
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese(III) porphyrin-catalyzed regioselective dual functionalization of C(sp3)–H bonds: the transformation of arylalkanes to 1,4-diketones†
Jakub
Sukiennik‡
a,
Audrey
Pranowo‡
a,
Sylwester
Domański
b and
Karolina
Hurej
 *a
*a
aDepartment of Chemistry, University of Wroclaw, F. Joliot-Curie 14, Wrocław 50383, Poland. E-mail: karolina.hurej@uwr.edu.pl
bPolbionica, L. Rydygiera 8, Warszawa 01793, Poland
First published on 3rd January 2023
Abstract
The first, direct way from arylalkanes to 1,4-dicarbonyl compounds has been shown. It makes obtaining these useful products more accessible and cheaper. Our method is based on a one-pot reaction with excellent regioselectivity, mild conditions, and water as the main solvent. A plausible reaction mechanism has also been proposed.
Regioselective C–H functionalization represents a powerful approach to the synthesis of complex molecules.1 Previous studies have introduced metalloporphyrins as effective catalysts in such reactions2 and as more general valuable tools in C–H and C–C activation.3,4 The activity and selectivity of these complexes strongly depend on the central metal and stereo-electronic features of porphyrin ligands. Under the current protocols, specific substrates bearing one inherently most reactive C–H bond react with high selectivity, while others typically react unselectively, leading to a mixture of products.5 The control of regioselectivity for such unactivated substrates remains challenging due to the relatively low reactivity of current catalysts, which require high temperatures.1
Until now, the vast majority of the functionalizations of C–H bonds have been achieved thanks to the use of popular catalysts based on heavier transition metals such as rhodium, iridium, palladium, and ruthenium. The preference for the use of heavier metals over the related congeners of the fourth period of the elemental system, emerges probably due to the relatively stronger M–H and M–C bonds they form. Unfortunately, the 4d and 5d noble metals are typically toxic and expensive. Therefore, there is a growing demand for the development of methods based on the use of more environmentally friendly 3d transition metals, including manganese.5–13
One of the interesting classes of compounds, to which so far no direct synthetic way, when starting from alkanes/arylalkanes, has been reported are 1,4-dicarbonyl compounds. 1,4-Diketones are common synthetic precursors for bioactive 5-membered heterocycles such as furans,14 thiophenes,15 pyrroles16 and cyclopentenones.17,18
Due to the importance of 1,4-carbonyl compounds, many synthetic routes have been developed. Some of the examples include the Stetter reaction of aldehydes and α,β-unsaturated carbonyl derivatives19–21 and its Sila-Stetter variant22 as well as coupling between aromatic enones and aryl acyl chlorides,23 Michael addition of β-dicarbonyl nucleophiles to α,β-unsaturated compound, oxidative homocoupling of 1,3-dicarbonyl compounds,24 and oxidative cross-coupling of silyl enol ethers25,26 or vinylarenes with ketones.27 Another promising approach to obtaining 1,4-diketones is the multicomponent reaction of alkylglyoxals, 1,3-dicarbonyl compounds, and nucleophiles, which Fu and Gu recently reported.28 Some other interesting synthetic protocols are photochemical or electrochemical oxidation of ketones29–31 as well as oxidative cleavage of alkenes by oxygen and non-heme manganese catalyst.10
Despite remarkable progress in elaborating many routes for the synthesis of 1,4-diketones, most of these methods can be characterized by limitations related to the multi-step procedures, low product yield, expensive catalysts, or harsh reaction conditions. Therefore, we have developed a complete methodology for one-pot oxidation of arylalkanes to 1,4-diketones under mild conditions with high site-selectivity, using water as the main solvent and the most basic manganese(III) porphyrin complex as a catalyst.
