
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-sorting of two imine-based metal complexes: balancing kinetics and thermodynamics in constitutional dynamic networks†
Jean-François
Ayme
ab and
Jean-Marie
Lehn
 *ab
*ab
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
bLaboratoire de Chimie Supramoléculaire, Institut de Science et d'Ingénierie Supramoléculaires, Université de Strasbourg, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: lehn@unistra.fr
First published on 17th December 2019
Abstract
A major hurdle in the development of complex constitutional dynamic networks (CDNs) is the lack of strategies to simultaneously control the output of two (or more) interconnected dynamic processes over several species, namely reversible covalent imine bond formation and dynamic metal–ligand coordination. We have studied in detail the self-sorting process of 11 constitutional dynamic libraries containing two different amines, aldehydes and metal salts into two imine-based metal complexes, having no overlap in terms of their compositions. This study allowed us to determine the factors influencing the fidelity of this process (concentration, electronic and steric parameters of the organic components, and nature of the metal cations). In all 11 systems, the outcome of the process was primarily determined by the ability of the octahedral metal ion to select its pair of components from the initial pool of components, with the composition of the weaker tetrahedral complex being imposed by the components rejected by the octahedral metal ions. Different octahedral metal ions required different levels of precision in the “assembling instructions” provided by the organic components of the CDN to guide it towards a sorted output. The concentration of the reaction mixture, and the electronic and steric properties of the initial components of the library were all found to influence the lifetime of unwanted metastable intermediates formed during the assembling of the two complexes.
Introduction
Rivalling with the intricacy of biological processes in the construction and the exploitation of complex networks of molecules requires the development of new synthetic strategies to control the organization of large sets of molecules having a wide compositional and interactional diversity.1 To this end, the notions of orthogonal self-assembly2 and self-sorting3–7 are cornerstones of these strategies, especially when applied within the context of constitutional dynamic chemistry (CDC), based on molecules capable of adapting their constitution by exchange of their reversibly linked components.1l–n,8,9By addressing chemical systems at both molecular and supramolecular levels, CDC provides a convenient way to organize complex dynamic systems and consequently to control their properties. Constitutional dynamic libraries (CDLs) operating via the reversible condensation of amine- and 2-formylpyridine-containing components into dynamic imine-based constituents acting as ligands for transition metal cations have unlocked the path toward architectures that would otherwise be inaccessible by traditional synthetic means and/or capable of displaying complex properties (e.g. feedback loops, adaptation, etc.).7e–i,9b–e,10,11 It was shown that the simultaneous assembly of several types of such constitutional dynamic architectures within the same reaction mixture allowed for the emergence of new properties going beyond those of each individual architecture,1n,6d,9d,e,11a–d,q highlighting a link between increased compositional diversity and diversification of the displayed properties. However, to date, the compositional diversities of “poly-architecture” constitutional dynamic systems are limited. The systems reported in the literature involve architectures which always have at least one or two reagents in common (i.e. organic components and/or type of metal cations),3,6,7 thus limiting their potential in terms of compositional diversification and properties. One major hurdle in the development of more diverse systems is the lack of strategies to simultaneously control the outcome of two (or more) interconnected dynamic processes over several architectures, namely reversible covalent imine bond formation and dynamic metal–ligand coordination.
Here we investigate the simultaneous generation of 11 different pairs of fully non-identical imine-based metal complexes via the self-sorting of their six initial building blocks. The study provides insights into the factors influencing the fidelity and the rate of such self-sorting processes and points at the interplay between thermodynamic driving forces and kinetic traps in the self-assembly of constitutional dynamic systems.
Results and discussion
Rationale and initial design
Factors such as the denticity, the steric bulk and the electronic properties of a ligand can be exploited to tune its binding strength with a given type of transition metal, as each transition metal has well-defined coordination preferences due to its unique electronic makeup.12 The formation of a given imine-containing ligand constituent from a library of amine and aldehyde components can be selectively promoted via its preferential binding to one type of transition metal, if this constituent offers the best coordination environment for this metal cation out of all the possible combinations of the initial components.1l–n,8,9 The same principle can be extended to the simultaneous but selective generation of two (or more) imine-containing ligand constituents if two (or more) types of transition metals having different coordination preferences are used.9e,fA difference between metals of tetrahedral and octahedral coordination geometries exploitable by appropriate ligand design is that the former can be considered to involve two orthogonal planes containing two donor atoms whereas the latter involves two orthogonal planes with three donor atoms. Thus, by choosing amine and aldehyde reactants giving either a planar bidentate or a planar tridentate chelate by imine condensation, selectivity results in that full coordination of the former forms a tetrahedral species while that of the latter yields an octahedral one. When the donor atoms of both ligands are similar (or identical) and the concentrations of both are the same, the higher denticity of the tridentate ligand should lead to a chelate-type binding13 strongly favouring its binding to a metal ion of octahedral coordination geometry. For such reasons, the condensation of aniline derivative 1 (Fig. 1) with 2-formylpyridine was expected to give a ligand suited to coordination of the tetrahedral Cu(I) center, while condensation of the aminoquinoline derivative 2 was expected to give a ligand suited to coordination of the octahedral Fe(II) center.
