
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical modification of the adeno-associated virus capsid to improve gene delivery†
Mathieu
Mével
 *a,
Mohammed
Bouzelha
a,
Aurélien
Leray
ac,
Simon
Pacouret
a,
Mickael
Guilbaud
a,
Magalie
Penaud-Budloo
a,
Dimitri
Alvarez-Dorta
c,
Laurence
Dubreil
b,
Sébastien G.
Gouin
c,
Jean Philippe
Combal
d,
Mirja
Hommel
e,
Gloria
Gonzalez-Aseguinolaza
de,
Véronique
Blouin
a,
Philippe
Moullier
a,
Oumeya
Adjali
a,
David
Deniaud
*c and
Eduard
Ayuso
*a
*a,
Mohammed
Bouzelha
a,
Aurélien
Leray
ac,
Simon
Pacouret
a,
Mickael
Guilbaud
a,
Magalie
Penaud-Budloo
a,
Dimitri
Alvarez-Dorta
c,
Laurence
Dubreil
b,
Sébastien G.
Gouin
c,
Jean Philippe
Combal
d,
Mirja
Hommel
e,
Gloria
Gonzalez-Aseguinolaza
de,
Véronique
Blouin
a,
Philippe
Moullier
a,
Oumeya
Adjali
a,
David
Deniaud
*c and
Eduard
Ayuso
*a
aINSERM UMR 1089, Université de Nantes, CHU de Nantes, 44200 Nantes, France. E-mail: mathieu.mevel@univ-nantes.fr; eduard.ayuso@univ-nantes.fr
bPanTher-UMR 703, INRA-ONIRIS, Atlanpole-Chanterie, 44307 Nantes, France
cLUNAM Université, CEISAM, Chimie Et Interdisciplinarité, Synthèse, Analyse, Modélisation, UMR CNRS 6230, UFR des Sciences et des Techniques, 44322 Nantes, France. E-mail: david.deniaud@univ-nantes.fr
dVivet Therapeutics SAS, Paris, France
eGene Therapy and Regulation of Gene Expression Program, CIMA, FIMA, University of Navarra, Navarra Institute for Health Research (IdisNA), Pamplona, Spain
First published on 9th December 2019
Abstract
Gene delivery vectors based on adeno-associated virus (AAV) are highly promising due to several desirable features of this parent virus, including a lack of pathogenicity, efficient infection of dividing and non-dividing cells and sustained maintenance of the viral genome. However, the conclusion from clinical data using these vectors is that there is a need to develop new AAVs with a higher transduction efficiency and specificity for relevant target tissues. To overcome these limitations, we chemically modified the surface of the capsid of AAV vectors. These modifications were achieved by chemical coupling of a ligand by the formation of a thiourea functionality between the amino group of the capsid proteins and the reactive isothiocyanate motif incorporated into the ligand. This strategy does not require genetic engineering of the capsid sequence. The proof of concept was first evidenced using a fluorophore (FITC). Next, we coupled the N-acetylgalactosamine ligand onto the surface of the AAV capsid for asialoglycoprotein receptor-mediated hepatocyte-targeted delivery. Chemically-modified capsids also showed reduced interactions with neutralizing antibodies. Taken together, our findings reveal the possibility of creating a specific engineered platform for targeting AAVs via chemical coupling.
Introduction
Adeno-associated virus (AAV) is a small non-enveloped single stranded DNA (ssDNA) virus whose capsid is composed of 60 viral proteins (VP1, VP2 and VP3 with an average ratio of around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:10).
:1:10).
Viral vectors derived from AAV have become the tool of choice for gene transfer, mainly because of their greater in vivo efficiency compared to other vectors, their tropism for a broad variety of tissues, and their excellent safety profile.1 Therapeutic efficacy following AAV vector gene transfer has been reported in several preclinical studies and, over the past decade, some of these approaches have been successfully transferred to clinical practice, leading to exciting results in the field of gene therapy.2 Market approval of two AAV-based gene therapy products (Luxturna® and Zolgensma®) in Europe and the USA constitutes additional evidence that the field is progressing from proof-of-concept studies toward clinical development.3–5
Nonetheless, clinical human trials have shed light on the limitations of using AAV vectors as therapeutics due to their broad tropism, which results in transgene expression in off-target tissues.1 It is also well recognized that host- and vector-related immune challenges need to be overcome for long-term gene transfer.6,7
Most gene therapy applications to date have used the serotype 2 (AAV2) due to its high in vivo level of transduction in a wide range of mammalian post-mitotic cells, including muscle cells, hepatocytes and neurons.8–12 This serotype has also been used for gene transfer to the muscle and liver in clinical trials for hemophilia B13 and has been approved to be used in the retina for the treatment of Leber congenital amaurosis.5 The discovery of naturally occurring AAV isolates (>100 serotypes)14 in humans and animals species and genetic modification of the capsid of these AAV serotypes using molecular tools15–17 resulted in promising results in preclinical animal models and phase I/II clinical trials, which foster exciting clinical translation in the near future. However, their therapeutic index remains low, which implies that high concentrations have to be administered (>1014 AAV particles per kg) with the risk of adverse effects such as immunogenicity and toxicity.18,19 Moreover, a large part of the human population is also seropositive for AAV and has developed neutralizing antibodies (NAb) impairing gene delivery.20 Thus, the current technology delivers recombinant AAV with a perfectible low therapeutic index.
Recently, the grafting of a functionalized RGD peptide onto the capsid of a genetically modified AAV to specifically target tumor cells was reported.21,22 The results showed the possibility of attaching a ligand onto the capsid of AAV in order to improve its transduction efficiency for tumor cells. However, this approach required the introduction of an azide moiety into the AAV capsid by mutating the VP3 sequence and introducing unnatural amino acids. Consequently, the manufacturing of genetically modified AAV with this specific coupling functionality remains very demanding because it requires optimization of each step of production and purification, followed by complex characterization of the particles.
An alternative approach is to develop engineered AAV through chemical strategies without the need to modify the amino acid composition of the AAV capsid. Lysine residues on the viral surface are most commonly exploited as a molecular anchor for conjugating amine-reactive molecules. For example, amine-reactive taxol has already been conjugated to the amino groups of the AAV surface, as confirmed qualitatively by dot blot analysis.23 The resulting taxol-AAV particles did not effectively eradicate cancer cells in vitro, possibly due to the limited drug conjugation and its low release from the virus.23 PEGylated AAV particles via amine functionalities have also been developed to protect the virus from neutralization and enable significant levels of gene expression upon re-administration without compromising the patient's immune system.24,25 While these examples from the literature are very promising and encouraging, there is still the need of improvement to increase the therapeutic index of AAVs.
The liver remains a main target for gene therapy. Successful long-term gene transfer to the liver has the potential to treat not only various plasma protein deficiencies, including hemophilia, but also metabolic disorders such as ornithine transcarbamylase (OTC) deficiency,26 methylmalonic acidaemia,27 familial hypercholesterolaemia syndromes, and the biochemical effects of a number of lysosomal storage disorders. Receptor-mediated endocytosis is one of the most attractive approaches to deliver drugs to specific cell types, particularly hepatocytes. The asialoglycoprotein receptor (ASGP-R), highly expressed on the hepatocytes, is a carbohydrate-binding protein that recognizes and binds N-acetylgalactosamine (GalNAc) or galactose residues. The efficient endocytosis of appropriate ligands, i.e. GalNAc, has long been a validated strategy in medicinal chemistry for liver-specific drug and gene delivery.28
Here, we have used targeting ligands with a reactive isothiocyanate coupling functionality for chemical modification of the AAV capsid. The proof of concept was first validated with the fluorophore FITC to find the optimal conditions (buffer and pH) of covalent coupling on the surface of the AAV capsid, avoiding the degradation of the virus during the process. Next, we developed a viable and easy method to chemically modify the AAV with hepatocyte-targeting ligands (GalNAc) and demonstrated a significant increase in hepatocyte transduction in vitro compared to the non-modified AAV. Interestingly, when injected into mice, chemically modified AAV generated less anti-capsid antibodies compared to natural AAV2 vectors. This versatile bioconjugation method will be of high interest to tune AAV capsid immunoreactivity and tropism in multiple therapeutic applications.
Results and discussion
We first investigated the optimal buffer and pH conditions for bioconjugation, which are key factors impacting both the viral vector stability and the chemical coupling yields. Classically, for the modification of amino groups of proteins by nucleophilic addition, it is recommended to work with Na2CO3/NaHCO3 buffer at pH = 9.3. The AAV2 capsid is not altered when formulated in the following buffers: Na2CO3/NaHCO3 pH = 9.3 (ref. 29–31), PBS,32,33 dPBS23 or HEPES at pH = 7.4 (ref. 24). However, it is well established that buffer pH and composition can affect infectivity. In this work, we first screened different buffers and showed that incubation of AAV2 in TBS buffer at pH = 9.3 for 4 h had no harmful effect on the transduction efficiency of HEK293 cells measured by green forming units (GFU), contrary to the use of PBS or, to a lesser extent, dPBS (Fig. S1-A†). Surprisingly, the infectivity of AAV2 in Na2CO3/NaHCO3 buffer at pH = 9.3, Fig. S1-B,† was reduced in a log scale compared to AAV2 in TBS buffer at the same pH.These results clearly demonstrate that the impact of the buffer composition is an important factor to consider in developing a technological platform for AAV capsid modifications with chemical compounds.
Using the available crystallographic data on the AAV2 structure, we have determined the surface exposed amino acid residues on the capsid and the number of reactive functionalities available for chemical modifications. We focused our study on lysine residues bearing the reactive primary amino group involved in chemical coupling. Crystallographic data are only available for the VP3 subunit and not for VP1 and VP2.34 VP2 and VP3 share a common C-terminal amino-acid sequence with VP1 and are shortened by 67 and 137 residues, respectively, in their N-terminus part compared to VP1. Thus, only the amino acids located in the VP1/VP2/VP3 overlapping sequence were incorporated into our analysis. Nonetheless, VP1/2 N-terminal domains are thought to be inside the capsid under physiological conditions, potentially limiting their accessibility for chemical modification.
According to the AAV2 sequence, there are 18 lysine residues (out of 34) located in VP3, eight of them being exposed on the capsid external surface (Fig. S2†). Considering that there are 60 VP3 proteins per particle, this gives a total of 480 amino groups potentially accessible for chemical coupling on the AAV2 capsid.
Fluorescein isothiocyanate (FITC) was first used to prove the feasibility of covalent grafting on the capsid surface. We anticipated that isothiocyanate-armed compounds would form a covalent thiourea bond with the amino groups of the lysine on the AAV surface.
Fluorescein, a chemically related fluorophore without the reactive isothiocyanate functionality, was used as a control to prove the covalent linkage of FITC and to exclude the physical adsorption of the molecule on the AAV2 surface (Fig. 1A and B).
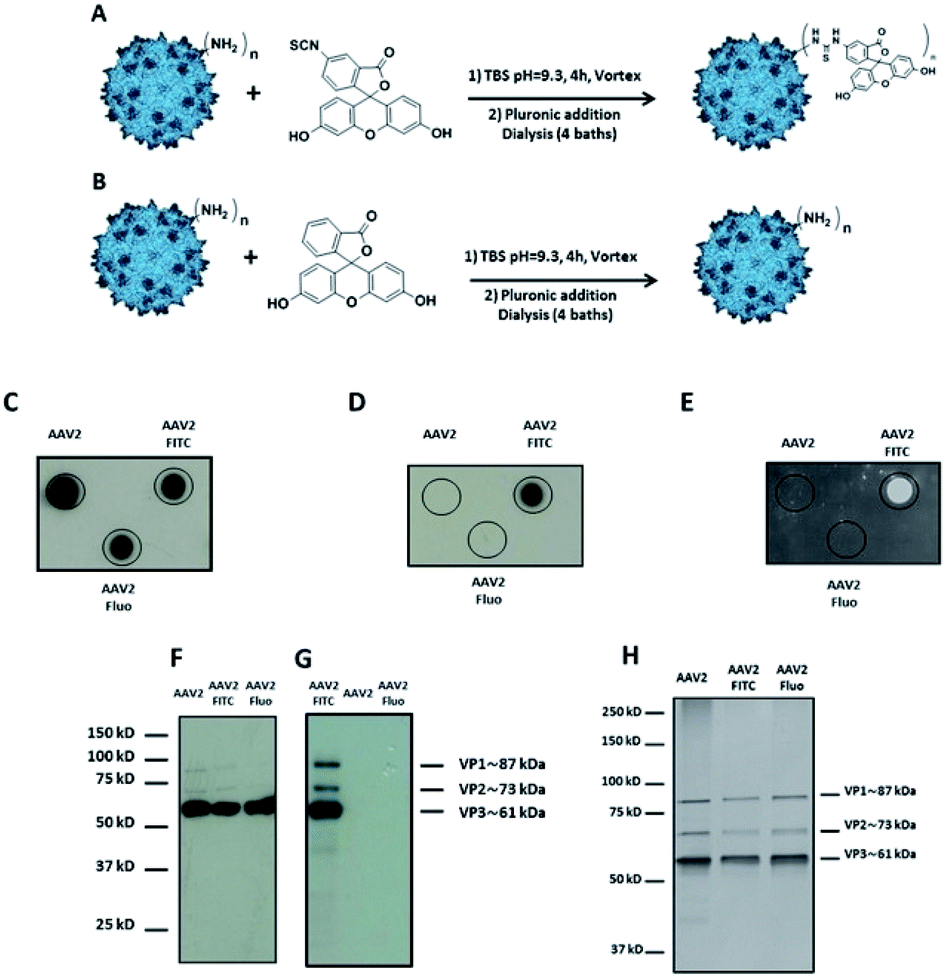 | ||
| Fig. 1 Covalent coupling of FITC onto the capsid of AAV2 via primary amino groups. (A) 1012 vg of AAV2-GFP vectors were added to a solution of FITC (3 × 105 eq.) in TBS buffer (pH 9.3) and incubated for 4 h at RT. (B) The procedure was carried out with fluorescein (3 × 105 eq.) as a control. (C–E) 109 vg of each condition was analyzed by dot blot using the A20 antibody that recognizes the assembled capsid (C) or using an anti-FITC antibody (D) or by direct fluorescence emission (E). (F and G) 5 × 108 vg of the same samples were analyzed by western blot using a polyclonal antibody to detect denatured AAV capsid proteins (F) or using an anti-FITC antibody (G). (H) 1010 vg of each condition was analyzed by silver nitrate staining. | ||
Dot blot analysis using the A20 antibody, which recognizes assembled AAV capsids, indicated that AAV particles remained intact after undergoing the reaction and subsequent dialysis (Fig. 1C). Notably, positive FITC and negative fluorescein dots (Fluo) demonstrated the covalent coupling of FITC onto the virus capsids and not its adsorption (Fig. 1D and E). Indeed, dialysis treatment of the samples and extensive washing during dot blot experiments should remove free molecules that are not covalently bond to the capsid. Western blot analysis was performed to further confirm the successful conjugation of FITC to the AAV capsid subunits. A polyclonal antibody was used to detect the denatured capsid proteins. As subunits are incorporated into the AAV capsid in a 1:1:10 ratio (VP1:VP2:VP3), a more intense VP3 band compared to VP1 and VP2 was observed under all the conditions tested. This also indicated that the AAV2 capsid subunits remained intact after undergoing the reaction and subsequent dialysis against dPBS + pluronic (Fig. 1F). An anti-FITC antibody was used to detect the presence of FITC covalently linked to the AAV capsid subunits. As shown in Fig. 1G, the capsid subunits from AAV2 and AAV2 incubated with fluorescein did not yield any positive bands. However, AAV2 incubated with FITC showed positive bands at appropriate molecular weights of VP1, VP2, and VP3, unambiguously demonstrating the covalent coupling of FITC onto the three subunits of the AAV capsid.
In order to visualize all the proteins and to confirm that there was no degradation during the chemical process, silver staining of the different conditions was also carried out.
As shown in Fig. 1H, the expected ratio of VP1:VP2:VP3 was observed under all the conditions tested.
All the analytical methods used in this study proved the covalent coupling of FITC onto the surface of the AAV2 capsid while maintaining the integrity of the virus capsid. Furthermore, we showed that TBS buffer can be used for the chemical modification of amino groups on the AAV capsid by the formation of a thiourea bond.
It is well established that genetic mutations of specific surface-exposed amino acids have an impact on AAV2 infectivity. Li et al. showed that the substitution of surface-exposed lysine residues increased the transduction efficiency of genetically modified AAV2 for murine hepatocytes in vivo.35 Lee et al.24 and Le et al.25 reported that chemical modifications of surface-exposed amino groups with PEG derivatives decreased the interaction of the virus with neutralizing antibodies. Thus, genetic or chemical modifications of the AAV2 capsid have a clear impact on different key parameters, like the trafficking of the modified virus and the interaction with neutralizing antibodies.
Nonetheless, none of these studies have evaluated the possibility of modulating the density of molecules conjugated on the surface of AAV2. Indeed, fine-tuning the molecule coating could be critical in improving the therapeutic index of the particles. AAV2 binds to its primary receptor heparan sulfate using a basic cluster of amino acids (R484, R487, K532, R585, and R588) located at the threefold axis of symmetry. Moreover, Patel et al. have identified a subset of five peptides on the AAV2 capsid, which potentially reconstitute a single neutralizing epitope, and three of these peptides are composed by at least one lysine.20 Thus, chemical modification of these essential amino groups could significantly impact trafficking and binding interactions with neutralizing antibodies.
Different molar ratios (from 3 × 105 to 1.5 × 107 eq.) of FITC were incubated with AAV2 in TBS following the same experimental procedure as above (Fig. 2). Fluorescein was also used at the highest molar ratio as a negative control. The reactivity with a polyclonal antibody indicated that the binding epitopes of the AAV2 capsid subunits remained accessible in denatured samples (i.e. western blot) at the higher FITC molar ratios used of 1.5 × 107 (Fig. 2A). This shows that high FITC conjugation did not disrupt the recognition of the polyclonal antibody with the AAV capsid. The capsid subunits from native AAV2 and AAV2 incubated with fluorescein at the highest ratio did not yield positive bands after incubation with the anti-FITC antibody (Fig. 2B). The enhanced band intensity for the higher molecular ratio (3 × 106 and 1.5 × 107) compared to 3 × 105 clearly showed that the number of FITC molecules covalently linked to AAV proportionally increased (Fig. 2B). It is therefore possible to tune the coating density of a chemical at the AAV surface, which may be highly relevant for improving its targeting capacity and PK/PD profile.
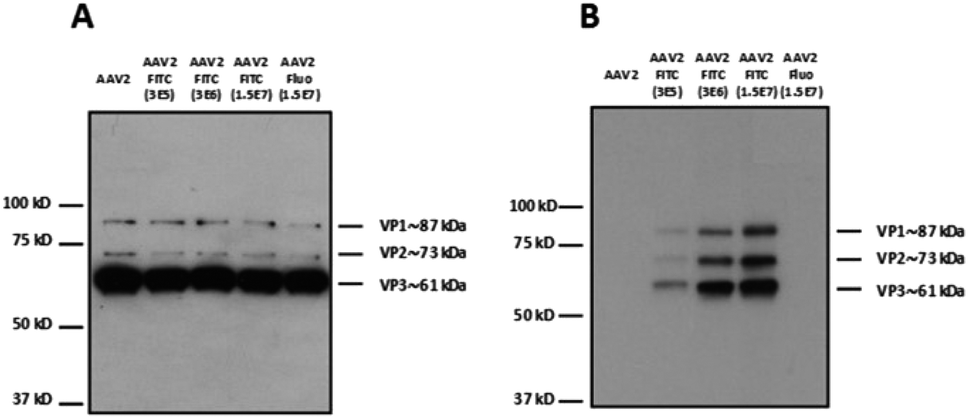 | ||
| Fig. 2 Modulation of the number of FITC molecules on the capsid of AAV. (A) 5 × 108 vg of the samples were analyzed by western blot using a polyclonal antibody to detect denatured AAV capsid proteins or (B) an anti-FITC antibody to detect VP capsid proteins chemically modified with FITC molecules. | ||
To examine whether the chemically modified virus remained infectious, AAV2-FITC and native AAV2 carrying a GFP reporter gene were used to infect HEK293 cells. As shown in Table 1, all the viral nanoparticles tested were infectious after the chemical process. However, the highest molar ratios of FITC used during the coupling led to a reduced infectivity on HEK293 cells as evidenced by the higher vg per GFU ratio. The high FITC payload may shield the basic amino acid cluster recognizing the heparan sulfate receptor involved in AAV2 binding. This highlights the importance of a balanced density of coating to confer new properties to AAV while preserving its infectivity.
| vg per mL | GFU per mL | vg per GFU | |
|---|---|---|---|
| Native AAV2 | 4.60 ± 0.21 × 1012 | 5.40 ± 0.18 × 109 | 84.6 ± 4.0 |
| AAV2-FITC (3 × 105 eq.) | 1.10 ± 0.0071 × 1011 | 1.10 ± 0.54 × 109 | 121.7 ± 52.7 |
| AAV2-FITC (3 × 106 eq.) | 2.20 ± 0.014 × 1011 | 6.20 ± 3.3 × 108 | 449.8 ± 221.8* |
| AAV2-FITC (1.5 × 107 eq.) | 1.10 ± 0.071 × 1011 | 1.20 ± 0.47 × 108 | 999.8 ± 377.1*** |
| AAV2 + fluo (1.5 × 107 eq.) | 2.20 ± 0.071 × 1011 | 1.80 ± 0.41 × 109 | 122.6 ± 24.0 |
Next, we studied the intracellular trafficking of AAV2-FITC particles in HeLa cells by confocal microscopy. The FITC fluorescence intensity was directly detected (green, λ = 525 nm) while the AAV capsid was labeled with the primary A20 conformational antibody followed by the AI647 secondary antibody (red). All appropriate controls are shown in Fig. S3.† As expected, the assembled capsid of AAV2 was only detected by the A20 antibody (Fig. 3B). The presence of both FITC and the assembled capsid was detected for the lower FITC coating conditions (3 × 105 molar ratio), as shown in Fig. 3F. The colocalization of the two fluorescent signals confirmed the integrity of the AAV2 capsid, the presence of the FITC ligand on the AAV surface, the recognition of the capsid by the A20 antibody and the infectivity (i.e. cellular entry) of these nanoparticles. For the 3 × 106 and 1.5 × 107 molar ratio conditions, the presence of FITC was clearly detected by confocal analysis (Fig. 3G and J) showing effective chemical coupling. However, the assembled capsid was not (or barely) recognized by the A20 antibody. These results are in full accordance with the decreased infectivity observed at a higher FITC payload. This further confirms that the chemical masking of amino groups by FITC molecules within surface-exposed domains could alter the antigenicity of the AAV2-FITC vectors. Such a chemically shielded AAV could potentially evade pre-existing neutralizing antibodies in the human population while being infectious. Altogether, these interesting results demonstrate the possibility of modifying the target properties of AAV using chemical modifications at different molar ratios during conjugation.
 | ||
| Fig. 3 Confocal imaging of modulated AAV2-FITC. (A–C) HeLa cells transduced with AAV2 and incubated with A20 primary antibody and Al647 secondary antibody. Red fluorescence from A20 immunolabeling (B) was detected. (D–F) HeLa cells transduced with AAV2-3 × 105 FITC and incubated with A20 primary antibody and Al647 secondary antibody. Green FITC fluorescence (D) and red fluorescence from A20 immunolabeling (E) were detected. Colocalisation of FITC and A20-Al647 (F). (G–I) HeLa Cells were transduced with AAV2-3 × 106 FITC and incubated with A20 primary antibody and Al647 secondary antibody. Green FITC fluorescence from AAV2-3 × 106 FITC was detected (G) whereas the detection of red fluorescence from A20 immunolabeling was very low (H). (J–L) HeLa cells were transduced with AAV2-1.5 × 107 FITC and incubated with A20 primary antibody and Al647 secondary antibody. Green FITC fluorescence from AAV2-1.5 × 107 FITC was detected (J) whereas the detection of red fluorescence from A20 immunolabeling was very low (L). Cell nuclei were counterstained with DAPI (blue); scale bars: 25 μm for FITC and A20 images; 5 μm for merged images. All the controls are detailed in the ESI.† | ||
Then, our efforts were focused on improving AAV infectivity for a specific cell type. ASGP-R, overexpressed on hepatocytes, exhibits a high affinity for GalNAc,28 and this sugar is widely used as a carbohydrate-targeting unit for specific liver delivery. Here, we designed three GalNAc ligands with a terminal isothiocyanate (compounds 4 and 5) or alkyl group (compound 8). 4 and 5 were developed to covalently coat the AVV2 surface and 8 lacking the NCS group was used as a control in the assay.
The chemical synthesis of alkylisothiocyanate (Alk-NCS) 4 and arylisothiocyanate (Ar-NCS) 5 is shown in Scheme 1. The azide derivative 136 was reduced with Pd/C under a hydrogen atmosphere in the presence of APTS to form compound 2 in a quantitative yield. After deprotection of the acetyl groups with the basic resin IRN78, the isothiocyanate 4 was obtained with 88% yield by adding 1,1′-thiocarbonyldi-2(1H)-pyridone. The Ar-NCS compound 5 was obtained with a good yield of 85% by reacting compound 3 with an excess of p-phenylene diisothiocyanate (Scheme 1A). The control compound 8 was obtained in two steps after glycosylation of the oxazolidinone 636 with 2-(2-ethoxyethoxy)ethanol followed by deprotection of the acetyl groups (Scheme 1B). All chemical compounds were characterized by NMR, HPLC and mass spectroscopy (Fig. S4–S26†).
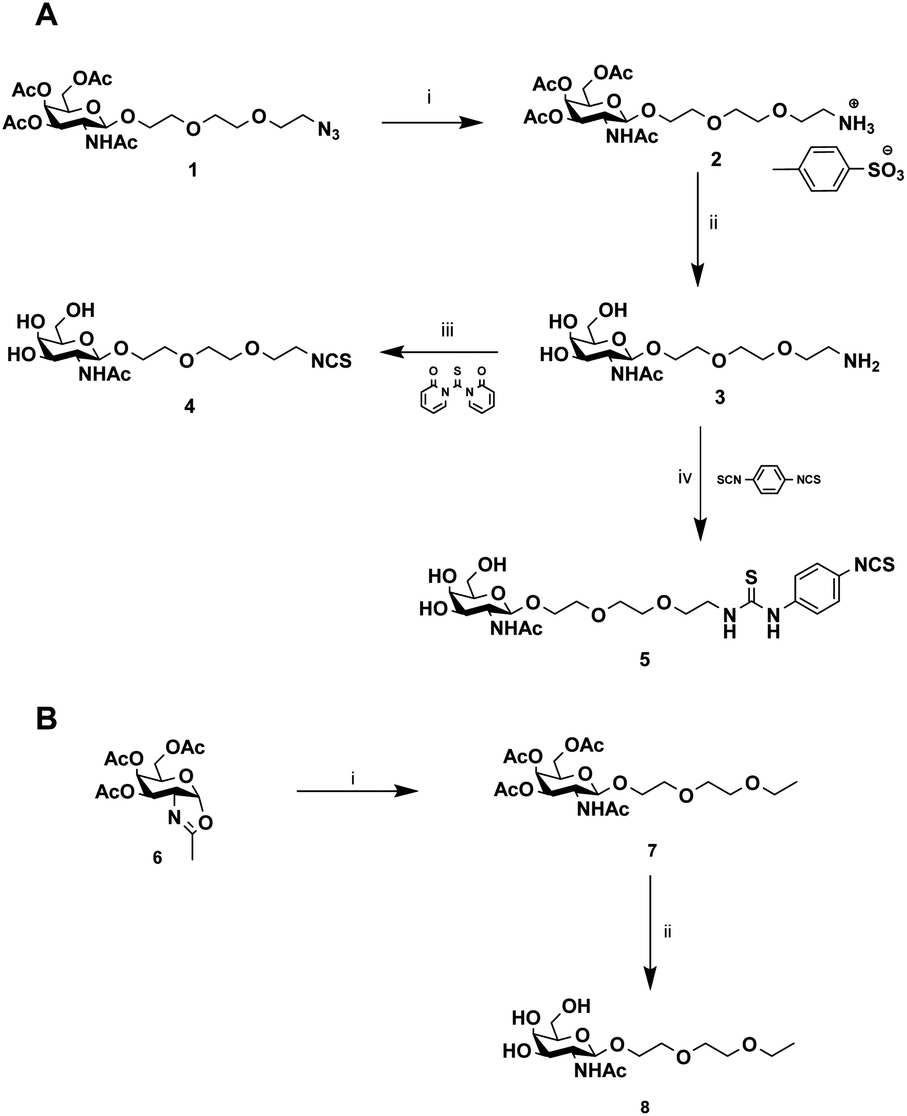 | ||
| Scheme 1 Synthesis of GalNAc derivatives with Alk-NCS, Aryl-NCS coupling functionality and without the coupling functionality. (A) (i) MeOH, APTS, H2, Pd–C (100%), (ii) MeOH/H2O, IRN78 (77%), (iii) DMF, 1,1′-thiocarbonyldi-2(1H)-pyridone (88%), (iv) DMF, p-phenylene diisothiocyanate (85%). (B) (i) DCM, 2-(2-ethoxyethoxy)ethanol, molecular sieves (73%), (ii) MeOH/H2O, IRN78 (77%). | ||
The same coupling conditions that were optimized in FITC experiments were used to assess the relative anchoring abilities of compounds 4 and 5 on the AAV2 capsid (Fig. 4A).
 | ||
| Fig. 4 Identification of the reactive function for the covalent coupling of GalNAc ligands on the capsid of AAV2 via primary amino groups. (A) 1012 vg of AAV2-GFP vectors were added to a solution of compound 4 or compound 5 (3 × 105 eq.) in TBS buffer (pH 9.3) and incubated for 4 h at RT. The same experimental procedure was followed for compound 8 (3 × 105 eq.) in TBS at pH 9.3 as a control. (B and C) AAV2 control and samples of AAV2 vectors incubated with GalNAc ligands in TBS buffer (AAV2 + 4, AAV2 + 5 and AAV2 + 8) were analyzed by dot blot. 1010 vg of each condition was analyzed using the A20 antibody that recognizes the intact capsid (B) or using soybean-FITC lectin that recognizes the N-acetylgalactosamine sugar (C). | ||
The coupling reaction of the sugar on the AAV surface was first studied by dot blot analysis using soybean lectin which binds selectively to GalNAc residues37 and the A20 antibody. The positive dots with the A20 antibody, which recognized the entire capsid, indicated that AAV2 remained intact after the coupling procedure with 4, 5 or 8 and subsequent dialysis (Fig. 4B). Positive signals with soybean lectin were only observed when AAV2 was co-incubated with compound 5 revealing that Ar-NCS but not Alk-NCS allowed an efficient coupling with the amino groups on the AAV2 capsid (Fig. 4C). Consistent with previous results obtained with fluorescein, no detection was observed with compound 8 that lacked the NCS group. Thus, compound 5 was covalently attached, and not physically adsorbed, to AAV2.
In order to modulate the number of GalNAc ligands on the capsid surface, two different molar ratios of 5 (3 × 105 and 3 × 106) were used. 8 was used only at the highest molar ratio (3 × 106) to probe the absence of adsorption of 5 onto the capsid surface.
For western blot analyses, the use of a capsid-specific polyclonal antibody indicated that VP capsid subunits remained intact regardless of the molar ratios used (Fig. 5A). Importantly, the band intensity clearly showed that the number of molecules from 5 covalently attached to the three VPs increased when the molar ratio was switched from 3 × 105 to 3 × 106. These results are in agreement with western blot using soybean lectin, which gave more intense bands for the higher stoichiometry of 5 used during the coupling procedure (Fig. 5B).
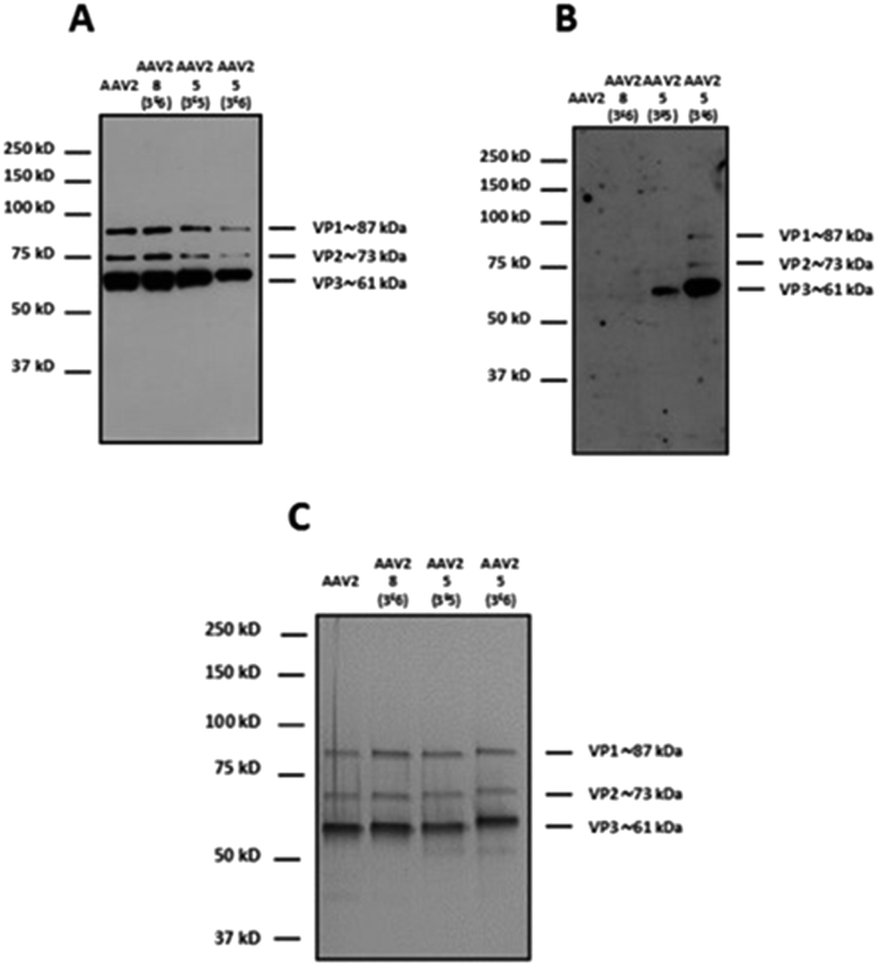 | ||
| Fig. 5 Modulation of the number of GalNAc molecules on the capsid of AAV vectors. 1012 vg of AAV2-GFP vectors were added to a solution of 5 (3 × 105 and 3 × 106 eq.) in TBS buffer (pH 9.3) and incubated for 4 h at RT. The same experimental procedure was followed but substituting 5 by 8 (3 × 106 eq.) as a control. 5 × 108 vg of the samples was analyzed by western blot using a polyclonal antibody against the capsid proteins to detect VP1, VP2 and VP3 proteins (A) or using FITC-soybean lectin (B). (C) 1010 vg of each condition was analyzed by silver nitrate staining. The capsid protein molecular weight is indicated at the right of the images according to a protein ladder. | ||
In contrast, the capsid subunits from AAV2 and AAV2 incubated with 8 at the highest ratio did not yield positive bands after incubation with the specific lectin indicating that no coupling occurred without the NCS functionality (Fig. 5B). To evaluate the purity and integrity of these GalNAc-AAV2 complexes, silver staining of the different samples was carried out.