We have started the optimization process by screening ruthenium catalysts based on the porphyrinoid skeleton due to their high reactivity in C(sp3)–H bond hydroxylation and oxidation reactions (Fig. 1 and Table 1).2,32,33 First, we checked the ruthenium(II) complex of tetraphenylporphyrin and its analogous derivative from the carbaporphyrinoid group – ruthenium(II) meta-benziporphyrin. In both cases, the substrate conversion was essentially similar in the range of 20%, but the cat. 1 oxidized the substrate mainly to a α-monoketone, while cat. 2 to a benzyl alcohol derivative.
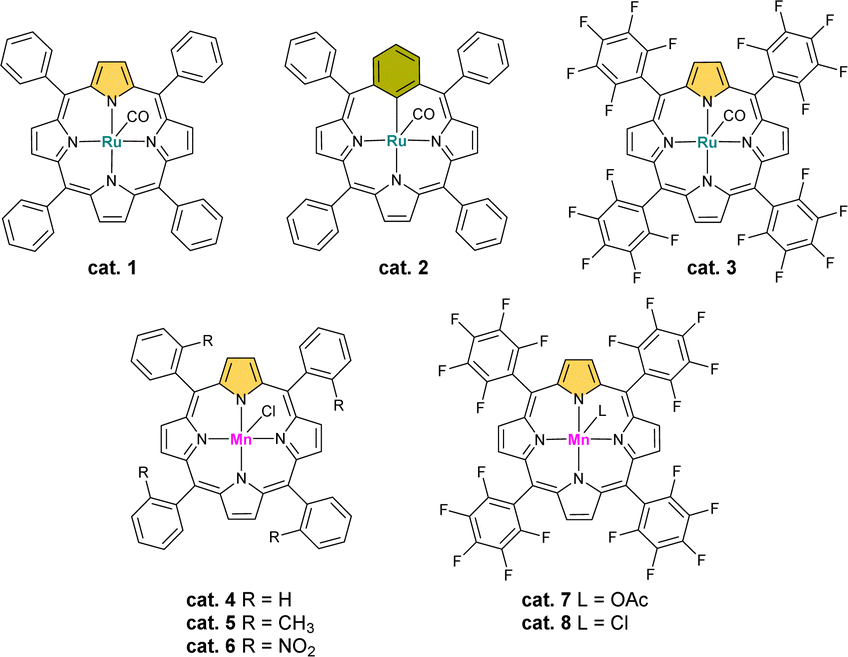 | ||
| Fig. 1 Tested ruthenium(II) and manganese(III) porphyrin catalysts. | ||
|
|
|||
|---|---|---|---|
| Lp. | Conditions | α-Monoketone | Diketone |
| 1. | Standard | 26% | 54% |
| 2. | w/o catalyst | Traces | Traces |
| 3. | cat. 1–2/cat. 4–6 instead of cat. 8 | Several percent | n.d. |
| 4. | cat. 3 instead of cat. 8 | 24% | Traces |
| 5. | cat. 7 instead of cat. 8 | 26% | 54% |
| 6. | w/o oxidant 1 (PIDA) | n.d. | n.d. |
| 7. | w/o oxidant 2 (Oxone®) | 53% | 42% |
| 8. | MnCl2(H2O)4 | n.d. | n.d. |
Because of the crucial role of the variable nature of meso-aryl substituents and the fact that electron-poor porphyrin derivatives are often more useful in C–H oxidation reactions,2 we also tested cat. 3, which gave trace amounts of the expected product. Then we decided to replace the metal ion with the target manganese(III) and tested the model reactions with the cat. 4–8. The last two have been the most promising, showing the 1,4-diketone as the main product. Changing the axial ligand did not affected the efficiency of the reaction.
Next, we examined the oxidants for C(sp3)–H bond oxidation of 1-(4-bromophenyl)pentane. We have noticed that the efficiency of the reaction depends on substituents on the oxidant's phenyl ring and the pKa of released acid, which is created during the final transformation of the oxidant (Table S6, ESI†). Nevertheless, we have obtained the highest yield by oxidizing with (diacetoxyiodo)benzene (PIDA).
