
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-assisted in vivo polymerisation of conjugated oligomer based conductors†
Gwennaël
Dufil
 a,
Daniela
Parker
a,
Jennifer Y.
Gerasimov
a,
Thuc-Quyen
Nguyen
b,
Magnus
Berggren
ac and
Eleni
Stavrinidou
*ac
a,
Daniela
Parker
a,
Jennifer Y.
Gerasimov
a,
Thuc-Quyen
Nguyen
b,
Magnus
Berggren
ac and
Eleni
Stavrinidou
*ac
aLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, SE-60174, Norrköping, Sweden. E-mail: eleni.stavrinidou@liu.se
bCenter for Polymers and Organic Solids, Department of Chemistry and Biochemistry, Materials, and Physics, University of California, Santa Barbara, CA 93106, USA
cWallenberg Wood Science Center, Linköping University, SE-60174, Norrköping, Sweden
First published on 13th March 2020
Abstract
Conjugated polymers conduct both electronic and ionic carriers and thus can stimulate and translate biological signals when used as active materials in bioelectronic devices. Self- and on-demand organization of the active material directly in the in vivo environment can result in the seamless integration of the bioelectronic interface. Along that line, we recently demonstrated spontaneous in vivo polymerization of the conjugated oligomer ETE-S in the vascular tissue of plants and the formation of conducting wires. In this work, we elucidate the mechanism of the in vivo polymerization of the ETE-S trimer and demonstrate that ETE-S polymerizes due to an enzymatic reaction where the enzyme peroxidase is the catalyst and hydrogen peroxide is the oxidant. ETE-S, therefore, represents the first example of a conducting polymer that is enzymatically polymerized in vivo. By reproducing the reaction in vitro, we gain further insight on the polymerization mechanism and show that hydrogen peroxide is the limiting factor. In plants the ETE-S triggers the catalytic cycle responsible for the lignification process, hacks this biochemical pathway and integrates within the plant cell wall, forming conductors along the plant structure.
Introduction
Electronic interfacing with living systems has been the main objective within the rising research area of organic bioelectronics.1,2 Biomedical applications have been prime targets in this field, therefore much effort has been devoted to developing materials and devices that translate and stimulate ionic signalling in the biological milieu, while at the same time cause a minimal immune reaction from the target organism. Conducting polymers have been proved ideal for bio-interfacing as they support transport and close coupling between both electronic and ionic carriers,3 thus providing signal transduction between the inherently ionic biological world and human-made technology. In addition, organic chemistry offers an unlimited toolbox to build pi-conjugated systems functionalized for specific binding and interactions with the biological targets.Enhancing the compatibility and integration of the bioelectronic interface has been achieved by using thin and conformable substrates or by encapsulating the rigid device within a soft matrix. Still, these strategies result in electrodes and devices that present limitations with respect to conformation and adaption to the multiscale and complex architectures of tissues. One way to overcome the limitation of substrates is to deposit the active materials directly onto the tissue. For example, Martin and co-workers demonstrated in vivo electropolymerization of poly(3,4-ethylenedioxythiophene) (PEDOT) within the brain of a mouse.4 The polymer formed a cloud-like electrode within the brain tissue without causing any major negative effects on the brain morphology and function. Along the same line, we demonstrated self-organization of PEDOT-Sulfonate (PEDOT-S) within the xylem vascular tissue of plants.5 The polymer was uptaken from a solution and then formed hydrogel-like conducting wires along the xylem tissue, the water transport vessels of the plant. Furthermore, we advanced the in vivo functionalization by designing a new molecule, the 4-(2-(2,5-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)thiophen-3-yl)ethoxy)butane-1-sulfonate (ETE-S).6 The ETE-S molecule was not only uptaken and distributed in the vascular tissue of the plant, but, it also polymerized in vivo without the external aid of oxidants or any physical stimulation. The ETE-S based conducting wires in the vascular tissue had conductivity of 10 S cm−1, specific capacitance of 20 F g−1 and were used as the electrodes for the construction of a supercapacitor within the plant structure. We hypothesized that the polymerization was driven by the natural physicochemical environment of the plant that acted as a template and catalyst for the polymerization reaction. Indeed, reactive oxygen species and enzymes are highly present in plants, but their involvement in the ETE-S polymerization has not been demonstrated.
Over the past two decades, the use of enzymes as catalysts in polymerization reactions has received significant attention as it offers a simple synthetic route at mild reaction conditions which is also environmentally benign.7,8In vitro polymerization of conducting polymers has been achieved with the use of oxidoreductase enzymes, usually peroxidases and laccases,9 enzymes which catalyse electron transfer from electron donors to electron acceptor molecules.10 The enzymatic polymerization of aniline11 has been extensively explored but only a few studies demonstrate enzyme catalysed polymerization of Pyrrole and EDOT. Polypyrrole12 and PEDOT13–15 are successfully polymerized only at low pH (2). Due to the higher oxidation potential of EDOT and Pyrrole polymerization at higher pH (∼4) is possible only with the use of mediators.16,17
In this work, we attribute the in vivo polymerization of the conjugated oligomer ETE-S in plants6 to an enzymatically assisted polymerization. While conducting polymers are widely used in bioelectronics and their enzymatic polymerization has been demonstrated in in vitro conditions, the in vivo environment has not been utilized so far for the fabrication of the bioelectronic interface. ETE-S, therefore, represents the first example of a conducting polymer that is enzymatically polymerized in vivo. We demonstrate that the ETE-S molecule triggers the peroxidative cycle of the plant cell wall that is regulated by peroxidases, where upon the ETE-S polymerizes and incorporates into the plant structure. Furthermore, we investigate which parameters affect the in vitro polymerization that, to the best of our knowledge, was performed previously only in the case of polyaniline.18 Using the natural biochemical environment one can then form conductors and electronic components that are seamlessly integrated within the living tissue. Bioelectronic interfaces can thus be designed to track the native structure and to create communication pathways with external devices or augment non-native functionalities.
