
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering and reprogramming the cyclization regioselectivity in bifurcation of indole alkaloid biosynthesis†
Zhuo
Wang‡
 ab,
Yiren
Xiao‡
ab,
Song
Wu
ab,
Jianghua
Chen
c,
Ang
Li
d and
Evangelos C.
Tatsis
*ae
ab,
Yiren
Xiao‡
ab,
Song
Wu
ab,
Jianghua
Chen
c,
Ang
Li
d and
Evangelos C.
Tatsis
*ae
aNational Key Laboratory of Plant Molecular Genetics, CAS Center for Excellence in Molecular Plant Sciences, Shanghai Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai, 200032, China. E-mail: etatsis@cemps.ac.cn; evangelos.tatsis@jic.ac.uk
bUniversity of Chinese Academy of Sciences, Beijing, China
cKey Laboratory of Tropical Plant Resources and Sustainable Use, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Mengla County, China
dState Key Laboratory of Bioorganic and Natural Product Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China
eCEPAMS – CAS-JIC Centre of Excellence for Plant and Microbial Sciences, China
First published on 28th September 2022
Abstract
The metabolism of monoterpene indole alkaloids (MIAs) is an outstanding example of how plants shape chemical diversity from a single precursor. Here we report the discovery of novel enzymes from the Alstonia scholaris tree, a cytochrome P450, an NADPH dependent oxidoreductase and a BAHD acyltransferase that together synthesize the indole alkaloid akuammiline with a unique methanoquinolizidine cage structure. The two paralogous cytochrome P450 enzymes rhazimal synthase (AsRHS) and geissoschizine oxidase (AsGO) catalyse the cyclization of the common precursor geissoschizine and they direct the MIA metabolism towards to the two structurally distinct and medicinally important MIA classes of akuammilan and strychnos alkaloids, respectively. To understand the pathway divergence, we investigated the catalytic mechanism of the two P450 enzymes by homology modelling and reciprocal mutations. Upon conducting mutant enzyme assays, we identified a single amino acid residue that mediates the space in active sites, switches the enzymatic reaction outcome and impacts the cyclization regioselectivity. Our results represent a significant advance in MIA metabolism, paving the way for discovery of downstream genes in akuammilan alkaloid biosynthesis and facilitating future synthetic biology applications. We anticipate that our work presents, for the first time, insights at the molecular level for plant P450 catalytic activity with a significant key role in the diversification of alkaloid metabolism, and provides the basis for designing new drugs.
Akuammilan alkaloids are a structurally diverse class of monoterpene indole alkaloids (MIAs) isolated mainly from plants of the Apocynaceae family across different parts of the world.1–4Akuammilan alkaloids exhibit a broad range of biological activities, varying from antidiabetic,2,5 antibacterial, anti-inflammatory, and antimalarial activities,1–4 while bis-indole alkaloids with akuammilan structures exhibit anticancer activity similar to vinblastine.6,7Akuammilans like rhazimol (1) and akuammiline (2) are considered to be the bioactive ingredients of traditional Chinese medicine application Deng-tai-ye which is based on extracts from leaves and bark of the Alstonia scholaris tree to treat diseases and conditions of the upper respiratory tract like cough, catarrhal fever, chronic bronchitis or asthma.1,4,8,9 To access their medicinal properties, numerous studies have been reported on the organic synthesis of various akuammilan alkaloids by several research groups.10–16
The elucidation of biosynthetic pathways17 can help to access scarce but medicinally important natural products through metabolic engineering and synthetic biology applications. The entry point to akuammilan biosynthesis is the transformation of geissoschizine (3) through intramolecular cyclization and bond formation between C7 and C16 of geissoschizine for the synthesis of aldehyde rhazimal (4). Despite rhazimal being assumed to be an intermediate18 in the biosynthesis of vinblastine, it remains totally cryptic how plants synthesize akuammilan alkaloids. Here, we report the discovery of cytochrome P450, oxidoreductase and acetyltransferase enzymes that control the biosynthesis of rhazimal, rhazimol and akuammiline (Fig. 1). Furthermore, based on homology modelling, mutants revealed that a key amino acid residue in the active site of the newly discovered cytochrome P450 conveys intramolecular cyclization regioselectivity and controls the divergence between the akuammiline and vinblastine biosynthesis.