The temperature also plays an important role. Cooling and heating the reaction mixture above and below 25 °C negatively affected the product yield. This occurs due to a α-monoketone formation at low temperatures, which could not convert to the final product. In contrast, higher temperatures favored a side reactivity (e.g., oxidative cleavage of C(1)–C(2) bond, giving benzoic acid derivatives). Despite the quantitative conversion of the substrate, the total amount of α-monoketone and diketone decreased with increasing temperature (see Table S3, ESI†). We have observed the same trend by increasing the reaction time above 24 hours (see Table S4, ESI†).
Another crucial factor is the proper amount of acetonitrile. Increasing the amount of solvent to 250 μL resulted in a nearly four-fold decrease in yield (see Table S5, ESI†). In addition, while we added 1000 μl of acetonitrile, the product was practically unobservable. An opposite effect was noted with water, added as the main solvent. The increase in its amount has not influenced the reaction efficiency, while the reduction to a volume of 250 μL slightly decreased the yield.
With the optimized protocol ready, we decided to test the scope and limitations of this procedure. The best results were obtained for reactions with amylbenzene derivatives, in which the substrates contained a substituent in the para position – 45–63% (2a–2f). The exception is the reaction with 4-pentylbenzaldehyde, as the product undergoes additional oxidation – the p-formyl group is transformed into a carboxyl group (2g – 31%) 3-fluoropentylbenzene (2h – 39%) or 1,3-difluoro-5-pentylbenzene (2i – 45%) gave similar or slightly worse product yields than in the case of 1a–1f. Expansion of the substrate with additional aryl rings also gave slightly substandard results, within the range of 21–34% (2j–2m, see Fig. 2). Additionally, we have obtained an adequate yield – 45% – in the reaction with 4-chloro-4′-n-pentylbenzophenone.
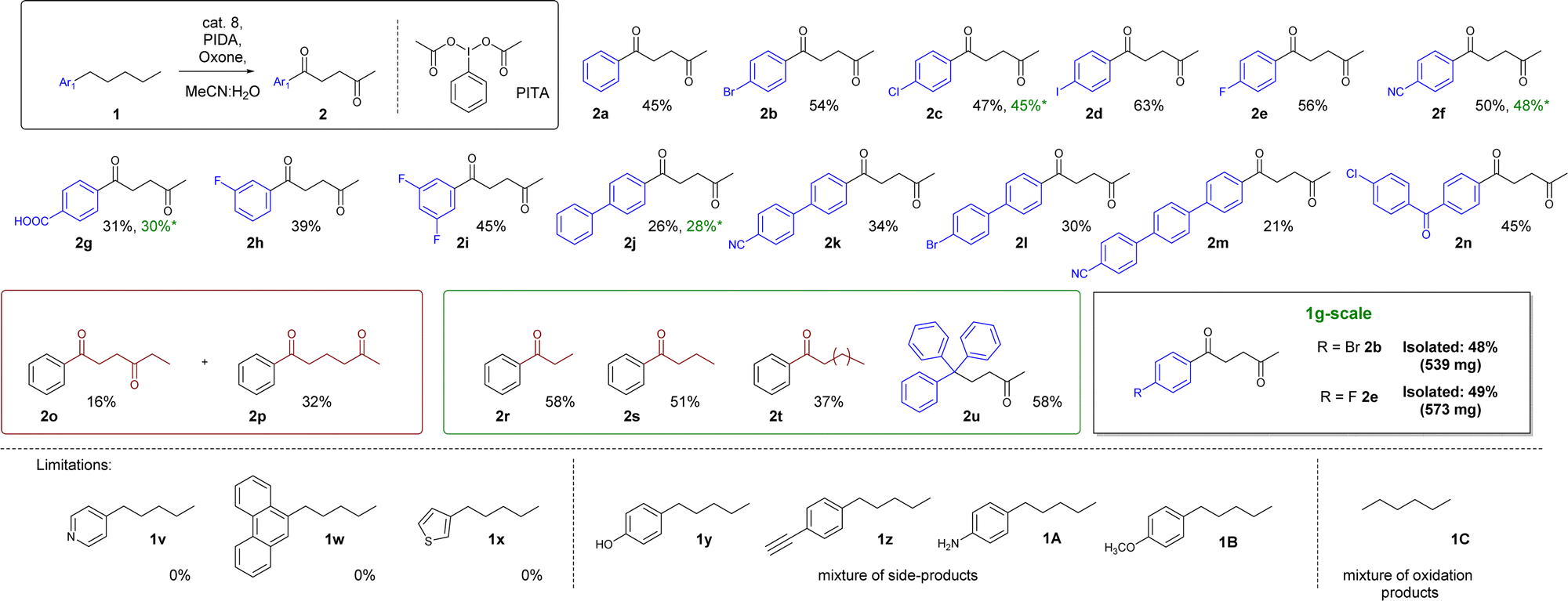 | ||