As shown in Fig. 5C and as expected, the ratio VP1:VP2:VP3 remained intact after undergoing the reaction and subsequent dialysis. The molecular weight of each VP also seemed to increase with increasing GalNAc loading.
As the aggregation of recombinant AAV2 is a well-known phenomenon, the particle size and dispersity of the modified and non-modified AAV2 were measured by dynamic light scattering (DLS) (Table 2) and visualized by transmission electron microscopy (Fig. 6). As shown in Table 2, all samples analyzed showed >90% of the particles around 26–29 nm, proving that the GalNAc residues did not promote AAV aggregation. This was also confirmed by TEM analyses as shown in Fig. 6. Taken together, these data evidenced the feasibility of grafting a targeting ligand at different densities on the AAV capsid surface without impacting its integrity.
| Samples | Size (nm) | Percentage of population |
|---|---|---|
| Native AAV2 | Test 1: 26.2 | >99% |
| Test 2: 26.4 | >99% | |
| AAV2 + 5 (3 × 105) | Test 1: 25.6 | >95% |
| Test 2: 25.2 | >96% | |
| AAV2 + 5 (3 × 106) | Test 1: 28.0 | >92% |
| Test 2: 28.6 | >91% | |
| AAV2 + 8 (3 × 106) | Test 1: 26.0 | >96% |
| Test 2: 25.4 | >96% |
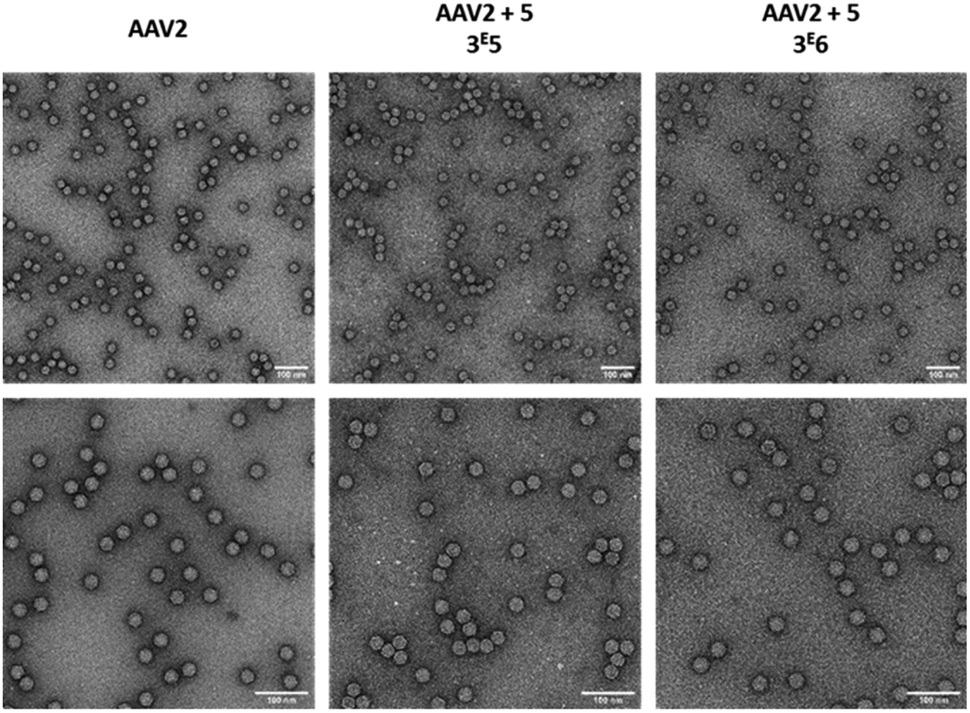 | ||
| Fig. 6 Electron microscopy images of GalNAc-AAV2 particles. Unmodified AAV2 vectors (left) and AAV2 vectors modified with 3 × 105 (middle) or 3 × 106 equivalent (right) of 5 were negatively stained with uranyl acetate and examined by transmission electron microscopy at a ×50 000 (upper panel) or ×80 000 (lower panel) nominal magnification. Scale bars: 100 nm. | ||
To evaluate the efficiency of GalNAc functionalized AAV2, the transduction of these modified particles was assessed using primary mouse hepatocytes. Of note, it has been shown that native AAV2 has a very low transduction level on this cell type.38 In order to prove a mechanism involving GalNAc and ASGP-R internalization in hepatocytes, AAV2 was also modified with 11, a structural analog of 5 bearing a mannose, which does not bind ASGP-R. The synthesis of 11 and the negative control 14 and the validation of the covalent coupling is described in Fig. S27.† As shown in Fig. 7, the chemically modified AAV2 capsid with GalNAc (ligand 5) had a strong effect on the transduction efficiency. The percentage of GFP positive cells dramatically increased from 5% with native AAV2 to 12% with AAV2 + 5 at 3 × 105 eq. and to 28% with AAV2 + 5 at 3 × 106 eq. The dose effect clearly showed the significant impact of the GalNAc ligands on the transduction efficiency. The percentage of GFP positive cells around 3% obtained with AAV2 + 11 (3 × 106) prove unambiguously that the higher transduction efficiency is not related to a physicochemical change of the AAV2 surface but that it was receptor mediated. These data suggest that GalNAc-modified AAV2 interacts with the ASGP-R receptor on the cell surface, inducing higher internalization via ASGP-R-promoted endocytosis which leads to enhanced protein expression.
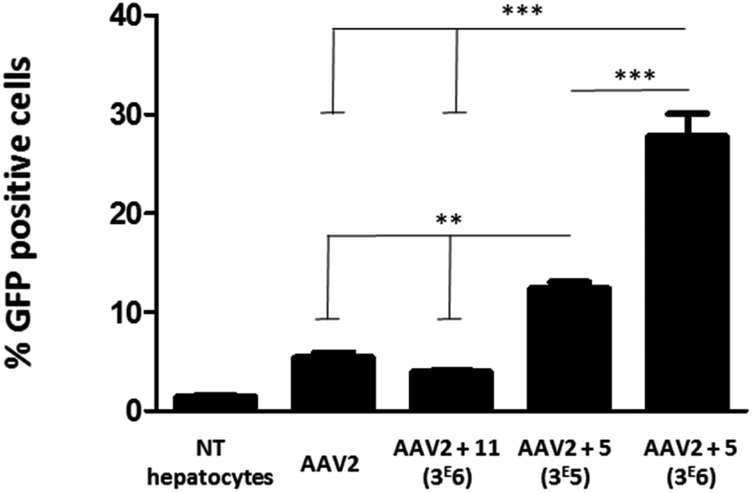 | ||
| Fig. 7 Transduction of primary mouse hepatocytes with native AAV2 and AAV2 vectors chemically modified with GalNAc and mannose ligands. Primary mouse hepatocytes (1.7 × 105 cells per well) were incubated in P24 plates and transduced with the native AAV2 control, AAV2 + 11 (3 × 106), AAV2 + 5 (3 × 105) and AAV2 + 5 (3 × 106) at a MOI of 105. All AAV vectors encoded for GFP. The percentage of GFP positive cells was measured by FACS analysis 72 h after the transduction. Non-transduced cells (NT hepatocytes) were used as a control for the fluorescence background. Three replicates of each condition were analyzed by ANOVA test (***p < 0.001, **p < 0.01). Data are represented as mean ± SD. | ||
Furthermore, the intra-cellular trafficking of the vector seemed unaffected by the chemical modification. It is tempting to speculate that the phospholipase A2 domain from VP1, which is crucial for endosomal escape,39 has not been chemically modified or the chemical ligands do not interfere in this process.
To test the specificity of these vectors in non-hepatocyte cell lines, we transduced HEK293 with native AAV2 and GalNac modified capsids. The chemical coupling of 3 × 105 and 3 × 106 eq. of GalNac ligands onto the AAV2 capsids reduced the transduction efficacy in HEK293 (i.e. increase in the vg per GFU ratio) in a dose dependent manner akin to results presented in Table 2 for AAV2-FITC (data not shown).
To test the transduction efficiency and immunogenicity of AAV2-GalNAc conjugates in vivo, mice were injected with either unmodified AAV2 carrying the eGFP transgene or GalNAc-AAV2 at a molar ratio of 3 × 105 and 3 × 106.
A control group received PBS only. Twenty-one days after vector administration, the animals were sacrificed, organs were extracted and the relative GFP mRNA expression levels were measured (Fig. 8A). In AAV2 and GalNAc-AAV2 (3 × 105) treated animals, the levels of GFP mRNA expression were within the same range whereas in the GalNAc-AAV2 (3 × 106) group the mRNA expression was lower, suggesting that the density of the GalNAc coating was too high to allow critical transduction in vivo. In addition, a small biodistribution study was performed to investigate the tropism of the vectors across multiple organs of the body (Fig. 8B). The vg copies in all tissues were very low, with more presence of the vector in the spleen and to a very low level also in the adrenal glands. At a molar ratio of 3 × 106, vg copy numbers in the spleen were reduced compared to unmodified AAV2. To study the immunogenicity of these vectors, total and neutralizing antibodies against AAV2 were measured in the serum of mice twenty-one days after administration (Fig. 8C and D). Interestingly, coating AAV2 capsids with GalNAc resulted in a drastic inhibition of antibody formation, likely due to an epitope shielding effect.
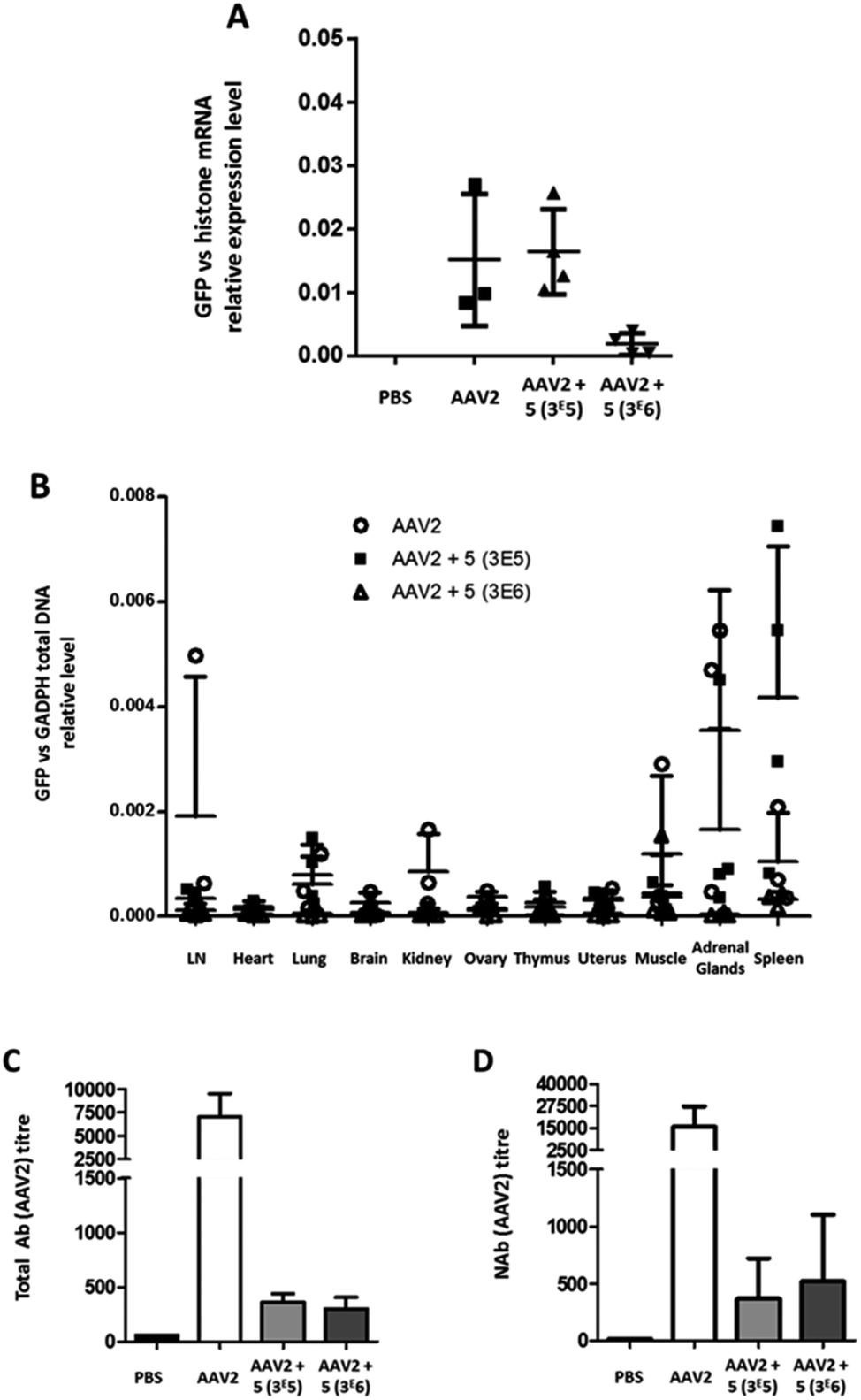 | ||
| Fig. 8 Liver transduction profile of GalNAc-AAV2 and development of anti-AAV2 antibodies. Groups of mice were injected with unmodified AAV2 eGFP or GalNAc-AAV2 carrying a GFP transgene at molar ratios of 3 × 105 or 3 × 106. A control group received PBS only. Twenty-one days after administration, the mice were sacrificed and organs were extracted. (A) The GFP mRNA expression levels in liver samples were normalized against endogenous histone mRNA expression. In addition, a biodistribution analysis was performed (B). Total (C) and neutralizing (D) antibody levels in serum were determined. Medians and range (A and B) or means and standard deviation (C and D) are shown. N = 2–6 animals per group. | ||
In contrast to in vitro data, this study in mice showed that the transduction efficiency of GalNAc-AAV2 in vivo was not superior to that of unmodified AAV2. However, the significant reduction (<1 log) of neutralizing antibody production with GalNac-AAV2 (3 × 105) while maintaining mRNA expression is a promising result.
The chemical bioconjugation developed here may be of broad interest for the capsid modification of virtually all AAV serotypes. To test this hypothesis, we have incubated AAV8 vectors with 3 × 105 eq. of the GalNAc ligands (compound 5) using the same buffers and conditions as described previously for AAV2.
Dot blot, western blot and silver staining unequivocally showed that AAV8 capsids are chemically modified as efficiently as AAV2 capsids (Fig. S28†). The same experiment has also been done with AAV3b, also showing the chemical modification of this serotype (Fig. S29†).
The transduction efficiency and immunogenicity of AAV8-GalNAc conjugates were then tested in vivo. The AAV8 serotype was selected for this study because this serotype has a much better liver tropism in mice than AAV3b. Groups of mice were injected with either unmodified AAV8 carrying an eGFP transgene or the conjugate of molar ratio 3 × 105. A control group received PBS only. Ten or twenty-one days after vector administration, the animals were sacrificed, organs were extracted and the relative eGFP mRNA expression levels were measured (Fig. 9A and B). While at ten days post administration, a statistically significant improvement in transgene expression could be observed for GalNAc-AAV8 compared to unmodified AAV8, despite the variability of samples. However, no differences were observed twenty-one days post vector administration. On the other hand, modifying AAV8 with GalNAc led to a small reduction of vector genome copies in the spleen when compared to unmodified AAV8 (Fig. 9C).
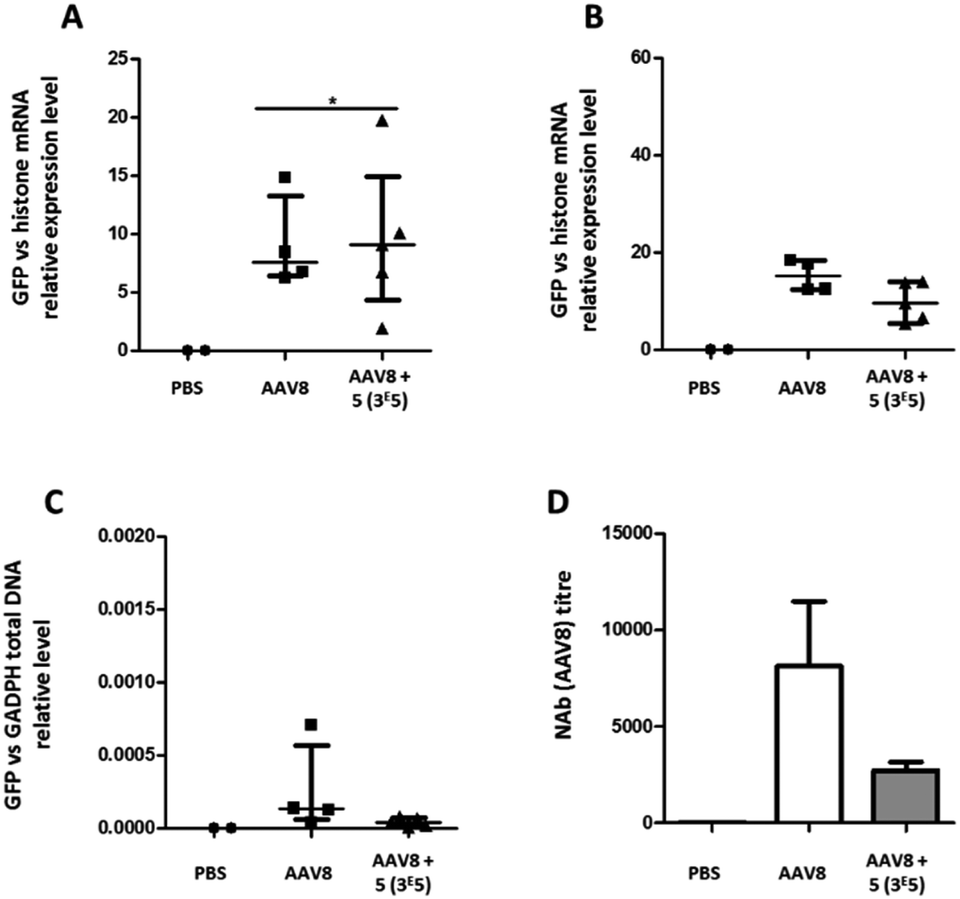 | ||
| Fig. 9 Liver transduction profile of GalNAc-AAV8 and development of anti-AAV8 antibodies. Groups of mice were injected with unmodified AAV8 eGFP or GalNAc-AAV8 carrying a GFP transgene at a molar ratio of 3 × 105. A control group received PBS only. Ten (A) and 21 days (B) after administration mice were sacrificed and organs extracted. (A and B) The GFP mRNA expression levels in liver samples were normalized against endogenous histone mRNA expression. GFP total DNA normalized to GAPDH in the spleen (C). Neutralizing antibody levels in serum were determined (D). Medians and range (A–C) or means and standard deviation (D) are shown. N = 2–6 animals per group. | ||
Importantly, in agreement with previous observations with AAV2, coating AAV8 with GalNAc led to a reduction of serotype-specific neutralizing antibodies (Fig. 9D), which opens the possibility to facilitate the re-administration of these vectors.
The presence of neutralizing antibodies (NAbs) against AAV in the human population is one of the main hurdles for AAV-mediated gene therapy. Today, patients' high NAb titers are excluded for clinical trials and NAbs generated upon rAAV administration block the possibility of readministration. Thus, there is a real need to overcome these limitations.
We previously observed with AAV2-FITC that surface modification of the capsid strongly impacted the interaction with A20 antibodies (Fig. 3). Hence, we assessed whether GalNAc shielding could also decrease the interaction of the modified AAV2 particles with neutralizing factors. To this end, GalNAc modified and non-modified AAV2 both encoding the beta-galactosidase protein (LacZ gene) were designed (Fig. S30†) and incubated with a pool of non-human primate serums known to neutralize rAAV2 transduction.
Neutralizing titers were determined as the highest dilution of serum that inhibited more than 50% of the AAV transduction signal. Non modified AAV2 LacZ was neutralized at a serum dilution of 1/640 (Fig. 10A) whereas GalNac-AAV2 + 5 (3 × 105) and (3 × 106) were neutralized at lower dilutions of serum (1/160 and 1/320 respectively – Fig. 10B and C). These data indicate reduced interactions with neutralizing factors opening interesting perspectives to avoid the detection of AAV2 by the immune system and potentially open the possibility to include patients that would be excluded in other clinical trials using non-modified AAV.
 | ||
| Fig. 10 Neutralizing assays of AAV2 and GalNAc-AAV2 on HeLa cells. HeLa cells were incubated with AAV2 LacZ (A), GalNac-AAV2 LacZ (3 × 105) (B) or GalNac-AAV2 LacZ (3 × 106) (C) in the presence of various dilutions of AAV2-neutralizing serum (1/20 to 1/5120). After measurement of beta-galactosidase activity, neutralizing titers were expressed as the highest dilution of serum that inhibited more than 50% (red police) of AAV2, GalNac-AAV2 LacZ (3 × 105) or GalNac-AAV2 (3 × 106) signals without incubation with neutralizing serum (IC50). | ||
Conclusion
In conclusion, we have successfully developed a method to chemically modulate the surface of AAV with specific ligands. Lysine on the AAV capsid surface was efficiently altered with phenylisothiocyanate anchors (i.e. covalent coupling) to coat the surface of AAV with specific ligands. Lysine on the AAV capsid surface was efficiently altered with phenylisothiocyanate anchors (i.e. covalent coupling) to coat the virus in a dose-dependent manner, without the need for genetic engineering of the capsid proteins.The density of FITC or GalNac ligands on the surface of the AAV vectors was shown to impact the transduction efficacy and ability to escape neutralizing antibodies, which opens interesting perspectives for improving their therapeutic index. The bioconjugation reaction could be performed with a similar efficiency on two other AAV serotypes, accounting for broad applicability. Although this proof-of-concept study was realized with AAVs carrying a marker gene, it is anticipated that therapeutically relevant AAV vectors can be chemically modified in a similar manner to be tested in animal models of disease and ultimately in clinical applications.
Of note, the bioconjugation step described herein is conducted with purified AAV, which means that it does not interfere with previously optimized production and purification protocols for well-known AAV serotypes. Moreover, we do not foresee difficulties to scale up this process to an industrial scale since the volume of purified vectors is usually <1 L and the coupling reaction takes place at room temperature for 4 h and can be accommodated easily under GMP conditions.
We foresee that the protocol developed here will be of great interest to not only fine-tune AAV tropism and enhance cell targeting in specific tissues, but also to decrease interactions with neutralizing antibodies. This procedure could help improve AAV selectivity and gene transduction efficiency, which are long standing issues limiting the scope of AAV in clinical trials.
Ethical statement
The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra and all experiments were performed in accordance with relevant institutional and national Spanish guidelines.Conflicts of interest
M. M., D. D., are E. A. are inventors on a patent including the technology described in this manuscript. JPC and GGA are employees of Vivet Therapeutics.Acknowledgements
The authors thank all the staff of the Vector Core at the University Hospital of Nantes (http://umr1089.univ-nantes.fr) for production of the AAV vectors and for technical assistance. We are grateful to Nicolas Jaulin for helping with flow cytometry analysis and Sandy Douthe for her contribution to the quality control of the vector stocks. Analysis and purification of chemical compounds were performed at the Chromatography Platform of CEISAM laboratory – UMR 6230 CNRS/UN, Université de Nantes. The authors thank the MRic Transmission Electron Microscopy Facility (University of Rennes, CNRS, Inserm, BIOSIT – UMS 3480, US_S 018, F-35000 Rennes, France) for its technical assistance. This research was supported by the Fondation d'Entreprise Thérapie Génique en Pays de Loire, the Centre Hospitalier Universitaire (CHU) of Nantes, the Institut National de la Santé et de la Recherche Médicale (INSERM) and the University of Nantes, by a grant from the French National Agency for Research called “Investissements d’Avenir” Equipex Arronax Plus n ANR-11-EQPX-0004 and by Vivet Therapeutics SAS.Notes and references
- C. Zincarelli, S. Soltys, G. Rengo and J. E. Rabinowitz, Mol. Ther., 2008, 16, 1073–1080 CrossRef CAS PubMed.
- F. Mingozzi and K. A. High, Nat. Rev. Genet., 2011, 12, 341–355 CrossRef CAS PubMed.
- H. Buning, EMBO Mol. Med., 2013, 5, 1–3 CrossRef.
- D. Gaudet, J. Methot, S. Dery, D. Brisson, C. Essiembre, G. Tremblay, K. Tremblay, J. de Wal, J. Twisk, N. van den Bulk, V. Sier-Ferreira and S. van Deventer, Gene Ther., 2013, 20, 361–369 CrossRef CAS.
- S. Russell, J. Bennett, J. A. Wellman, D. C. Chung, Z. F. Yu, A. Tillman, J. Wittes, J. Pappas, O. Elci, S. McCague, D. Cross, K. A. Marshall, J. Walshire, T. L. Kehoe, H. Reichert, M. Davis, L. Raffini, L. A. George, F. P. Hudson, L. Dingfield, X. Zhu, J. A. Haller, E. H. Sohn, V. B. Mahajan, W. Pfeifer, M. Weckmann, C. Johnson, D. Gewaily, A. Drack, E. Stone, K. Wachtel, F. Simonelli, B. P. Leroy, J. F. Wright, K. A. High and A. M. Maguire, Lancet, 2017, 390, 849–860 CrossRef CAS.
- F. Mingozzi and K. A. High, Blood, 2013, 122, 23–36 CrossRef CAS PubMed.
- E. Basner-Tschakarjan and F. Mingozzi, Front. Immunol., 2014, 5, 350 Search PubMed.
- B. K. Kaspar, D. Erickson, D. Schaffer, L. Hinh, F. H. Gage and D. A. Peterson, Mol. Ther., 2002, 5, 50–56 CrossRef CAS PubMed.
- B. K. Kaspar, B. Vissel, T. Bengoechea, S. Crone, L. Randolph-Moore, R. Muller, E. P. Brandon, D. Schaffer, I. M. Verma, K. F. Lee, S. F. Heinemann and F. H. Gage, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2320–2325 CrossRef CAS PubMed.
- R. W. Herzog, Methods Mol. Biol., 2004, 246, 179–194 CAS.
- S. Takeda, M. Takahashi, H. Mizukami, E. Kobayashi, K. Takeuchi, Y. Hakamata, T. Kaneko, H. Yamamoto, C. Ito, K. Ozawa, K. Ishibashi, T. Matsuzaki, K. Takata, Y. Asano and E. Kusano, Nephron Exp. Nephrol., 2004, 96, e119–e126 CrossRef CAS PubMed.
- L. Tenenbaum, A. Chtarto, E. Lehtonen, T. Velu, J. Brotchi and M. Levivier, J. Gene Med., 2004, 6(suppl 1), S212–S222 CrossRef CAS PubMed.
- C. S. Manno, A. J. Chew, S. Hutchison, P. J. Larson, R. W. Herzog, V. R. Arruda, S. J. Tai, M. V. Ragni, A. Thompson, M. Ozelo, L. B. Couto, D. G. Leonard, F. A. Johnson, A. McClelland, C. Scallan, E. Skarsgard, A. W. Flake, M. A. Kay, K. A. High and B. Glader, Blood, 2003, 101, 2963–2972 CrossRef CAS PubMed.
- G. Gao, L. H. Vandenberghe, M. R. Alvira, Y. Lu, R. Calcedo, X. Zhou and J. M. Wilson, J. Virol., 2004, 78, 6381–6388 CrossRef CAS PubMed.
- E. Zinn, S. Pacouret, V. Khaychuk, H. T. Turunen, L. S. Carvalho, E. Andres-Mateos, S. Shah, R. Shelke, A. C. Maurer, E. Plovie, R. Xiao and L. H. Vandenberghe, Cell Rep., 2015, 12, 1056–1068 CrossRef CAS PubMed.
- M. A. Kotterman and D. V. Schaffer, Nat. Rev. Genet., 2014, 15, 445–451 CrossRef CAS PubMed.
- D. Grimm, J. S. Lee, L. Wang, T. Desai, B. Akache, T. A. Storm and M. A. Kay, J. Virol., 2008, 82, 5887–5911 CrossRef CAS PubMed.
- J. R. Mendell, S. Al-Zaidy, R. Shell, W. D. Arnold, L. R. Rodino-Klapac, T. W. Prior, L. Lowes, L. Alfano, K. Berry, K. Church, J. T. Kissel, S. Nagendran, J. L'Italien, D. M. Sproule, C. Wells, J. A. Cardenas, M. D. Heitzer, A. Kaspar, S. Corcoran, L. Braun, S. Likhite, C. Miranda, K. Meyer, K. D. Foust, A. H. M. Burghes and B. K. Kaspar, N. Engl. J. Med., 2017, 377, 1713–1722 CrossRef CAS PubMed.
- C. Hinderer, N. Katz, E. L. Buza, C. Dyer, T. Goode, P. Bell, L. K. Richman and J. M. Wilson, Hum. Gene Ther., 2018, 29, 285–298 CrossRef CAS PubMed.
- M. Moskalenko, L. Chen, M. van Roey, B. A. Donahue, R. O. Snyder, J. G. McArthur and S. D. Patel, J. Virol., 2000, 74, 1761–1766 CrossRef CAS PubMed.
- C. Zhang, T. Yao, Y. Zheng, Z. Li, Q. Zhang, L. Zhang and D. Zhou, Biomaterials, 2016, 80, 134–145 CrossRef CAS PubMed.
- R. E. Kelemen, R. Mukherjee, X. Cao, S. B. Erickson, Y. Zheng and A. Chatterjee, Angew. Chem., Int. Ed. Engl., 2016, 55, 10645–10649 CrossRef CAS PubMed.
- F. Wei, K. I. McConnell, T. K. Yu and J. Suh, Eur. J. Pharm. Sci., 2012, 46, 167–172 CrossRef CAS PubMed.
- G. K. Lee, N. Maheshri, B. Kaspar and D. V. Schaffer, Biotechnol. Bioeng., 2005, 92, 24–34 CrossRef CAS PubMed.
- H. T. Le, Q. C. Yu, J. M. Wilson and M. A. Croyle, J. Controlled Release, 2005, 108, 161–177 CrossRef CAS PubMed.
- D. Moscioni, H. Morizono, R. J. McCarter, A. Stern, J. Cabrera-Luque, A. Hoang, J. Sanmiguel, D. Wu, P. Bell, G. P. Gao, S. E. Raper, J. M. Wilson and M. L. Batshaw, Mol. Ther., 2006, 14, 25–33 CrossRef CAS PubMed.
- N. Carrillo-Carrasco, R. J. Chandler, S. Chandrasekaran and C. P. Venditti, Hum. Gene Ther., 2010, 21, 1147–1154 CrossRef CAS PubMed.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed.
- J. S. Bartlett, R. Wilcher and R. J. Samulski, J. Virol., 2000, 74, 2777–2785 CrossRef CAS PubMed.
- W. Ding, L. N. Zhang, C. Yeaman and J. F. Engelhardt, Mol. Ther., 2006, 13, 671–682 CrossRef CAS PubMed.
- K. I. Joo, Y. Fang, Y. Liu, L. Xiao, Z. Gu, A. Tai, C. L. Lee, Y. Tang and P. Wang, ACS Nano, 2011, 5, 3523–3535 CrossRef CAS PubMed.
- E. D. Horowitz, M. S. Weinberg and A. Asokan, Bioconjugate Chem., 2011, 22, 529–532 CrossRef CAS.
- P. J. Xiao and R. J. Samulski, J. Virol., 2012, 86, 10462–10473 CrossRef CAS.
- Q. Xie, W. Bu, S. Bhatia, J. Hare, T. Somasundaram, A. Azzi and M. S. Chapman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10405–10410 CrossRef CAS PubMed.
- B. Li, W. Ma, C. Ling, K. Van Vliet, L. Y. Huang, M. Agbandje-McKenna, A. Srivastava and G. V. Aslanidi, Hum. Gene Ther: Methods., 2015, 26, 211–220 CrossRef CAS PubMed.
- P. C. Rensen, S. H. van Leeuwen, L. A. Sliedregt, T. J. van Berkel and E. A. Biessen, J. Med. Chem., 2004, 47, 5798–5808 CrossRef CAS PubMed.
- R. Zhou, X. Wang, H. Liu, L. Guo, Q. Su, H. Wang, T. Vasiliadis, W. Ho and J. Li, J. Virol., 2018, 92, e01720-17 CrossRef PubMed.
- H. Nakai, S. Fuess, T. A. Storm, S. Muramatsu, Y. Nara and M. A. Kay, J. Virol., 2005, 79, 214–224 CrossRef CAS PubMed.
- R. Popa-Wagner, M. Porwal, M. Kann, M. Reuss, M. Weimer, L. Florin and J. A. Kleinschmidt, J. Virol., 2012, 86, 9163–9174 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04189c |
| This journal is © The Royal Society of Chemistry 2020 |