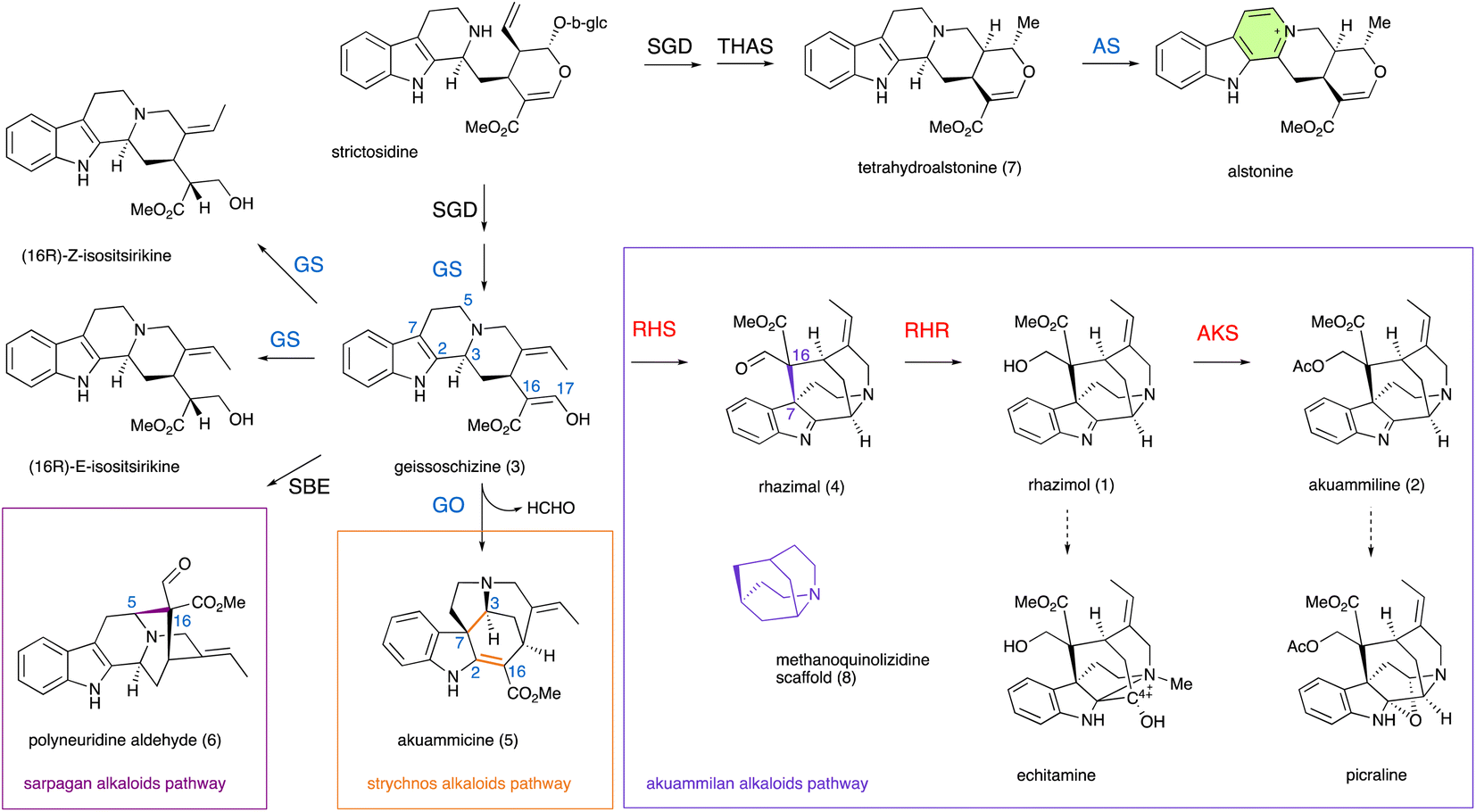 | ||
| Fig. 1 Schematic overview of monoterpene indole alkaloid (MIA) metabolic pathways in the Apocynaceae family. Strictosidine and its reduced aglycon geissoschizine generated by the activity of strictosidine glucosidase (SGD) and alcohol dehydrogenase geissoschizine synthase (GS), are the precursors of different important classes of MIAs. P450 geissoschizine oxidase (GO, CYP71D1V1) catalyzes the formation of a bond between C2 and C16 (highlighted in orange) followed by the breaking of the C2–C3 bond, the formation of a new C3–C7 bond (highlighted in orange) and breaking of the C16–C17 bond and the loss of the formaldehyde group for the synthesis of a strychnos scaffold of akuammicine. P450 sarpagan bridge enzyme (SBE, CYP71AY4) catalyzes the formation of a bond between C5 and C16 (highlighted in magenta) for the synthesis of a sarpagan scaffold of polyneuridine aldehyde. The paralog P450 enzyme alstonine synthase (AS, CYP71AY1) catalyzes the aromatization of a tetrahydroalstonine piperidine ring. Until the present work the enzymes that catalyze the formation of the bond between C7 and C16 (highlighted in purple) for the synthesis of the methanoquinolizidine scaffold and later steps in metabolism of akuammilan alkaloids were unknown. Newly discovered functionally characterized enzymes in this study are shown in red, while enzymes with known activity that are identified in this study are shown in light blue. Dashed arrows represent unknown (multiple) enzymatic steps. | ||
To identify genes involved in biosynthesis of rhazimol (1) and akuammiline (2), the two key central precursors of akuammilan alkaloid biosynthetic pathways (Fig. 1), we performed RNA sequencing to build a cDNA library of A. scholaris (Table S1†) and we mapped it to the publicly available19 genome of A. scholaris. We used synteny, phylogeny and homology to mine the genome and transcriptome database and identify candidate genes. Functional characterization was based on enzyme assays of heterologously produced proteins following the analysis with LCMS for the identification of enzyme products. Previous studies on MIA metabolism have demonstrated the examples of intramolecular cyclizations of geissoschizine (3) to establish new structural scaffolds (Fig. 1 and S1†). Catharanthus roseus GO (CYP71D1V1) catalyses the oxidative cyclization and rearrangement of a beta carboline scaffold of geissoschizine including bond formations between C2–C16 and C3–C7, breaking of the bond between C2–C3 and the loss of a HCHO group (breaking of the C16–C17 bond) to the synthesis of strychnos alkaloid akuammicine (5), highlighting the entry point into metabolic pathways of strychnos, iboga and aspidosperma alkaloids (Fig. 1 and S1†).20 The SBE (CYP71AY4) from Rauwolfia serpentina catalyses the oxidative cyclization of geissoschizine between C5–C16 to generate polyneuridine aldehyde (6) marking a distinctive transition into sarpagan and ajmalan alkaloid metabolism (Fig. 1 and S1†).21 We hypothesized that geissoschizine transformation into rhazimal involving the formation of a bond between C7–C16 (Fig. 1,2A, and S1†)1,4 can be catalyzed by a P450 paralogue to GO and SBE (CYP71 family). Thus, the first step was to confirm the synthesis of geissoschizine in A. scholaris. With the syntenic analysis of the genomes of A. scholaris and C. roseus (Fig. S2†) we identified A. scholaris geissoschizine synthase AsGS and by enzyme assays with a purified heterologously expressed protein, we confirmed that AsGS is functionally equivalent to CrGS20 (Table S2 and Fig. S3–S6†).