Fig. 2 Reaction scope of the oxidation of arylalkanes (standard conditions: cat. 8 (1.1%mol), Oxone® (0.29 equiv.), PIDA (13 equiv.), substrate (0.1 mmol, 1 equiv.), MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:37), 298 K, 24 h); *1 mmol-scale. :H2O (1:37), 298 K, 24 h); *1 mmol-scale. | ||
In the case of substrates with a shorter aliphatic chain, such as propylbenzene, or butylbenzene, only the benzyl position was oxidized. A different effect was observed for hexylbenzene, which gave a mixture of 3 products – a α-monoketone and two diketones, oxidized in the 1,4 and 1,5 positions in the ratio of 1:2, respectively. On the other hand, substrates with a longer aliphatic chain, namely phenyldecane, operate as 2r, or 2s, which produce the α-monoketone 2t as the main product (Fig. 2). Substrates 1v–1x and 1C showed the importance of the phenyl ring in the oxygenation process. It probably plays a crucial role in stabilizing the first activation of benzyl positions’ presumably properly aligning the catalytic center. In the case of simple alkanes (such as 1C), we have observed a lack of selectivity. Compounds containing pyridine, thiophene, or phenanthrene remain unreactive in this reaction. In turn, 4-pentylphenol 1y, 1-ethynyl-4-pentylbenzene 1z, 4-pentylaniline 1A and 1-metoxy-4-pentylbenzene 1B produced a mixture of side-products with a reaction centered at the para-substituent instead of the alkyl chain. What is important, we also tested the 1 mmol-scale for 2c, 2f, 2g, 2j and 1 g scale for 2b and 2e, which worked similarly efficiently.
Based on this information, initially, we postulated that the presence of a ketone in the benzyl position plays a crucial role in the selectivity determining step. However, another substrate excluded this hypothesis 1,1,1-triphenylpentane, when subjected to the reaction with optimized conditions, surprisingly afforded 5,5,5-triphenylpentan-2-one 2u as the major product of this reaction. This stipulated us to take another look at the proposed reaction mechanism (Fig. 3).