Results and discussion
Hydrogen peroxide and peroxidases are highly involved in the defence mechanism of the plant19–21 and therefore consist of the prime suspects with respect to catalysing the polymerization of ETE-S. Consequently, we sought to investigate, via UV-Vis absorption spectroscopy, whether the presence of H2O2 or peroxidase triggers the polymerization of ETE-S in vitro. An aqueous solution of ETE-S, 0.2 mg ml−1, was prepared and then mixed with H2O2 at a molar ratio of H2O2vs. ETE-S equal to 0.5 and with pH 5–6. The solution was let to react for 30 min and, then, we acquired the UV-Vis absorption spectra. As seen in Fig. 1a the absorption spectra of the ETE-S solution before and after 30 min of the addition of H2O2 are identical indicating that there has not been any polymerization. Even if the solution of ETE-S with H2O2 is let to react for 15 hours still no change is observed (Fig. S1, ESI†). The same results are obtained when we add peroxidase enzyme (HRP, horseradish peroxidase) to an ETE-S solution in the absence of H2O2. However, when both peroxidase and H2O2 are added to the ETE-S solution the ETE-S monomer peak diminishes and a broad absorption feature arises around 800 nm. The feature around 800 nm corresponds to the doped state of the p(ETE-S) as it was previously demonstrated from our group with a combination of experimental and theoretical evidence.22 Spectroelectrochemistry performed on electropolymerized p(ETE-S) revealed that the oxidized state of the polymer corresponds to the absorption band at 800 nm. At the same time electron paramagnetic resonance spectrometry, EPR revealed that the charge carriers in the doped state are bipolarons. The experimental evidence in that work were supported by DFT and DFT time depended calculations.22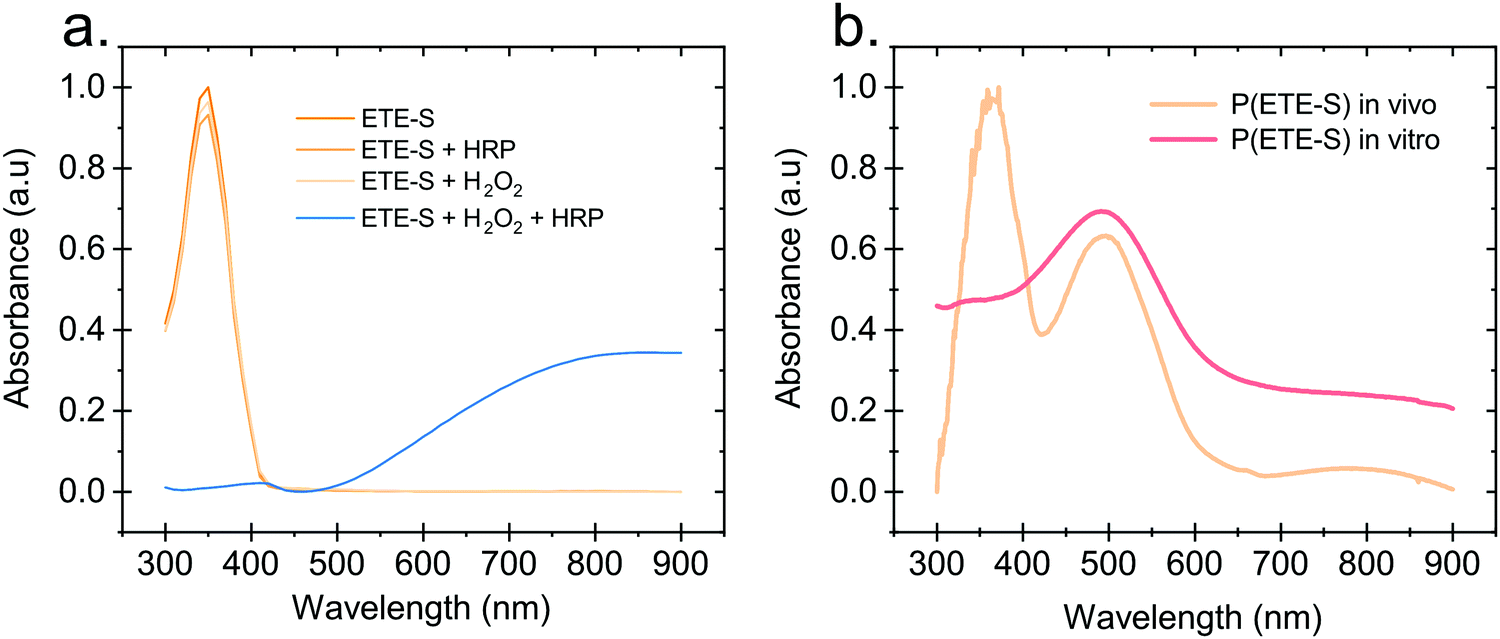 | ||
| Fig. 1 In vitro polymerization of ETE-S. (a) Absorption spectra of ETE-S (360 μM), ETE-S (360 μM) with HRP 5 U ml−1, ETE-S (360 μM) with H2O2 (180 μM), ETE-S (360 μM) with H2O2 (180 μM) and HRP 5 U ml−1. (b) Absorption spectra of DMSO extract of in vivo formed p(ETE-S) in rose xylem and in vitro formed p(ETE-S) with ETE-S (360 μM), H2O2 (180 μM) and HRP 5 U ml−1. | ||
Next, we compared the optical properties of the polymer formed via the enzymatic protocol in vitro (as described above) with the resulting material formed when ETE-S was introduced into the plant. Rose cuttings were immersed in ETE-S aqueous solution for 24 hours and then the functionalized xylem was collected and analysed. In order to extract the polymer formed in the plant, the functionalized xylem tissue was mechanically crushed in DMSO as previously described.6 Similarly, the polymer