We constructed a phylogenetic tree with A. scholaris P450s from the CYP71 family and the reported P450s involved in MIA metabolism from the plants of the Apocynaceae family (Fig. S7†), and through phylogeny, we selected P450 candidates to screen for RHS activity. The cytochrome P450 gene 45522_t with 56.5% amino acid sequence identity (Table S3†) to CrAS (CYP71AY1) showed no enzymatic activity towards geissoschizine. When microsomal protein 45522_t was tested against tetrahydroalstonine (7), the 45522_t enzyme demonstrated alstonine synthase activity and therefore was characterised as A. scholaris alstonine synthase AsAS (Fig. S8†). AsAS showed no oxidative activity towards ajmalicine (Fig. S9†) for the synthesis of serpentine.22 The A. scholaris gene 43978_t has 90.2% sequence identity to CrGO and demonstrated GO activity in microsome assays with the synthesis of akuammicine (m/z 323.17 at 8.1 min) in similar way like CrGO (Fig. S10†), and then 43978_t was identified as A. scholaris geissoschizine oxidase AsGO. Unexpectedly, AsGO also displayed AS activity (Fig. S11†) when it was assayed with tetrahydroalstonine but showed no activity towards ajmalicine (Fig. S9†).
The microsomal protein fraction harbouring 46693_t which shares 61.8% sequence identity to CrGO, showed activity against geissoschizine with a major peak detected at 6.6 min with m/z 351 and major fragment m/z 291 in MS2 spectra when the enzymatic reaction mixture was quenched with methanol (Fig. 2 and S12†). When the enzyme assay of microsomal protein 46693_t was quenched with NaBH4, the peak at 6.6 min with m/z 351 disappeared and a new peak appeared with m/z 353 and with a retention time 6.2 min. The MS and MS2 spectra of the new compound eluted at 6.2 min, were identical to synthetic rhazimol23 (Fig. 2B and S12†) indicating that the 46693_t microsomal protein catalyses the transformation of geissoschizine to rhazimal (4), while the addition of NaBH4 reduces the aldehyde function to alcohol. To our surprise, in enzyme assays of 46693_t, the microsomal protein showed some GO activity towards geissoschizine with the synthesis of akuammicine (m/z 323.17 at 8.1 min) detected (Fig. S12†). When the 46693_t microsomal protein was assayed with ajmalicine or tetrahydrolastonine, no enzymatic activity was observed (Fig. S9 and S13†). Since 46693_t is the first ever reported enzyme that demonstrated rhazimal synthase activity, hereafter, it will be referred to as A. scholaris rhazimal synthase AsRHS. Beyond the crucial role of AsRHS acting as a gateway into akuammilan alkaloid metabolism, AsRHS transforms the β-carboline geissoschizine into methanoquinolizidine rhazimal, significantly increasing the complexity of the chemical scaffold by the formation of an elusive polycyclic structure.
As the next step in the biosynthetic pathway, a reduction is expected to convert the aldehyde rhazimal (4) to alcohol rhazimol (1) (Fig. 2). A similar transformation is catalysed by the NADPH dependent oxidoreductase enzyme Redox2 (ref. 25) in the Catharanthus roseus metabolic pathway of vinblastine, transforming aldehyde (9) into the alcohol stemmadenine (10) (Fig. S14†). The oxidoreductase gene 15881_t from A. scholaris has a syntenic relationship with CrRedox2 sharing 80.2% sequence identity at the amino acid level (Fig. S12 and Table S2†). To assay the enzymatic activity for reduction of rhazimal, geissoschizine was incubated with microsomal protein AsRHS to generate rhazimal and the purified 15881_t protein was added after 30 minutes. The enzyme assays analysed by LCMS showed a new peak eluted at 6.2 min (Fig. 2C and S15†) identified as rhazimol, based on comparison of retention time, MS and MS2 spectra to synthetic rhazimol.23 Therefore, 15881_t shows rhazimal reductase (RHR) activity and will be referred to as AsRHR1. The syntenic relationship between CrRedox2-AsRHR1, and biochemical data reveal a new example of enzyme neo-functionalization in MIA metabolism.26 The oxidoreductases in MIA metabolism display substrate promiscuity and catalyse the conversion of aldehydes to primary alcohols like the example of CrGS which reduces the keto–enol function of geissoschizine to isoistsirikines20 (Fig. 1). Since we could not test the functional relevance of AsRHR1 in planta e.g. by VIGS experiments, we expanded our search for enzymes with RHR activity. Using the sequences of reported reductases in MIA biosynthetic pathways20,25,27–29 for homology search in the A. scholaris genome, we identified 13 additional alcohol dehydrogenase/reductase (ADH) enzymes to assay for RHR activity (Fig. S16, Table S2†) as we assayed AsRHR1. LCMS analysis of enzyme assays revealed that only ADH10 (33135_t), the closest homologue to AsRHR1 (Table S2 and Fig. S16–S18†), had RHR activity (Fig. 2C, S15, and S18†) and thereafter ADH10 will be referred as AsRHR2.