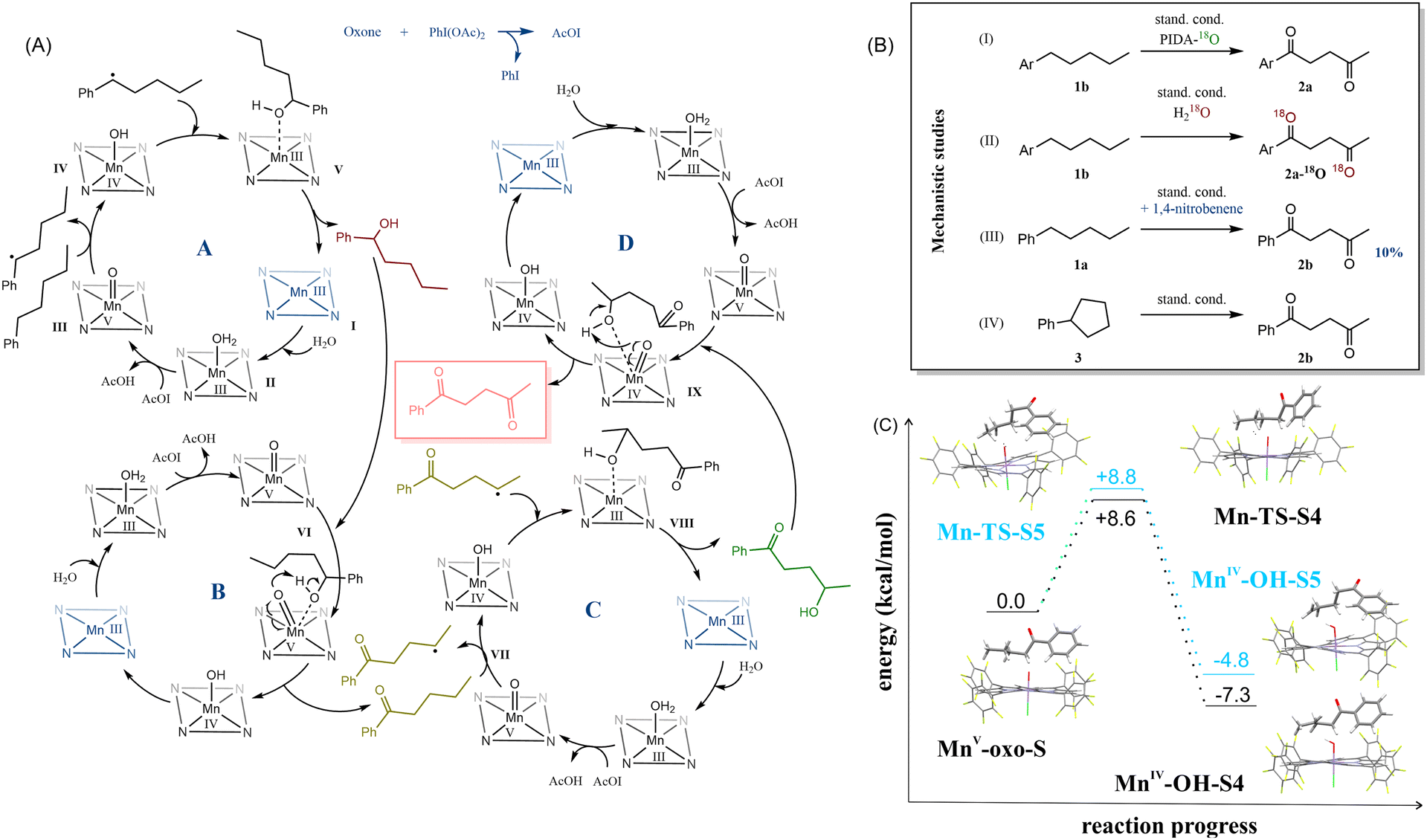 | ||
| Fig. 3 (A) The postulated mechanism, (B) mechanistic studies (standard conditions: cat. 8 (1.1%mol), Oxone® (0.29 equiv.), PIDA (13 equiv.), MeCN:H2O (1:37), 298 K, 24 h), and (C) DFT studies focused on steps MnV-oxo-S → Mn-TS-S (VII) → MnIV-OH-S. | ||
The first examined issue was the origin of the oxygen atoms in the final products (Fig. 3B, I–II). Based on the literature,2,34 we have assumed our main oxidant – (diacetoxyiodo)benzene as an oxygen source. Thus, we have prepared it from iodobenzene and 18O-acetic acid.35 After testing the model reaction with 18O-(diacetoxyiodo)benzene, we haven't noticed any 18O-enriched product. Therefore, we have used 18O-water, instead of typically adding regular H2O. The reaction proceeded at a slower rate – after 24 h, < 20% conversion of the substrate was observed, but a small portion of the α-monoketone and diketone contained 18O atoms built in their structures (see Fig. S3, ESI†). Based on the collected information, we proposed a mechanism for this reaction (Fig. 3A).