formed in vitro was first allowed to dry and then dissolved in DMSO. In Fig. 1b, we show the absorption spectra of the DMSO extraction of the polymer formed in vivo in the rose xylem and the polymer formed in vitro via the enzymatic protocol. In both cases, we observe a peak at 500 nm that corresponds to the neutral polymer with a minimum of 4 ETE-S units as defined by DFT calculations, spectroelectrochemistry and EPR.6,22 While in the in vitro formed polymer there is no presence of the monomer, in the plant extract the monomer peak is still present suggesting that the polymerization in vivo is incomplete. In addition, the polymer in both cases is mostly in its undoped form. When water is added into the polymer-DMSO solution, some polymer converts to its doped state as shown by the broad absorption feature at 800 nm (Fig. S2, ESI†). These observations point out towards a doping mechanism that recently has been reported by Yurash et al.25 The presence of water results in the protonation of the thiophene unit of the polymer. Upon protonation, a cation is formed in the conjugated backbone of the polymer. A neutral polymer chain can then undergo a single electron transfer with the protonated polymer giving one polymer chain with a positive polaron and another polymer chain with an unpaired electron.25,26 All the above findings suggest that the ETE-S can be polymerized with the peroxidase enzyme acting as a catalyst in the presence of hydrogen peroxide in physiological pH.
In Fig. 2, we illustrate the proposed mechanism of ETE-S polymerization following the classical scheme of the peroxidative cycle in plants.27 Hydrogen peroxide oxidizes the peroxidase to an active enzymatic intermediate, called compound I. Compound I then oxidizes one ETE-S molecule and converts to compound II. Then, the peroxidase returns to its initial state by oxidizing a second ETE-S molecule and releasing water. The ETE-S radicals can then couple to form ETE-S dimers. Further polymerization can occur either by ETE-S oligomers entering the peroxidative cycle or through radical transfer between the ETE-S radical to ETE-S oligomers. The radicals then take part in oxidative coupling to form larger oligomers.
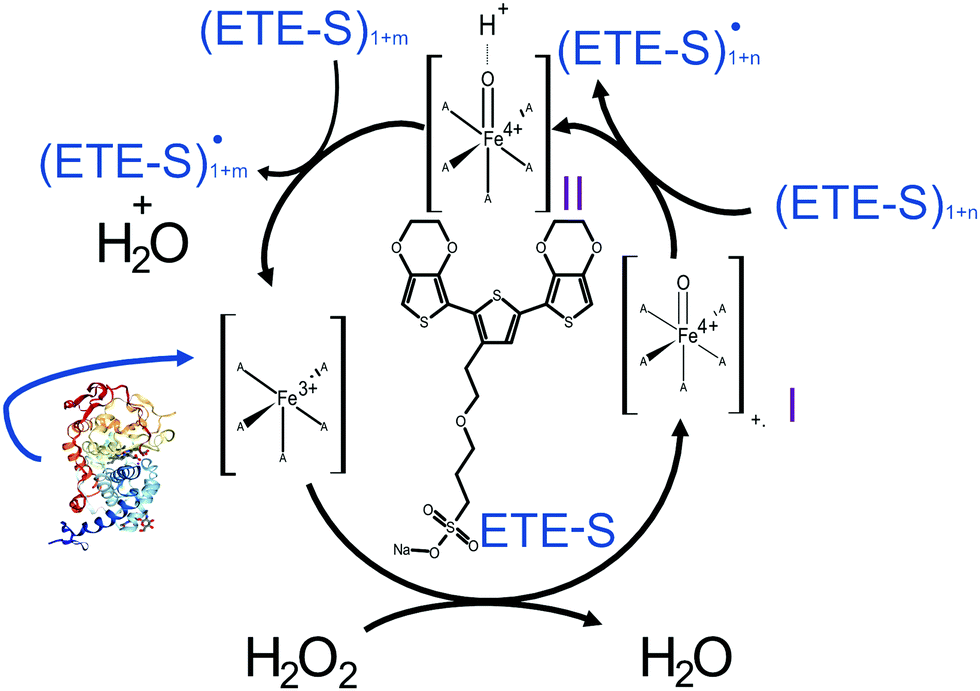 | ||
| Fig. 2 Proposed mechanism of the ETE-S polymerization under the plant's peroxidative cycle with the peroxidase being the catalyst and H2O2 the oxidant for the polymerization reaction. Peroxidase image vizualized with NGL viewer.23,24 | ||
In order to gain further insight into the polymerization reaction, we investigated the effect of the relative H2O2 and ETE-S concentrations on polymer formation. We performed in vitro polymerization by varying the amount of H2O2 with respect to ETE-S while keeping constant the HRP concentration to 5 U ml−1 (Fig. 3a). The solution was allowed to react for 24 hours before acquiring the UV-Vis spectra. We observe that for H2O2/ETE-S < ∼0.5, H2O2 is a limiting factor for the reaction as not all ETE-S is polymerized, while for H2O2/ETE-S > 0.5, all ETE-S polymerizes (Fig. 3b). Therefore, at lower H2O2 concentration, insufficient radicals are formed, thus limiting the polymerization. By increasing the concentration of H2O2 we reach an optimal point where all ETE-S is polymerized. This indicates that the H2O2 concentration is the limiting factor for the polymerization reaction suggesting that this could be the origin of the incomplete polymerization observed in the plant extract (Fig. 1b), although other factors cannot be excluded.
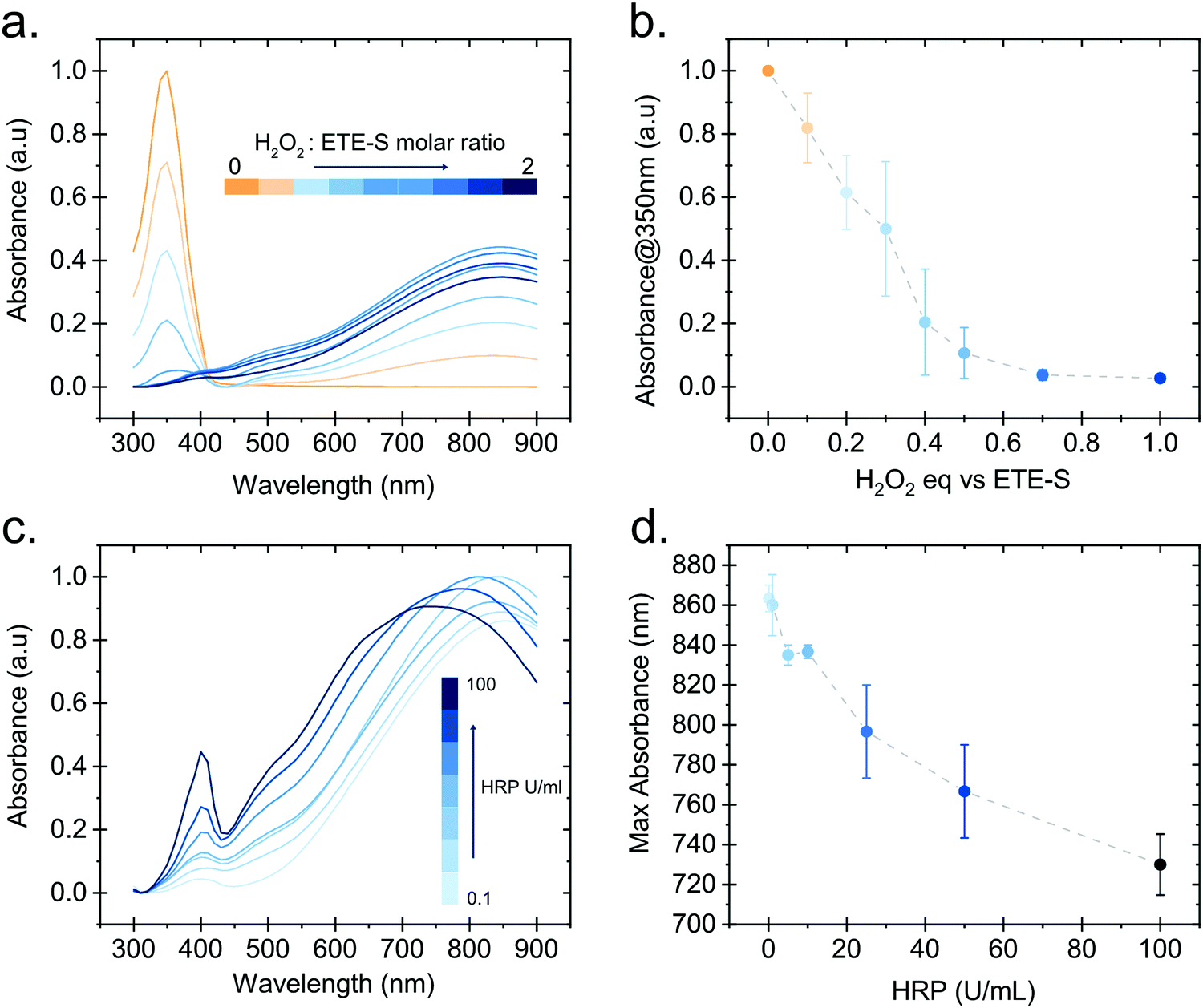 | ||
| Fig. 3 Effect of H2O2 and HRP concentration on the ETE-S in vitro polymerization. (a) Absorption spectra of p(ETE-S) formed in vitro with 5 U ml−1 HRP and various molar equivalents of H2O2 to ETE-S. (b) Absorbance at 350 nm for the spectra shown in (a) as a function of the molar equivalents of H2O2 to ETE-S. (c) Absorption spectra of p(ETE-S) formed in vitro for various HRP concentrations. (d) Wavelength corresponding to the highest absorbance value in (c) as a function of the HRP concentration. For (b) and (d) n = 3. | ||
Next, we examined how the enzyme concentration is influencing the polymerization reaction. By keeping the H2O2/ETE-S molar ratio equal to the optimal value, being 0.5, we varied the enzyme concentration. The UV-Vis spectra were taken after 1 hour of reaction (Fig. 3c). For the HRP activity above 1 U ml−1, the reaction is completed within 5 min, while for HRP = 0.1 U ml−1 the reaction is completed after 30 min (Fig. S3, ESI†). Apart from influencing the reaction kinetics, HRP also affects the polymerization product. For HRP ≥ 25 U ml−1 we observe a blue shift in the absorption peak of the polymer (Fig. 3d). This can be explained by the formation of shorter oligomeric chains in high enzyme concentrations. As we described, the polymerization occurs by combining ETE-S radicals or by radical transfer to oligomeric units. At high HRP concentrations, more ETE-S radicals will be formed resulting in a higher probability of radical coupling and the formation of oligomers. While at lower HRP concentrations less ETE-S radicals will be formed, therefore the polymerization is likely to happen with radical transfer to polymeric chains rather than radical monomer coupling.28 The shift in the absorption spectra could also be a result of polymer aggregation.
To further confirm our findings of the involvement of HRP and H2O2 in the in vivo polymerization, we functionalized plant roots with ETE-S by modifying the protocol that was developed for the functionalization of the vascular system in roses.6 Plant roots do not have a cuticle and the epidermal cells are therefore exposed, allowing the polymerization to be present directly on their surface. We choose bean plants as our model system. A root of an intact bean plant was immersed into a 1 mg ml−1 aqueous solution of ETE-S (1.8 mM) of pH 5–6. After 24 hours of incubation, the root was completely covered with the polymer, Fig. 4a.
 | ||
| Fig. 4 Amplex Red conversion to resorufin in the presence of HRP and H2O2. Top view and cross section of (a) bean root after 24 hours in 1 mg ml−1 ETE-S. (b) Bean root after Amplex Red staining. (c) Boiled bean root after Amplex Red staining. Scalebar represents 100 μm. | ||
At the same time, we stained roots with Amplex Red, a dye that under a peroxidase/peroxide reaction converts into resorufin, a molecule with an intense red color. Fig. 4b shows roots after staining with the Amplex Red for 3 hours. It can be seen that the roots acquire a red coloration on the epidermis and around cell walls. The staining covers the whole epidermis while its presence around the cell wall is more intense. To confirm that the coloration occurs only when both HRP and H2O2 are present, we boiled a root in water for 30 min to denature the enzymes. Indeed, Fig. 4c shows that the boiled roots do not acquire any color. Even if we add H2O2 at the boiled roots, no coloration is observed (Fig. S4, ESI†) because the Amplex Red requires the presence of both H2O2 and the active peroxidase. From Fig. 4, we observe that the ETE-S polymer and Amplex Red staining are localized in the same areas of the plant suggesting that the polymerization occurs where both H2O2 and HRP are present simultaneously.
Furthermore, we performed fluorescence microscopy to observe the polymer on the functionalized cross section of the root (Fig. 5). We used two filter sets, DAPI (337 nm, 447 em) and FTIC (482 nm, 536 nm) that correspond to the spectra of the ETE-S and the neutral p(ETE-S).6 Non-functionalized cross sections exhibit auto fluorescence while functionalized roots do not exhibit any fluorescence, in either of the two filter sets tested. However, the cross sections appear to be darker in the fluorescence mode at the areas where polymerization occurred. This indicates that the polymer is in its oxidized state and that the ETE-S molecule has polymerized. From both the brightfield images as well as the fluorescent images, it is clear that the polymerization is localized around the cell wall.
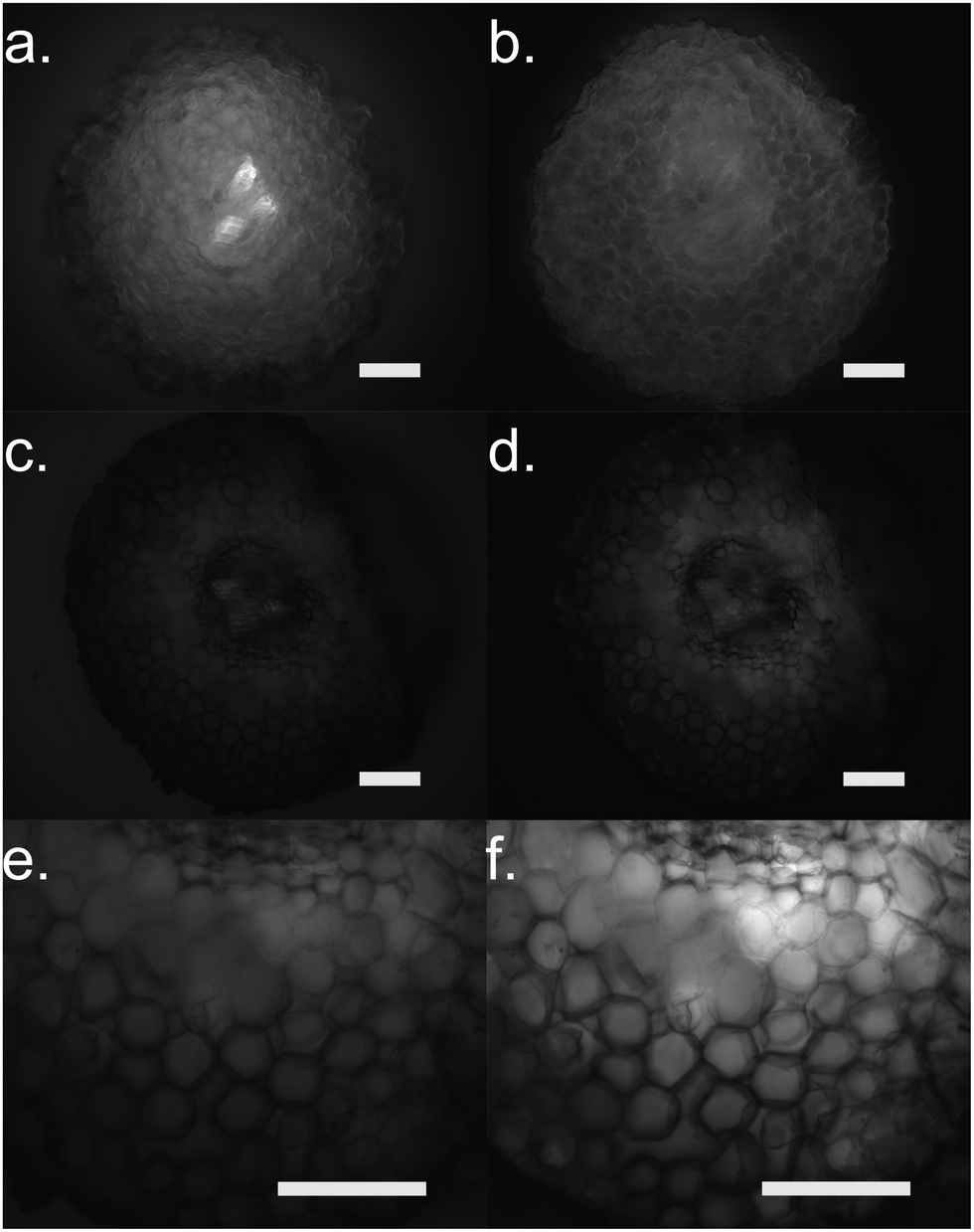 | ||
| Fig. 5 Fluorescence microscopy of root cross sections after functionalization with ETE-S, 1 mg ml−1 for 2 hours. (a), (c) and (e) DAPI mode for (a) control root and (c and e) functionalized root with (c) same exposure settings as control and (e) longer exposure as control. (b), (d) and (f) FTIC mode for (b) control root and (d and f) functionalized root with (d) same exposure settings as control (f) and longer exposure as control. Scale bar represents 100 μm. | ||
Conclusions
Peroxidases are highly localized in the plant cell wall since they are vital for tuning cell wall density with the help of H2O2. Two distinct catalytic cycles exist – the hydroxylic and peroxidative cycles – responsible for decreasing and increasing the cell wall density, respectively. The peroxidative cycle crosslinks cell wall compounds including the formation of lignin and suberin within the cell wall, forming a physical barrier that stops cell elongation and protects from external aggressors. Our findings suggest that the ETE-S triggers the peroxidative cycle of the plant, enters into the cycle and polymerizes along the cell wall. Why ETE-S triggers the peroxidative cycle of the plant is not yet understood and in order to pinpoint this at the molecular level further studies are needed.In this work, we elucidate the mechanism of in vivo polymerization of ETE-S. We demonstrate that ETE-S can be polymerized in vitro at physiological pH (5–6) through enzymatic polymerization catalysed by HRP in the presence of H2O2. ETE-S has relatively low oxidation potential (Fig. S5, ESI†) and this can be a reason for the effective enzymatic polymerization. In vivo this polymerization route occurs naturally in the plant cell wall as part of the plant's defence response and lignification process. Our findings suggest that indeed the ETE-S is localized in the plant cell wall. This work paves the way for rational design of materials that can be distributed into living systems to augment functionality. In the future, by tuning the molecular structure, backbone and side chains, one can target specific binding but also target the polymerization route in the in vivo environment.
Methods
UV-Vis absorption spectrometry characterization of the enzymatic polymerisation
A hydrogen peroxide (≥30%, Sigma Aldrich) stock solution of 10 mM was prepared freshly before mixing with an ETE-S stock solution of 1.8 mM. Horseradish Peroxidase type VI-A (Sigma Aldrich) aliquots of 1 mg ml−1 (≥250 U ml−1) were freshly prepared before use and added to the mixture of H2O2/ETE-S. The final concentrations of the reagents were: ETE-S 360 μM, H2O2 180 μM and HRP 5 U ml−1. The pH of the solutions prior and after the reaction were between pH 5–6. The absorption spectra were recorded from 300 to 900 nm (step 10 nm) using a microplate reader (BioTek, Synergy H1). For the H2O2 dependence investigation, different amounts of H2O2 from 0.1 to 2 equivalent vs. ETE-S concentration of 360 μM were added. The plate was mixed for 30 s and spectra were acquired 24 hours after the preparation of the samples. HRP activity was kept constant at 5 U ml−1. For the HRP dependence investigation, the molar ratio ETE-S/H2O2 was kept constant at 0.5 with a concentration of ETE-S at 360 μM. The HRP activity was varied from 0.1 U ml−1 to 100 U ml−1. The plate was mixed for 30 s and spectra were acquired 1 hour after the preparation of the samples.In vivo and in vitro formed p(ETE-S) extraction and characterization
The protocol for extraction of p(ETE-S) from the plant xylem tissue is the same as the one described previously.6 Briefly Rose cuttings were immersed in 1 mg ml−1 ETE-S aqueous solution for 24 h. The stem was collected and the functionalized xylem was dissected in small pieces. Using a mortar and pestle the functionalized xylem was mechanically crushed in DMSO in order to dissolve the polymer and the resulting solution was collected and analyzed. As a control, the same procedure was done on xylem tissue from stems that did not absorbed ETE-S. The UV-Vis spectra (300–1300 nm, step 1 nm) of sample and control were taken using a PerkinElmer Lambda 900 spectrometer. To acquire the optical signature of the polymer formed in vivo the contribution of the control (plant tissue) was subtracted from the sample. In order to compare the spectra with the in vitro synthesized p(ETE-S), the polymer was first let to dry and then dissolved in DMSO after mechanical crashing using a mortar and a pestle. The UV-Vis absorption spectra were acquired using the microplate reader (BioTek, Synergy H1), 300 to 900 nm (step 10 nm).Growth of bean plants
Bean (Phaseolus vulgaris) seeds were purchased from Impecta, Sweden. The seeds were inoculated in a 1/3 mix commercial bleach solution/DI water with constant stirring for 5 min, followed by 3 × 5 min of rising in DI water. Seeds were germinated in a MS/MES (Duchefa Biochemie) (0.215 g/0.05 g in 100 ml DI water) medium agar gel pH = 5.7 in the dark with a RH ∼60% and at ∼25 °C. After one week of germination, the plants were transferred to a liquid plant growth medium from Plantagen (Flytende Plantenaering), Sweden with the same growth conditions and a 12 hours day/12 hours night cycle. Beans were used after one week of growth.ETE-S functionalisation of bean roots
A solution of 1 mg ml−1 ETE-S was prepared in DI water. A root from the bean was isolated and immersed inside an Eppendorf tube for 24 hours while the plant was suspended in a commercial agar medium (SeedThru gel). After 24 hours the root was cut from the bean for further analysis. The pH of the ETE-S solution prior and after functionalization of the roots remained the same and between 5–6.Amplex red assay
Fresh 10 mM aliquots of Amplex Red (Thermofisher) were prepared in DMSO and added to 20 ml DI water to obtain a final concentration of 50 μM. Fresh roots were cut from a bean plant and incubated in Amplex Red and ETE-S. In order to denaturate the enzymes roots were boiled for 30 min in water and then were stained with Amplex Red with and without 100 μM H2O2. The samples were then incubated for 3 hours in an opaque Petri dish and then imaged via a Stereomicroscope (Nikon SM Z1500).Fluorescence microscopy
Functionalized and non-functionalized roots were cut using a sterile scalpel and deposited on a glass slide. The samples were immobilized under a coverslip using DAKO mounting medium (Agilent) to keep the fluorescence intact and avoid drying. Images were acquired using a fluorescence microscope (fluorescence/water dipping Microscope Nikon Ni-E). DAPI filter (ex: 377 nm em: 447 nm) and FITC filter (ex: 482 nm em: 536 nm) from Semrock. Images in Fig. 5a–d were obtained using an S Plan Fluor ELWD x20(NA:0.45, WD8.2; Nikon) and images in Fig. 5e and f were obtained using a TU Plan ELWD ×50 (NA:0.6, WD11; Nikon) lens.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to thank Roger Gabrielsson (Linköping University) for the synthesis of ETE-S. This work was supported by the European Union's Horizon 2020 research and innovation programme under grant agreement no. 800926 (FET-OPEN-HyPhOE), by the Swedish Research Council (VR) and the Wallenberg Wood Science Center (KAW 2018.0452) Additional funding was provided by the Knut and Alice Wallenberg Foundation, the Swedish Foundation for Strategic Research (SSF), The European Research Council (ERC) project e-NeuroPharma 834677 and the Swedish Government Strategic Research Area in Materials Science on Advanced Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009-00971). ES thanks L'Oreal-Unesco For Women in Science (FWIS) and TQN thanks the Department of Energy (DE-SC0017659) for the support.References
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009 CrossRef CAS PubMed.
- E. Zeglio, A. L. Rutz, T. E. Winkler, G. G. Malliaras and A. Herland, Adv. Mater., 2019, 31, 1806712 CrossRef PubMed.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2019, 1 Search PubMed.
- S. M. Richardson-Burns, J. L. Hendricks and D. C. Martin, J. Neural Eng., 2007, 4 DOI:10.1088/1741-2560/4/2/L02.
- E. Stavrinidou, R. Gabrielsson, E. Gomez, X. Crispin, O. Nilsson, D. T. Simon and M. Berggren, Sci. Adv., 2015, 1 DOI:10.1126/sciadv.1501136.
- E. Stavrinidou, R. Gabrielsson, K. P. R. Nilsson, S. K. Singh, J. F. Franco-Gonzalez, A. V. Volkov, M. P. Jonsson, A. Grimoldi, M. Elgland, I. V. Zozoulenko, D. T. Simon and M. Berggren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2807 CrossRef CAS PubMed.
- S. I. Shoda, H. Uyama, J. I. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307 CrossRef CAS PubMed.
- Z. G. Peter, Soft Matter, 2011, 7, 316 RSC.
- A. V. Karamyshev, S. V. Shleev, O. V. Koroleva, A. I. Yaropolov and I. Y. Sakharov, Enzyme Microb. Technol., 2003, 33, 556 CrossRef CAS.
- G. V. Otrokhov, O. V. Morozova, I. S. Vasil’eva, G. P. Shumakovich, E. A. Zaitseva, M. E. Khlupova and A. I. Yaropolov, Biochemistry, 2013, 78, 1539 CAS.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71 CrossRef CAS.
- M. R. Nabid and A. A. Entezami, J. Appl. Polym. Sci., 2004, 94, 254 CrossRef CAS.
- V. Rumbau, J. A. Pomposo, A. Eleta, J. Rodriguez, H. Grande, D. Mecerreyes and E. Ochoteco, Biomacromolecules, 2007, 8, 315 CrossRef CAS PubMed.
- T. Sikora, R. Marcilla, D. Mecerreyes, J. Rodriguez, J. A. Pomposo and E. Ochoteco, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 306 CrossRef CAS.
- L. Duan, Y. Zhao, F. Guo, W. Liu, C. Hou and Z. Ni, Polym. Adv. Technol., 2014, 25, 896 CrossRef CAS.
- S. Nagarajan, J. Kumar, F. F. Bruno, L. A. Samuelson and R. Nagarajan, Macromolecules, 2008, 41, 3049 CrossRef CAS.
- G. Shumakovich, G. Otrokhov, I. Vasil’Eva, D. Pankratov, O. Morozova and A. Yaropolov, J. Mol. Catal. B: Enzym., 2012, 81, 66 CrossRef CAS.
- K. Junker, G. Zandomeneghi, Z. Guo, R. Kissner, T. Ishikawa, J. Kohlbrecher and P. Walde, RSC Adv., 2012, 2, 6478 RSC.
- N. C. Veitch, Phytochemistry, 2004, 65, 249 CrossRef CAS PubMed.
- F. Passardi, C. Cosio, C. Penel and C. Dunand, Plant Cell Rep., 2005, 24, 255 CrossRef CAS PubMed.
- T. Kawano, Plant Cell Rep., 2003, 21, 829 CrossRef CAS PubMed.
- A. V. Volkov, S. K. Singh, E. Stavrinidou, R. Gabrielsson, J. F. Franco-Gonzalez, A. Cruce, W. M. Chen, D. T. Simon, M. Berggren and I. V. Zozoulenko, Adv. Electron. Mater., 2017, 3 DOI:10.1002/aelm.201700096.
- A. S. Rose, A. R. Bradley, Y. Valasatava, J. M. Duarte, A. Prlic and P. W. Rose, Bioinformatics, 2018, 34, 3755 CrossRef CAS PubMed , PBD ID: 1GZA.
- K. Fukuyama, K. Sato, H. Itakura, S. Takahashi and T. Hosoya, J. Biol. Chem., 1997, 272, 5752 CrossRef CAS PubMed.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T.-Q. Nguyen, Nat. Mater., 2019, 1 Search PubMed.
- C.-K. Mai, H. Zhou, Y. Zhang, Z. B. Henson, T.-Q. Nguyen, A. J. Heeger and G. C. Bazan, Angew. Chem., Int. Ed., 2013, 52, 12874 CrossRef CAS PubMed.
- F. Passardi, C. Penel and C. Dunand, Trends Plant Sci., 2004, 9, 534 CrossRef CAS PubMed.
- K. Ryu, J. P. McEldoon, A. R. Pokora, W. Cyrus and J. S. Dordick, Biotechnol. Bioeng., 1993, 42, 807 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00212g |
| This journal is © The Royal Society of Chemistry 2020 |