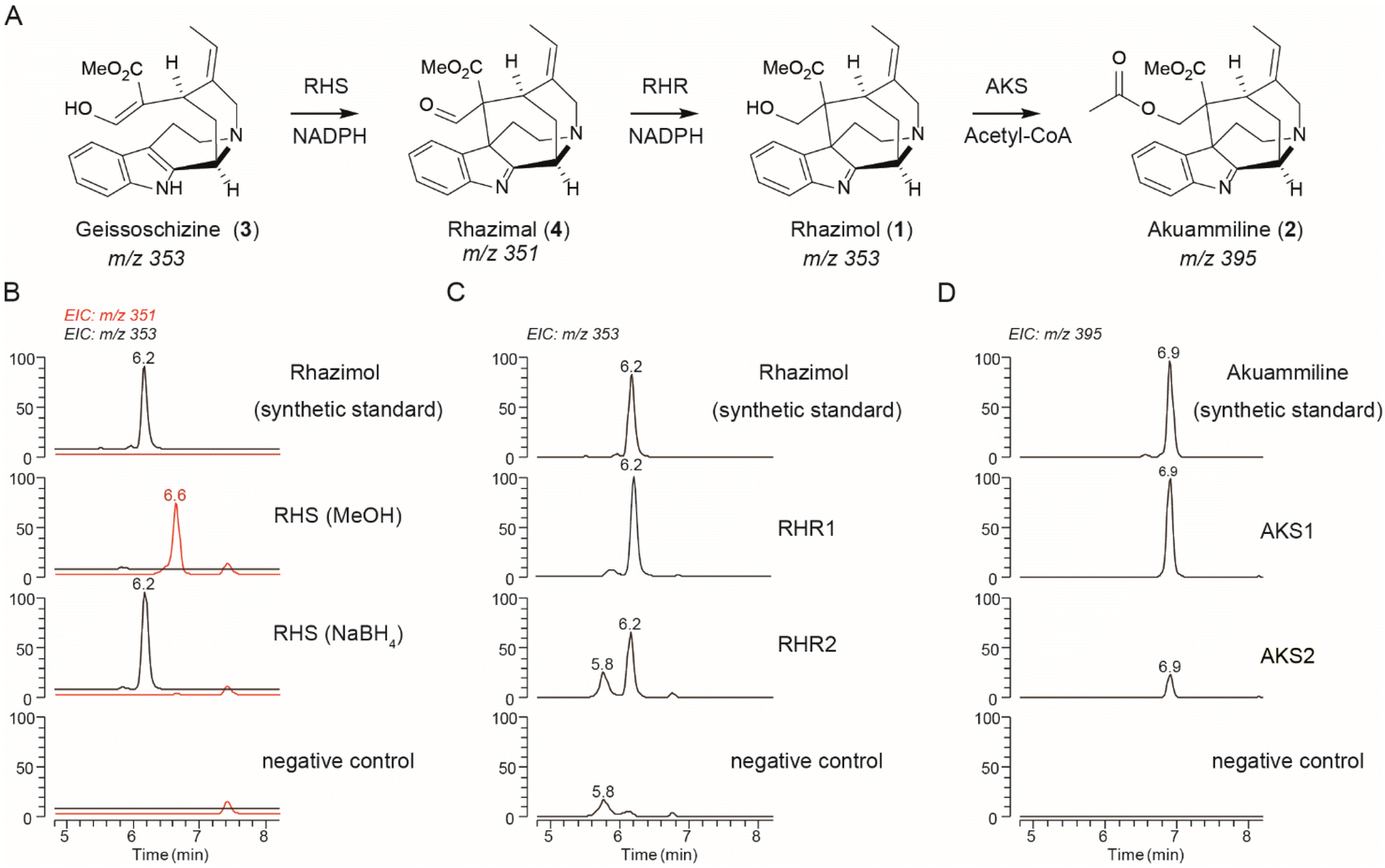 | ||
| Fig. 2 Discovery of akuammiline biosynthetic pathway genes. (A) The methanoquinolizidine structure and the biosynthetic pathway of akuammiline. (B) LCMS chromatograms at m/z 351 (highlighted in red) and 353 showing the in vitro catalytic activity of AsRHS using geissoschizine as the substrate and quenched either by MeOH or NaBH4. (C) LCMS chromatograms at m/z 353 showing the in vitro catalytic activity of AsRHR1 and AsRHR2 by incubation together with geissoschizine and AsRHS. (D) LCMS chromatograms at m/z 395 showing the in vitro catalytic activity of AKS1 and AKS2 using synthetic rhazimol24 as the substrate. Denatured enzymes were used for negative controls, the peak at m/z 353 with a retention time of 5.9 min corresponds to the substrate (geissoschizine). RHS: rhazimal synthase; RHR: rhazimal reductase; AKS: akuammiline synthase. | ||
The eponym of the akuammilan alkaloid group refers to akuammiline, since it was the first isolated compound with this structural scaffold.4 To close the pathway to akuammiline an acetyltransferase is required (Fig. 1, 2, and S1†). We identified 9 BAHD enzymes in the A. scholaris genome (Fig. S19 and Table S4†) based on homology to previously reported BAHD acetyltransferases in MIA metabolism.25,30,31 The BAHD candidates were assayed by incubation with rhazimol and acetyl-CoA and enzyme assays were analysed by LCMS (Fig. S20†). BAHD3 and BAHD5 exhibited acetyltransferase activity with a new peak appearing at 6.9 min with m/z 395 and based on agreement with retention time, MS and MS2 spectra of synthetic akuammiline10 this peak was identified as akuammiline (Fig. 2D and S21†). As a result, the enzymes BAHD3 (43565_t) and BAHD5 (37058_t) were renamed A. scholaris akuammiline synthase AsAKS1 and AsAKS2, respectively. Both AsAKS1 and AsAKS2 have a rather low sequence identity with the other reported BAHD enzymes (Table S4, Fig. S19, S22†) and share only 28.5% sequence identity between them indicating a different evolutionary lineage compared to that of known BAHD acetyltransferases in MIA metabolism.
To confirm that the three enzymes work together in accordance with the enzyme assay results, we assembled the akuammiline pathway in Saccharomyces cerevisiae by co-expressing AsRHS, AsRHR1-2, and AsAKS1 enzymes. Yeast strains were incubated with geissoschizine, and we screened them by LCMS for rhazimal, rhazimol and akuammiline as metabolic products of yeast (Fig. S23 and Table S5†). The presence of rhazimal and rhazimol was confirmed in all yeast strains, indicating that reductases endogenous to yeast can reduce the aldehyde function of rhazimal to alcohol, whereas akuammiline was detected only in yeast strains expressing the AsAKS1 enzyme indicating the specificity of the enzymatic transformation.