In cycle A the first step is water coordination to I to create II and then the active form III of catalyst MnV–oxo is prepared. A probable high-reactivity low-spin d2 complex is in line with the previous studies.36 Subsequently, hydrogen atom transfer reduces MnV–oxo to MnIV–OHIV and produces a radical substrate intermediate. We confirmed the radical character of this process using 1,4-dinitrobenzene as a radical scavenger (Fig. 3B, III) and 3 as a substrate (Fig. 3B, IV). In the first case, the reaction efficiency dropped to 10%, and in the second one – we observed the same main product as with previously-used amylbenzene instead of cyclopentylbenzene. As a result of cycle A, the 1-phenylpentan-1-ol is prepared. In cycle B, the alcohol is oxidized to the ketone, and the catalyst is regenerated (IV → I → II → III). Finally, α-monoketone goes to cycle C, which undergoes similar processes as the substrate in cycle A. The high regioselectivity that was observed in our reaction may result from the hydrophobic effect.37 A minimal amount of acetonitrile (Table S5, ESI†), surrounded by water molecules, forces the catalyst and the substrate to form a supramolecular complex, whose formation is driven by the hydrophobic effect. The formed associate can align the chain over the reaction center to expose the distant parts of it, after the benzylic position is already oxidized. This may also be the reason for when a higher amount of a less polar solvent is used, the yield drops (Table S5, ESI†). As a result of the last cycle D, 1,4-diketones are formed.
We have closely examined part of the crucial cycle C, using DFT calculations (Fig. 3). In Fig. 3C, the black line marks the path where position 4 is activated, and the blue line - position 5. The energy differences between the transition states are minimal (approx. 0.2 kcal mol−1), but the difference in radicals stability was visible (MnIV-OH-S4vs.MnIV-OH-S5). We have postulated that the activation can occur at both positions, but the substrate intermediate is rearranged into a more stable radical MnIV-OH-S4. In the case of hexylbenzene, the energy differences between the radicals formed at positions 4, 5, and 6 were also relatively small (approx. 3–4 kcal mol−1). However, the energy differences increased when comparing the next step – the radical forms of the substrate with MnIV-OH, which was the most stable form of the radical, localized at position 5 (see Fig. S16, ESI†).
In conclusion, a dual C(sp3)–H oxidation method of arylalkanes has been successfully developed. The direct, straightforward pathway allows obtaining the 1,4-dicarbonyl compounds, which can be important synthetic intermediates in preparing five-membered heterocycles. Furthermore, our protocol uses (diacetoxyiodo)benzene, manganese(III) porphyrin catalyst, and water as the main solvent, making it a cheap and environmentally friendly method. Furthermore, our approach shows that complex processes can be carried out directly and regioselectively in an appropriate solvent mixture, using simple and readily available catalysts and cheap substrates. In addition, we presented postulated mechanistic pathways based on the control experiments.
Financial support from the National Science Center (Grants 2019/35/D/ST4/00361) is kindly acknowledged. The Wrocław Supercomputer Centre (KDM WCSS) is kindly acknowledged for sharing computation resources necessary for DFT calculations (Grant 329). Publication of this article was supported by the Excellence Initiative – Research University program for the University of Wrocław.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef PubMed.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- K. Hurej, M. Pawlicki, L. Szterenberg and L. Latos-Grazynski, Angew. Chem., Int. Ed., 2016, 55, 1427–1431 CrossRef PubMed.