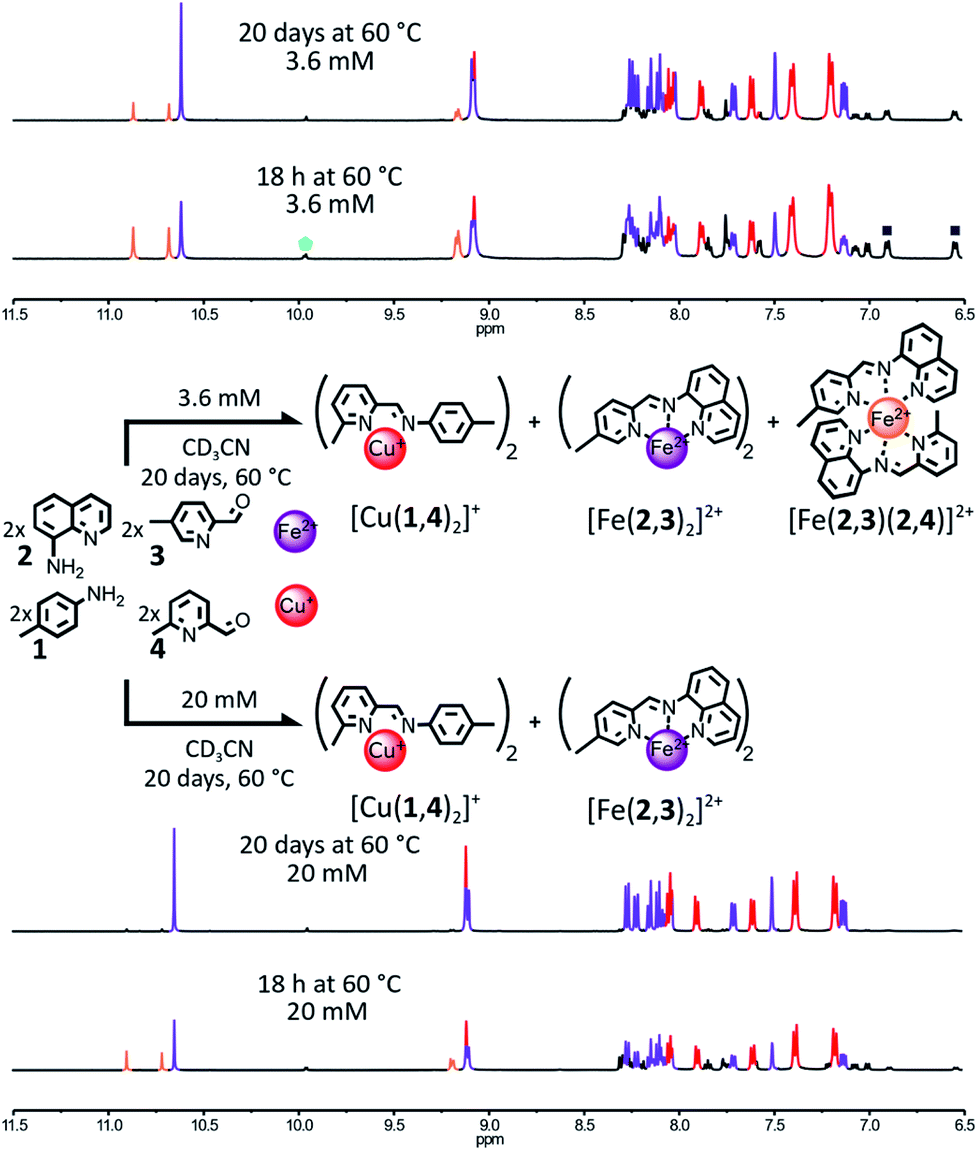 | ||
Fig. 1 Simultaneous generation of complexes [Cu(1,4)2]+ and [Fe(2,3)2]2+ through the self-sorting of their initial reactants. For clarity, the components 1 and 3 and the Cu(I) ions not incorporated into a complex have been omitted from the representation. Top – Reaction conditions: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:4:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), 3.6 mM, CD3CN, 60 °C, up to 20 days. Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h and 20 days at 60 °C. Bottom – Reaction conditions: 1:2:3:4:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), 20 mM, CD3CN, 60 °C, up to 20 days. The partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h and 20 days at 60 °C. Diagnostic signals of the complexes are colour coded, [Cu(1,4)2]+ in red and [Fe(2,3)2]2+ in purple and [Fe(2,3)(2,4)]2+ in orange. One of the diagnostic signals of the free aldehydes 3 and 4 is highlighted by a green pentagon, and two of the diagnostic signals of the free amine 1 are highlighted by grey squares. :2:3:4:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), 3.6 mM, CD3CN, 60 °C, up to 20 days. Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h and 20 days at 60 °C. Bottom – Reaction conditions: 1:2:3:4:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), 20 mM, CD3CN, 60 °C, up to 20 days. The partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h and 20 days at 60 °C. Diagnostic signals of the complexes are colour coded, [Cu(1,4)2]+ in red and [Fe(2,3)2]2+ in purple and [Fe(2,3)(2,4)]2+ in orange. One of the diagnostic signals of the free aldehydes 3 and 4 is highlighted by a green pentagon, and two of the diagnostic signals of the free amine 1 are highlighted by grey squares. | ||
The pairing of Fe(II) with one of the two derivatives of 2-formylpyridine can be inhibited by manipulating the steric hindrance around the coordination site of its pyridine. The coordination of the three nitrogen nuclei of 2,2′:6′2′′-terpyridine-like ligands—such as the ligands obtained by the condensation of aminoquinoline 2 with derivatives of 2-formylpyridine—to an octahedral metal ion is known to induce a “pinching” of these ligands resulting in a contraction of the distance between their positions 6 and 6′′. Any congestion in these positions will destabilize the resulting complex.14 Consequently, Fe(II) should favor the inclusion of 2-formylpyridine 3 (Fig. 1) in its coordination sphere as it bears less steric hindrance than its peer 4.
This rationale was tested by mixing components 1, 2, 3 and 4 in a 2:2:2:2 ratio in the presence of 1 eq. of Fe(BF4)2 and 1 eq. of Cu(BF4) in CD3CN (Fig. 1). After 18 h at 60 °C, the 1H NMR spectrum of the crude mixture (Fig. 1, upper part) was not consistent with the complete self-sorting of the initial reagents into the expected complexes [Cu(1,4)2]+ and [Fe(2,3)2]2+; the notation (n,m) refers to the imine-based constituent generated by the condensation of amine n with aldehyde m (in no specific order). Three different sets of signals were observed. These could be assigned to the homoleptic complexes [Cu(1,4)2]+ and [Fe(2,3)2]2+ along with the heteroleptic complex [Fe(2,3)(2,4)]2+. The homo- and heteroleptic Fe(II) complexes formed in a 0.9:1 ratio. The homoleptic complex [Fe(2,4)2]2+ containing two sterically hindered pyridine ligands 4 was not observed, attesting to its lower stability compared to the other two Fe(II) complexes.
Upon further heating of the reaction at 60 °C (Fig. 1, upper part, and ESI, Fig. S51†), the 1H NMR signals of [Fe(2,3)(2,4)]2+ started to fade. The disappearance of the heteroleptic complex was correlated with an increase in the population of [Cu(1,4)2]+ and [Fe(2,3)2]2+, hinting that the formation of [Fe(2,3)(2,4)]2+ is under kinetic control. However, after 20 days at 60 °C, [Fe(2,3)(2,4)]2+ was still present in the reaction mixture, with a homo-to heteroleptic Fe(II) complex ratio of 1:0.25. Under these conditions (60 °C, 20 days) this system is not under thermodynamic control, resulting in an incomplete self-sorting process. Thermodynamic control over the assembling of the system could be re-established by concentrating the reaction mixture. At 20 mM (up from 3.6 mM), [Fe(2,3)(2,4)]2+ was fully converted into [Cu(1,4)2]+ and [Fe(2,3)2]2+ after 20 days at 60 °C, with the 1H NMR spectrum of the reaction mixture being a superimposition of the spectra of the isolated complexes [Cu(1,4)2]+ and [Fe(2,3)2]2+ (Fig. 1, lower part and ESI, Fig. S54†).
To probe the driving forces governing the selective generation of [Cu(1,4)2]+ and [Fe(2,3)2]2+ at 20 mM, the formation of each one of the two complexes was attempted by mixing 1 eq. of the appropriate metal salt with a 2:2:2:2 mixture of 1, 2, 3, 4 in CD3CN at 60 °C (ESI, Fig. S55 and S56†). The two reaction mixtures were monitored by 1H NMR spectroscopy over the course of 20 days.
When Fe(II) was added to the organic components, the affinity of Fe(II) for the imine ligand (2,3) was strong enough to ensure the exclusive formation of one Fe(II) complex after 18 h at 60 °C, [Fe(2,3)2]2+ (ESI, Fig. S56†), whereas when Cu(I) was added alone to the four organic components, [Cu(1,4)2]+ did not form. Instead, an ill-defined Cu(I) complex was obtained (ESI, Fig. S55†). This result shows that [Cu(1,4)2]+ is not the thermodynamically most stable Cu(I) complex attainable from the initial library of components but instead constituent (1,4) is imposed upon Cu(I) by default via the ligand selection of Fe(II). Consequently, the formation of the thermodynamic product [Fe(2,3)2]2+ is the main driving force of this self-sorting processes. Similar results were obtained at 3.6 mM (ESI, Fig. S52 and S53†).
This system exemplifies how agonistic amplification between two constituents of a constitutional dynamic network can be exploited to force the expression of a product.1l–n,8,9 It also confirms the validity of our initial design principles as, at a concentration of 20 mM, two fully non-identical metal complexes could be generated via the self-sorting of their initial components. However, it also exposed one major shortcoming of this strategy. At a lower concentration range (2.7–3.6 mM), the slower rearrangement of an intermediate generated under kinetic control (namely, the heteroleptic complex [Fe(2,3)(2,4)]2+) trapped the system out-of-equilibrium—and thus out of a sorted outcome—for a length of time limiting the applicability of this type of system. This shortcoming could be addressed by refining the design of the initial library of components so that the rearrangement of this intermediate would be accelerated or its formation prevented.
Influence of the electronic parameters of the initial components on the fidelity of the self-sorting process
The influence of the electronic properties of the organic components on the output of their self-assembly in the presence of Fe(II) and Cu(I) was probed by modifying the electron-donating ability of the 4-substituent of aniline 1 or of the 6-substituent of 2-formylpyridine 4.The use of a more electron-rich aniline residue should generate a more stable Cu(I) complex15 and as complexes [Fe(2,3)(2,4)]2+ and [Cu(1,4)2]+ share the same aldehyde component 4, the formation of a more stable Cu(I) complex may hamper the formation of [Fe(2,3)(2,4)]2+. This hypothesis was tested by replacing the amine component 4 with the more electron rich N,N-dimethylaminoaniline 5 in the initial library of components (Fig. 2).
 | ||
| Fig. 2 Top – Simultaneous generation of complexes [Cu(4,5)2]+ and [Fe(2,3)2]2+ through the self-sorting of their initial reactants. Reaction conditions: 1:3:4:5:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C, diagnostic signals of the complexes are colour coded, [Cu(4,5)2]+ in red, [Fe(2,3)2]2+ in purple, one of the diagnostic signals of the free aldehyde 3 is highlighted by a grey circle and one of the diagnostic signals of the free aldehyde 4 is highlighted by a green pentagon. | ||
When components 2, 3, 4, 5, Cu(BF4) and Fe(BF4)2 were mixed in a 2:2:2:2:1:1 ratio in CD3CN, the self-sorting of the system was clearly apparent in the 1H NMR spectrum of the crude mixture. After 3 days at 60 °C, only the diagnostic signals of the two homoleptic complexes [Fe(2,3)2]2+ and [Cu(4,5)2]+ were observed in the spectrum. The two complexes were generated in an almost perfect 1:1 ratio. The use of the electron rich aniline 5 did not inhibit the generation of the kinetic intermediate [Fe(2,3)(2,4)]2+. When the self-assembly of [Fe(2,3)2]2+ and [Cu(4,5)2]+ was followed over time by 1H NMR, the diagnostic signals of [Fe(2,3)(2,4)]2+ were immediately detectable upon mixing of the reactants (ESI, Fig. S58†). After 90 min of heating at 60 °C, the amount of [Fe(2,3)(2,4)]2+ in solution reached its peak. From this point onward, [Fe(2,3)(2,4)]2+ slowly faded whereas the population of [Fe(2,3)2]2+ and [Cu(4,5)]+ kept on increasing for the next 60 h (ESI, Fig. S60†). After 60 h at 60 °C, only the diagnostic signals of the two homoleptic complexes remained visible in the 1H NMR spectra of the crude reaction mixture (ESI, Fig. S59†). While 5 did not prevent the formation of [Fe(2,3)(2,4)]2+, it considerably enhanced the rate of redistribution of its components.
The formation of [Fe(2,3)(2,4)]2+ could also be impeded by making the sterically hindered 2-formylpyridine 4 a poorer ligand. To do so, the methyl group of 4 was replaced with a slightly bigger and electron withdrawing CF3 group (Fig. 3). The addition of 1 eq. of Fe(BF4)2 and 1 eq. of Cu(BF4) to a 2:2:2:2 mixture of components 1, 2, 3, 6 in CD3CN prompted a rapid self-sorting of the reactants into a 1:1 mixture of complexes [Fe(2,3)2]2+ and [Cu(1,6)2]+ in 18 h at 60 °C (see Fig. 3). The binding abilities of 6 were sufficiently weakened by the introduction of the CF3 group so that any Fe(II) complex containing it was swiftly converted into [Fe(2,3)2]2+. However, the improvement of the rate of the self-sorting process came at the expense of the stability of the Cu(I) complex generated (ESI, Section 2.2.9†).
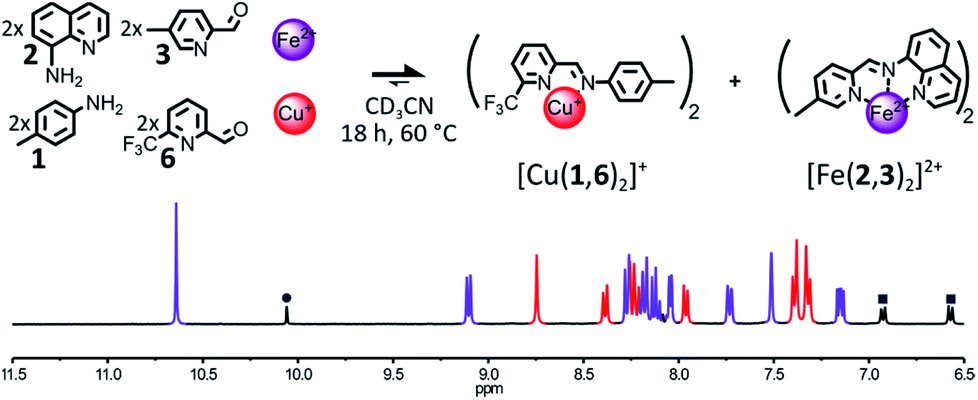 | ||
| Fig. 3 Top – Generation of complexes [Cu(1,6)2]+ and [Fe(2,3)2]2+ through the self-sorting of their initial reactants. For clarity, the components 1 and 6 and the Cu(I) ions not incorporated in a complex are omitted from the representation. Reaction conditions: 1:2:3:6:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C, the diagnostic signals of the complexes are colour coded, [Cu(1,6)2]+ in red and [Fe(2,3)2]2+ in purple, one of the diagnostic signals of the free aldehyde 6 is highlighted by a grey circle and diagnostic signals of the free aniline 1 are highlighted by grey squares. | ||
Influence of the steric properties of the initial components on the self-sorting process
The influence of the steric hindrance borne by the organic components on the outcome of their self-assembly in the presence of Fe(II) and Cu(I) was investigated by modulating the bulkiness of the substituents ortho to the nitrogens of the pyridine nucleus of aminoquinoline 2 and 2-formylpyridine 4. The incorporation of a phenyl ring in place of the methyl group of 2-formylpyridine 4 yielded the bulkier aldehyde 7 (Fig. 4). In most cases, the enhanced steric congestion brought by the phenyl ring of 7 was sufficient to prevent the subsistence of the heteroleptic complex [Fe(2,3)(2,7)]2+ at equilibrium.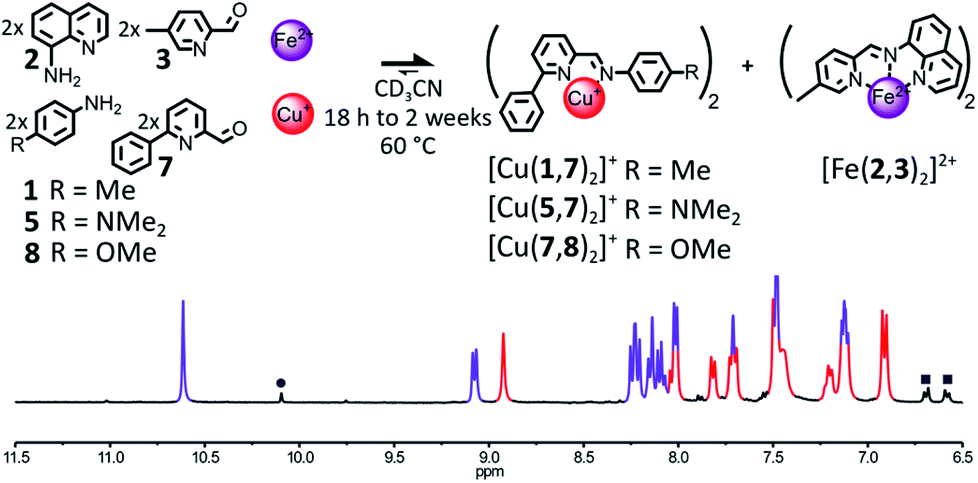 | ||
| Fig. 4 Top – Generation of complexes [Cu(X,7)2]+ (X = 1 or 5 or 8) and [Fe(2,3)2]2+ through the self-sorting of their initial reactants. When amine 1 was used, trace amounts of the heteroleptic complex [Fe(2,3)(2,7)]2+ were still observable even after 2 weeks at 60 °C. Reaction conditions: 1 or 5 or 8:2:3:7:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h to 2 weeks. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C. The diagnostic signals of the complexes are colour coded, [Cu(7,8)2]+ in red and [Fe(2,3)2]2+ in purple; one of the diagnostic signals of the free aldehyde 7 is highlighted by a grey circle and two of the diagnostic signals of aniline 8 are highlighted by grey squares. | ||
When a 2:2:2:2 mixture of 2, 3, 7 and 8 in CD3CN was treated with 1 eq. of Cu(BF4) and 1 eq. of Fe(BF4)2, [Cu(7,8)2]+ and [Fe(2,3)2]2+ were the only two complexes observable in the 1H NMR of the reaction mixture after 18 h at 60 °C (1:1 ratio, Fig. 4). A similar result was obtained when aniline 5 was used in place of 8 (ESI, Fig. S64†). During the monitoring of the organization of these two libraries by 1H NMR at 60 °C, both self-sorting processes were found to operate via the generation under kinetic control of the heteroleptic complex [Fe(2,3)(2,7)]2+ (ESI, Fig. S65, S66 and S69, S70†). [Fe(2,3)(2,7)]2+ was immediately generated following the addition of Cu(I) and Fe(II) to the libraries 2, 3, 7, 8 or 2, 3, 5, 7. Its concentration quickly peaked after 6 min and 1 h, respectively, before gradually declining for the remaining of the experiment. In both cases after 13 h, most of the [Fe(2,3)(2,7)]2+ had rearranged and the two desired homoleptic complexes had mostly reached their final concentrations. The incorporation of a phenyl ring in place of the methyl group of 4 did not prevent the formation of [Fe(2,3)(2,7)]2+, but significantly increased the rate of its rearrangement. Thermodynamic equilibrium was reached in only 13 h in the case of the library 2, 3, 5, 7 compared to 3 days in the case of the library 2, 3, 4, 5.
When Cu(I) and Fe(II) were added alone to the library 2, 3, 5, 7, [Fe(2,3)2]2+ could form whereas [Cu(5,7)2]+ could not, indicating that, here again, the generation of [Fe(2,3)2]2+ is the main driving force of the self-sorting process (ESI, Fig. S67†).
When the electron poorer aniline 1 was used in place of 5 or 8 in the initial library of components, the heteroleptic complex [Fe(2,3)(2,7)]2+ was still observable in the reaction mixture after 2 weeks of heating at 60 °C (ESI, Fig. S62 and S63†), yielding [Fe(2,3)(2,7)]2+, [Fe(2,3)2]2+ and [Cu(1,7)2]+ in a 0.08:1:0.86 ratio. As free aldehyde 1 was also observed in solution, in a proportion corresponding to that of the missing Cu(I) complex, the lower amount for [Cu(1,7)2]+ compared to [Fe(2,3)2]2+ could be explained by the partial hydrolysis of imine (1,7) due to prolonged heating. By itself, the use of the bulkier 2-formylpyridine 7 did not accelerate significantly the redistribution of the components of [Fe(2,3)(2,7)]2+ into [Cu(1,7)2]+ and [Fe(2,3)2]2+. Even in the presence of 7, the assistance of a derivative of aniline more basic than 1 (such as 5 or 8) is required to obtain a time efficient self-sorting process.
The destabilization of the Fe(II) heteroleptic complex needs not originate solely from the impairment of the binding abilities of a single one of its components. Instead, milder destabilizing features could be spread through several of its components, so that each one of these features would have a lower effect on the binding abilities of the component bearing it, but the concurrent incorporation of several of them into the same complex would be disfavored. If 6-methylpyridine 4 is reintroduced into the initial library of components in place of 7, the formation of [Fe(2,3)(2,4)]2+ could be hindered by introducing a methyl group ortho to the nitrogen atom of aminoquinoline 2, such as in 9 (Fig. 5).
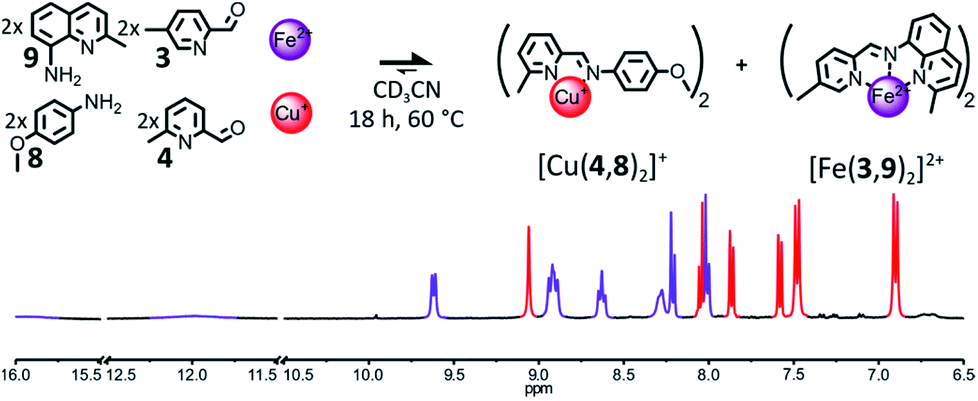 | ||
| Fig. 5 Top – Generation of complexes [Cu(4,8)2]+ and [Fe(3,9)2]2+ through the self-sorting of their initial reactants. Reaction conditions: 3:4:8:9:Cu(BF4):Fe(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C. The diagnostic signals of the complexes are colour coded, [Cu(4,8)2]+ in red and [Fe(3,9)2]2+ in purple. | ||
An equimolar mixture of components 3, 4, 8 and 9 (2:2:2:2) was found to self-sort into the two homoleptic complexes [Cu(4,8)2]+ and [Fe(3,9)2]2+ after being treated with 1 eq. of Cu(BF4) and 1 eq. of Fe(BF4)2 (Fig. 5) in CD3CN and left to react at 60 °C overnight. The 1H NMR signals of the two complexes were the only ones visible in the spectrum of the crude reaction mixture after 18 h. The two complexes were generated in the expected 1:1 ratio. Some of the 1H NMR signals of [Fe(3,9)2]2+ were strongly shifted downfield compared to those of [Fe(2,3)2]2+, indicating that [Fe(3,9)2]2+ must adopt a more distorted octahedral coordination geometry due to the bulkier aminoquinoline 9.
The incorporation of both the sterically demanding components aminoquinoline 9 and 2-formylpyridine 7 into the initial library markedly improved the rate of the self-sorting process by preventing the generation of the heteroleptic complex [Fe(3,9)(7,9)]2+.
Upon the treatment of a 2:2:2:2 mixture of 3, 7, 8 and 9 in CD3CN with 1 eq. of Fe(II) and 1 eq. of Cu(I), the formation of the complexes [Fe(3,9)2]2+ and [Cu(7,8)2]+ was complete (in the sense that no further changes in the 1H NMR spectrum were detectable) after about 100 min of heating at 60 °C (ESI, Fig. S72–S74†). During the monitoring by 1H NMR of the self-sorting of [Fe(3,9)2]2+ and [Cu(7,8)2]+, the formation of the heteroleptic complex [Fe(3,9)(7,9)]2+ was not observed (ESI, Fig. S73†). The greater steric hindrance of aminoquinoline 9 compared to 2 is likely to disfavor the inclusion of 7 in the same Fe(II) complex, allowing for a quick establishment of the thermodynamic equilibrium state of the system.
A correlation between the prominence of the structural features guiding the self-assembly of the libraries and the rate of their self-sorting process became apparent (ESI, Fig. S75†). (i) When both sterically demanding components aminoquinoline 9 and 2-formylpyridine 7 were incorporated into the initial library, the generation of [Fe(3,9)2]2+ and [Cu(7,8)2]+ from 3, 7, 8 and 9 took 100 min at 60 °C. (ii) In the absence of aminoquinoline 9 the formation of [Fe(2,3)2]2+ and [Cu(7,8)2]+ from 2, 3, 7 and 8 took 800 min under similar conditions. (iii) In the absence of both bulky components, the self-sorting of [Fe(2,3)2]2+ and [Cu(4,5)2]+ from 2, 3, 4 and 5 took 60 h at 60 °C. Such influence of steric effects on the kinetics of a self-assembly process, allowing for quicker establishment of the thermodynamic equilibrium state, deserves further exploration in view of its interest for the design of complex dynamic systems.
Influence of the nature of the metal cation connectors on the self-sorting process
The influence of the coordination preferences of the metal cations triggering the self-sorting process on its fidelity was studied by altering the nature of either the tetracoordinated metal cation used or the hexacoordinated one. We have shown earlier that a library composed of components 2, 3, 7 and 8 was able to self-sort into [Cu(7,8)2]+ and [Fe(2,3)2]2+ when treated with the appropriate metal salts (Fig. 4). Now, the same library of components was treated with either a combination of Ag(I) and Fe(II) or a combination of Cu(I) and Zn(II).The addition of 1 eq. Ag(I) and 1 eq. Fe(II) to a 2:2:2:2 mixture of 2, 3, 7 and 8 in CD3CN resulted almost exclusively in the formation of the two expected homoleptic complexes [Ag(7,8)2]+ and [Fe(2,3)2]2+ in a 1:1 ratio after 18 h at 60 °C (Fig. 6). In the 1H NMR spectrum of the crude reaction mixture, only small traces of hydrolysis of the constituent (7,8) were observable besides the two complexes. Such an outcome corroborates our hypothesis that the formation of the octahedral complex is the main driving force of the self-sorting of the library. Thus, the nature of the tetracoordinated metal cation employed has little influence on the outcome of the self-sorting process and Ag(I) could be used in place of Cu(I). Conversely, the substitution of Fe(II) for Zn(II) was found to have more dramatic effects on the output of the self-sorting process.
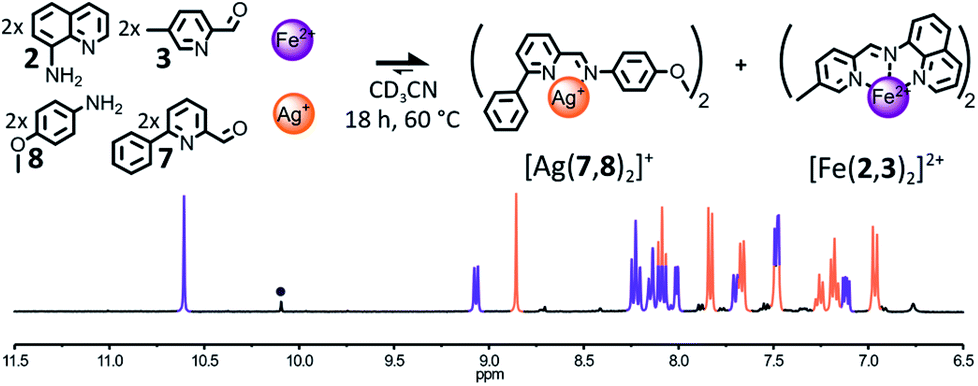 | ||
| Fig. 6 Top – Generation of complexes [Ag(7,8)2]+ and [Fe(2,3)2]2+ through the self-sorting of their initial reactants. Reaction conditions: 2:3:7:8:Ag(BF4):Fe(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C. The diagnostic signals of the complexes are colour coded, [Ag(7,8)2]+ in orange and [Fe(2,3)2]2+ in purple; one of the diagnostic signals of the free aldehyde 7 is designated by a grey circle. | ||
When 1 eq. of Zn(BF4)2 and 1 eq. of Cu(BF4) were mixed with an equimolar mixture of 2, 3, 7 and 8 (2:2:2:2) in CD3CN, the 1H NMR spectra of the crude reaction mixture revealed the formation of four different complexes after 18 h at 60 °C (Fig. 7A). Besides [Cu(7,8)2]+, the three Zn(II) complexes [Zn(2,7)2]2+, [Zn(2,3)(2,7)]2+ and [Zn(2,3)2]2+ were obtained in a 0.07:0.27:1 ratio. Further heating of the reaction mixture at 60 °C for up to 5 days did not change this outcome (ESI, Fig. S78†). The reduced fidelity of this self-sorting process could be explained by the more flexible and accommodating coordination of d10 Zn(II) compared to low-spin d6 Fe(II).12 Thus, the disparities in energy between [Zn(2,7)2]2+, [Zn(2,3)(2,7)]2+ and [Zn(2,3)2]2+—induced by the steric and electronic properties of 2, 3 and 7—became insufficient to guide the system towards the exclusive generation of [Zn(2,3)2]2+. Indeed, when 1 eq. of Zn(II) was added to a 2:2:2 mixture of 2, 3, 7 (ESI, Fig. S79 and S80†), [Zn(2,7)2]2+, [Zn(2,3)(2,7)]2+ and [Zn(2,3)2]2+ were generated in a 0.05:0.21:1 ratio after 2 days at 60 °C. This outcome did not change upon further heating of the mixture at 60 °C for up to 6 days. In contrast, when the bulkier aminoquinoline 9 was used in place of 2, the addition of 1 eq. of Zn(II) to a 2:2:2 mixture of 3, 7, 9 yielded almost exclusively [Zn(3,9)2]2+, after 2 days at 60 °C (ESI, Fig. S82†). These results indicate that stronger assembly instructions in the initial components of the system can offset the more accommodating coordination of Zn(II). When an equimolar mixture of 3, 7, 8, 9 (2:2:2:2) was treated with 1 eq. of Zn(BF4)2 and 1 eq. of Cu(BF4) in CD3CN, the fidelity of the self-sorting process was largely restored (Fig. 7B). After 18 h at 60 °C, the diagnostic signals of complexes [Cu(7,8)2]+ and [Zn(3,9)2]2+ dominated the 1H NMR spectrum of the crude reaction mixture. [Zn(3,9)(7,9)]2+ was still observed in the reaction mixture but in a lower 0.1:1 ratio with [Zn(3,9)2]2+. [Zn(7,9)2]2+ was not detectable. The above systems highlight the correlation existing between the narrowness of the coordination preferences of the metal ions used to drive the self-assembly of a system and the amount of instructions required in the organic components of this system to guide it towards a specific outcome.
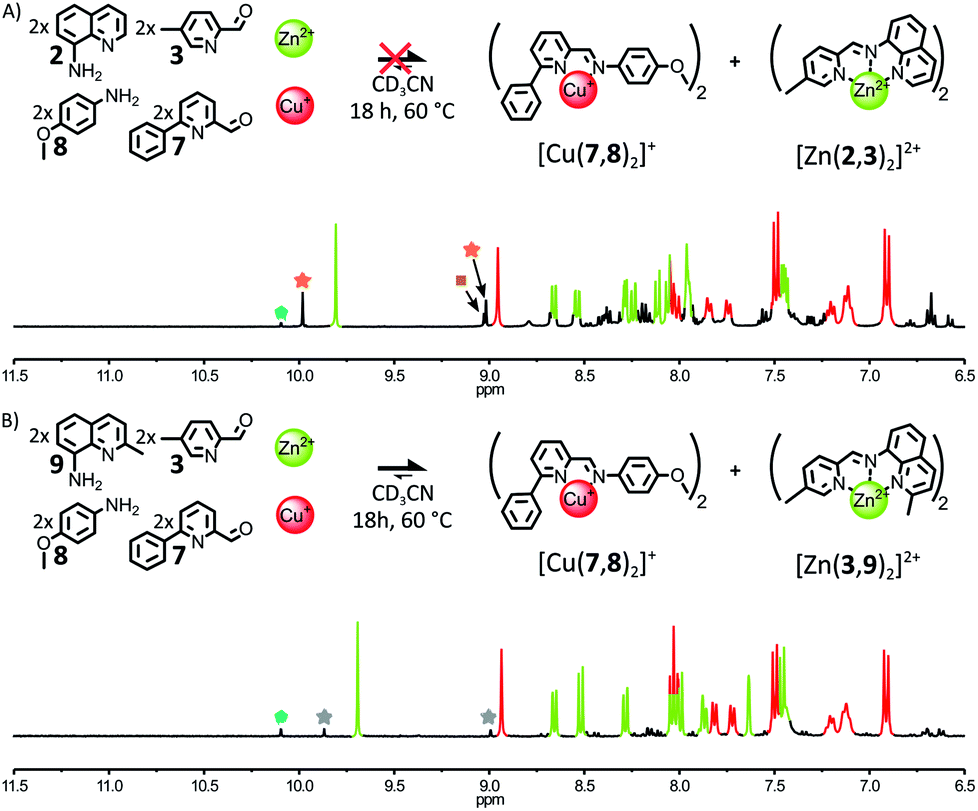 | ||
| Fig. 7 (A) Top – Attempted generation of complexes [Cu(7,8)2]+ and [Zn(2,3)2]2+ through the self-sorting of their initial reactants. Reaction conditions: 2:3:7:8:Cu(BF4):Zn(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C; the diagnostic signals of the complexes are colour coded, [Cu(7,8)2]+ in red and [Zn(2,3)2]2+ in green. Two of the diagnostic signals of [Zn(2,3)(2,7)]2+ are highlighted by orange stars, one of the diagnostic signals of [Zn(2,7)2]2+ is highlighted by a brown square and one of the diagnostic signals of the free aldehyde 7 is highlighted by a green pentagon. (B) Top – Generation of complexes [Cu(7,8)2]+ and [Zn(3,9)2]2+ through the self-sorting of their initial reagents. Reaction conditions: 3:7:8:9:Cu(BF4):Zn(BF4)2 (2:2:2:2:1:1), CD3CN, 60 °C, 18 h. Bottom – Partial 1H NMR spectrum (400 MHz, CD3CN, 297 K) of the crude reaction mixture after 18 h at 60 °C; the diagnostic signals of the complexes are colour coded, [Cu(7,8)2]+ in red and [Zn(3,9)2]2+ in green. Two of the diagnostic signals of [Zn(3,9)(7,9)]2+ are highlighted by grey stars and one of the diagnostic signals of the free aldehyde 7 is highlighted by a green pentagon. | ||
Conclusions
The unique coordination preferences of tetrahedral and octahedral metal cations can be exploited to drive the self-sorting of two amine components and two 2-formylpyridine components into two fully non-identical metal complexes of imine ligands.The fidelity of the self-sorting of the two metal complexes was determined by the capacity of the octahedral metal ion to select its pair of components from the initial pool of reactants, with the ligand of the weaker tetrahedral metal ions being dictated by the components discarded by the octahedral metal ions. The present study stresses the pivotal role of the kinetic intermediates of the various processes involved in the generation of the thermodynamic products of metal ion-driven dynamic covalent systems in reaching a given self-sorted outcome. The concentration, the electronic properties and the steric properties of the initial components of the system were all found to be crucial parameters to manipulate for avoiding kinetic trapping during the sorting of the system. The present study also illustrates how modest alterations of the coordination and/or structural features guiding the self-assembly of the systems can tip the balance between a sorted and disordered final output.
We anticipate that the information gleaned from our investigation will facilitate the design and the manipulation of complex CDNs of molecules having a wide compositional and interactional diversity, an important step towards rivalling with the mastery of biological systems at organizing and exploiting dynamic networks of molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the ERC (Advanced Research Grant SUPRADAPT 290585). JFA acknowledges the financial support by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 749351. JFA gratefully acknowledges Prof. Jack Harrowfield, Dr Jean-Louis Schmitt and Cyril Antheaume for extensive discussions.Notes and references
- For selected resources on the self-organization of chemical systems: (a) G. M. Whitesides and R. F. Ismagilov, Science, 1999, 284, 89–92 CrossRef CAS PubMed; (b) G. M. Whitesides and B. A. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed; (c) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed; (d) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed; (e) D. Newth and J. Finnigan, Aust. J. Chem., 2006, 59, 841–848 CrossRef CAS; (f) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (g) M. Schmittel and K. Mahata, Angew. Chem., Int. Ed., 2008, 47, 5284–5286 CrossRef CAS PubMed; (h) J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed; (i) M. D. Ward and P. R. Raithbyb, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC; (j) G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC; (k) T. Kosikova and D. Philp, Chem. Soc. Rev., 2017, 46, 7274–7305 RSC; (l) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed; (m) J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed; (n) J.-F. Ayme and J.-M. Lehn, Adv. Inorg. Chem., 2018, 71, 3–78 CrossRef CAS.
- For selected general resources on orthogonal self-assembly: (a) W. T. S. Huck, R. Hulst, P. Timmerman, F. C. J. M. van Veggel and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1997, 36, 1006–1008 CrossRef CAS; (b) V. Goral, M. I. Nelen, A. Eliseev and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1347–1352 CrossRef CAS PubMed; (c) M. L. Saha, S. De, S. Pramanikw and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC; (d) X.-Y. Hu, T. Xiao, C. Lin, F. Huang and L. Wang, Acc. Chem. Res., 2014, 47, 2041–2051 CrossRef CAS PubMed; (e) A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC; (f) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- For selected general resources on self-sorting: (a) R. Kramer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef CAS; (b) S. J. Rowan, D. G. Hamilton, P. A. Brady and J. K. M. Sanders, J. Am. Chem. Soc., 1997, 119, 2578–2579 CrossRef CAS; (c) A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed; (d) K. Osowska and O. Š. Miljanić, Synlett, 2011, 1643–1648 CAS; (e) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (f) M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC; (g) Q. Ji, R. C. Lirag and O. Š. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC; (h) Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC; (i) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (j) C.-W. Hsu and O. Š. Miljanić, in Dynamic Covalent Chemistry: Principles, Reactions, and Applications, ed. W. Zhang and Y. Jin, Wiley, 2017 Search PubMed; (k) M. Schmittel and S. Saha, Adv. Inorg. Chem., 2018, 71, 135–176 CrossRef CAS; (l) S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- For selected examples of self-sorting processes based on dynamic covalent bonds: (a) J. W. Sadownik and D. Philp, Angew. Chem., Int. Ed., 2008, 47, 9965–9970 CrossRef CAS PubMed; (b) B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154–3155 CrossRef PubMed; (c) P. Vongvilai and O. Ramström, J. Am. Chem. Soc., 2009, 131, 14419–14425 CrossRef CAS PubMed; (d) K. Osowska and O. Š. Miljanić, J. Am. Chem. Soc., 2011, 133, 724–727 CrossRef CAS PubMed; (e) Q. Ji and O. Š. Miljanić, J. Org. Chem., 2013, 78, 12710–12716 CrossRef CAS; (f) C. W. Hsu and O. Š. Miljanić, Angew. Chem., Int. Ed., 2015, 54, 2219–2222 CrossRef CAS PubMed; (g) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed.
- For selected examples of self-sorting processes based on metal complexes: (a) P. N. W. Baxter, J.-M. Lehn, A. DeCian and J. Fischer, Angew. Chem., Int. Ed., 1993, 32, 69–72 CrossRef; (b) D. L. Caulder and K. N. Raymond, Angew. Chem., Int. Ed., 1997, 36, 1440–1442 CrossRef CAS; (c) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS; (d) C. Addicott, N. Das and P. J. Stang, Inorg. Chem., 2004, 43, 5335–5338 CrossRef CAS PubMed; (e) Y. R. Zheng, H. B. Yang, K. Ghosh, L. Zhao and P. J. Stang, Chem.–Eur. J., 2009, 15, 7203–7214 CrossRef CAS PubMed; (f) A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed; (g) X. Lu, X. Li, K. Guo, T.-Z. Xie, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2014, 136, 18149–18155 CrossRef CAS PubMed; (h) W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285–8289 CrossRef CAS PubMed.
- For selected examples of self-sorting processes based on constitutional dynamic metal complexes: (a) D. Schultz and J. R. Nitschke, Angew. Chem., Int. Ed., 2006, 45, 2453–2456 CrossRef CAS PubMed; (b) M. Schmittel, M. L. Saha and J. Fan, Org. Lett., 2011, 13, 3916–3919 CrossRef CAS; (c) C. J. Campbell, D. A. Leigh, I. J. Vitorica-Yrezabal and S. L. Woltering, Angew. Chem., Int. Ed., 2014, 53, 13771–13774 CrossRef CAS PubMed; (d) A. Jiménez, R. A. Bilbeisi, T. K. Ronson, S. Zarra, C. Woodhead and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 4556–4560 CrossRef PubMed; (e) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Angew. Chem., Int. Ed., 2014, 53, 7823–7827 CrossRef CAS; (f) L. R. Holloway, M. C. Young, G. J. Beran and R. J. Hooley, Chem. Sci., 2015, 6, 4801–4806 RSC; (g) A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2015, 54, 5641–5645 CrossRef CAS PubMed; (h) C. A. Wiley, L. R. Holloway, T. F. Miller, Y. Lyon, R. R. Julian and R. J. Hooley, Inorg. Chem., 2016, 55, 9805–9815 CrossRef CAS PubMed; (i) L. R. Holloway, P. M. Bogie and R. J. Hooley, Dalton Trans., 2017, 46, 14719–14723 RSC.
- For selected examples on self-sorting processes involving supramolecular architectures built around more than one type of metal template: (a) X. Sun, D. W. Johnson, D. L. Caulder, R. E. Powers, K. N. Raymond and E. H. Wong, Angew. Chem., Int. Ed., 1999, 38, 1303–1307 CrossRef CAS; (b) F. Ibukuro, M. Fujita, K. Yamagushi and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 11014–11015 CrossRef CAS; (c) K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed; (d) K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed; (e) M. M. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed; (f) M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743–17746 CrossRef CAS; (g) M. L. Saha, N. Mittal, J. W. Bats and M. Schmittel, Chem. Commun., 2014, 50, 12189–12192 RSC; (h) W. J. Ramsay, F. T. Szczypinski, H. Weissman, T. K. Ronson, M. M. J. Smulders, B. Rybtchinski and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 5636–5640 CrossRef CAS PubMed; (i) F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed; (j) M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004–1010 RSC; (k) H. Sepehrpour, M. L. Saha and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 2553–2556 CrossRef CAS PubMed; (l) S. Gaikwad, M. L. Saha, D. Samanta and M. Schmittel, Chem. Commun., 2017, 53, 8034–8037 RSC.
- For selected general resources on constitutional dynamic chemistry: (a) J.-M. Lehn, Top. Curr. Chem., 2012, 322, 1–32 CAS; (b) Constitutional Dynamic Chemistry, Topics in Current Chemistry, ed. M. Barboiu, Springer, Berlin, 2012, vol. 322 Search PubMed.
- For selected recent examples of applications of constitutional dynamic chemistry: (a) N. Hafezi and J. -M. Lehn, J. Am. Chem. Soc., 2012, 134, 12861–12868 CrossRef CAS PubMed; (b) G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS PubMed; (c) J. Holub, G. Vantomme and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS PubMed; (d) S. Dhers, J. Holub and J.-M. Lehn, Chem. Sci., 2017, 8, 2125–2130 RSC; (e) G. Men and J.-M. Lehn, J. Am. Chem. Soc., 2017, 139, 2474–2483 CrossRef CAS PubMed; (f) P. Kovaricek, A. C. Meister, K. Flidrova, R. Cabot, K. Kovarickova and J.-M. Lehn, Chem. Sci., 2016, 7, 3215–3226 RSC; (g) O. Shyshov, R.-C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed; (h) H. Löw, E. Mena-Osteritz and M. von Delius, Chem. Sci., 2018, 9, 4785–4793 RSC.
- For selected examples of the complexity of the metallo-supramolecular architectures reachable from amine-and 2-formylpyridine-containing components: (a) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (b) J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS PubMed; (c) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed; (d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney and D. Schultz, J. Am. Chem. Soc., 2012, 134, 9488–9497 CrossRef CAS PubMed; (e) J. E. Beves, C. J. Campbell, D. A. Leigh and R. G. Pritchard, Angew. Chem., Int. Ed., 2013, 52, 6464–6467 CrossRef CAS PubMed; (f) C. S. Wood, T. K. Ronson, A. M. Belenguer, J. J. Holstein and J. R. Nitschke, Nat. Chem., 2015, 7, 354–358 CrossRef CAS PubMed.
- For selected examples of the functional potential of metallosupramolecular architecture built from amine-and 2-formylpyridine-containing components: potential for adaptation to stimuli, see ref. 9 and (a) V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684–687 CrossRef CAS PubMed; (b) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (c) B. S. Pilgrim, D. A. Roberts, T. G. Lohr, T. K. Ronson and J. R. Nitschke, Nat. Chem., 2017, 9, 1276–1281 CrossRef CAS; (d) D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC; (e) S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed; (f) L. R. Holloway, H. H. McGarraugh, M. C. Young, W. Sontising, G. J. O. Beran and R. J. Hooley, Chem. Sci., 2016, 7, 4423–4427 RSC; (g) D. A. Roberts, B. S. Pilgrim, G. Sirvinskaite, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 9616–9623 CrossRef CAS PubMed ; potential as molecular containers; (h) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed; (i) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC; (j) A. G. Salles Jr, S. Zarra, R. M. Turner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 19143–19146 CrossRef PubMed; (k) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC ; potential as biologically active molecules or as a mimic of biological molecules; (l) A. C. G. Hotze, N. J. Hodges, R. E. Hayden, C. Sanchez-Cano, C. Paines, N. Male, M.-K. Tse, C. M. Bunce, J. K. Chipman and M. J. Hannon, Chem. Biol., 2008, 15, 1258–1267 CrossRef CAS PubMed; (m) T. R. Cook, V. Vajpayee, M. H. Lee, P. S. Stang and K. W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed; (n) A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed; (o) R. Kaner, S. Allison, A. Faulkner, R. Phillips, D. Roper, S. Shepherd, D. Simpson, N. Waterfield and P. Scott, Chem. Sci., 2016, 7, 951–958 RSC ; potential as polymeric materials; (p) C.-F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007–5010 CrossRef CAS PubMed; (q) C.-F. Chow, S. Fujii and J.-M. Lehn, Chem.–Asian J., 2008, 3, 1324–1335 CrossRef CAS PubMed; (r) G. Schaeffer, E. Buhler, S. J. Candau and J.-M. Lehn, Macromolecules, 2013, 46, 5664–5671 CrossRef CAS; (s) S. Ulrich and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2240–2243 CrossRef CAS PubMed; (t) S. Ulrich, E. Buhler and J.-M. Lehn, New J. Chem., 2009, 33, 271–292 RSC; (u) S. Ulrich and J.-M. Lehn, J. Am. Chem. Soc., 2009, 131, 5546–5559 CrossRef CAS PubMed ; potential as anion binders; (v) I. A. Riddell, T. K. Ronson, J. K. Clegg, C. S. Wood, R. A. Bilbeisi and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 9491–9498 CrossRef CAS PubMed; (w) J.-F. Ayme, J. E. Beves, C. J. Campbell, G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, J. Am. Chem. Soc., 2015, 137, 9812–9815 CrossRef CAS PubMed; (x) R. A. Bilbeisi, T. Prakasam, M. Lusi, R. El-Khoury, C. Platas-Iglesias, L. J. Charbonnière, J.-C. Olsen, M. Elhabiri and A. Trabolsi, Chem. Sci., 2016, 7, 2524–2532 RSC.
- Comprehensive Coordination Chemistry II, ed, J. A. McCleverty and T. J. Meyer, Elsevier Ltd., 2004 Search PubMed.
- G. Schwarzenbach, Helv. Chim. Acta, 1952, 35, 2344–2363 CrossRef CAS.
- J.-F. Ayme, J. Lux, J.-P. Sauvage and A. Sour, Chem.–Eur. J., 2012, 18, 5565–5573 CrossRef CAS PubMed.
- D. Schultz and J. R. Nitschke, J. Am. Chem. Soc., 2006, 128, 9887–9892 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04988f |
| This journal is © The Royal Society of Chemistry 2020 |