The formation of the C7–C16 bond in geissoschizine by AsRHS is the entry-point to akuammilan alkaloid metabolism. AsRHS and AsGO as biocatalysts play a significant role in diversification of MIA metabolism and bifurcation between the akuammilan and strychnos biosynthetic pathways marking the formation of different structural scaffolds and different chemical spaces. AsRHS and AsGO enzymes share high (62.4%) sequence identity, but they display different enzyme promiscuity and a distinct product profile. These two enzymes can be used to explore the structural basis behind the regioselectivity of geissoschizine cyclizations and to gain insights into those critical transformations; thus, we focused on reciprocal mutations of AsRHS and AsGO. We used the crystal structure of Salvia miltiorrhiza ferruginol synthase (SmFS/CYP76AH1),32 a plant cytochrome P450 acting on diterpenoid metabolism, as a template to build up homology model structures33 of AsRHS and AsGO (Fig. 3A, D, and S24†). We aligned the amino acid sequences of AsRHS, AsGO with those of homologous P450 enzymes CrGO, CrAS, RsSBE, and AsAS (Fig. S25†). Additionally, in silico docking34 was performed to visualize the position of geissoschizine in AsRHS and AsGO and to explore the potential precatalytic binding modes (Fig. S26†). We examined the superimposed models of AsRHS, AsGO and CYP76AH1, and we identified 5 divergent amino acids that potentially convey the diversification of a common precursor geissoschizine towards akuammilan or strychnos scaffolds. Those 5 amino acids belonged to three different substrate recognition sites (SRSs) SRS1, SRS2 and SRS5. The SRSs are considered typical features of cytochrome P450s and they play a role in substrate interaction and catalytic activity.35,36 We conducted the reciprocal mutations of the 5 amino acid residues on both AsRHS and AsGO and the enzymatic activity of mutant enzymes was analysed by LC-tQMS for functional validation (Fig. S27–S30†).
The mutant enzyme AsGOV372F was the only mutant AsGO enzyme that we observed RHS activity in (Fig. 3B and S28†). The mutant AsRHSF372V displayed reduced RHS activity (Fig. S27†) while its GO activity seemed to be favoured (Fig. S29†) showing a significant differentiation in the ratio of catalytic outcome between rhazimol (1)/akuammicine (5) (Fig. S31†). The rest of the four reciprocal mutations of AsGO and AsRHS enzymes did not substantially change the enzymatic product profile (ratio (1)/(5)) (Fig. S31†) though they might affect the catalytic activity by a reduction of product yield (Tables S6–S7†). To further confirm our findings, we assayed the activity of double AsRHS mutants. We observed significant changes in the enzymatic product ratio (1)/(5), only between the enzymes with the F372V mutation (Fig. 3 and S31†). The experimental data showed that the reciprocal mutations on amino acids at position 372 change the enzymatic product profile and imply the key role of F372/V372 amino acids in regioselectivity of cyclization reactions on geissoschizine. The kinetic parameters (Km and vmax) were determined for AsRHS and AsGO and their mutants AsRHSF372V and AsGOV372F (Fig. S32†). Though both wild type enzymes have similar affinity to geissoschizine as the substrate, the AsRHSF372V mutant enzyme shows significantly reduced affinity towards geissoschizine. To rationalize our experimental findings, we looked at the molecular modelling data. The model structures (Fig. 3) demonstrate that the side chains of the two amino acids at position 372 occupy the space between the heme and the substrate (geissoschizine). Prediction of the active site pocket37 displayed the reduced space of active site pocket in AsRHS in comparison to AsGO. It can then be hypothesized that the different side chain sizes of the amino acids at position 372 change the placement of the substrate in the active site and affect the interactions between geissoschizine (substrate) and heme.
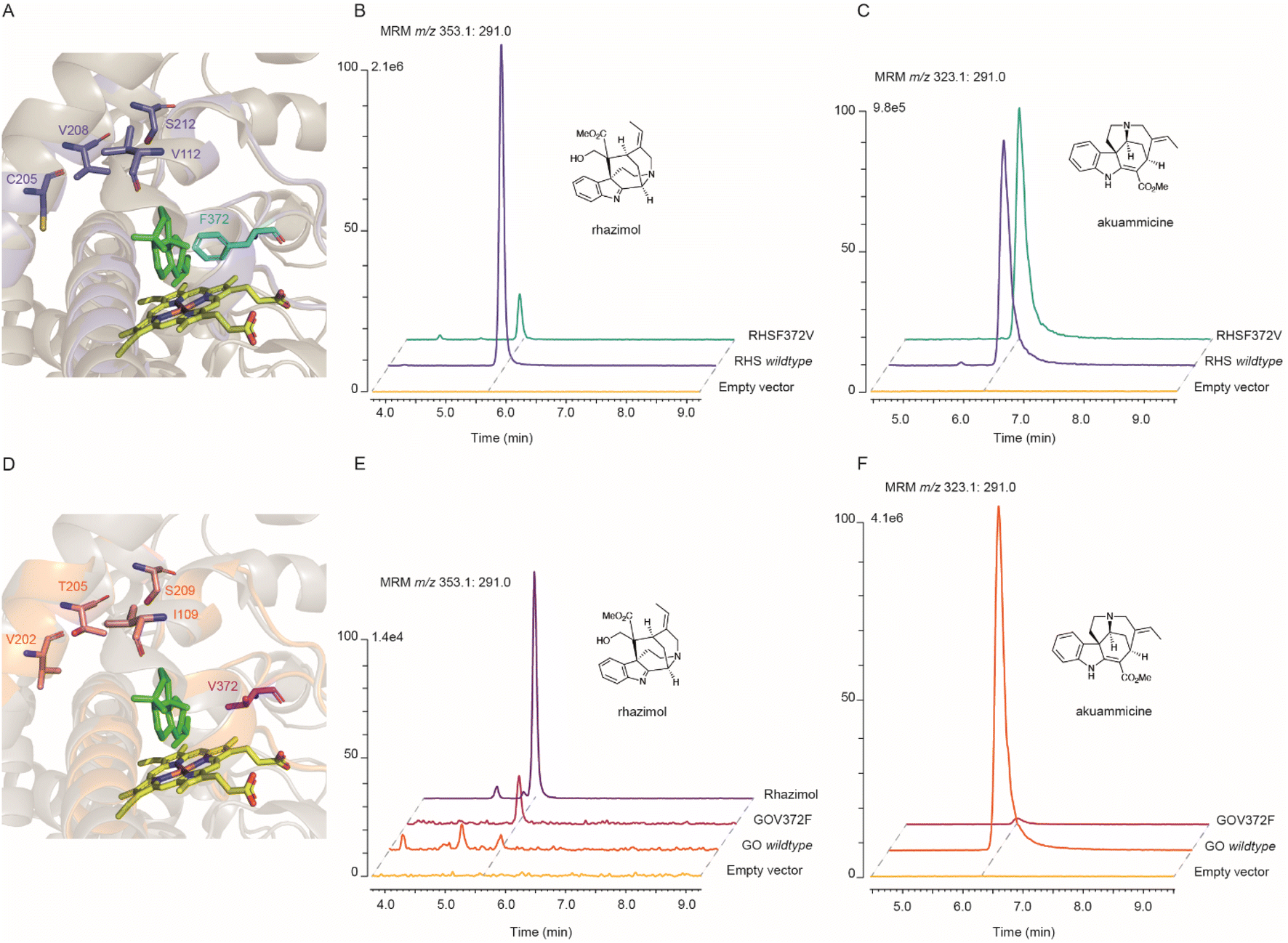 | ||
Fig. 3 Molecular modelling and mutagenesis on active sites of AsRHS and AsGO. (A and D) Overlay of the homology model structures of AsRHS and AsGO and the template X-ray structure 7CB9 of SmCYP76AH1 showing the P450 active site, with the heme (in yellow) and substrate milliradian (in green) from the template structure. The structural elements (SRSs and amino acids) from AsRHS and AsGO are depicted in purple and orange, respectively. The divergent amino acids selected for reciprocal mutations are shown in sticks. The key amino acid residues with side chains occupying the space between the heme and substrate that direct the enzyme assay outcome are highlighted in cyan (AsRHS F372) and magenta (AsGO V372). (B and C) Characterization of rhazimal synthase (B) and geissoschizine oxidase (C) activities by in vitro enzyme assays of the AsRHS wildtype, AsRHSF372V and the empty vector (control) using geissoschizine as the substrate and stopped by NaBH4; assays analyzed by LCMS using MRM chromatographic traces m/z 353.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m/z 291.0 for rhazimol and m/z 323.1:m/z 291.0 for akuammicine. (E and F) Characterization of rhazimal synthase (E) and geissoschizine oxidase (F) activities by in vitro enzyme assays of the AsGO wildtype, AsGOV372F and the empty vector (control) using geissoschizine as the substrate and stopped by NaBH4; assays analyzed by LCMS using MRM chromatographic traces m/z 353.1:m/z 291.0 for rhazimol and m/z 323.1:m/z 291.0 for akuammicine. The intensity of LCMS chromatograms in each panel was normalized to the intensity of the higher peak. :m/z 291.0 for rhazimol and m/z 323.1:m/z 291.0 for akuammicine. (E and F) Characterization of rhazimal synthase (E) and geissoschizine oxidase (F) activities by in vitro enzyme assays of the AsGO wildtype, AsGOV372F and the empty vector (control) using geissoschizine as the substrate and stopped by NaBH4; assays analyzed by LCMS using MRM chromatographic traces m/z 353.1:m/z 291.0 for rhazimol and m/z 323.1:m/z 291.0 for akuammicine. The intensity of LCMS chromatograms in each panel was normalized to the intensity of the higher peak. | ||
Most likely the formation of either akuammilan and strychnos scaffolds by RHS and GO, respectively, proceeds through a two-step mechanism in which the P450s first catalyse the oxidation of geissoschizine followed by a nucleophilic attack from the keto–enol function of geissoschizine, which serves as an excellent nucleophile,21 though it cannot be excluded that these cyclizations may proceed either through a radical mechanism or the formation of rhazimal as a precursor for strychnos scaffold as previously hypothesized.1,18,20 Based on our results, the amino acid residue at position 372 affects the substrate positioning and changes the distances of C-2 and C-7 relative to the heme and therefore controls the reaction and one enzyme may preferentially oxidise C2 (GO), and the other C7 (RHS) of geissoschizine, followed by the release of one H2O molecule and nucleophilic attack of C16 to the carbocation on C2 (GO) or C7(RHS) (Fig. 4 and S33†).
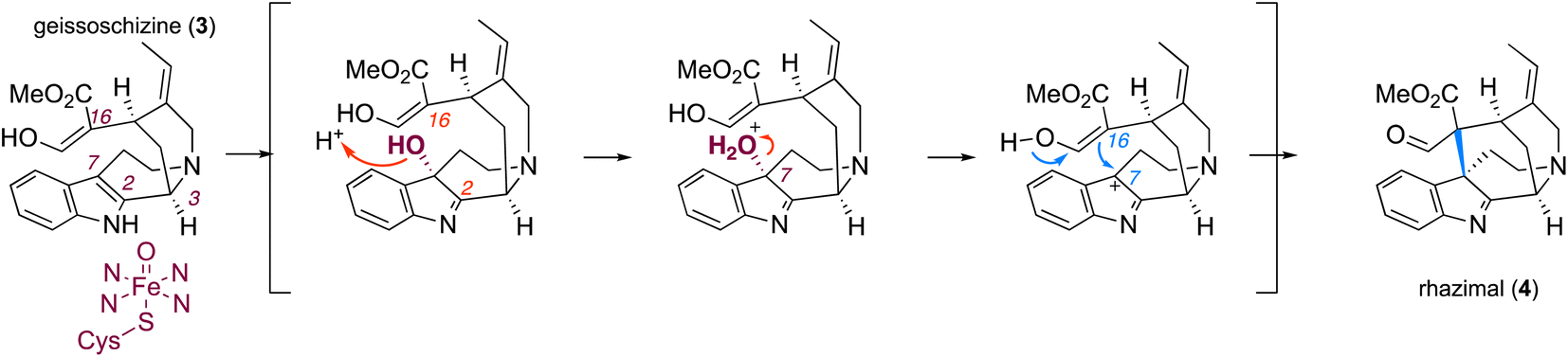 | ||
| Fig. 4 Proposed mechanism for catalytic activity of RHS transforming geissoschizine (3) to akuammilan alkaloid rhazimal (4) and the formation of a methanoquinolizidine scaffold. The plausible mechanism for formation of rhazimal (4) (methanoquinolizidine scaffold) through the oxidation/hydroxylation of geissoschizine on C7 followed by nucleophilic attack by C16. The newly formed bond C7–C16 is highlighted in blue. | ||
There is an increased interest in exploring the medicinal potential of akuammilan alkaloids;3,11,24 however, until the current work their biosynthesis was totally unknown. Here we report the discovery of five new enzymes with novel RHS, RHR and AKS activities that together synthesize akuammiline. These newly identified genes and the demonstrated pathway assembly in yeast can facilitate the future discovery of downstream steps in biosynthesis of akuammilan alkaloids through syntenic analysis of genomic resources and co-expression analysis of transcriptomic data.
Cyclization reactions that create new complex polycyclic scaffolds are generating chemical diversity in specialized metabolism; however, such enzymatic steps in plant specialized metabolism are poorly understood especially when they are catalysed by enzymes that are challenging to recombinantly express and purify such as plant P450 enzymes. We investigated the catalytic mechanism of the two paralogue P450 enzymes AsRHS and AsGO by homology modelling, site-directed mutagenesis, to identify a single amino acid residue that controls the catalysed cyclizations of P450s AsRHS and AsGO by switching the reaction outcome. Our work provides novel insights into the structural elements that control the regioselectivity on geissoschizine cyclizations and suggests a possible unifying mechanism for the formation of akuammilan and strychnos scaffolds. To our knowledge this is the first example of protein engineering and reprogramming of a plant P450 acting on enzymatic formation of new chemical scaffolds in alkaloid biosynthesis.
In conclusion, the discovery of these enzymes demonstrates the astonishing biochemistry that occurs in plants, and the current work will enable future metabolic engineering efforts to overproduce high-value chemicals.
Data availability
The sequence data of the reported genes for this study have been deposited in the National Center for Biotechnology Information (NCBI) database (GenBank accession numbers OM323326-OM323333).Conflicts of interest
The authors declare no conflict of interest.Author contributions
Y. X., Z. W. and E. C. T. designed the experiments and wrote the manuscript. Z. W. and Y. X. conducted the cloning, protein expression, enzyme assays, and construction of mutants. S. W. and E. C. T. collected the plant material. Y. X. and S. W. conducted the bioinformatic analysis. Y. X. performed homology modelling and molecular docking. J.C. traced and provided the resources (plant material). A. L. provided the synthetic rhazimol and akuammiline. All authors read and approved the final manuscript.Acknowledgements
The authors would like to thank the CEMPS Core Facility Center for the excellent support in metabolomics and especially Mrs Xiaoyan Xu and Mrs Shanshan Wang; Dr Tomasz Szmaja for his early work and preliminary results; staff and administration of Xishuangbanna Tropical Botanical Garden (XTBG) for access to A. scholaris plants. The work was financially supported by the National Natural Sciences Foundation of China, Research Fund for International Excellent Young Scientists, grant 32150610477; Strategic Priority Research Program of the Chinese Academy of Sciences grant XDB27020204; Chinese Academy of Sciences, International Partnership Program of CAS grant 153D31KYSB20160074; and National Key Laboratory of Plant Molecular Genetics Special Fund.References
- A. Ramírez and S. García-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef PubMed.
- T. Gaich and R. Eckermann, Synthesis, 2013, 45, 2813–2823 CrossRef.
- J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed. Engl., 2015, 54, 400–412 CAS.
- G. L. Adams and A. B. Smith 3rd, The Alkaloids: Chemistry and Biology, 2016, vol. 76, pp. 171–257 Search PubMed.
- S. E. Kearney, G. Zahoránszky-Kőhalmi, K. R. Brimacombe, M. J. Henderson, C. Lynch, T. Zhao, K. K. Wan, Z. Itkin, C. Dillon, M. Shen, D. M. Cheff, T. D. Lee, D. Bougie, K. Cheng, N. P. Coussens, D. Dorjsuren, R. T. Eastman, R. Huang, M. J. Iannotti, S. Karavadhi, C. Klumpp-Thomas, J. S. Roth, S. Sakamuru, W. Sun, S. A. Titus, A. Yasgar, Y.-Q. Zhang, J. Zhao, R. B. Andrade, M. K. Brown, N. Z. Burns, J. K. Cha, E. E. Mevers, J. Clardy, J. A. Clement, P. A. Crooks, G. D. Cuny, J. Ganor, J. Moreno, L. A. Morrill, E. Picazo, R. B. Susick, N. K. Garg, B. C. Goess, R. B. Grossman, C. C. Hughes, J. N. Johnston, M. M. Joullie, A. D. Kinghorn, D. G. I. Kingston, M. J. Krische, O. Kwon, T. J. Maimone, S. Majumdar, K. N. Maloney, E. Mohamed, B. T. Murphy, P. Nagorny, D. E. Olson, L. E. Overman, L. E. Brown, J. K. Snyder, J. A. Porco, F. Rivas, S. A. Ross, R. Sarpong, I. Sharma, J. T. Shaw, Z. Xu, B. Shen, W. Shi, C. R. J. Stephenson, A. L. Verano, D. S. Tan and Y. Tang, ACS Cent. Sci., 2018, 4, 1727–1741 CrossRef CAS PubMed; R. E. Taylor, R. J. Thomson, D. A. Vosburg, J. Wu, W. M. Wuest, A. Zakarian, Y. Zhang, T. Ren, Z. Zuo, J. Inglese, S. Michael, A. Simeonov, W. Zheng, P. Shinn, A. Jadhav, M. B. Boxer, M. D. Hall, M. Xia, R. Guha and J. M. Rohde, ACS Cent. Sci., 2018, 4, 1727–1741 CrossRef PubMed.
- J. S. Yeap, S. Navanesan, K. S. Sim, K. T. Yong, S. Gurusamy, S. H. Lim, Y. Y. Low and T. S. Kam, J. Nat. Prod., 2018, 81, 1266–1277 CrossRef CAS PubMed.