- K. Hurej, M. Pawlicki and L. Latos-Grazynski, Chem. – Eur. J., 2018, 24, 115–126 CrossRef PubMed.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef PubMed.
- G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef CAS PubMed.
- M. Guo, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2019, 141, 12187–12191 CrossRef CAS PubMed.
- G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513–9517 CrossRef CAS.
- Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005–10013 CrossRef CAS PubMed.
- N. Liu, X. Chen, L. Jin, Y.-F. Yang and Y.-B. She, Org. Chem. Front., 2021, 8, 1858–1866 RSC.
- S. Weber, L. F. Veiros and K. Kirchner, ACS Catal., 2021, 11, 6474–6483 CrossRef CAS PubMed.
- C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 20, 4789–4795 CrossRef CAS PubMed.
- V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301–307 CrossRef CAS.
- G. Minetto, L. F. Raveglia, A. Sega and M. Taddei, Eur. J. Org. Chem., 2005, 5277–5288 CrossRef CAS.
- B. K. Banik, S. Samajdar and I. Banik, J. Org. Chem., 2004, 69, 213–216 CrossRef CAS PubMed.
- C. C. Galopin, Tetrahedron Lett., 2001, 42, 5589–5591 CrossRef CAS.
- X. Ma, D. F. Dewez, L. Du, X. Luo, I. E. Markó and K. Lam, J. Org. Chem., 2018, 83, 12044–12055 CrossRef CAS PubMed.
- H. Stetter, Angew. Chem., Int. Ed. Engl., 1976, 15, 639–647 Search PubMed.
- M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632–2634 Search PubMed.
- M. M. D. Wilde and M. Gravel, Angew. Chem., Int. Ed., 2013, 52, 12651–12654 CrossRef CAS PubMed.
- A. E. Mattson, A. R. Bharadwaj and K. A. Scheidt, J. Am. Chem. Soc., 2004, 126, 2314–2315 CrossRef CAS PubMed.
- Y. Liu, Y. Li, Y. Qi and J. Wan, Synthesis, 2010, 4188–4192 CAS.
- J. Song, H. Zhang, X. Chen, X. Li and D. Xu, Synth. Commun., 2010, 40, 1847–1855 CrossRef CAS.
- M. D. Clift, C. N. Taylor and R. J. Thomson, Org. Lett., 2007, 9, 4667–4669 CrossRef CAS PubMed.
- E. Baciocchi, A. Casu and R. Ruzziconi, Tetrahedron Lett., 1989, 30, 3707–3710 CrossRef CAS.
- X.-W. Lan, N.-X. Wang, W. Zhang, J.-L. Wen, C.-B. Bai, Y. Xing and Y.-H. Li, Org. Lett., 2015, 17, 4460–4463 CrossRef CAS PubMed.
- J. Yang, F. Mei, S. Fu and Y. Gu, Green Chem., 2018, 20, 1367–1374 RSC.
- M. Mitani, M. Tamada, S.-i Uehara and K. Koyama, Tetrahedron Lett., 1984, 25, 2805–2808 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS.
- C. Wang, K. V. Shalyaev, M. Bonchio, T. Carofiglio and J. T. Groves, Inorg. Chem., 2006, 45, 4769–4782 CrossRef CAS PubMed.
- X. Chen, Q. Wang, H. Shen, G. Li, Y.-F. Yang and Y.-B. She, Org. Biomol. Chem., 2020, 18, 346–352 RSC.
- J.-H. In, S.-E. Park, R. Song and W. Nam, Inorg. Chim. Acta, 2003, 343, 373–376 CrossRef CAS.
- M. Iinuma, K. Moriyama and H. Togo, Synlett, 2012, 2663–2666 CAS.
- J. T. Groves, J. Lee and S. S. Marla, J. Am. Chem. Soc., 1997, 119, 6269–6273 CrossRef CAS.
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06126k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |