
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequence-sorted redox-switchable hetero[3]rotaxanes†
Marius
Gaedke‡
 a,
Henrik
Hupatz‡
a,
Felix
Witte
b,
Susanne M.
Rupf
c,
Clara
Douglas
a,
Hendrik V.
Schröder§
a,
Lukas
Fischer
a,
Moritz
Malischewski
c,
Beate
Paulus
b and
Christoph A.
Schalley
*a
a,
Henrik
Hupatz‡
a,
Felix
Witte
b,
Susanne M.
Rupf
c,
Clara
Douglas
a,
Hendrik V.
Schröder§
a,
Lukas
Fischer
a,
Moritz
Malischewski
c,
Beate
Paulus
b and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie der Freien Universität Berlin, Arnimallee 20, 14195 Berlin, Germany. E-mail: c.schalley@fu-berlin.de
bInstitut für Chemie und Biochemie der Freien Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
cInstitut für Chemie und Biochemie der Freien Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany
First published on 27th October 2021
Abstract
From a library of five crown ether macrocycles with different ring sizes and redox-active moieties, such as tetrathiafulvalene (TTF) and naphthalene dimiide (NDI), directional heterocircuit[3]rotaxanes were constructed. Using an axle with two binding sites with different steric accessibility, the concept of integrative self-sorting was applied to program the sequence of functional units in heteropseudo[3]rotaxanes. Depending on binding strength and ring size of the smaller macrocycles, different heteropseudo[3]rotaxane selectivities and stabilities were determined by 2D NMR spectroscopy and tandem mass spectrometry. A heteropseudo[3]rotaxane with rotaxane-like behaviour was isolated chromatographically, displaying electrochemically “frustrated” properties. A robust synthetic procedure was developed allowing the synthesis of four new hetero[3]rotaxanes incorporating specific sequences of functional units. Sequence pseudoisomeric rotaxanes which have the naphthalene diimide subunit at two different positions show distinct electrochemical properties. DFT calculations suggest that this differences could arise from a folding of the structure, in which the redox-active moieties stack with a stopper unit. This study presents a blueprint for the construction of hetero[3]rotaxanes with sequential control of the functional units along the track of the axle and paves the way to extend the functionality of mechanically interlocked molecules.
Introduction
Mechanically interlocked molecules (MIMs) have shown great potential as artificial molecular switches and motors performing various tasks at the nanoscopic level and rudimentarily mimicking functions of natural molecules.1–3 However, the molecular complexity and thereby accessible functions of MIMs are often limited by their symmetrical structures.4–7 The asymmetrical and directional structure of functional biomolecules derives not only from the centrochirality of their molecular building blocks originating from a defined structural pool of building blocks, such as DNA nucleotides or amino acids, but also from the specific sequences in which these building blocks are connected. This sequence induces for example the secondary and tertiary structure formation in proteins.8 The precise positional control of functional units in the natural assembly processes is remarkable. Only minor changes in the sequence can cause a significantly different structure and hence may change function.9 Therefore, the precise control over the directionality and sequence of subcomponents in molecular assemblies is an important goal when constructing functional molecules.Most MIMs described in the literature are [2]rotaxanes and some examples incorporate directional axles (Fig. 1, top left). Directional axles have been used to build sophisticated molecular machines which operate in a directional fashion. For example, so-called “ribosomal rotaxanes”, incorporate a sequence of amino acids on the track of a directional axle. This amino acid sequence is then sequentially connected to a macrocycle upon chemically induced and directional wheel translation.10–12
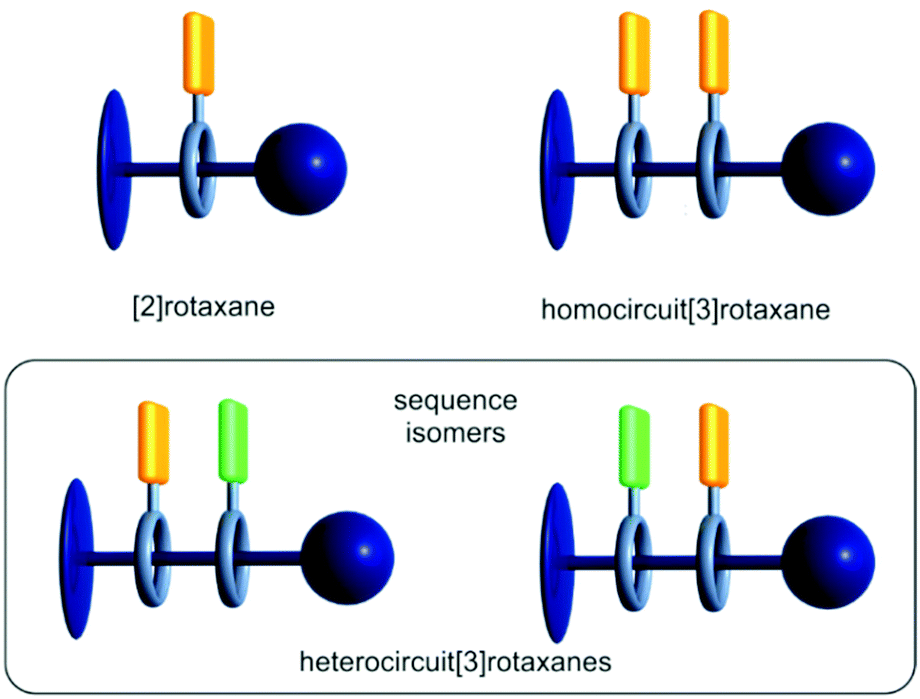 | ||
| Fig. 1 Schematic representation of [2] and [3]rotaxanes containing a directional axle (dark blue) and one or two macrocycles (grey) carrying functional units (orange and green). | ||
In a homocircuit[3]rotaxane, two identical macrocycles are threaded onto the axle and a directionality can again only be defined by the axle itself (Fig. 1, top right). Also, oligohomo[n]rotaxanes containing more than two macrocycles have been described.13–16 In particular, molecular pumps, where multiple macrocycles are threaded onto an axle bringing the system to a higher energy state, are fascinating examples for the application of directional homocircuit[n]rotaxanes.17–19
If the two threaded macrocycles are not identical, a more complex picture arises. In heterocircuit[3]rotaxanes (Fig. 1, bottom), the directionality is additionally described by the order of macrocycles on the directional axle, consequently, sequence isomers may exist.20–23 As a consequence of their unique structure, the isomer-specific synthesis of heterocircuit[3]rotaxanes is not trivial.24 Hence, only few examples of heterocircuitrotaxanes have been described yet.25 In 1995, the first heterorotaxane synthesis was reported by the group of Stoddart applying a sequential threading approach where a [2]rotaxane is synthesised first. In a second step, the second macrocycle is then threaded onto the axle.26 A decade passed with minor advancements in this field due to the synthetic obstacles. However, in the last 15 years the interest in heterocircuitrotaxanes increased and outstanding examples could be realised via new synthetic approaches.24,27–34 The group of Loeb, for example, utilised a sequential threading–clipping method yielding a [3]rotaxane facilitating a ring-through-ring molecular shuttling.35
Fundamentally different are syntheses using the concept of self-sorting. Here, no sequential addition of components defines the sequences, but instead all components are present in one solution, and find their positions in the final structure through the sorting of orthogonal binding motifs.36 Even though homorotaxanes or sequence isomers could form, the macrocycles and axles are structurally programmed to prefer one of the heterorotaxane isomers. The group of Goldup, for example, developed a kinetic self-sorting method to obtain heterocircuit[3]rotaxanes.37 In 2008, the concept of integrative self-sorting was introduced by us for constructing heterorotaxanes using sufficiently different crown/ammonium binding motifs.38,39 This principle allows to program sequences of two macrocycles on one axle, for example by descending ring size accompanying binding site selectivity.36 Later, it was also applied in the synthesis of a [c2]daisy-chain-containing hetero[4]rotaxane40 and a hetero[6]rotaxane.41 In a time-dependent mass spectrometric study of the self-sorting process, the crucial role of error correction and kinetic path selection was demonstrated.42 So far, all examples of heterorotaxanes have a narrow scope of macrocycle combinations and are mostly complex assemblies, but do not contain functional or stimuli-responsive units. The precise arrangement of functional units would pave the way to new molecular switches with emerging properties.
In our recent work on redox-active crown/ammonium complexes we observed that the combination of electron donor tetrathiafulvalene (TTF) and electron acceptor naphthalene diimide (NDI) in a divalent rotaxane yielded a donor–acceptor complex.43 Furthermore, homooligorotaxanes incorporating TTF-decorated crown ethers enabled the understanding of new switching modes in donor–donor assemblies, such as synchronised pirouetting motion of two macrocycles44 and an accordion-like motion of the crown ethers.15 In a study on TTF- and NDI-decorated crown ethers with different cavity sizes, crown[8] ethers revealed a preference for dibenzylammonium and crown[7] wheels for benzyl alkyl ammonium stations, suggesting integrative self-sorting for more functional wheels.45
Herein, we propose a blueprint to sequence-sorted heterocircuit[3]rotaxanes using one directional axle and a library of five macrocycles carrying three different types of functional units. This approach allows for unrestricted selection and combination of the functional units in rotaxane structure. We investigate the influence of different ring sizes and binding strengths of the crown ethers involved on the self-sorting equilibrium and heteropseudo[3]rotaxane properties. We apply these findings to the synthesis of four redox-switchable hetero[3]rotaxanes and study the influence of the sequence of functional units on the spectroelectrochemical properties of the rotaxane assemblies.
Results and discussion
Design of molecular building blocks
The macrocycle library consists of three types of functionalised crown[8] ethers and two types of functionalised crown[7] ethers (Fig. 2): while TTFC8 contains an electron-rich pro-aromatic tetrathiafulvalene with electron-donor property (yellow, “donor”), DBC8 and BC7 exhibit an aromatic system with no significant donor or acceptor properties (grey, “neutral”), and NDIC8 and NDIC7 incorporate an electron-poor aromatic system with electron-acceptor properties (green, “acceptor”). The axle Ax contains two secondary ammonium binding stations which are separated by a phenyl group to prevent threading of the crown[7] ethers to the dibenzyl ammonium station (Fig. 2).38 The directionality of the axle is not only described by the two binding stations, but also, the di-tert-butyl stopper. This stopper prevents any wheel threading from this end of the axle and is crucial for programming the wheel sequence. Weakly coordinating tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF24−) counterions are used to facilitate a stable threaded complex of the weaker binding crown ethers NDIC8, TTFC8 and NDIC7. NDIC7 and axle Ax are novel structures and were synthesised along the procedures given in the ESI (ESI section 1†). For NDIC7 we obtained a crystal structure (ESI section 8†). In the crystal, the dihedral angle between the phenyl group and the NDI is 70.01(11)°, which is considerably flatter than for NDIC845 (84.18(8)°), in order to facilitate π–π interactions between the NDIs of neighbouring NDIC7 in the solid state. The ring sizes of NDIC8, NDIC7 and TTFC8 are wider than those of DBC8 and BC7, respectively, as the 4.766(6) Å (NDIC7)/4.730(4) Å (NDIC8) distance of the two phenolic O atoms in the NDI-substituted macrocycles as well as the S–S distance of 3.3136(13) Å in TTFC846 are substantially larger than the O–O distance of 2.5642(5) Å in DBC8.47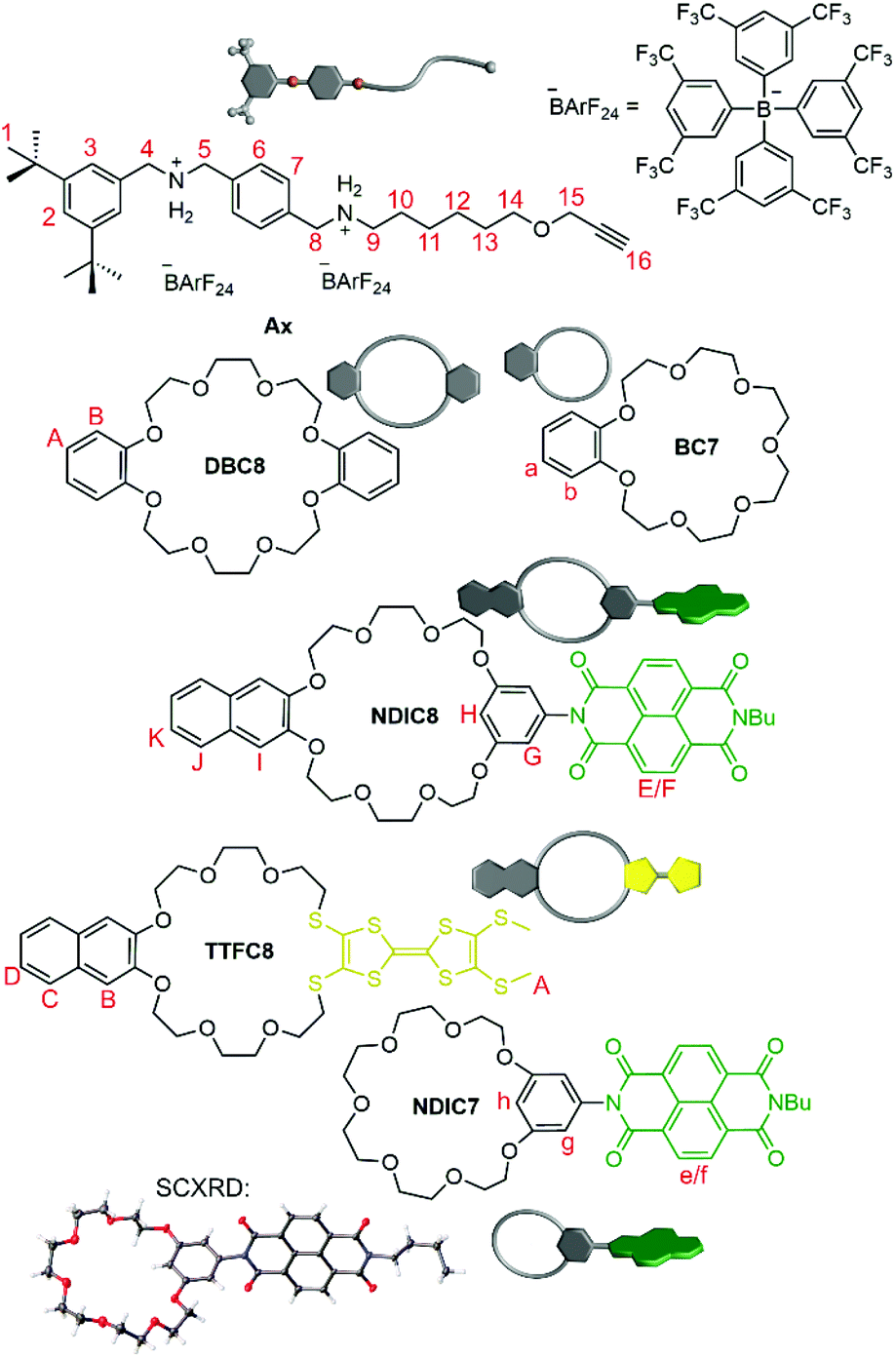 | ||
| Fig. 2 Molecular Axle Ax and crown ether macrocycles used in this work. Protons of crown[8] wheels are denoted with capital letters, whereas crown[7] ether protons are denoted with small letters. | ||
Recently, we have shown that the spectroelectrochemical properties of redox-active crown ethers are not significantly influenced by the crown ether size.45 This allows us to use the crown ether sizes to program the position of the functional unit on the axle without changing the spectroelectrochemical properties of the functional units. Therefore, the hetero[3]rotaxane combinations carrying DBC8/NDIC7 and NDIC8/BC7 are considered as “sequence pseudoisomers”.
The thermodynamics of pseudorotaxane formation has been studied for monovalent model axles by isothermal titration calorimetry, revealing a 10 times stronger binding for DBC8 and BC7 compared to NDIC8 and TTFC8, as well as, NDIC7, respectively (for binding data, see ESI Table S1†). Crown[8] ethers bind to the aliphatic ammonium slightly weaker than the crown[7] analogues.
Self-sorting of pseudorotaxanes
The intermediates and products of the self-assembly of equimolar mixtures of divalent axle and the two differently sized generic crown ethers C8 (pink) and C7 (blue) are shown schematically in Fig. 3a, where homopseudo[3]rotaxane PRC8C8 and pseudo[2]rotaxane PRC7 represent dead-ends on the way to the desired heteropseudo[3]rotaxane PRC8C7. Error-correction is necessary to overcome these dead-ends.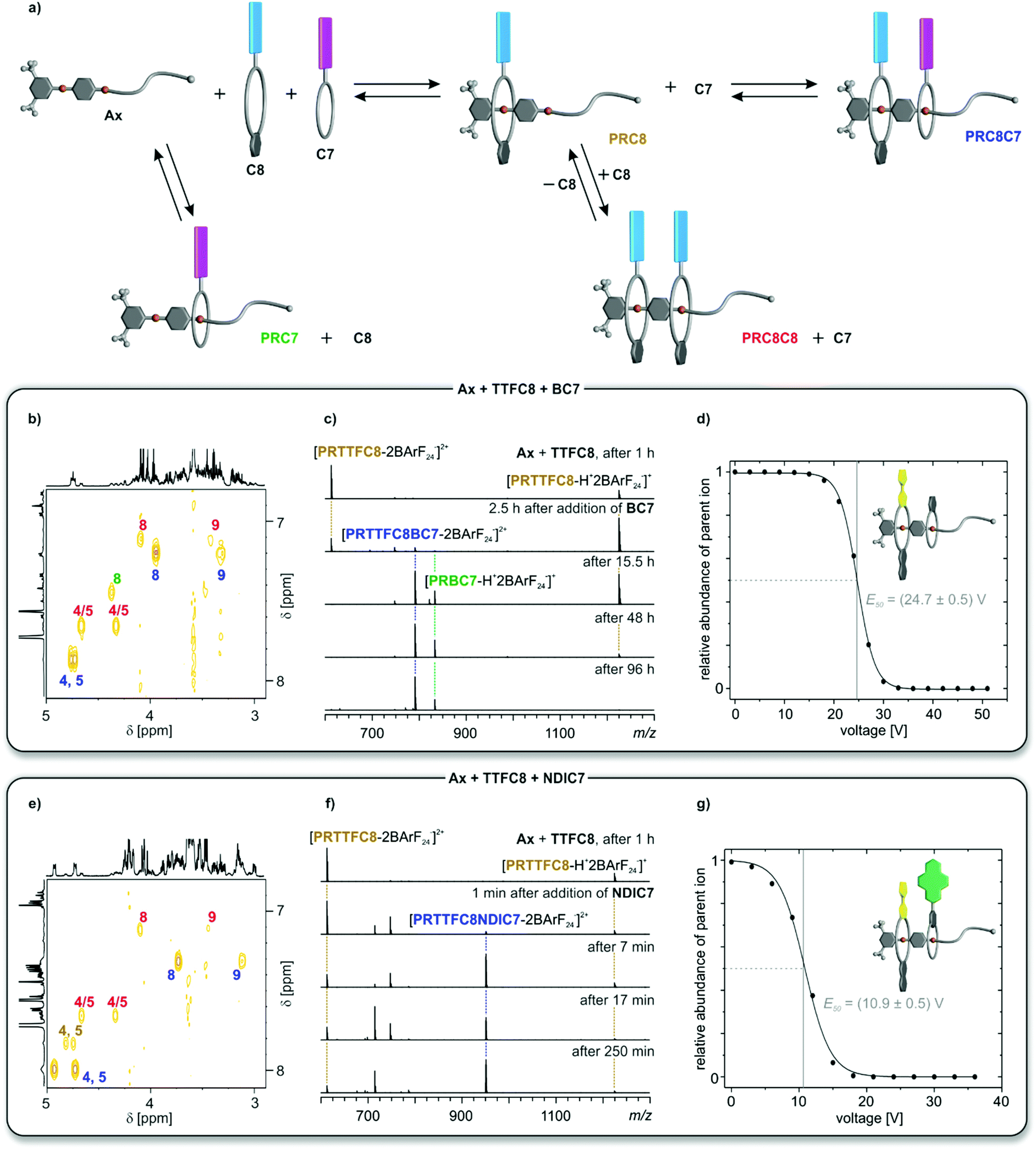 | ||
| Fig. 3 (a) Schematic representation of the self-sorting equilibrium for two generic crown ethers C8 (pink) and C7 (blue). Spectra in (b, c and d) use an equimolar solution of TTFC8, BC7 and Ax; and in (e, f and g) of TTFC8, NDIC7 and Ax. Signals of protons or ions for the heteropseudo[3]rotaxanes are labeled in blue, for the homopseudo[3]rotaxanes in red, for the pseudo[2]rotaxane PRTTFC8 in brown and to the crown[7] pseudo[2]rotaxanes in green. (b and e) Partial 1H,1H COSY spectra (700 MHz, CD2Cl2, 298 K, 5 mM, 16 d). For full 1H NMR spectra and integration see Fig. S24 and S26.† (c and f) Time-dependent ESI-MS spectra in CH2Cl2 at 293 K. Non-labeled peaks correspond to different crown ether ions (ESI section 4†). (d and g) Survival yield curves obtained for mass-selected ions of PRTTFC8BC7m/z 791 (d) and PRNDIC8BC7m/z 951 (g) obtained from CID experiments at increasing collision voltages. Solid lines represent a sigmoidal fitting to determine 50% survival yield voltages E50 (Table 1 and ESI section 4†). | ||
For all six combinations of one C8, one C7 and Ax, equimolar solutions were equilibrated for 16 days at room temperature after which no further change in the NMR spectra was observed. Although a full assignment of all signals in the highly complex 1H NMR spectra is not straightforward due to severe signal overlap (ESI section 3†), the four methylene signals (H(4), H(5), H(8) and H(9)) can be identified in the COSY spectra and are highly indicative for pseudorotaxane formation (Fig. 3b and e and ESI section 3†). For the mixture containing TTFC8, BC7 and Ax, three major species could be identified and assigned by the chemical shift of their methylene protons to the heteropseudo[3]rotaxane PRTTFC8BC7 (Fig. 3b, blue), homopseudo[3]rotaxane PRTTFC8TTFC8 (Fig. 3b, red) and pseudo[2]rotaxane PRBC7 (Fig. 3b, green). Changing the smaller macrocycle to the weaker binding NDIC7 a higher amount of heteropseudo[3]rotaxane PRTTFC8NDIC7 (Fig. 3e, blue) is observed. While the pseudo[2]rotaxane PRNDIC7 cannot be identified, a small fraction of pseudo[2]rotaxane PRTTFC8 (Fig. 3e, brown) is visible. The other four combinations show similar results (ESI section 3†). As calculated from the integrals of the methylene protons H(4) and H(5), the heteropseudo[3]rotaxanes are preferentially formed with amounts of 56–74% in all six cases (Table 1). For a non-self-sorting system with a directional axle a statistical amount for one specific heteropseudo[3]rotxane of 25% would be expected. The weaker binding macrocycle NDIC7 forms the corresponding heteropseudo[3]rotaxanes with higher selectivity as compared to BC7.
| Macrocycle combination | DBC8 | TTFC8 | NDIC8 | DBC8 | TTFC8 | NDIC8 |
|---|---|---|---|---|---|---|
| BC7 | BC7 | BC7 | NDIC7 | NDIC7 | NDIC7 | |
| a Calculated from signal integration in 1H NMR experiments (700 MHz, CD2Cl2, 298 K, 5 mM, 16 d). Amounts are given as percentages of all identified pseudorotaxane species (estimated error: 5%). b Qualitative estimation from time-dependent ESI-MS, when 90% of the maximum heteropseudo[3]rotaxane concentration is reached. c Estimated error amounts to 0.5 V. | ||||||
| Hetero[3]pseudorotaxane amounta | 58% | 56% | 57% | 72% | 70% | 74% |
| Threading timeframeb | Days | Days | Days | Hours | Hours | Hours |
E
50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
21.7 V | 24.7 V | 27.1 V | 9.4 V | 10.9 V | 11.4 V |
Due to the charge inherent in all pseudorotaxanes under study, electrospray ionisation mass spectrometry (ESI-MS) is a valuable additional analytical technique to study the self-sorting equilibrium (Fig. 3c and f and ESI section 4†). Due to differences in ionisation efficiencies, the overall abundances cannot be directly related to solution concentrations, yet a semiquantitative overview of the formation of the pseudorotaxanes with reaction time can certainly be obtained. A solution of Ax and TTFC8 was equilibrated for 1 h. In the ESI mass spectrum, pseudo[2]rotaxane PRTTFC8 can be observed in the two +1 and +2 charge states (Fig. 3c). The homopseudo[3]rotaxane PRTTFC8TTFC8 is not observed in the mass spectrum, as it forms PRTTFC8 rapidly upon dilution and ionisation due to the low thermodynamic and kinetic stability of PRTTFC8TTFC8. After addition of BC7, heteropseudo[3]rotaxane PRTTFC8BC7 forms only slowly on an hour time scale, as the threading of BC7 is a slow process. At the same time, also the dead-end pseudo[2]rotaxane PRBC7 is formed.
In marked contrast, hetero[3]pseudorotaxane PRTTFC8NDIC7 forms already within minutes upon addition of the wider NDIC7 to the solution of Ax and TTFC8. After 4 h virtually no changes are observed in the spectrum (Fig. 3f). The same experiments with the other crown[8] ethers revealed that the equilibrium for NDIC7-containing mixtures are reached after a couple of hours, while mixtures including BC7 take several days to equilibrate (Table 1 row 3 and ESI section 4†). This difference can be rationalised by the bigger ring size and thereby faster threading kinetics of NDIC7. Slower threading kinetics come together with a slower error correction process, as it can also be seen by the appearance of dead-end PRBC7 in the mass spectrum as well as the 1H NMR experiments. The different crown[8] ethers do not significantly alter the overall threading kinetics and heteropseudo[3]rotaxane PRC8C7 selectivity, as their threading is comparably fast.42
Collision-induced dissociation (CID) experiments with mass-selected heteropseudo[3]rotaxane ions were used to establish the sequence of the pseudorotaxanes and to exclude non-threaded binding (ESI section 4†). For all heteropseudo[3]rotaxanes incorporating NDIC7, the CID mass spectra showed the loss of the crown[7] ether as the first fragmentation step (ESI Fig. S39–S41†). In contrast, BC7 containing heteropseudo[3]rotaxane ions fragment by breaking of a covalent bond first (ESI Fig. S36–S38†). The 50% survival yield voltages E50 obtained from the CID mass spectra allow to compare the gas-phase stabilities of the heteropseudo[3]rotaxane assemblies (Fig. 3d and g, Table 1 and ESI Fig. S42†). PRTTFC8BC7 exhibits a significantly higher gas phase stability as compared to PRTTFC8NDIC7 (Fig. 3d and g). Also, E50 mainly is impacted by the threaded crown[7] ether (Table 1 row 4). Taking into account, that the isolated ions are doubly charged, the E1/2 values of all six heteropseudo[3]rotaxane ions are significantly higher than for a non-threaded crown/sec-ammonium complex.46 Overall, these observations evidence the desired sequence and threaded structures were achieved in all six heteropseudo[3]rotaxane combinations.
These results clearly show that the second, smaller macrocycles NDIC7 and BC7 dominate the thermodynamic and kinetic properties of the self-sorting equilibrium. The benefit of faster threading kinetics for a self-sorting process can be highlighted by the observation of a higher selectivity for the NDIC7 containing heteropseudo[3]rotaxanes. The higher binding constants achieved by using BArF24− counterions compared to previous studies38,39,42 where PF6− counterions were used, caused a higher kinetic barrier for the dethreading reaction of BC7, which is the crucial step for all error-correction processes, resulting in a comparably long equilibration time. Yet, the used conditions are robust and facilitate the formation of heteropseudo[3]rotaxanes for all combinations.
A cascade-stoppered pseudo[3]rotaxane
The slow self-sorting process together with the remarkably high gas phase stability of BC7-containing heteropseudo[3]rotaxanes motivated us to study PRTTFC8BC7 in a greater detail. Although a few exceptions are known,34,46 crown/ammonium pseudorotaxanes are usually not sufficiently stable to survive column chromatography. Therefore, it is surprising that the pseudorotaxane PRTTFC8BC7 was chromatographically isolated in 24% yield. For all other combinations of wheels under study here, the formed complexes dissociate on the silica column. Pseudo[3]rotaxanes with NDIC7 dissociated quickly. The two other BC7-containing pseudo[3]rotaxanes required more polar mobile phases, which undermine their stability so that they also dissociate during chromatography. The stronger binding wheel BC7 on the outer binding site acts as a stopper for the weaker binding wheel TTFC8 on the dibenzyl ammonium binding site. Pseudo[3]rotaxanes of two TTFC8 on Ax could not be isolated as they fell apart during the purification. This underlines the exceptional importance of the sequence in which the macrocycles are threaded on the one-side stoppered axle for the physico-chemical properties of the assembly.For comparison, the rotaxane RTTFC8BC7 was synthesised in 15% yield by adding 1.5 equiv. of 2,6-dimethoxybenzonitrile oxide stopper St1 and stirring for one day at 35 °C. Fig. 4 shows the 1H NMR shifts of (P)RTTFC8BC7 in comparison to the axle Ax. Characteristic shifts include benzylic methylene protons H(4/5) and H(9), which shift downfield (Δδ ≥ +0.4 ppm and +0.2 ppm) upon threading of TTFC8 and BC7. Additionally, methylene protons H(8) shift upfield by 0.3 ppm. Both is true for the pseudo[3]rotaxane PRTTFC8BC7 and the [3]rotaxane RTTFC8BC7. A strong downfield shift by 3.9 ppm of the former alkyne proton H(16) is observed, as it is incorporated into the newly formed isoxazole moiety. The methylene group H(15) adjacent to the former alkyne also shifts by 0.4 ppm downfield. Except for the protons on stopper St1 and H(15,16), the shifts of pseudorotaxane PRTTFC8BC7 and rotaxane RTTFC8BC7 are similar.
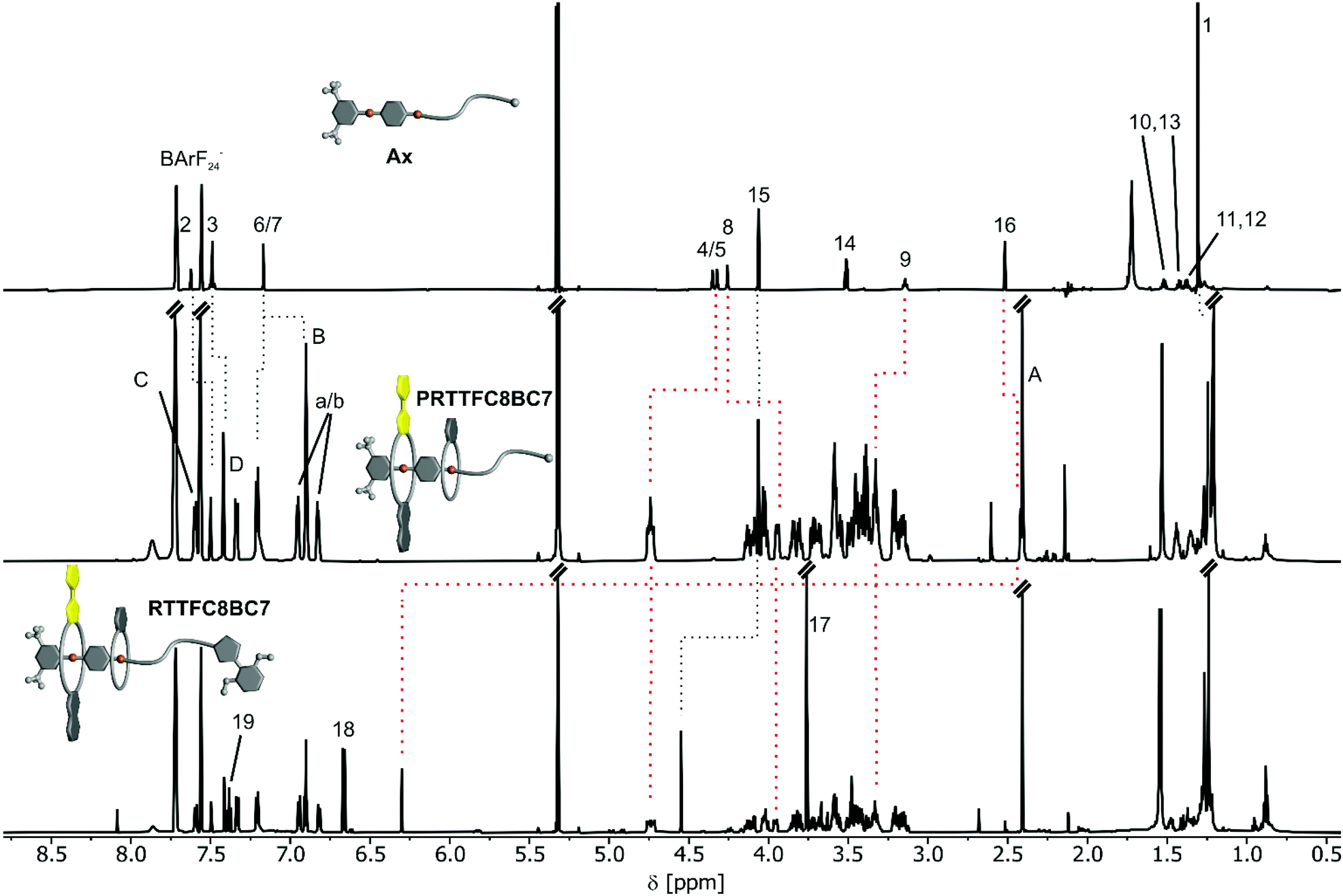 | ||
| Fig. 4 1H NMR comparison of free axle Ax, PRTTFC8BC7 and RTTFC8BC7. Characteristic shifts are highlighted by red dotted lines (ESI section 1.4† for full signal assignment). | ||
To test the stability in a more polar solvent, chromatographically isolated PRTTFC8BC7 was dissolved in acetonitrile in high dilution (1 μM). After 6 h a significant amount of threaded PRTTFC8BC7 could still be observed by ESI-MS (ESI Fig. S43†). It took multiple days to reach thermodynamic equilibrium in which virtually no complex has survived, evidencing the high kinetic stability and nearly rotaxane-like behaviour of PRTTFC8BC7.
Electrochemical characterisation of both species (P)RTTFC8BC7, by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) show similar results (Fig. 5). The two reversible oxidations of TTF are anodically shifted, as expected for wheel TTFC8 bound to an ammonium site.48 In previous work, we reported that the ammonium site is expelled upon oxidation due to coulombic repulsion with the oxidised TTF unit. Electrochemically induced dethreading results in a new signal for the TTF˙+/2+ redox couple of unbound TTFC8 at ∼1 V against decamethylferrocene/decamethylferrocenium (Fc*0/+).48 Sterically demanding end groups can kinetically hamper oxidation-induced dethreading efficiently.46 However, for both, pseudorotaxane and rotaxane, no dethreading is observed and redox potentials vary only marginally. This underlines the remarkable stability, which is exceptional for a non-interlocked crown/ammonium complex, and rotaxane-like behaviour of PRTTFC8BC7 as already observed in the CID measurements. The increased TTF oxidation potentials (+126 and 362 mV for first and second oxidation) together with its unability to dethread turn PRTTFC8BC7 it into an electrochemically “frustrated” pseudorotaxane that is kinetically trapped in a metastable state.
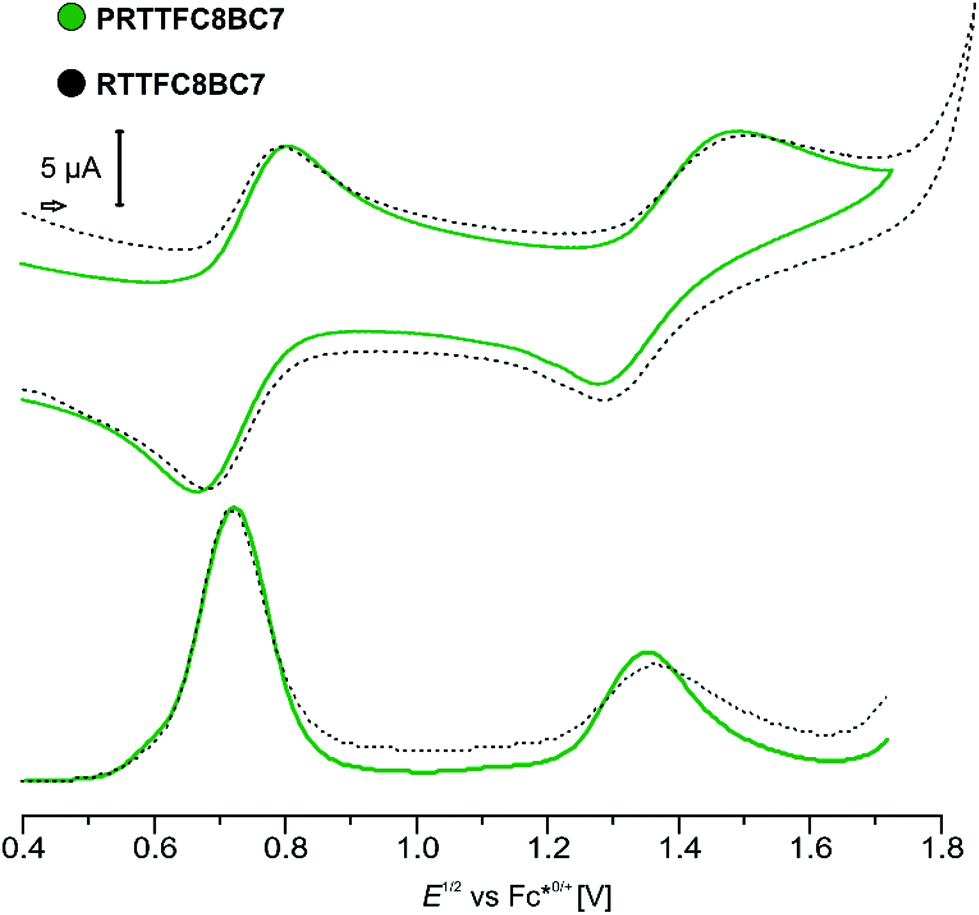 | ||
| Fig. 5 CV and DPV of (pseudo)hetero[3]rotaxanes (P)RTTFC8BC7 (1,2-dichloroethane with n-Bu4NBArF24 as the electrolyte (0.1 M) 1.5 mM analyte, 25 mV s−1 scan rate for CV and 225 mV modulation amplitude, 50 ms modulation time, 5 mV step potential and 0.5 s interval time for DPV). | ||
Hetero[3]rotaxane synthesis
The self-sorting experiments show pseudohetero[3]rotaxane formation to be prominent for all C8/C7 macrocycle combinations. We therefore developed a generally applicable synthetic procedure for the synthesis of the corresponding hetero[3]rotaxanes, which does not depend on a prior chromatographic purification of the pseudo[3]rotaxane precursors. The pseudorotaxane mixtures were treated with the 2,6-dimethoxybenzonitrile stopper St1. This reaction was optimised by using up to six equivalents of the smaller macrocycle relative to the axle to reduce homo[3]rotaxane formation (for detailed synthetic procedures and characterisation data see ESI section 1.4†). Afterwards, the excess of the smaller macrocycle was recycled. Fig. 6 shows the isolated hetero[3]rotaxanes which all feature the diagnostic 1H NMR shifts upon threading and rotaxanation as well as the diastereotopic splitting of the crown ether methylene groups as described in the previous chapter (ESI Fig. S14, S17 and S20†). The desired macrocycle sequence and the mechanically interlocked structure of the four hetero[3]rotaxanes was confirmed by CID experiments with mass-selected hetero[3]rotaxane ions, where at very high collision energies axle cleavage is observed as the major fragmentation pathway (ESI Fig. S49–S52†). The isolated examples consist of a “donor-neutral” (yellow-grey), “donor–acceptor” (yellow-green) and the pseudo sequence isomers “acceptor-neutral” (green-grey) and “neutral-acceptor” (grey-green) hetero[3]rotaxanes. The isolated yields compare very well to the majority of isolated heterorotaxanes in the literature.26,28,35,37 Additionally, the isolation of hetero[3]rotaxanes with different wheel combinations without self-sorting would certainly be a very tedious process.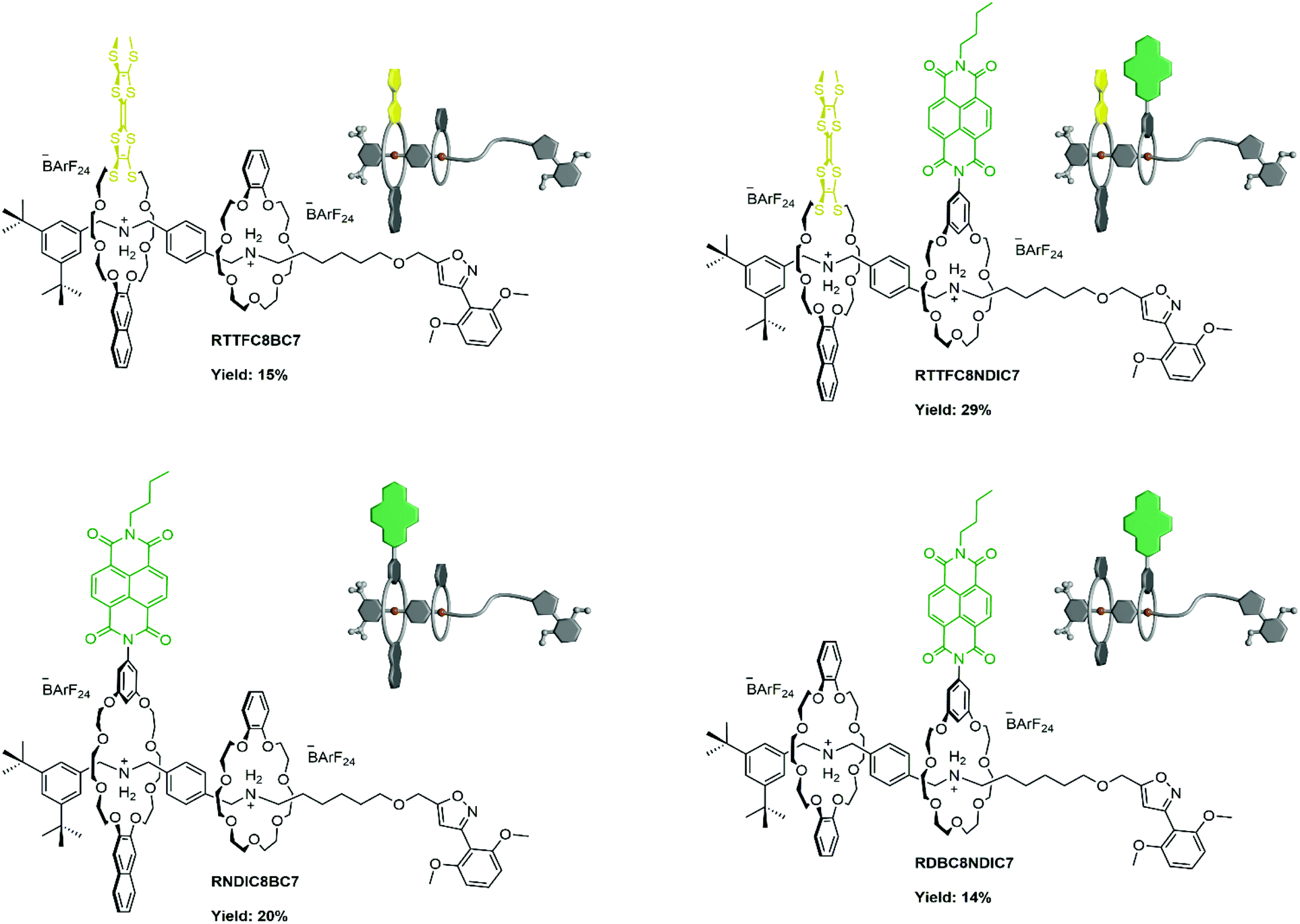 | ||
| Fig. 6 Hetero[3]rotaxanes synthesised from the pseudorotaxane mixtures and their isolated yields. | ||
Optoelectronic properties of sequence-sorted rotaxanes
A comparison between isolated hetero[3]rotaxanes and the free macrocycles TTFC8, NDIC7 or NDIC8 allow an assessment of the influence of the macrocycle sequence on the electrochemical properties. Fig. 7 summarises all peak potentials obtained by DPV measurements in 1,2-dichloroethane with n-Bu4NBArF24 as supporting electrolyte (ESI Table S2†). The potentials are referenced against the Fc*0/+ redox-couple. The peaks at positive potentials can be attributed to the two reversible redox waves, TTF(0/˙+) and TTF(˙+/2+). As previously shown,48 all species with TTFC8 bound to an axle are shifted anodically in a comparable fashion. The second macrocycle does not have strong impact on the oxidation of TTFC8 in the rotaxane as can be seen on the similar oxidation potentials of donor–acceptor rotaxane RTTFC8NDIC7 and donor-neutral rotaxane RTTFC8BC7.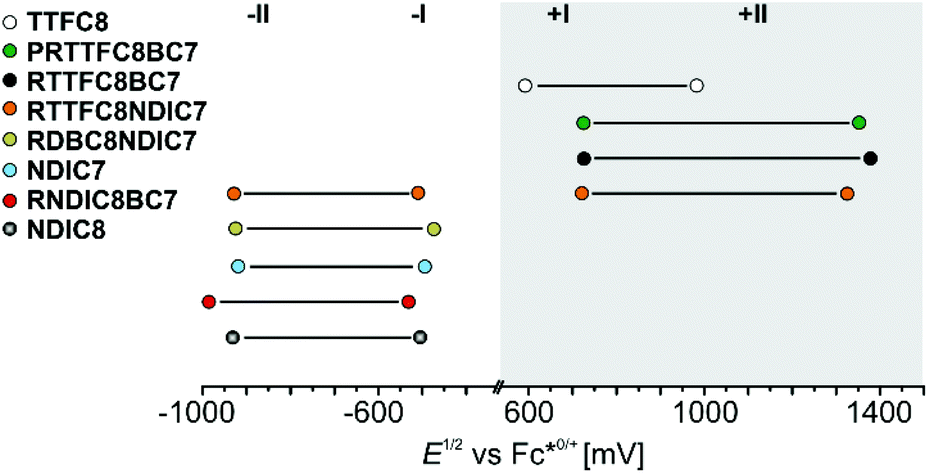 | ||
| Fig. 7 Correlation diagram of half-wave potentials for hetero[3]rotaxanes and free macrocycles determined by DPV (Table S2,† the error is estimated to be ±10 mV). | ||
In contrast, the peaks at negative potentials are the two reversible reductions NDI(0/˙−) and NDI(˙−/2−). For the free wheels NDIC7 and NDIC8 both reduction peaks are similar (ΔE1/2 = 11 mV for NDI(0/˙−) and ΔE1/2 = 16 mV for NDI(˙−/2−)), as expected from their similar structures. For [3]rotaxane RNDIC8BC7 a significant cathodic shift of 20 mV for NDI(0/˙−) and 55 mV for NDI(˙−/2−) compared to wheel NDIC8 is observed. In contrast, [3]rotaxanes RTTFC8NDIC7 and RDBC8NDIC7 only show minor differences in the reduction potentials (ΔE1/2 ≤ 10 mV for NDI(0/˙−) and ΔE1/2 ≤ 11 mV for NDI(˙−/2−)) compared to wheel NDIC7. Consequently, the position of the NDI-unit on the axle indeed has a significant impact on the electrochemical properties. For NDIC7 we could not determine a significant influence of the second larger macrocycle neither being neutral nor electron donor. The difference of electrochemical properties of the NDI sequence pseudoisomers RNDCI8BC7 and RDBC8NDIC7 presumably occurs as a consequence of the sequence rather than the macrocycle combination. Indeed, DFT calculations and conformational analysis at the PBEh-3c level49 using the COSMO50 solvation model provide a molecular structure for RNDIC8BC7, where the NDI-unit stacks with the dimethoxybenzene stopper (Fig. 8a and ESI section 7†).
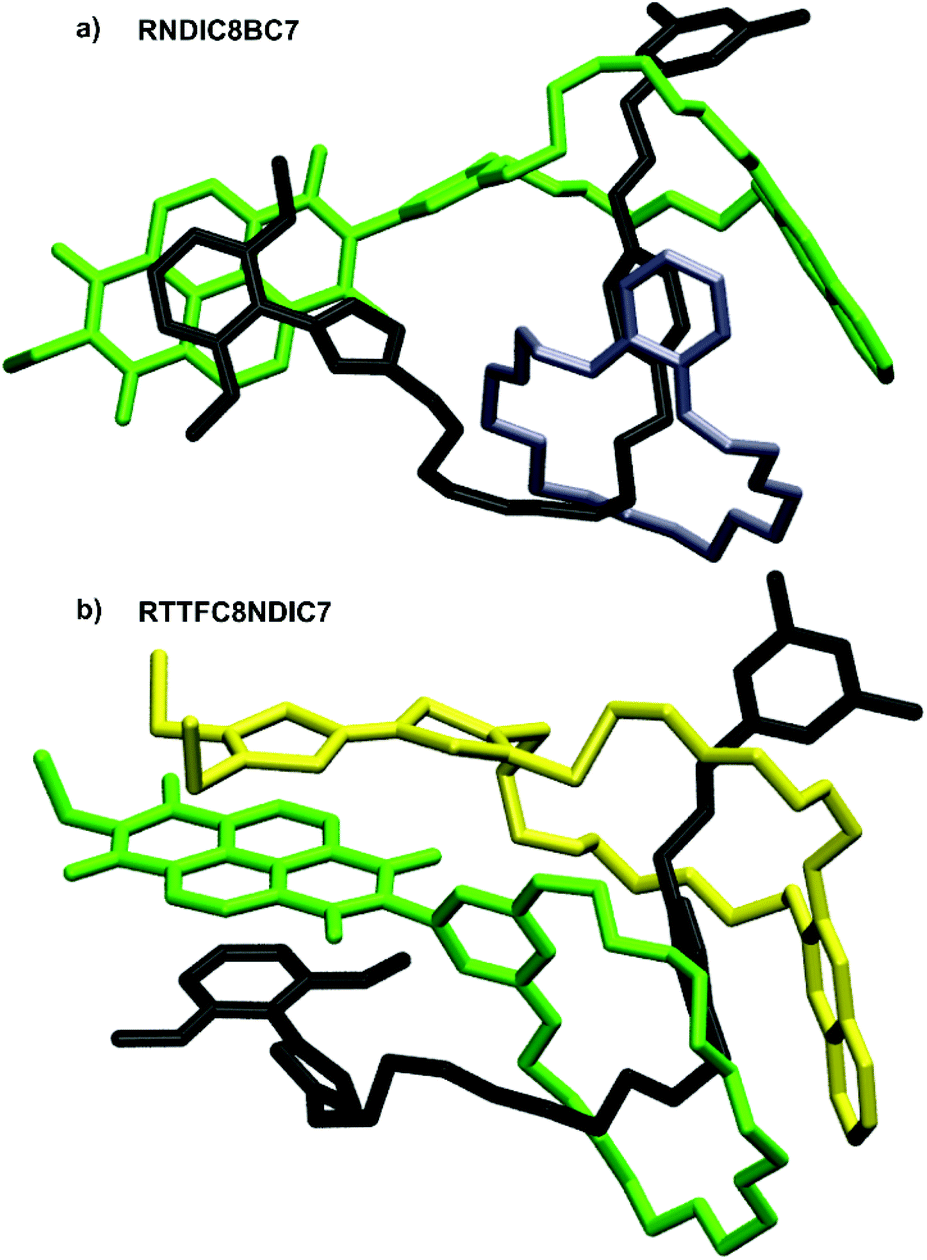 | ||
| Fig. 8 Calculated structures of (a) RNDIC8BC7 and (b) RTTFC8NDIC7, with the axle denoted in black, TTFC8 in yellow, BC7 in grey and NDIC7 as well as NDIC8 in green. | ||
For donor–acceptor hetero[3]rotaxane RTTFC8NDIC7 the calculated structure displays a stacked conformation of TTF- and NDI-unit as well as the dimethoxybenzene stopper (Fig. 8b and ESI section 7†). Even though it is rather difficult to understand the exact reason for the different potential shifts of the NDI-unit, the distance of NDI-bearing crown ether to the stopper unit seems to have an influence. Yet, spin-density plots are TTF-localised for the oxidised species and NDI-localised for the reduced species (ESI Fig. S60 and S61†). Chemical oxidation of RTTFC8NDIC7 shows similar bands in the UV/Vis spectra as those of the free wheel TTFC8 for the three redox states of TTF (ESI section 6†). Together with the electrochemical data, these observations demonstrate the spectroelectric properties of TTFC8 to be retained within the mechanically interlocked assembly.
Conclusions
We demonstrated how integrative self-sorting can be applied as a molecular programming language to dictate the sequence of redox-switchable units along a molecular track. For the first time, we integrated functional redox-switchable units, namely TTF and NDI, into hetero[3]rotaxanes using this strategy.The properties of the self-sorting process are dominated by the second macrocycle in the sequence. While the wider and weaker binding NDIC7 facilitates fast threading kinetics, as well as good hetero[3]rotaxane selectivity, the smaller and stronger binding BC7 results in slow threading kinetics and a slow error correction as well as in lower selectivities for the heteropseudo[3]rotaxane. The BC7-containing heteropseudo[3]rotaxanes show a surprising stability. The pseudorotaxane PRTTFC8BC7, could even be isolated by chromatography and thus shows rotaxane-like behaviour with BC7 being a pseudostopper. Upon TTF oxidation, an electrochemically “frustrated” pseudorotaxane forms, in which the larger wheel experiences charge repulsion with the axle ammonium station, but is unable to dethread.
With the synthesis of four new hetero[3]rotaxanes, we demonstrate that different functional units can be placed in different sequences on the axle, as long as one crown[8] ether and one crown[7] ether is used. While for each individual hetero[3]rotaxane combination more selective conditions such as counterion, solvent or equilibration time, can be found, our synthetic approach is general and robust and facilitates the synthesis of hetero[3]rotaxanes with varying crown ether sizes, binding strength and functional units. This concept is applicable to other functionalised crown ethers. By the ease to decorate crown ethers51 with various functional moieties, our approach represents a modular strategy to incorporate and combine a broad variety of such units using hetero[3]rotaxanes as the scaffold.
In our work the functional units were combined to generate a donor-neutral (RTTFC8BC7), a donor–acceptor (RTTFC8NDIC7), an acceptor-neutral (RNDIC8BC7) and a neutral-acceptor (RDBC8NDIC7) rotaxane. Studying the electrochemical properties of theses assemblies, we could show that the donor moiety (TTF) was marginally influenced. Instead, the acceptor moiety (NDI) showed shifted redox potentials depending on the position on the axle, probably depending on the availability of interactions to one of the stoppers. Regardless of their sequence, the redox-active units retain their reversible spectroelectrochemical properties upon incorporation into the mechanically interlocked structure. Using these results, highly directional functional molecules bearing different macrocycles can be envisioned and facilitate new switching modes or functions for rotaxane assemblies.
Author contributions
The project was developed by H. H. and M.G. H. H., M. G. and C. A. S. wrote the manuscript with main contributions from H. H. and M. G. The synthetic work was carried out by M. G., C. D., S. M. R., L. F. and H. H. with main contributions coming from M. G. H. H. conducted and evaluated all time dependent NMR and tandem MS experiments. M. G. performed further NMR, UV/Vis and CV/DPV experiments and prepared the single crystals. H. V. S. helped with data evaluation and conception. H. H. measured and analysed the ITC data. All computational work was done by F. W. and B. P. S. M. R. and M. M. measured and solved the SCXRD data. Both M. G. and H. H. contributed equally and have the right to list their name first in their CV. All Authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sebastian Müller for help with the synthesis. We are grateful for funding from the Deutsche Forschungsgemeinschaft (DFG; project number 434455294) and acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. Furthermore, F. W. and B. P. acknowledge the DFG for funding (PA 1360/16-1) and the North-German Supercomputing Alliance (Norddeutscher Verbund für Hoch- und Höchstleistungsrechnen) for providing computational resources.Notes and references
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, Molecular Pumps and Motors, J. Am. Chem. Soc., 2021, 143, 5569–5591 CrossRef CAS PubMed.
- L. Zhang, V. Marcos and D. A. Leigh, Molecular machines with bio-inspired mechanisms, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397–9404 CrossRef CAS.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Great expectations: can artificial molecular machines deliver on their promise?, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi and A. Credi, Making and Operating Molecular Machines: A Multidisciplinary Challenge, ChemistryOpen, 2018, 7, 169–179 CrossRef CAS.
- C. A. Schalley, K. Beizai and F. Vögtle, On the Way to Rotaxane-Based Molecular Motors: Studies in Molecular Mobility and Topological Chirality, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS.
- N. Badi and J. F. Lutz, Sequence control in polymer synthesis, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- M. Kosloff and R. Kolodny, Sequence-similar, structure-dissimilar protein pairs in the PDB, Proteins, 2008, 71, 891–902 CrossRef CAS.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Sequence-specific peptide synthesis by an artificial small-molecule machine, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, Efficient assembly of threaded molecular machines for sequence-specific synthesis, J. Am. Chem. Soc., 2014, 136, 5811–5814 CrossRef CAS PubMed.
- C. M. Wilson, A. Gualandi and P. G. Cozzi, A rotaxane turing machine for peptides, ChemBioChem, 2013, 14, 1185–1187 CrossRef CAS PubMed.
- A. J. Avestro, D. M. Gardner, N. A. Vermeulen, E. A. Wilson, S. T. Schneebeli, A. C. Whalley, M. E. Belowich, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Gated electron sharing within dynamic naphthalene diimide-based oligorotaxanes, Angew. Chem., Int. Ed., 2014, 53, 4442–4449 CrossRef CAS PubMed.
- D. Sluysmans, S. Hubert, C. J. Bruns, Z. Zhu, J. F. Stoddart and A. S. Duwez, Synthetic oligorotaxanes exert high forces when folding under mechanical load, Nat. Nanotechnol., 2018, 13, 209–213 CrossRef CAS.
- H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Accordion-Like Motion in Electrochemically Switchable Crown Ether/Ammonium Oligorotaxanes, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 CrossRef.
- H. Y. Zhou, Q. S. Zong, Y. Han and C. F. Chen, Recent advances in higher order rotaxane architectures, Chem. Commun., 2020, 56, 9916–9936 RSC.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, An artificial molecular pump, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS.
- C. Pezzato, M. T. Nguyen, D. J. Kim, O. Anamimoghadam, L. Mosca and J. F. Stoddart, Controlling Dual Molecular Pumps Electrochemically, Angew. Chem., Int. Ed., 2018, 57, 9325–9329 CrossRef CAS.
- S. Amano, S. D. P. Fielden and D. A. Leigh, A catalysis-driven artificial molecular pump, Nature, 2021, 594, 529–534 CrossRef CAS.
- H. Y. Au-Yeung and A. W. H. Ng, Mechanical Interlocking of Macrocycles in Different Sequences, Synlett, 2020, 31, 309–314 CrossRef CAS.
- A. M. Fuller, D. A. Leigh and P. J. Lusby, Sequence isomerism in [3]rotaxanes, J. Am. Chem. Soc., 2010, 132, 4954–4959 CrossRef CAS PubMed.
- E. A. Neal and S. M. Goldup, Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis, Chem. Commun., 2014, 50, 5128–5142 RSC.
- C. Talotta, C. Gaeta, Z. Qi, C. A. Schalley and P. Neri, Pseudorotaxanes with self-sorted sequence and stereochemical orientation, Angew. Chem., Int. Ed., 2013, 52, 7437–7441 CrossRef CAS PubMed.
- E. A. Wilson, N. A. Vermeulen, P. R. McGonigal, A. J. Avestro, A. A. Sarjeant, C. L. Stern and J. F. Stoddart, Formation of a hetero[3]rotaxane by a dynamic component-swapping strategy, Chem. Commun., 2014, 50, 9665–9668 RSC.
- X. Q. Wang, W. J. Li, W. Wang and H. B. Yang, Heterorotaxanes, Chem. Commun., 2018, 54, 13303–13318 RSC.
- D. B. Amabilino, P. R. Ashton, M. Bělohradský, F. M. Raymo and J. F. Stoddart, The controlled self-assembly of a [3]rotaxane incorporating three constitutionally different components, J. Chem. Soc., Chem. Commun., 1995, 7, 747–750 RSC.
- J. E. Lewis, J. Winn, L. Cera and S. M. Goldup, Iterative Synthesis of Oligo[n]rotaxanes in Excellent Yield, J. Am. Chem. Soc., 2016, 138, 16329–16336 CrossRef CAS.
- X. Zhao, X. K. Jiang, M. Shi, Y. H. Yu, W. Xia and Z. T. Li, Self-assembly of novel [3]- and [2]rotaxanes with two different ring components: donor-acceptor and hydrogen bonding interactions and molecular-shuttling behavior, J. Org. Chem., 2001, 66, 7035–7043 CrossRef CAS.
- A. Joosten, Y. Trolez, V. Heitz and J. P. Sauvage, Use of cleavable coordinating rings as protective groups in the synthesis of a rotaxane with an axis that incorporates more chelating groups than threaded macrocycles, Chem. – Eur. J., 2013, 19, 12815–12823 CrossRef CAS PubMed.
- X. Hou, C. Ke and J. F. Stoddart, Cooperative capture synthesis: yet another playground for copper-free click chemistry, Chem. Soc. Rev., 2016, 45, 3766–3780 RSC.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Quantitative emergence of hetero[4]rotaxanes by template-directed click chemistry, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS.
- C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, Pillar[5]arene as a co-factor in templating rotaxane formation, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Tunable solid-state fluorescent materials for supramolecular encryption, Nat. Commun., 2015, 6, 6884 CrossRef.
- M. A. Soto, F. Lelj and M. J. MacLachlan, Programming permanent and transient molecular protection via mechanical stoppering, Chem. Sci., 2019, 10, 10422–10427 RSC.
- K. Zhu, G. Baggi and S. J. Loeb, Ring-through-ring molecular shuttling in a saturated [3]rotaxane, Nat. Chem., 2018, 10, 625–630 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Integrative self-sorting: a versatile strategy for the construction of complex supramolecular architecture, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- E. A. Neal and S. M. Goldup, A Kinetic Self-Sorting Approach to Heterocircuit [3]Rotaxanes, Angew. Chem., Int. Ed., 2016, 55, 12488–12493 CrossRef CAS PubMed.
- W. Jiang, H. D. Winkler and C. A. Schalley, Integrative self-sorting: construction of a cascade-stoppered hetero[3]rotaxane, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS.
- W. Jiang and C. A. Schalley, Molecular recognition and self-assembly special feature: Integrative self-sorting is a programming language for high level self-assembly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS.
- X. Fu, Q. Zhang, S. J. Rao, D. H. Qu and H. Tian, One-pot synthesis of a [c2]daisy-chain-containing hetero[4]rotaxane via a self-sorting strategy, Chem. Sci., 2016, 7, 1696–1701 RSC.
- S. J. Rao, Q. Zhang, J. Mei, X. H. Ye, C. Gao, Q. C. Wang, D. H. Qu and H. Tian, One-pot synthesis of hetero[6]rotaxane bearing three different kinds of macrocycle through a self-sorting process, Chem. Sci., 2017, 8, 6777–6783 RSC.
- W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, Monitoring self-sorting by electrospray ionization mass spectrometry: formation intermediates and error-correction during the self-assembly of multiply threaded pseudorotaxanes, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed.
- H. V. Schröder, H. Hupatz, A. J. Achazi, S. Sobottka, B. Sarkar, B. Paulus and C. A. Schalley, A Divalent Pentastable Redox-Switchable Donor-Acceptor Rotaxane, Chem. – Eur. J., 2017, 23, 2960–2967 CrossRef.
- H. V. Schröder, A. Mekic, H. Hupatz, S. Sobottka, F. Witte, L. H. Urner, M. Gaedke, K. Pagel, B. Sarkar, B. Paulus and C. A. Schalley, Switchable synchronisation of pirouetting motions in a redox-active [3]rotaxane, Nanoscale, 2018, 10, 21425–21433 RSC.
- H. Hupatz, M. Gaedke, H. V. Schröder, J. Beerhues, A. Valkonen, F. Klautzsch, S. Müller, F. Witte, K. Rissanen, B. Sarkar and C. A. Schalley, Thermodynamic and electrochemical study of tailor-made crown ethers for redox-switchable (pseudo)rotaxanes, Beilstein J. Org. Chem., 2020, 16, 2576–2588 CrossRef CAS PubMed.
- M. Gaedke, H. Hupatz, H. V. Schröder, S. Suhr, K. F. Hoffmann, A. Valkonen, B. Sarkar, S. Riedel, K. Rissanen and C. A. Schalley, Dual-stimuli pseudorotaxane switches under kinetic control, Org. Chem. Front., 2021, 8, 3659–3667 RSC.
- I. R. Hanson, D. L. Hughes and M. R. Truter, Crystal and molecular structure of 6,7,9,10,12,13,20,21,23,24,26,27-dodecahydrodibenzo[b,n][1,4,7,10,13,16,19,22]octaoxacyclotetracosin (dibenzo-24-crown-8), J. Chem. Soc., Perkin Trans. 2, 1976, 972 RSC.
- H. V. Schröder, S. Sobottka, M. Nössler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Impact of mechanical bonding on the redox-switching of tetrathiafulvalene in crown ether-ammonium [2]rotaxanes, Chem. Sci., 2017, 8, 6300–6306 RSC.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, Consistent structures and interactions by density functional theory with small atomic orbital basis sets, J. Chem. Phys., 2015, 143, 054107 CrossRef PubMed.
- A. Klamt and G. Schüürmann, COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Nicoli, M. Baroncini, S. Silvi, J. Groppi and A. Credi, Direct synthetic routes to functionalised crown ethers, Org. Chem. Front., 2021, 8, 5531–5549 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data, results from isothermal titration calorimetry, NMR and UV/Vis spectroscopy, tandem mass spectrometry, electrochemical measurements, crystallography and theoretical calculations. CCDC 2047286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01553b |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA. |
| This journal is © the Partner Organisations 2022 |