- C. H. Tan, J. S. Yeap, S. H. Lim, Y. Y. Low, K. S. Sim and T. S. Kam, J. Nat. Prod., 2021, 84, 1524–1533 CrossRef CAS PubMed.
- Z. Yang, L. Sun, C. Liang, Y. Xu, J. Cao, Y. Yang and J. Gu, J. Sep. Sci., 2016, 39, 2652–2660 CrossRef CAS.
- M. S. Khyade, D. M. Kasote and N. P. Vaikos, J. Ethnopharmacol., 2014, 153, 1–18 CrossRef CAS PubMed.
- W. Li, Z. Chen, D. Yu, X. Peng, G. Wen, S. Wang, F. Xue, X. Y. Liu and Y. Qin, Angew. Chem., Int. Ed. Engl., 2019, 58, 6059–6063 CrossRef CAS PubMed.
- E. Picazo, L. A. Morrill, R. B. Susick, J. Moreno, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2018, 140, 6483–6492 CrossRef CAS.
- M. W. Smith, Z. Zhou, A. X. Gao, T. Shimbayashi and S. A. Snyder, Org. Lett., 2017, 19, 1004–1007 CrossRef CAS PubMed.
- R. Eckermann, M. Breunig and T. Gaich, Chemistry, 2017, 23, 3938–3949 CrossRef CAS PubMed.
- J. Moreno, E. Picazo, L. A. Morrill, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 1162–1165 CrossRef CAS PubMed.
- R. Eckermann, M. Breunig and T. Gaich, Chem. Commun., 2016, 52, 11363–11365 RSC.
- E. S. Andreansky and S. B. Blakey, Org. Lett., 2016, 18, 6492–6495 CrossRef CAS PubMed.
- L. Caputi, J. Franke, S. C. Farrow, K. Chung, R. M. E. Payne, T. D. Nguyen, T. T. Dang, I. Soares Teto Carqueijeiro, K. Koudounas, T. Dugé de Bernonville, B. Ameyaw, D. M. Jones, I. J. C. Vieira, V. Courdavault and S. E. O'Connor, Science, 2018, 360, 1235–1239 CrossRef CAS PubMed.
- S. Benayad, K. Ahamada, G. Lewin, L. Evanno and E. Poupon, Eur. J. Org. Chem., 2016, 2016, 1494–1499 CrossRef CAS.
- S. Cheng, M. Melkonian, S. A. Smith, S. Brockington, J. M. Archibald, P. M. Delaux, F. W. Li, B. Melkonian, E. V. Mavrodiev, W. Sun, Y. Fu, H. Yang, D. E. Soltis, S. W. Graham, P. S. Soltis, X. Liu, X. Xu and G. K. Wong, Gigascience, 2018, 7, 1–9 CrossRef PubMed.
- E. C. Tatsis, I. Carqueijeiro, T. Dugé de Bernonville, J. Franke, T. T. Dang, A. Oudin, A. Lanoue, F. Lafontaine, A. K. Stavrinides, M. Clastre, V. Courdavault and S. E. O'Connor, Nat. Commun., 2017, 8, 316 CrossRef PubMed.
- T. T. Dang, J. Franke, I. S. T. Carqueijeiro, C. Langley, V. Courdavault and S. E. O'Connor, Nat. Chem. Biol., 2018, 14, 760–763 CrossRef CAS PubMed.
- K. Yamamoto, D. Grzech, K. Koudounas, E. A. Stander, L. Caputi, T. Mimura, V. Courdavault and S. E. O'Connor, Plant Physiol., 2021, 187, 846–857 CrossRef CAS PubMed.
- X. Zhang, B. N. Kakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed., 2019, 58, 6053–6058 CrossRef CAS PubMed.
- X. Zhang, B. N. Kakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed. Engl., 2019, 58, 6053–6058 CrossRef CAS PubMed.
- Y. Qu, M. Easson, R. Simionescu, J. Hajicek, A. M. K. Thamm, V. Salim and V. De Luca, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3180–3185 CrossRef CAS PubMed.
- J. Franke, J. Kim, J. P. Hamilton, D. Zhao, G. M. Pham, K. Wiegert-Rininger, E. Crisovan, L. Newton, B. Vaillancourt, E. Tatsis, C. R. Buell and S. E. O'Connor, Chembiochem, 2019, 20, 83–87 CrossRef CAS PubMed.
- A. Stavrinides, E. C. Tatsis, L. Caputi, E. Foureau, C. E. Stevenson, D. M. Lawson, V. Courdavault and S. E. O'Connor, Nat. Commun., 2016, 7, 12116 CrossRef CAS PubMed.
- A. Stavrinides, E. C. Tatsis, E. Foureau, L. Caputi, F. Kellner, V. Courdavault and S. E. O'Connor, Chem. Biol., 2015, 22, 336–341 CrossRef CAS PubMed.
- Y. Qu, M. L. Easson, J. Froese, R. Simionescu, T. Hudlicky and V. De Luca, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6224–6229 CrossRef CAS PubMed.
- A. Bayer, X. Ma and J. Stöckigt, Bioorg. Med. Chem., 2004, 12, 2787–2795 CrossRef CAS PubMed.
- B. St-Pierre, P. Laflamme, A. M. Alarco and V. De Luca, Plant J., 1998, 14, 703–713 CrossRef CAS PubMed.
- C. Shi, M. Gu, Z. Chen, X. Huang, J. Guo, L. Huang, J. Deng, K. He, L. Zhang, L. Huang and Z. Chang, Biochem. Biophys. Res. Commun., 2021, 582, 125–130 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–w303 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS.
- A. Seifert and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2009, 74, 1028–1035 CrossRef CAS PubMed.
- Ł. Gricman, C. Vogel and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2015, 83, 1593–1603 CrossRef PubMed.
- A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann and M. Rarey, J. Chem. Inf. Model., 2012, 52, 360–372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03612f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |