
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir and water stable germacarbonyl compounds†‡
Pritam
Mahawar
 ,
Pratima
Shukla
,
Prakash
Chandra Joshi
,
Dharmendra
Singh
,
Hemant
Kumar
,
Goutam
Mukherjee§
and
Selvarajan
Nagendran
*
,
Pratima
Shukla
,
Prakash
Chandra Joshi
,
Dharmendra
Singh
,
Hemant
Kumar
,
Goutam
Mukherjee§
and
Selvarajan
Nagendran
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110 016, India. E-mail: sisn@chemistry.iitd.ac.in
First published on 22nd September 2022
Abstract
Germacarbonyl compounds are the germanium analogs of carbonyl compounds requiring an inert atmosphere for stability. Making these compounds survive the ambient conditions was not feasible given the lability of the Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E bonds (E = O, S, Se, Te). However, the first examples of germacarbonyl compounds synthesized under ambient conditions by taking advantage of dipyrromethene ligand stabilization are detailed here; the isolated compounds are thiogermanone 3, selenogermanone 4, thiogermacarboxylic acid 6, selenogermacarboxylic acid 7, thiogermaester 9, selenogermaester 10, thiogermaamide 12, and selenogermaamide 13 with GeE bonds (E = S, Se). Compounds 12 and 13 can react under atmospheric conditions with copper(I) halides offering air and water stable monomeric 14–15 and dimeric 16–19 copper(I) complexes (halide = Cl, Br, I). Apart from just binding, selectivity was also observed; thiogermaamide 12 and selenogermaamide 13 bind CuCl and CuBr, respectively, when treated with a mixture of copper(I) halides.
E bonds (E = O, S, Se, Te). However, the first examples of germacarbonyl compounds synthesized under ambient conditions by taking advantage of dipyrromethene ligand stabilization are detailed here; the isolated compounds are thiogermanone 3, selenogermanone 4, thiogermacarboxylic acid 6, selenogermacarboxylic acid 7, thiogermaester 9, selenogermaester 10, thiogermaamide 12, and selenogermaamide 13 with GeE bonds (E = S, Se). Compounds 12 and 13 can react under atmospheric conditions with copper(I) halides offering air and water stable monomeric 14–15 and dimeric 16–19 copper(I) complexes (halide = Cl, Br, I). Apart from just binding, selectivity was also observed; thiogermaamide 12 and selenogermaamide 13 bind CuCl and CuBr, respectively, when treated with a mixture of copper(I) halides.
Introduction
Inspired by the variety and usefulness of carbonyl compounds, such as aldehydes, ketones, amides, esters, carboxylic acids, acid halides, and acid anhydrides in organic chemistry, the synthesis of their heavier analogs constitutes an essential aspect of the modern main group chemistry.1 Thermodynamic and kinetic stabilizations are essential for isolating these compounds in a stable form as long as air and moisture are avoided.1 The examples of heavy ketones are shown in Chart 1.2 Silanone i and germanones ii–iii [LL′MO] were isolated through the reactions of the corresponding NHC-silylene and germylene adducts [LL′M] with N2O, respectively (L = [CH{(CCH2)(CMe)(NDip)2}], L′ = [{(Me)CN(R)}2C], Dip = 2,6-iPr2C6H3; M = Si, R = Me (i); M = Ge, R = Me (ii), iPr (iii)).2a,b The reactions of pentacoordinate silane [(C11H8N(Me2)SiH2Ph)] with elemental sulphur and selenium resulted in silanethione and silaneselenone ([(C11H8N(Me2)Si(E)Ph)]; E = S (iv) and Se (v)).2c The desulphurization and deselenation of tetrathiogermolane and tetraselenogermolane ([Tbt(Tip)Ge(E)4]; E = S and Se), gave germanethione and germaneselenone ([Tbt(Tip)GeE]; E = S (vi) and Se (vii)), respectively.2d,e Germatellurones ([Tbt(R)Ge(Te)]; R = Tip (viii), Dis (ix)) were synthesized by the oxidation of the corresponding kinetically stabilized germylenes [Tbt(R)Ge] with elemental tellurium.2f The desulphurization of tetrathiostannolane [Tbt(Ditp)Sn(S)4] by PPh3 afforded stannanethione [Tbt(Ditp)SnS] (x).2g Stannaneselenone and stannanetellurone ([L2SnE]; E = Se (xi), E = Te (xii)) were isolated through the reaction of alkyl stannylene [L2Sn] with elemental selenium and tellurium (L = CH(SiMe3)C9H6N-8).2h These seminal studies have spurred interest in heavy carbonyl compounds; a variety of reports on synthesis and characterization is found in contemporary literature.1–3 However, there is no example of a heavy carbonyl compound that is stable in air and water to the best of our knowledge.
 | ||
| Chart 1 Examples of heavy ketones. | ||
With the objective to develop air and water stable low-valent main group chemistry, we were looking at the possibility of making air and water stable heavy carbonyl compounds. Overcoming various challenges, we successfully isolated air and water stable germacarbonyl compounds with GeE bonds (E = S, Se). Consequently, the synthesis of the first examples of air and water stable thiogermanone DPMGe(S)Ph (3), selenogermanone DPMGe(Se)Ph (4), thiogermacarboxylic acid DPMGe(S)OH (6), selenogermacarboxylic acid DPMGe(Se)OH (7), thiogermaester DPMGe(S)OEt (9), selenogermaester DPMGe(Se)OEt (10), thiogermaamide DPMGe(S)N(TMS)2 (12), and selenogermaamide DPMGe(Se)N(TMS)2 (13) are reported (DPM = dipyrrinate). Further described are the reactions of compounds 12 and 13 with Cu(I)X (X = Cl, Br, I) to afford thiogermaamide and selenogermaamide stabilized copper(I) complexes (DPMGe(E)N(TMS)2→CuCl; E = S (14), Se (15) and [DPMGe(E)N(TMS)2→CuX]2; E = S; X = Br (16), I (17) and E = Se; X = Br (18), I (19)) that are air and water stable. All the reactions offering these copper complexes were conducted under ambient conditions using non-dried solvents. Intriguing is the discovery of selectivity involved in the reactions of compounds 12 and 13 with a mixture of Cu(I)X (X = Cl, Br, I); the former and latter bind only with CuCl and CuBr, respectively.
Synthesis and spectra
With the knowledge that dipyrrinate stabilized monochlorogermylenes are air and water stable,4a,b we studied the utility of DPMGeCl (1) to afford air and water stable thiogermaacid and selenogermaacid chlorides. The treatment of compound 1 with excess elemental sulphur/selenium in toluene (12 h, rt) gave no product. At a high temperature (60 °C), the desired thiogermaacyl and selenogermaacyl chlorides were formed along with an inseparable unidentified side product. It is anticipated that the –I effect of chlorine may be the reason for this result; therefore, compounds with other functional groups were reacted with chalcogens. Phenyl germylene DPMGePh (2) was synthesized in 95% yield as an air and water stable solid through the reaction of germylene 1 with phenyl lithium at −20 °C in toluene for 12 h (see ESI; Scheme S1‡). As the handling of phenyl lithium requires an inert atmosphere, phenyl germylene 2 was synthesized under a nitrogen atmosphere using a dried solvent. As anticipated, the reactions of compound 2 under ambient conditions with stoichiometric amounts of elemental sulphur and selenium occurred smoothly in toluene at room temperature for 1 h to afford thiogermanone DPMGe(S)Ph (3) and selenogermanone DPMGe(Se)Ph (4) in 95% and 93% yields (Scheme 1). THF and DCM as solvents instead of toluene did not offer germanones 3 and 4 cleanly. As phenyl germylene 2 did not react with elemental tellurium at room temperature or high (60 °C) temperature, tellurogermanone was not isolable. Similarly, the reactions of compound 2 with nitrous oxide, N-(methyl)morpholine-N-oxide, and pyridine N-oxide also did not occur, prohibiting the synthesis of germanone with a GeO bond. A possible reason for this could be the bulkiness of the mesityl groups protecting the germylene center. Monoanionic N-heterocyclic ligand stabilized germylenes have offered germanones indirectly, which means that germylene reacted with N2O to form a μ-oxo dimer and the reaction of this dimer with a suitable Lewis acid afforded germanone.4c,d Considering this aspect, it is anticipated that the reaction of germylene 2 with an oxygen transfer agent does not occur due to the steric effect posed by the mesityl groups for the μ-oxo dimer formation. Concerning the reaction with elemental tellurium, the large size of tellurium may prohibit its interaction with the germanium(II) center heavily guarded by bulky mesityl groups.
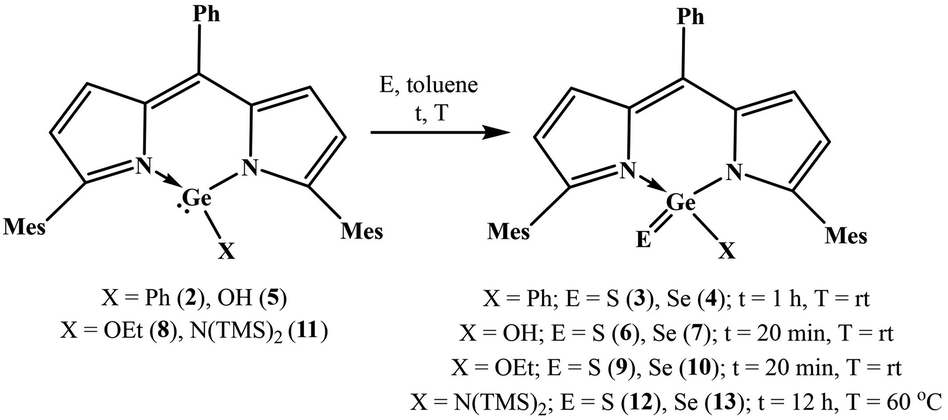 | ||
| Scheme 1 Synthesis of germacarbonyl compounds. | ||
The synthesis of thiogermaaldehyde and selenogermaaldehyde was tried; this requires a germylene hydride precursor. The reactions of monochlorogermylene 1 with various hydride sources, such as NaBH4, LiAlH4, K-selectride, and NaH, did not result in the anticipated germylene hydride. The reactions of germylene hydroxide DPMGeOH4a (5) with elemental sulphur and selenium at room temperature in toluene were checked to isolate thiogermacarboxylic and selenogermacarboxylic acids. These reactions afforded thiogermacarboxylic acid DPMGe(S)OH (6) and selenogermacarboxylic acid DPMGe(Se)OH (7) in 95% and 96% yields after 20 min (Scheme 1). Similarly, under the same reaction conditions, thiogermaester DPMGe(S)OEt (9) and selenogermaester DPMGe(Se)OEt (10) were also synthesized from germylene ethoxide DPMGeOEt4a (8) in 97% and 96% yields (Scheme 1). Finally, the synthesis of thiogermaamide and selenogermaamide was tried; the required aminogermylene 11 was obtained in 97% yield through the reaction of monochlorogermylene 1 with LiN(TMS)2 at −20 °C for 12 h in toluene (see ESI; Scheme S2‡). The reactions of aminogermylene 11 with excess amounts of elemental sulphur and selenium in toluene at 60 °C for 12 h resulted in thiogermaamide DPMGe(S)N(TMS)2 (12) and selenogermaamide DPMGe(Se)N(TMS)2 (13) in 95% and 94% yields (Scheme 1). The steric crowding due to the bulky N(TMS)2 group of germylene 11 may justify the high-temperature requirement to form thiogermaamide 12 and selenogermaamide 13.
Compounds 3–4, 6–7, 9–10, and 12–13 are the first examples of air and water stable heavy carbonyl compounds (Table 1); this stability reveals the ability of the bulky DPM ligand to protect the polar GeE bonds (E = S, Se). The air and water stability of these germacarbonyl compounds was monitored by 1H NMR spectroscopy (see ESI; Fig. S7, S8, S11, S12, S16, S19, S20, S24, S25, S28, S29, S38, S39, S43, and S44‡). The air stability was checked for up to 10 days and it was found that all the compounds were stable. Concerning water stability, the germacarbonyl compounds 3, 4, 9, 10, 12, and 13 are stable in water for 2, 4, 3, 5, 2, and 5 days, respectively (Table 1; the indicated stability refers to the duration for which the compounds show no detectable sign of decomposition). The thiogermacarboxylic and selenogermacarboxylic acids displayed poor water stability; selenogermacarboxylic acid 7 is stable for 6 h, while thiogermacarboxylic acid 6 is not stable and produces DPMH (2%) after just 10 min of water addition. It is anticipated that two electronegative atoms, such as oxygen and S/Se attached to germanium, are responsible for this observation. These atoms make germanium more electrophilic; therefore, compounds 6 and 7 are more reactive toward water than the other compounds. Among all the germacarbonyl compounds, selenogermacarbonyl compounds are more stable than the corresponding thiogermacarbonyl compounds, perhaps due to the stronger GeSe bond in selenogermacarbonyl compounds than the GeS bond in thiogermacarbonyl compounds (Table 1)3e,h–j Theoretical calculations on thiogermanone 3, selenogermanone 4, thiogermaamide 12, and selenogermaamide 13, offer evidence for this assumption; the Wiberg bond index (WBI) for the GeS bond in compounds 3 (1.457) and 12 (1.419) is marginally lower than that of compounds 4 (1.484) and 13 (1.439) with a GeSe bond.
| Compound | Air stabilitya (days) | Water stabilityb (day(s)) |
|---|---|---|
| a Air stability was checked for up to 10 d only; therefore, they may be stable for a considerable period beyond this 10 d. For example, our experience with compounds 13 and 16 reveals that they did not start to decompose even after one month of storage under ambient conditions. b Formation of 1–2% of DPMH was seen after the specified period of water stability. | ||
| DPMGe(S)Ph (3) | 10 | 2 |
| DPMGe(Se)Ph (4) | 10 | 4 |
| DPMGe(S)OH (6) | 10 | Not stable |
| DPMGe(Se)OH (7) | 10 | 0.25 |
| DPMGe(S)OEt (9) | 10 | 3 |
| DPMGe(Se)OEt (10) | 10 | 5 |
| DPMGe(S)N(TMS)2 (12) | 10 | 2 |
| DPMGe(Se)N(TMS)2 (13) | 10 | 5 |
| [DPMGe(S)N(TMS)2→CuCl] (14) | 10 | 0.125 |
| [DPMGe(S)N(TMS)2→CuBr]2(16) | 10 | 1 |
| [DPMGe(S)N(TMS)2→CuI]2 (17) | 10 | 3 |
| [DPMGe(Se)N(TMS)2→CuCl] (15) | 10 | 0.125 |
| [DPMGe(Se)N(TMS)2→CuBr]2 (18) | 10 | 0.50 |
| [(DPMGe(Se)N(TMS)2→CuI]2 (19) | 10 | 2 |
Furthermore, to explain the observed stability of the germacarbonyl compounds, the NPA charges of the atoms in the GeE bond and the nature of the HOMO of dipyrrinate stabilized thiogermaamide 12 (E = S) and selenogermaamide 13 (E = Se) were analyzed and compared with those of aminotroponiminate and amidinate stabilized thio- and selenogermaamides (see computational details in the ESI‡). As no significant differences were seen, it was concluded that these electronic properties could not explain the observed air and water stability of dipyrrinate compounds with GeS/Se bonds. Therefore, it is anticipated that the steric protection offered by the mesityl groups of the dipyrrinate ligand may provide air and water stability. To test this, the isolation of PhDPMGeS(N(TMS)2) with phenyl groups at the α and α′ positions of the DPM ligand instead of the mesityl groups was tried. Surprisingly, it was not possible to synthesize the required germylene precursor (PhDPMGeCl) by reacting the in situ generated PhDPMLi with GeCl2·(1,4-dioxane) until now. This result highlights the mesityl groups' role in offering stability.
The successful isolation of air and water stable germacarbonyl compounds prompted us to examine their reactivity under ambient conditions. Considering the presence of σ-donor chalcogen atoms (S, Se) in the germacarbonyl compounds 3–4, 6–7, 9–10, and 12–13, we started to scrutinize their ability to stabilize transition metal complexes.3c,f,g,5 The reactions of compounds 3–4, 6–7, and 9–10 with excess amounts of Cu(I)X at room temperature for 1 h did not result in the desired complexes; the reactants remained unreacted (X = Cl, I). However, the reaction of thiogermaamide DPMGe(S)N(TMS)2 (12) with an equimolar amount of Cu(I)Cl at room temperature in toluene for 30 min resulted in a monomeric thiogermaamide stabilized copper(I) chloride complex [DPMGe(S)N(TMS)2→CuCl] (14) in 89% yield (see ESI; Scheme S3‡). In contrast, its reactions with other copper(I) halides (Cu(I)Br and Cu(I)I) in toluene at room temperature for 30 min resulted in dimeric thiogermaamide stabilized copper(I) complexes [DPMGe(S)N(TMS)2→CuBr]2 and [DPMGe(S)N(TMS)2→CuI]2 with a Cu2X2 core in 94% and 90% yields, respectively (X = Br (16) and I (17)) (see ESI; Scheme S3‡). Similarly, equimolar reactions of selenogermaamide DPMGe(Se)N(TMS)2 (13) with Cu(I)Cl and Cu(I)X (X = Br, I) in toluene for 30 min at room temperature afforded monomeric and dimeric selenogermaamide stabilized copper(I) halides complexes [DPMGe(Se)N(TMS)2→CuCl] (15; yield 95%) and [DPMGe(Se)N(TMS)2→CuX]2 (X = Br (18; yield 92%), I (19; yield 94%)), respectively (see ESI; Scheme S4‡). The thiogermaamide and selenogermaamide stabilized monomeric (14, 15) and dimeric copper complexes (16–17, 18–19) represent the first examples of germacarbonyl compound stabilized copper(I) halide complexes. The polar GeS/Se bond of germacarbonyl compounds should become further polarized after forming complexes with copper halides; this anticipation is supported by the decreased WBI values of the GeS/Se bond(s) in complexes 14 (1.135) and 19 (1.205) compared to those of their precursors 12 (1.419) and 13 (1.439), respectively. The electron-donating and bulky nature of the (Me3Si)2N substituent in compounds 12 and 13 is expected to stabilize the largely polarized GeS/Se bond(s) of Cu(I) halide complexes more efficiently.
Thiogermaamide 12 and selenogermaamide 13, apart from reacting independently with CuX (X = Cl, Br, I), showed a novel aspect of selective binding towards a particular copper halide when a mixture of copper halides is present (Scheme 2). The reaction of thiogermaamide 12 with an equimolar mixture of CuX (X = Cl, Br, I) in toluene for 15 min at room temperature exclusively gave compound 14 by reacting with CuCl only (Scheme 2). In contrast, selenogermaamide 13, under the same reaction conditions, reacted selectively with CuBr and gave compound 18 (Scheme 2). Even when thiogermaamide 12 was reacted with a mixture of CuX containing one equivalent of copper chloride and an excess of copper bromide and copper iodide (three equivalents each), it reacted only with copper chloride affording copper chloride complex 14 (Scheme 2). The result was the same for selenogermaamide 13; its reaction with a mixture of CuX salts containing copper chloride, copper bromide, and copper iodide in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 gave selectively copper bromide complex 18 (Scheme 2). Pearson's HSAB principle may better explain the observed selectivity. Among compounds 12 and 13, the GeS bond of thiogermaamide 12 is more polarized than that of selenogermaamide 13 (vide supra). The NPA charge on the sulphur (−0.826) of compound 12 is higher than that on the selenium (−0.685) of compound 13 (see computational details in the ESI‡). These factors suggest that the softness of the sulphur in compound 12 is less than that of compound 13's selenium atom. For the copper(I) halides, copper(I) has the least softness when attached to chlorine (see computational details in the ESI‡). Considering all these aspects, it is anticipated that the softness of sulphur in compound 12 closely matches the softness of copper(I) in CuCl rather than the copper(I) atom of CuBr/CuI. Extending the same argument to compound 13, the softness of its selenium matches the copper(I)'s softness in CuBr. Furthermore, compounds 12 and 13 did not react with AgX (X = Cl, Br, I) and AuX (Cl, I).
:1:3 gave selectively copper bromide complex 18 (Scheme 2). Pearson's HSAB principle may better explain the observed selectivity. Among compounds 12 and 13, the GeS bond of thiogermaamide 12 is more polarized than that of selenogermaamide 13 (vide supra). The NPA charge on the sulphur (−0.826) of compound 12 is higher than that on the selenium (−0.685) of compound 13 (see computational details in the ESI‡). These factors suggest that the softness of the sulphur in compound 12 is less than that of compound 13's selenium atom. For the copper(I) halides, copper(I) has the least softness when attached to chlorine (see computational details in the ESI‡). Considering all these aspects, it is anticipated that the softness of sulphur in compound 12 closely matches the softness of copper(I) in CuCl rather than the copper(I) atom of CuBr/CuI. Extending the same argument to compound 13, the softness of its selenium matches the copper(I)'s softness in CuBr. Furthermore, compounds 12 and 13 did not react with AgX (X = Cl, Br, I) and AuX (Cl, I).
 | ||
| Scheme 2 Selective complexation of thiogermaamide 12 and selenogermaamide 13 with CuCl and CuBr, respectively. | ||
Interestingly, compounds 14–19 are the first examples of germacarbonyl compound stabilized transition metal complexes that are air and water stable. This feature was achievable due to the favorable steric protection and electronic stabilization offered by the bulky dipyrrinate ligand to the GeE→Cu moieties in these complexes. Akin to the methodology followed with germacarbonyl compounds, these copper(I) complexes' stability was studied using 1H NMR spectroscopy (see ESI; Fig. S49, S50, S54, S55, S60, S61, S65, S66, S70, S71, S76, and S77‡). The complexes were stable in air up to the monitored period of 10 days. Regarding water stability, thiogermaamide stabilized copper(I) complexes 14, 16, and 17 were stable for 3 h, 1 day, and 3 days, respectively. It is explicit from the data that moving from chloride to iodide, the water stability increases. The same trend is seen for the selenogermaamide stabilized copper(I) complexes 15, 18, and 19; they were stable for 3 h, 12 h, and 2 days, respectively (Table 1).
The compounds 3–4, 6–7, 9–10, and 12–13 are well soluble in toluene, tetrahydrofuran, dichloromethane, and chloroform. The thiogermaamide and selenogermaamide stabilized copper(I) complexes 14–19 have bad solubility in tetrahydrofuran and toluene. The thiogermaamide stabilized copper(I) complexes 14, 16, and 17 are well soluble in dichloromethane; however, their selenium analogs 15, 18, and 19 are poorly soluble. The newly synthesized compounds 2–4, 6–7, and 9–19 were characterized in the solution state through multinuclear NMR spectroscopic techniques (1H, 13C, 29Si, 77Se). In the 1H NMR spectra of compounds 3–4, 6–7, 9–10, and 12–13, almost all the resonances are slightly downfield shifted compared to those of their germylene precursors 2, 5, 8, and 11, respectively. This shift is due to increase in the germanium atoms' formal oxidation state from +2 (in compounds 2, 5, 8, and 11) to +4 (in compounds 3–4, 6–7, 9–10, and 12–13) owing to their attachment to an electronegative sulphur/selenium atom. The OH proton of thiogermacarboxylic acid 6 and selenogermacarboxylic acid 7 resonated at 1.77 and 1.79 ppm, respectively, which was downfield shifted compared to that of germylene hydroxide 5 (1.21 ppm). The trimethylsilyl protons of aminogermylene 11 were seen as two singlets (−0.46 and −0.25 ppm); in comparison, these protons of thiogermaamide (−0.05 ppm) 12 and selenogermaamide (0.02 ppm) 13 appeared as a broad singlet. Almost all the resonances of thio- and selenogermaamide stabilized copper(I) complexes 14, 16, 17 and 15, 18, 19 showed downfield shifts compared to those of thiogermaamide 12 and selenogermaamide 13, respectively. This effect is due to the donation of a lone pair of electrons from the sulphur/selenium atom of the GeE bond to the copper atom (E = S/Se). Akin to compounds 12 and 13, the trimethylsilyl protons of the monomeric 14–15 and dimeric 16–19 copper(I) halide complexes resonate as a broad singlet (between −0.06 and 0.01 ppm). In the 13C NMR spectra of compounds 2 (23 signals), 3 (22) 4 (21), 6 (18), 7 (16), 9 (22), 10 (21), 11 (16), 12 (21), 13 (20), 14 (21), 15 (25), 16 (21), 17 (20), 18 (19), and 19 (20) different number of signals were observed. In the 29Si NMR spectra of compounds 11–19, except germylene 11 that gave two resonances at −3 and 2 ppm, all the other compounds showed a single resonance (−21.8 (12), −21.9 (13), −21.8 (14), −21.9 (15), −21.8 (16), −21.9 (17), −21.9 (18), and −21.9 ppm (19)). As the selenium resonances of compounds 4 (−386 ppm), 7 (−340 ppm), 10 (−379 ppm), 13 (−178 ppm), 15 (−237 ppm), 18 (−228 ppm), and 19 (−235 ppm) are in between the resonances of (H3Ge)2Se (−612 ppm) with a Ge–Se single bond6 and [Tbt(Tip)Ge(Se)] (vii) (940.6 ppm)2e having an electronically unperturbed GeSe double bond, their GeSe bonds should be polarized with partial positive and negative charges on the germanium and selenium atoms, respectively (see ESI; Table S2‡). Despite such polarization, it is interesting to see them as air and water stable compounds, which should be attributed to the kinetic and thermodynamic stabilizations the bulky DPM ligand bestowed. In the IR spectra of compounds 6 and 7, the hydroxyl group's stretching band was seen at 3612.69 and 3612.05 cm−1, respectively; in comparison, the OH stretching band of compound 5 was detected at 3627 cm−1 (Fig. S80 and S81; see ESI‡). The UV-vis spectra of thiogermacarbonyl compounds 3, 6, 9, and 12 (Fig. S82‡), selenogermacarbonyl compounds 4, 7, 10, and 13 (Fig. S83‡), and thio/selenogermaamide stabilized copper(I) complexes 14, 18, and 19 (Fig. S84‡) were recorded in toluene at room temperature. All these compounds showed an absorption maximum in the visible region between 505 and 525 nm (Table S3‡). Preliminary theoretical studies on germacarbonyl compounds 12 and 13 showed that the absorptions are essentially due to 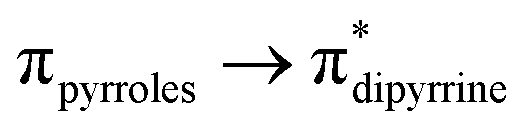 (∼82%) and
(∼82%) and 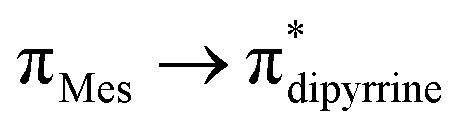 (∼15%) transitions. A computational study on copper complex 18 revealed that the observed absorption maximum is due to multiple transitions;
(∼15%) transitions. A computational study on copper complex 18 revealed that the observed absorption maximum is due to multiple transitions; 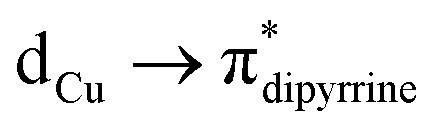 (34.3%) and
(34.3%) and  (14.1%) transitions contribute majorly, and all other transitions have below 5% contributions.
(14.1%) transitions contribute majorly, and all other transitions have below 5% contributions.
X-ray crystal structures of compounds 2–4, 9, 11–14, 16–17, and 19
Molecular structures of germylenes (2 and 11), germacarbonyl compounds (3, 4, 9, 12, and 13), and metal complexes (14 (Fig. 1), 16, 17, and 19 (Fig. 2)) were confirmed by single-crystal X-ray diffraction analysis. The Ge–X bond in compounds 3 (1.928(6) Å; X = CPh), 4 (1.933(2) Å; X = CPh), 12 (1.843(3) Å; X = NN(TMS)2), and 13 (1.837(7) Å; X = NN(TMS)2) is shorter compared to the corresponding bond in compounds 2 (2.001(2) Å; X = CPh) and 11 (1.924(2) Å; X = NN(TMS)2). This effect is due to the higher electrophilicity of the germanium atom in compounds 3, 4, and 12–13 than that in germylenes 2 and 11; the electrophilicity is increased by the electronegative chalcogen atom doubly bonded to germanium. The GeS bonds in thiogermanone 3 (2.052(2) Å), thiogermaester 9 (2.058(5) Å), and thiogermaamide 12 (2.062(1) Å) are shorter than that in aminotroponimine ligand stabilized thiogermanone LGe(S)Ph (xix) (2.102(7) Å),3g thiogermaester LGe(S)OtBu (xvi) (2.076(1) Å),3j and thiogermaamide LGe(S)N(SiMe3)2 (xviii) (2.083(1) Å), respectively3d (L = (iBu)2ATI; ATI = aminotroponimine). Furthermore, the GeS bond of compound 3 is much shorter than the Ge–S single bond (2.239(1) Å)7 in compound [{(TMS)2C(2-py)}{(TMS)C(2-py)}]GeS(TMS), and is slightly longer than the unperturbed GeS bond (2.049(3) Å) in the kinetically stabilized thiogermanone Tbt(Tip)GeS (vi).2d These comparisons may indicate that the polarization in the GeS bond of compound 3 is in between that of compounds vi and xix. A similar trend was seen for the selenium analogs 4 and 13. The GeSe bond of compounds 4 (2.195(3) Å) and 13 (2.194(1) Å) is shorter than that in ATI ligand stabilized selenogermanone (xiii) (2.235(4) Å)3g and selenogermaamide (xvii) (2.222(1) Å),3d respectively. The GeSe bond of compound 4 is much shorter than the Ge–Se single bond (2.433(1) Å) in compound [Tbt(Mes)GeSe]2 and marginally longer than the GeSe bond (2.180(2) Å) in the kinetically stabilized selenogermanone [Tbt(Tip)GeSe] (vii).2e
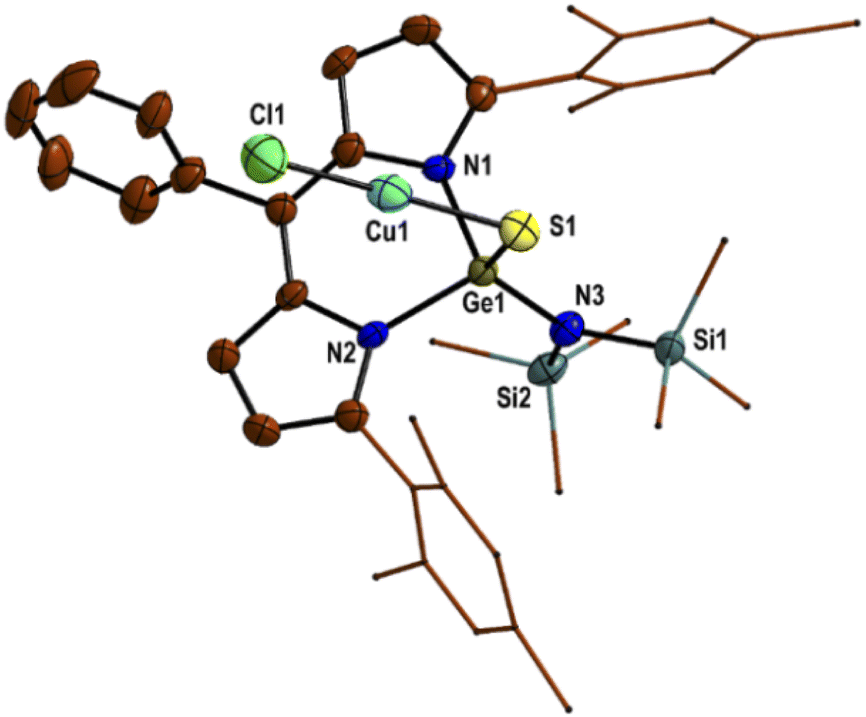 | ||
| Fig. 1 The molecular structure of thiogermaamide stabilized copper(I) chloride complex 14 with thermal ellipsoids at a 40% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ge(1)–S(1) 2.132(7), Ge(1)–N(1) 1.934(1), Ge(1)–N(2) 1.938(1), Ge(1)–N(3) 1.831(1), S(1)–Cu(1) 2.143(8), Cu(1)–Cl(1) 2.087(2); N(3)–Ge(1)–N(1) 112.2(5), N(3)–Ge(1)–N(2) 112.0(5), N(1)–Ge(1)–N(2) 96.6(4), N(3)–Ge(1)–S(1) 116.30(4), and S(1)–Cu(1)–Cl(1) 178.04(2). Data collection temperature: 100 K. | ||
Due to the coordination of the sulphur atom of the GeS bond with Lewis acid (CuCl/CuBr/CuI), the GeS bond of thiogermaamide stabilized metal complexes 14 (2.132(7) Å), 16 (2.101(7) Å), and 17 (2.103(8) Å) is elongated compared to that in thiogermaamide 12 (2.062(1) Å) (Fig. 1) (see ESI; Fig. S90, S93 and S94‡). A similar trend is seen in selenogermaamide stabilized copper complex 19; its GeSe bond (2.234(6) Å) is longer than that of compound 13 (2.194(1) Å) (Fig. 2) (see ESI; Fig. S91‡). In compound 14, the copper atom is dicoordinate with a sulphur and chlorine atom; it has a linear geometry apparent from the S–Cu–Cl bond angle of 178.04° (Fig. 1). The complexes 16, 17, and 19 (Fig. 2) have a planar dimeric Cu2X2 (X = Br, I) core; the copper atoms are tricoordinate with the sum of bond angles around them, equalling 360°. The Cu⋯Cu distance in compounds 16 (2.725(5) Å), 17 (2.699(8) Å), and 19 (2.581(8) Å) is less than the sum of the van der Waals radii of two copper atoms (2.80 Å) and indicates the presence of cuprophilic interaction (Fig. 2) (see ESI; Fig. S93 and S94‡).
 | ||
| Fig. 2 The molecular structure of selenogermaamide stabilized copper(I) iodide complex 19 with thermal ellipsoids at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ge(1)–Se(1) 2.234(6), Ge(1)–N(1) 1.928(2), Ge(1)–N(2) 1.931(3), Ge(1)–N(3) 1.853(3), Se(1)–Cu(1) 2.349(5), Cu(1)–I(1) 2.566(5), Cu(1)–I(1) 2.632(4), Cu1–Cu1 2.581(8); N(3)–Ge(1)–N(1) 110.98(2), N(3)–Ge(1)–N(2) 105.98(2), N(1)–Ge(1)–N(2) 94.50 (1), Ge(1)–Se(1)–Cu(1) 102.18(2), I(1)–Cu(1)–I(1) 120.47(2), and Se(1)–Cu(1)–I(1) 133.05(2). Data collection temperature: 100 K. | ||
Conclusions
The first examples of germacarbonyl compounds 3–4, 6–7, 9–10, and 12–13 that are stable under ambient conditions were synthesized and structurally characterized. Though thiogermanone 3, selenogermanone 4, thiogermacarboxylic acid 6, selenogermacarboxylic acid 7, thiogermaester 9, and selenogermaester 10 did not bind with copper(I) halides, thiogermaamide 12 and selenogermaamide 13 did react under ambient conditions providing copper(I) complexes (14–19) that are also stable outside inert atmospheres. The air and water stabilities of these germacarbonyl compounds and copper(I) complexes were studied using 1H NMR spectroscopy; the stability of these compounds is due to the precise thermodynamic and kinetic stabilizations provided by a bulky dipyrromethene ligand. Uniquely, selective binding of thiogermaamide 12 and selenogermaamide 13 towards Cu(I)Cl and Cu(I)Br was noticed when they were reacted with a mixture of Cu(I)X, respectively (X = Cl, Br, I).Data availability
The experimental and computational data associated with this article are provided in the ESI.‡Author contributions
P. M. carried out the experimental studies and drafted the manuscript. P. S. and P. C. J. helped P. M. during (a) dipyrromethene synthesis and (b) monitoring the air and water stability of compounds reported in the manuscript. D. S. assisted P. M. during the crystallographic studies on compounds 14, 16, and 17. H. K. assisted P. M. with the UV-vis spectroscopic studies. P. M. and P. S. carried out the theoretical calculations; G. M. helped them analyze the computational data. S. N. corrected the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dedicated to Prof. Anil J. Elias. P. M. and P. S. thank IIT Delhi for their research fellowships. P. C. J. and D. S. thank CSIR, New Delhi, India, for research fellowships. H. K. thanks MHRD, New Delhi, India, for a Prime Minister's Research Fellowship (PMRF). S. N. thanks SERB, DST, New Delhi, India, for funding (EMR/2017/005519) and DST-FIST for establishing a single-crystal X-ray diffraction facility (SR/FST/CSII-027/2014) in the Department of Chemistry, IIT Delhi.Notes and references
- For examples, see, (a) Y. K. Loh and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 8626–8648 CrossRef CAS PubMed; (b) A. Hanft and C. Lichtenberg, Eur. J. Inorg. Chem., 2018, 3361–3373 CrossRef CAS; (c) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS; (d) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (e) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (f) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS; (g) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS; (h) R. Okazaki and N. Tokitoh, Acc. Chem. Res., 2000, 33, 625–630 CrossRef CAS; (i) P. P. Power, Chem. Rev., 1999, 99, 3463–3503 CrossRef CAS; (j) M. C. Kuchta and G. Parkin, Coord. Chem. Rev., 1998, 176, 323–372 CrossRef CAS.
- (a) Y. Xiong, S. Yao and M. Driess, J. Am. Chem. Soc., 2009, 131, 7562–7563 CrossRef CAS PubMed; (b) S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 6466–6468 RSC; (c) P. Arya, J. Boyer, F. Carr, R. Corriu, G. Lanneau, J. Lapasset, M. Perrot and C. Priou, Angew. Chem., Int. Ed., 1989, 28, 1016–1018 CrossRef; (d) N. Tokitoh, T. Matsumoto, K. Manmaru and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 8855–8856 CrossRef CAS; (e) T. Matsumoto, N. Tokitoh and R. Okazaki, Angew. Chem., Int. Ed., 1994, 33, 2316–2317 CrossRef; (f) N. Tokitoh, T. Matsumoto and R. Okazaki, J. Am. Chem. Soc., 1997, 119, 2337–2338 CrossRef CAS; (g) M. Saito, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 2004, 126, 15572–15582 CrossRef CAS; (h) W.-P. Leung, W.-H. Kwok, L. T. C. Law, Z.-Y. Zhou and T. C. W. Mak, Chem. Commun., 1996, 505–506 RSC.
- (a) N. Parvin, N. Sen, P. V. Muhasina, S. Tothadi, P. Parameswaran and S. Khan, Chem. Commun., 2021, 57, 5008–5011 RSC; (b) X. Zhao, T. Szilvási, F. Hanusch and S. Inoue, Chem.–Eur. J., 2021, 1–5 Search PubMed; (c) D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS; (d) N. Parvin, S. Pal, S. Khan, S. Das, S. K. Pati and H. W. Roesky, Inorg. Chem., 2017, 56, 1706–1712 CrossRef CAS; (e) I. Alvarado-Beltran, A. Rosas-Sánchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CrossRef CAS PubMed; (f) R. K. Siwatch, S. Karwasara, M. K. Sharma, S. Mondal, G. Mukherjee, G. Rajaraman and S. Nagendran, Organometallics, 2016, 35, 429–438 CrossRef CAS; (g) S. Karwasara, D. Yadav, C. K. Jha, G. Rajaraman and S. Nagendran, Chem. Commun., 2015, 51, 4310–4313 RSC; (h) B. Li, Y. Li, N. Zhao, Y. Chen, Y. Chen, G. Fu, H. Zhu and Y. Ding, Dalton Trans., 2014, 43, 12100–12108 RSC; (i) D. Yadav, R. K. Siwatch, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2014, 53, 10054–10059 CrossRef CAS; (j) R. K. Siwatch, D. Yadav, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2014, 53, 5073–5079 CrossRef CAS PubMed; (k) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS; (l) S. Sinhababu, R. K. Siwatch, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2012, 51, 9240–9248 CrossRef CAS PubMed; (m) R. K. Siwatch and S. Nagendran, Organometallics, 2012, 31, 3389–3394 CrossRef CAS; (n) L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Nat. Chem., 2012, 4, 361–365 CrossRef CAS PubMed; (o) M. Kirchmann, T. Gädt, F. M. Schappacher, R. Pöttgen, F. Weigend and L. Wesemann, Dalton Trans., 2009, 1055–1062 RSC; (p) W.-P. Leung, K.-H. Chong, Y.-S. Wu, C.-W. So, H.-S. Chan and T. C. W. Mak, Eur. J. Inorg. Chem., 2006, 2006, 808–812 CrossRef; (q) T. Iwamoto, K. Sato, S. Ishida, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2006, 128, 16914–16920 CrossRef CAS PubMed; (r) I. Saur, G. Rima, H. Gornitzka, K. Miqueu and J. Barrau, Organometallics, 2003, 22, 1106–1109 CrossRef CAS; (s) S. R. Foley, G. P. A. Yap and D. S. Richeson, J. Chem. Soc., Dalton Trans., 2000, 1663–1668 RSC.
- (a) P. Mahawar, M. K. Wasson, M. K. Sharma, C. K. Jha, G. Mukherjee, P. Vivekanandan and S. Nagendran, Angew. Chem., Int. Ed., 2020, 59, 21377–21381 CrossRef CAS; (b) C. K. Jha, S. Karwasara and S. Nagendran, Chem.–Eur. J., 2014, 20, 10240–10244 CrossRef CAS PubMed; (c) S. Sinhababu, D. Yadav, S. Karwasara, M. K. Sharma, G. Mukherjee, G. Rajaraman and S. Nagendran, Angew. Chem., Int. Ed., 2016, 55, 7742–7746 CrossRef CAS; (d) M. K. Sharma, S. Sinhababu, P. Mahawar, G. Mukherjee, B. Pandey, G. Rajaraman and S. Nagendran, Chem. Sci., 2019, 10, 4402–4411 RSC.
- For examples, see, (a) S. Yadav, R. Kumar, K. V. Raj, P. Yadav, K. Vanka and S. S. Sen, Chem.–Asian J., 2020, 15, 3116–3121 CrossRef CAS PubMed; (b) S. Sinhababu, M. K. Sharma, P. Mahawar, S. Kaur, V. K. Singh, A. Paliwal, D. Yadav, H. K. Kashyap and S. Nagendran, Dalton Trans., 2019, 48, 16366–16376 RSC.
- H. C. E. McFarlane and W. McFarlane, in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987, pp. 417–435 Search PubMed.
- G. Ossig, A. Meller, C. Brönneke, O. Müller, M. Schäfer and R. Herbst-Irmer, Organometallics, 1997, 16, 2116–2120 CrossRef CAS.
Footnotes |
| † P. Mahawar, P. Shukla, P. C. Joshi, D. Singh and S. Nagendran, ChemRxiv, 2021, DOI: 10.26434/chemrxiv-2021-m8793. This content is a preprint and has not been peer-reviewed. |
| ‡ Electronic supplementary information (ESI) available: Experimental section and molecular structure determination of compounds 2–4, 9, 11–14, 16–17, and 19 (PDF). CIFs for compounds 2–4, 9, 11–14, 16–17, and 19, are deposited with the Cambridge Structural Database (CSD). CCDC 2116996 (2), 2116997 (3), 2116998 (4), 2117002 (9), 2116999 (11), 2117005 (12), 2117004 (13), 2117001 (14), 2117000 (16), 2117006 (17), and 2117003 (19). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01869a |
| § Present address: PharmCADD, 12F, 331, Jungang-daero, Dong-gu, Busan, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2022 |