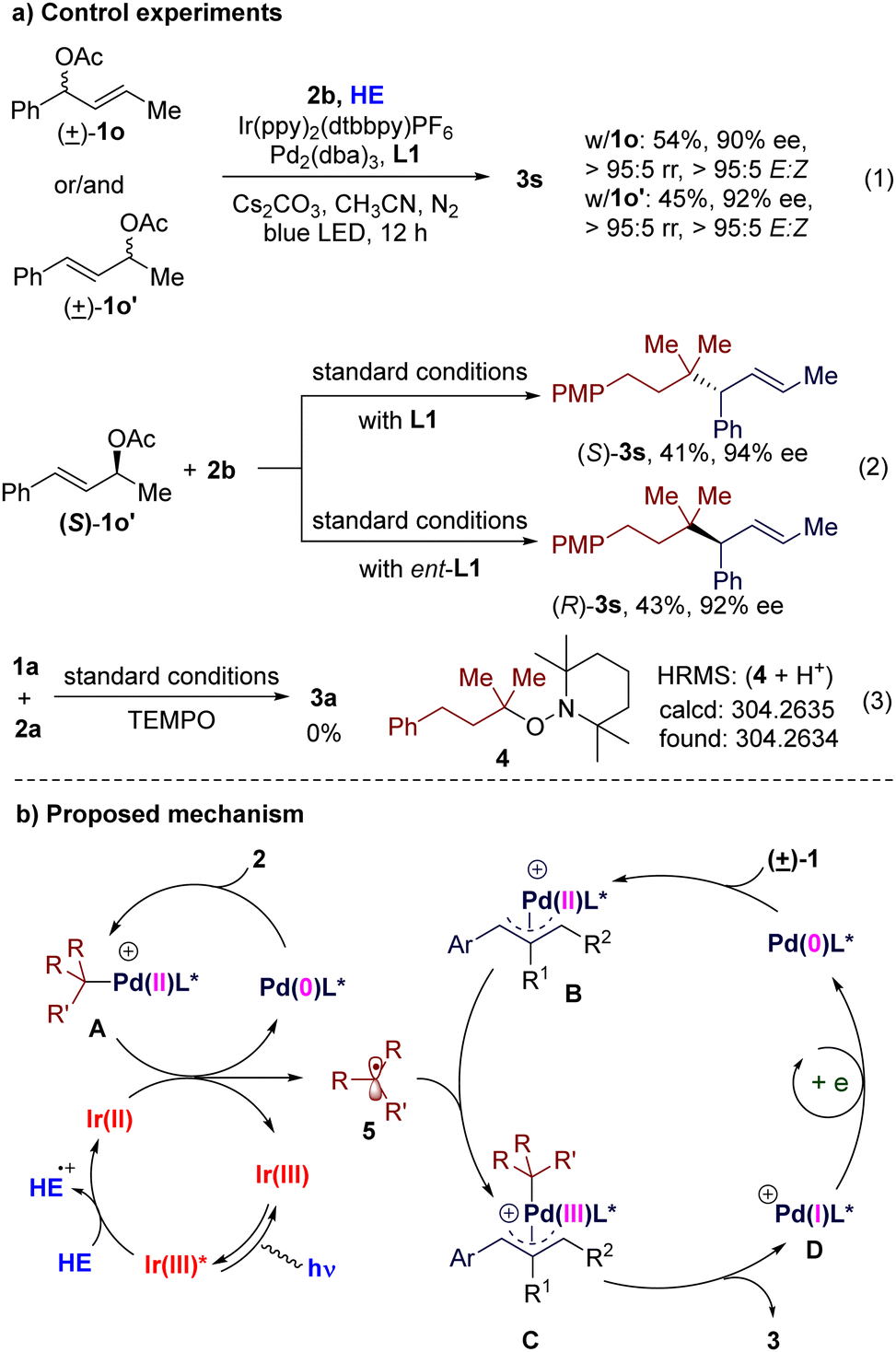 | ||
| Scheme 3 Mechanism studies and proposal. | ||
Based on these results and previously published works on photoredox/Pd co-catalysis,12–14 a plausible mechanism is proposed (Scheme 3(b)). An excited-state photocatalyst Ir(III)* is formed by the absorption of visible light, which is reductively quenched by HE to give a low-valent Ir(II) complex. Oxidative addition of alkyl bromide 2 to Pd(0) gives alkyl-Pd(II) species A. The β-H elimination side product of A could be detected by GC-MS, which supports the existence of A. Reduction of A by the Ir(II) complex regenerates Ir(III) and Pd(0), together with an alkyl radical 5. Meanwhile, Pd(0) oxidatively adds to the allylic acetate 1 to give a Pd-π-allyl species B. Then B traps the alkyl radical 5 to generate the Pd(III) complex C. Reductive elimination of C gives the allylic alkylation product 3 and a Pd(I) species D. Finally single-electron reduction of D by the Ir(II) complex or the HE radical cation regenerates Pd(0). Alternatively, an excited-state palladium catalysis pathway cannot be ruled out completely at this stage (for more detailed discussions, see Fig. S6 in ESI†).15 However, the necessity of a photocatalyst suggests that the cooperative photoredox and palladium catalysis pathway might be the major route.
In summary, we have described a highly regio- and enantioselective reductive C(sp3)–C(sp3) cross-coupling of allylic acetates with tertiary/secondary alkyl bromides through cooperative palladium and photoredox catalysis, and Hantzsch ester is used as the reductant. This dual catalytic protocol allows a direct and stereoselective construction of C(sp3)–C(sp3) bonds enantioselectively. This mechanistically novel strategy expands the scope of the traditional transition metal-catalyzed asymmetric allylic alkylation reactions and enantioselective reductive cross-coupling reactions.
We thank the National Natural Science Foundation of China (21971110, 21732003, and 22001120), and the Natural Science Foundation of Jiangsu Province (BK20200297) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews on reductive couplings, see: (a) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem. – Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (b) T. Moragas, A. Correa and R. Martin, Chem. – Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed; (d) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (e) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 43 CrossRef PubMed; (f) X. Pang, P.-F. Su and X.-Z. Shu, Acc. Chem. Res., 2022, 55, 2491–2509 CrossRef CAS PubMed; (g) M. J. Goldfogel, L. Huang and D. J. Weix, Nickel Catalysis in Organic Synthesis, 2020, pp.183–222 Search PubMed.
- For selected reviews on conventional transition metal-catalyzed cross-coupling reactions, see: (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- For selected reviews on stereoselective reductive couplings, see: (a) E. L. Lucas and E. R. Jarvo, Nat. Rev. Chem., 2017, 1, 0065 CrossRef; (b) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed.
- For selected reviews on C(sp3)–C(sp3) couplings, see: (a) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (b) F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 8347–8349 CrossRef CAS PubMed; (c) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (d) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC; (e) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- For selected examples of reductive C(sp3)–C(sp3) cross-couplings, see: (a) X. Qian, A. Auffrant, A. Felouat and C. Gosmini, Angew. Chem., Int. Ed., 2011, 50, 10402–10405 CrossRef CAS PubMed; (b) Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Chem. – Eur. J., 2012, 18, 808–812 CrossRef CAS PubMed; (c) H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022–4029 RSC; (d) J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem. – Eur. J., 2014, 20, 15334–15338 CrossRef CAS PubMed; (e) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef CAS PubMed; (f) A. B. Sanford, T. A. Thane, T. M. McGinnis, P.-P. Chen, X. Hong and E. R. Jarvo, J. Am. Chem. Soc., 2020, 142, 5017–5023 CrossRef CAS PubMed.
- For selected examples of stereoselective reductive C(sp3)–C(sp3) cross-couplings, see: (a) E. J. Tollefson, L. W. Erickson and E. R. Jarvo, J. Am. Chem. Soc., 2015, 137, 9760–9763 CrossRef CAS PubMed; (b) L. W. Erickson, E. L. Lucas, E. J. Tollefson and E. R. Jarvo, J. Am. Chem. Soc., 2016, 138, 14006–14011 CrossRef CAS PubMed; (c) H. Fu, J. Cao, T. Qiao, Y. Qi, S. J. Charnock, S. Garfinkle and T. K. Hyster, Nature, 2022, 610, 302–307 CrossRef CAS PubMed.
- For selected examples of reductive cross-couplings with tertiary alkyl halides, see: (a) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS PubMed; (b) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed; (c) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490–14497 CrossRef CAS PubMed.
- For selected reviews on metallaphotoredox catalysis, see: (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (c) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- For selected examples of metallaphotoredox-catalyzed reductive couplings, see: (a) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012–4015 CrossRef CAS PubMed; (b) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (c) R. T. Smith, X. Zhang, J. A. Rincón, J. Agejas, C. Mateos, M. Barberis, S. García-Cerrada, O. de Frutos and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 17433–17438 CrossRef CAS PubMed; (d) F. Song, F. Wang, L. Guo, X. Feng, Y. Zhang and L. Chu, Angew. Chem., Int. Ed., 2020, 59, 177–181 CrossRef CAS PubMed; (e) T. J. Steiman, J. Liu, A. Mengiste and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7598–7605 CrossRef CAS PubMed.
- For selected reviews on the enantioselective metallaphotoredox catalysis, see: (a) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, Sci. China: Chem., 2020, 63, 637–647 CrossRef CAS; (b) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS PubMed; (c) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC.
- For selected examples of metallaphotoredox-catalyzed enantioselective reductive couplings, see (a) H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172–5177 CrossRef CAS PubMed; (b) S. H. Lau, M. A. Borden, T. J. Steiman, L. S. Wang, M. Parasram and A. G. Doyle, J. Am. Chem. Soc., 2021, 143, 15873–15881 CrossRef CAS PubMed; (c) P. Zheng, P. Zhou, D. Wang, W. Xu, H. Wang and T. Xu, Nat. Commun., 2021, 12, 1646 CrossRef CAS PubMed; (d) H. Wang, P. Zheng, X. Wu, Y. Li and T. Xu, J. Am. Chem. Soc., 2022, 144, 3989–3997 CrossRef CAS PubMed.
- H.-H. Zhang, M. Tang, J.-J. Zhao, C. Song and S. Yu, J. Am. Chem. Soc., 2021, 143, 12836–12846 CrossRef CAS PubMed.
- (a) H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef CAS PubMed; (b) H.-H. Zhang, J.-J. Zhao and S. Yu, ACS Catal., 2020, 10, 4710–4716 CrossRef CAS; (c) X. Shen, L. Qian and S. Yu, Sci. China: Chem., 2020, 63, 687–691 CrossRef CAS; (d) C. Song, H.-H. Zhang and S. Yu, ACS Catal., 2022, 12, 1428–1432 CrossRef CAS; (e) X. Bai, L. Qian, H.-H. Zhang and S. Yu, Org. Lett., 2021, 23, 8322–8326 CrossRef CAS PubMed.
- For selected examples of photoredox/Pd co-catalysis, see: (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed; (b) S. B. Lang, K. M. O’Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606–13609 CrossRef CAS PubMed; (c) J. Xuan, T.-T. Zeng, Z.-J. Feng, Q.-H. Deng, J.-R. Chen, L.-Q. Lu, W.-J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2015, 54, 1625–1628 CrossRef CAS PubMed; (d) C. Zhou, P. Li, X. Zhu and L. Wang, Org. Lett., 2015, 17, 6198–6201 CrossRef CAS PubMed; (e) K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467–9470 CrossRef CAS PubMed; (f) J. Zheng, A. Nikbakht and B. Breit, ACS Catal., 2021, 11, 3343–3350 CrossRef CAS.
- For selected reviews on excited-state palladium catalysis, see: (a) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed; (b) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (c) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06705f |
| This journal is © The Royal Society of Chemistry 2023 |