
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linking genomics and metabolomics to chart specialized metabolic diversity
Justin J. J.
van der Hooft
 a,
Hosein
Mohimani
b,
Anelize
Bauermeister
c,
Pieter C.
Dorrestein
defg,
Katherine R.
Duncan
*h and
Marnix H.
Medema
*a
a,
Hosein
Mohimani
b,
Anelize
Bauermeister
c,
Pieter C.
Dorrestein
defg,
Katherine R.
Duncan
*h and
Marnix H.
Medema
*a
aBioinformatics Group, Wageningen University, Wageningen, The Netherlands. E-mail: marnix.medema@wur.nl
bComputational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA
cDepartamento de Farmacologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, SP, Brazil
dCollaborative Mass Spectrometry Innovation Center, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA, USA
eCenter for Microbiome Innovation, University of California San Diego, La Jolla, CA, USA
fDepartment of Pharmacology, School of Medicine, University of California San Diego, La Jolla, CA, USA
gDepartment of Pediatrics, University of California San Diego, La Jolla, CA, USA
hStrathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK. E-mail: katherine.duncan@strath.ac.uk
First published on 12th May 2020
Abstract
Microbial and plant specialized metabolites constitute an immense chemical diversity, and play key roles in mediating ecological interactions between organisms. Also referred to as natural products, they have been widely applied in medicine, agriculture, cosmetic and food industries. Traditionally, the main discovery strategies have centered around the use of activity-guided fractionation of metabolite extracts. Increasingly, omics data is being used to complement this, as it has the potential to reduce rediscovery rates, guide experimental work towards the most promising metabolites, and identify enzymatic pathways that enable their biosynthetic production. In recent years, genomic and metabolomic analyses of specialized metabolic diversity have been scaled up to study thousands of samples simultaneously. Here, we survey data analysis technologies that facilitate the effective exploration of large genomic and metabolomic datasets, and discuss various emerging strategies to integrate these two types of omics data in order to further accelerate discovery.
 Justin J. J. van der Hooft | Justin van der Hooft is an Assistant Professor of Bioinformatics at Wageningen University, The Netherlands. He obtained a BSc (2004) and MSc (2007) in Molecular Sciences (Wageningen University). In 2012, he obtained his PhD at the Biochemistry and Bioscience groups in Wageningen. He then moved to Glasgow, UK, for postdoctoral positions with Alan Crozier and Michael Barrett, subsequently. In 2017, he took up a shared postdoctoral position between Marnix Medema and Pieter Dorrestein. In January 2020, he started his own group in Wageningen that will develop computational metabolomics methodologies to decompose complex metabolite mixtures into their (sub)structures and apply these workflows to study plant and microbiome-associated metabolites and the food metabolome. |
 Hosein Mohimani | Hosein Mohimani is an Assistant Professor at the Computational Biology Department, Carnegie Mellon University. He received his BSc from Sharif University of Technology, Iran, and PhD from the University of California, San Diego, working with Dr Pavel Pevzner. At CMU, his group develops error-tolerant approaches to connect metagenomics and metabolomics datasets for discovering novel small molecule natural products. He is a recipient of an Alfred P. Sloan research fellowship and a NIH Director's New Innovator Award. |
 Anelize Bauermeister | Anelize Bauermeister obtained her BSc in Chemistry (2008) and MSc in Microbiology (2010) from University of Londrina, and PhD from University of São Paulo (2015), exploring mass spectrometry approaches to guide the isolation and characterization of microbial metabolites with anticancer properties. In 2016, she started a postdoctoral position at Institute of Biomedical Science, University of São Paulo with Dr Leticia Costa-Lotufo for investigation of marine natural products employing mass spectrometry-based metabolomics. She is currently sharing a postdoctoral position between University of São Paulo, Brazil and University of California, San Diego with Dr Pieter Dorrestein. |
 Pieter C. Dorrestein | Pieter C. Dorrestein is a Professor at the University of California – San Diego and is trained as a chemist at Northern Arizona University (BS) and then Cornell University (PhD). He is currently the Director of the Collaborative Mass Spectrometry Innovation Center and a Co-Director, Institute for Metabolomics Medicine in the Skaggs School of Pharmacy & Pharmaceutical Sciences, and Departments of Pharmacology and Pediatrics. He is also a key member of the Scripps Institution of Oceanography Center for Marine Biotechnology and Biomedicine and the UCSD Center for Microbiome Innovation. Dorrestein has been pioneering the development of mass spectrometry methods to study the chemical ecological crosstalk between populations of microorganisms, including host interactions for agricultural, diagnostic and therapeutic applications. |
 Katherine R. Duncan | Katherine Duncan is an Assistant Professor (Chancellor's Fellow) in Drug Discovery at the University of Strathclyde. She obtained a Chemistry MChem (University of Aberdeen) with International Placement (Florida Atlantic University) in 2005. After three years in industry, she completed a PhD in Biomedical Sciences (University of Prince Edward Island, 2012, international scholarship) followed by two Postdoctoral Fellowships at Scripps Institution of Oceanography, UCSD in Marine Biomedicine and at The Scottish Marine Institute in Marine Biotechnology. In 2016 she started her own research group at the University of Strathclyde, studying marine microbial natural products with a focus on characterizing the biological and environmental boundaries of microbial metabolism to accelerate antibiotic discovery from marine microorganisms. |
 Marnix H. Medema | Marnix Medema is an Assistant Professor of Bioinformatics at Wageningen University, The Netherlands. He obtained a Biology BSc (Radboud University Nijmegen, 2006) and a Biomolecular Sciences MSc (University of Groningen, 2008). In 2013, he completed his PhD with Eriko Takano and Rainer Breitling in Groningen; during this period, he was also a visiting fellow with Michael Fischbach at the University of California, San Francisco. Following a postdoc at the Max Planck Institute for Marine Microbiology in Bremen, Germany, he joined Wageningen University in 2015. There, his group develops computational methodologies to unravel natural product biosynthesis using omics data, and applies these methods to the study of molecular interactions in microbiomes. |
Key learning points1. Natural product discovery is transitioning from single strains to environmental strain collections and microbiomes, enabled by large-scale multi-omics data.2. Network analysis of genomes and metabolomes provides a bird's eye perspective on biosynthetic diversity and facilitates prioritizing key novel metabolites also in relation to relevant metadata (samples, activities, phenotypes). 3. Chemical substructures and modifications of natural products can be predicted from both genome and metabolome data, but more reliably when integrating both types of omics data. 4. Matching of metabolites to gene clusters enables producers to be identified and facilitates studies of their biosynthesis and ecological function. 5. Increasing the amount of publicly available paired multi-omics data along with additional algorithmic development will make structural characterization of natural products high-throughput. |
1. Introduction
Virtually all forms of life have the capacity to produce specific molecules that set them apart from others, and that allow them to cope with the distinct challenges they face in their native environments. These specialized metabolites (also known as natural products) facilitate a wide variety of mechanisms for chemical warfare, communication, nutrient acquisition or stress protection. Chemically, these metabolites belong to a diverse range of classes, including peptides, polyketides, flavonoids, terpenes and saccharides. The large chemical space available and the incredible variety and dynamic nature of ecological interactions and selective pressures have driven organisms across the tree of life to produce the hundreds of thousands of structurally varied metabolites that we know of today.Naturally, this abundance has been leveraged extensively as a valuable resource for drug discovery. Many antibiotics, chemotherapeutics, and other drugs are either natural products themselves or are inspired by them. Recently, a variety of antibiotics have been discovered that provide a new arsenal to combat multidrug-resistant superbugs. Additionally, natural products have been used as crop protection agents and as ingredients for manufacturing of foods, cosmetics, dyes, and many other products.
Recently, another area of interest has emerged for specialized metabolites: they are key mediators of molecular interactions in microbiomes and function as a ‘chemical language’ that underlies many microbiome-associated phenotypes. For example, suppression of fungal diseases by plant microbiota has been linked to the biosynthesis of lipopeptides like thanamycin in the rhizosphere, produced by specific strains of Pseudomonas,1 while the endosphere microbiome hosts additional biosynthetic pathways that are crucial for disease suppression.2 In the human microbiome, staphylococci producing the nonribosomal peptide lugdunin have been shown to impair colonization by their pathogenic relative Staphylococcus aureus,3 while N-acyl amides produced by diverse gut bacteria have been shown to modulate host metabolism.4
Traditionally, the majority of natural product discovery has been driven by bioactivity-guided fractionation of chemical extracts from individual microbes and plants. This has enabled the cataloguing of many thousands of metabolites and elucidation of their structures, which provides much of the knowledgebase on natural product diversity available today. However, the lag in acquiring and incorporating key analytical information and the associated high rate of rediscovery renders this approach less effective.
With the advent of DNA sequencing, it became clear that the genomes of natural product producing-organisms encode Biosynthetic Gene Clusters (BGCs) for the production of many metabolites that had never been observed in the laboratory. In bacteria and fungi, the majority of biosynthetic pathways are encoded in BGCs; in plants, a considerable number of pathways (but clearly not all) show signs of genomic clustering as well. These observations led to the emergence of genome mining as a technology to identify the biosynthetic pathways for both known and unknown metabolites.5
Additionally, methodological advances in untargeted metabolomics and data analysis of tandem mass-spectrometric (MS/MS) data have enabled comprehensive analysis of molecular components in metabolic extracts.6 This has made it possible to identify natural products in complex extracts, which would otherwise have remained ‘hidden’. In microbiomes, these developments have contributed greatly to the identification of metabolites for which it would be very hard to isolate sufficient quantities for structure elucidation by classical approaches.
Since then, the development of technologies for genomic and metabolomic data acquisition as well as computational tools for their analysis has accelerated rapidly. Thus, driven by these advances, the starting point and scale of natural product discovery is now shifting from single organisms to collections of many organisms, as well as natural communities such as microbiomes. It is becoming increasingly feasible to acquire metabolomics data for a large series of bacterial strains7 or plants.8 Similarly, genome sequencing and assembly technologies have undergone several successive revolutions, which has allowed the sequencing of thousands of genomes9 and the reconstruction of hundreds of thousands of draft genome assemblies from metagenomes (ref. 10 and references therein).
In recent years, our labs and others have developed tools to analyze such large-scale genomic and metabolomic data from a bird's eye perspective, through computational networking approaches that facilitate visualization and analysis of data from hundreds or thousands of organisms,11,12 and through algorithms that predict chemical (sub)structures from omics data. These advances are unlocking a range of potential new approaches to not only mine genomes and metabolomes at large scales separately, but also in an integrated manner. In this tutorial review, we will outline the key technologies that have been developed for genome and metabolome mining, and present our vision on how these can be combined in the future for integrative omics-based discovery of metabolites and their BGCs, elucidation of natural product structures and identification of their biological activities and ecological functions.
2. Genome mining
The process of genome mining (Fig. 1) entails a number of steps, including genome assembly and annotation, the identification of biosynthetic genes and gene clusters, prediction of natural product structures from sequence, and comparative genomic analysis to identify similarities and differences between organisms.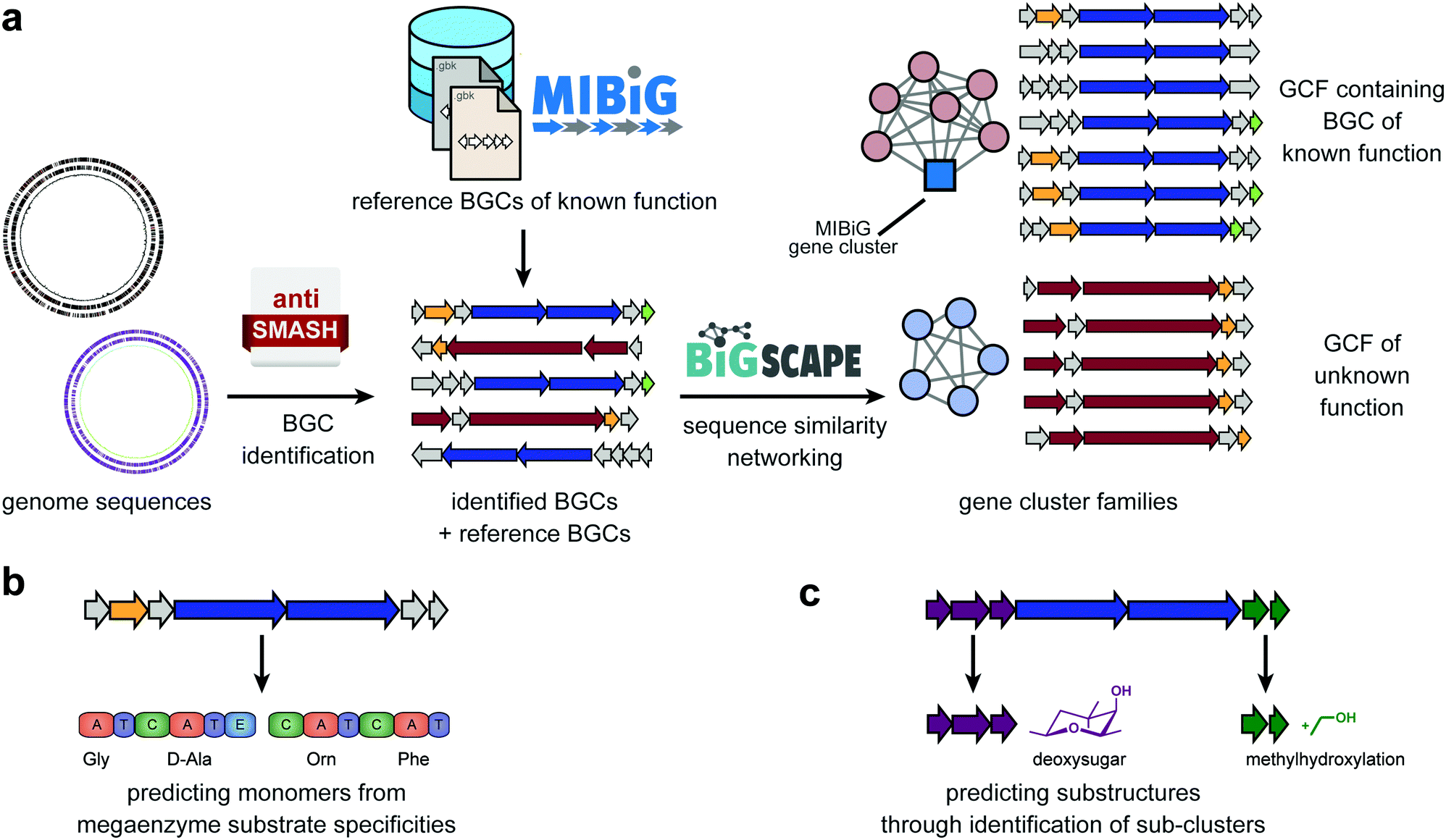 | ||
| Fig. 1 Computational approaches to mine genomes for metabolic diversity. (a) Biosynthetic Gene Clusters (BGCs) can be automatically identified in genome sequences using antiSMASH or related tools. Subsequently, they can be dereplicated using databases for BGCs of known function, such as MIBiG. Sequencing similarity networking can identify groups of similar BGCs across large datasets; the grouping of reference BGCs can aid to annotate the resulting gene cluster families (GCFs). Two strategies can be employed to predict (partial) chemical structures from these gene clusters: (b) monomers of peptides and polyketides, along with their order, can be predicted using machine learning algorithms for substrate specificity prediction in adenylation (A) domains of nonribosomal peptide synthetases (NRPSs) or acyltransferase (AT) domains of polyketide synthases, combined with the analysis of the domain architecture of the whole enzymatic assembly line. (c) Identification of sub-clusters that are known to be responsible for the biosynthesis of specific chemical moieties (e.g., a deoxysugar) or chemical modification (e.g., a methylhydroxylation) can be used to predict additional structural features of the metabolic product(s) of a BGC. | ||
Assembled and annotated genome sequences normally constitute the raw material for genome mining. It is important to realize that the quality of the assembly and annotation can have major effects on the outcome of any genome-based analysis. For example, in ‘fragmented’ genome assemblies containing many small contigs, BGCs are likely to be broken up into many pieces across contigs. In fact, due to their repetitive organization, genes encoding modular polyketide synthase (PKS) and nonribosomal synthetase (NRPS) assembly lines are very often localized at contig breaks. Frequently, some of the gene cluster pieces will be on contigs that are so small that they are not picked up by BGC identification algorithms.
Obtaining fully contiguous BGC sequences from metagenome data is particularly challenging. While microbial communities are clearly an untapped resource for natural product discovery, it is difficult to assemble an unorganized pool of next-generation sequencing reads from hundreds or thousands of—sometimes highly similar—organisms into contigs that each correspond to regions from the same source genome. BiosyntheticSPAdes13 is a metagenome assembler specialized for the assembly of BGCs from metagenomes, and utilizes the structure of the assembly graph, which provides a clue on how to combine multiple contigs into segments encoding long BGCs. On BGCs from the MiBIG dataset, BiosyntheticSPAdes correctly assembled roughly twice more BGCs into a single contig compared to previous assembly algorithms.
Besides the contiguity of assemblies, their accuracy is of course at least equally important. Misassemblies regularly occur, especially when assembling genomes with low-coverage short-read data. When this happens within a BGC, it can for example lead to the skipping or ‘duplication’ of NRPS or PKS modules, especially when they are highly similar in sequence. Alternatively, it can lead to ‘swaps’ that obscure the true order of genes or protein domains. Sometimes, BGCs are broken up into separate pieces that appear to be located in different genomic regions. Long-read technologies provided by Pacific Biosciences and Oxford Nanopore Technologies have their own problems, as higher error rates can sometimes lead to the introduction of spurious frameshifts that break up genes into multiple pieces or lead to annotation of premature stops.
After assembly, the annotation of the start and stop coordinates of genes within the genome constitutes a critical step. For bacterial genomes, this is largely a solved problem, with modern gene prediction tools being able to identify ∼99% of genes, and with correct start sites for >90% of them. Still, there are cases when genome annotations can still miss crucial genes. For example, biosynthetic pathways for ribosomally synthesized and posttranslationally modified peptides (RiPPs) often involve tiny peptide precursor-encoding genes that can be as small as 20–30 base pairs; these genes are often missed by even the best gene prediction algorithms. For this reason, RiPP genome mining algorithms like RODEO14 scan the six-frame translation of regions annotated as ‘intergenic’ within prioritized genomic loci to find such short genes. In fungi, plants and most other eukaryotes, the presence of introns makes gene prediction even more challenging. For these organisms, mapping transcriptome data to the genomes is often required to identify coding sequences accurately. However, since many genes involved in specialized metabolism are not expressed under typical conditions, these genes suffer from frequent misannotation. This can be partly solved by using pooled transcriptome samples from a variety of biotic and abiotic stress conditions.
Several tools have been developed for the identification of BGCs in genome sequences. Among these, antiSMASH, for example, identifies BGCs in bacterial and fungal genomes by a rule-based system that looks for specific combinations of enzymatic domains (detected using profile Hidden Markov Models [pHMMs]) being encoded within the same genomic neighbourhood. It currently contains detection rules for 52 different classes of BGCs, which have all undergone careful manual curation.15 The concept was extended to mining plant genomes as well, albeit with slightly different types of rules, in order to deal with distinguishing real gene clusters from tandem repeats that are commonly found in plant genomes.16 For publicly available bacterial genomes and metagenomes, pre-computed antiSMASH runs are available through the antiSMASH-DB and IMG-ABC databases;17,18 this not only averts the need to wait for antiSMASH web servers to analyze these genomes, but also facilitates the possibility to search all genomes for BGCs with specific characteristics of interest.
In the past decades, many BGCs have been experimentally linked to specific natural products with characterized chemical structures. To enable efficient access to this knowledge, a community standard was introduced: the Minimum Information about a Biosynthetic Gene cluster (MIBiG).19 This standard covers a range of metadata, including the genomic coordinates of the BGC, the chemical structure of its product(s), but also e.g. the functions of enzymes encoded in the cluster. Cross-links are also provided with the Natural Products Atlas (NPAtlas),20 a database of microbial natural product structures, and the ‘Global Natural Product Social molecular networking’ (GNPS) knowledgebase that stores metabolomic data (for more discussion, see next section).11 Because all MIBiG data are stored according to a standardized ontology, they can easily be searched, and the dataset can be used as reference data for annotation purposes; within antiSMASH, this allows identifying which BGCs are highly similar (and likely functionally equivalent) to a BGC of known function (Fig. 1a). All MIBiG data are stored in an online data repository, which currently contains around 2000 validated gene cluster–molecule pairs.21
Still, given the enormous biosynthetic diversity found in nature, the vast majority of BGCs in publicly available genomes will not be closely related to any MIBiG reference gene cluster. Several computational methods have emerged that allow predicting the (core) chemical structures of their products de novo. These methods are guided by mechanistic insight into the enzymatic mechanisms involved in producing these metabolites. For example, modular PKSs and NRPSs constitute an ‘assembly line’ comprised of enzymatic modules that each integrate a monomer (e.g., an amino acid) into a growing chain that is released at the end, before being cyclized and/or tailored through additional modifying enzymes. The modules of these PKSs and NRPSs contain specific domains that are involved in selecting which monomers are incorporated: acyltransferase (AT) domains for PKSs and adenylation (A) domains for NRPSs. The residues lining their active sites largely confer this substrate specificity. Hence, various algorithms, ranging from simple motif matching to sophisticated machine-learning models, have been developed to predict substrate specificities from sequence information (Fig. 1b). SANDPUMA, for example, predicts A domain specificities through phylogenetic, heuristic motif-matching, support-vector machine and pHMM algorithms, and then uses ensemble supervised machine learning to combine these into a consensus prediction.22 Combining individual module-level predictions, tools like antiSMASH15 and PRISM23 then provide a prediction of the sequence of monomers that are incorporated into the core polyketide or peptide scaffold. PRISM also attempts to predict post-assembly-line cyclization and tailoring reactions, by e.g. providing all combinatorial possibilities of reactions that are chemically feasible, given the initial predicted core scaffold. This often leads to large combinatorial explosions of possibilities, however, and effective methods to distinguish the most likely actual structures from less likely ones based on sequence data alone have not yet been reported. For BGCs that are not closely related to gene clusters with known products, predicting the full structure of their products is very challenging, and predicting a core scaffold and/or a list of predicted chemical features and modifications from sequence will often be more realistic. This is illustrated by the fact that for most BGC classes outside modular PKS and NRPS biosynthetic systems, few structure prediction tools exist yet, even for assessing the core scaffold. Nonetheless, prediction of chemical features can also be done without attempting to predict the full structure, e.g. through the identification of sub-clusters within BGC: groups of genes that occur across multiple different BGCs and in each of these contexts encode the biosynthesis of a specific substructure that is part of the final products of each of these BGCs (Fig. 1c). AntiSMASH identifies such sub-clusters through comparative genomic analysis with annotated sub-clusters of known functions from reference BGCs, but recently, an initial method for de novo identification of sub-clusters based on statistical association of gene families across BGCs has also been established.24 The fact that even sub-clusters that are not functionally annotated are likely responsible for the production of specific chemical substructures opens up interesting opportunities for matching with metabolomic data.
The upscaling of (meta)genome sequencing has led to hundreds of thousands of genomes being sequenced and/or being reconstructed from metagenomes. Given the enormous diversity of BGCs across these genomes, this provides the potential for expanding genome mining endeavors to unprecedented proportions. Yet, ‘traditional’ analyses of individual genomes using e.g. antiSMASH are not suitable for this kind of analysis, as manually vetting thousands of outputs and predictions for the corresponding BGCs would take years. For this reason, sequence similarity networking approaches have been developed that facilitate systematically mapping relationships between thousands of BGCs at once, and grouping them into gene cluster families (GCFs): sets of BGCs with similar gene content that encode the production of identical or highly similar molecules. This type of approach, originally developed in parallel by multiple research groups (reviewed in ref. 5), has recently been formalized, accelerated and streamlined in the BiG-SCAPE software12 (Fig. 1a). BiG-SCAPE takes as input BGCs directly taken from antiSMASH and MIBiG, and uses optimized combinations of various metrics to group BGCs at multiple levels. Specifically, it includes measures for the degree to which protein domains are shared between a pair of BGCs, synteny conservation and amino acid sequence identity. The resulting interactive network visualization makes it possible to instantly identify GCFs with known members from MIBiG, which may lead to the identification of BGCs for novel congeners, or GCFs with no MIBiG reference BGCs, which may represent biosynthetic pathways that are novel or have gone unnoticed by researchers in the field. The potential of this principle was shown previously by Cimermancic et al., who identified taxonomically widespread BGCs for aryl polyenes that are found in thousands of bacterial species but had been almost entirely overlooked.25 On top of the sequence similarity networking, phylogenetic analysis offers an additional technique to map biosynthetic diversity; the CORASON algorithm, which has also been integrated with BiG-SCAPE, makes it straightforward to construct multi-locus phylogenies of BGCs within and across GCFs; this allows systematically mapping evolved biosynthetic variation by identifying, e.g., clade-specific variation in BGC content in the form of unique tailoring enzymes or unique changes to the core scaffold biosynthetic enzymes. Navarro-Muñoz et al. showed the potential of the combined use of BiG-SCAPE and CORASON to this end, by identifying three novel clades of detoxin BGCs that encoded specific modifications to this natural product scaffold.12
It should be noted that the sequence similarity networking technique comes with several caveats. For example, networks can be constructed at a range of different cut-offs. Many of these cut-offs produce networks that ‘make sense’, but each of them tell a different story. When the goal is to identify a group of BGCs that should be linked to one specific molecule, a stricter threshold would be required than in attempts to link a group of BGCs to a class of molecules, or to a biological activity or microbiome-associated phenotype of interest. In many cases, it is therefore a good practice to repeat the analysis at multiple cut-offs, and compare the resulting networks and GCF groupings carefully. This gives a more complete picture than a single cut-off will, as choosing a single cut-off will always carry a large degree of arbitrariness.
An additional consideration is how to define GCFs from the graph. Taking complete connected components as GCFs has the danger of combining unrelated BGCs into one family: e.g., if gene cluster A is related to the first half of gene cluster B and the second half of gene cluster B is related to gene cluster C, cluster A may not have any similarity to cluster C. Yet, performing graph clustering on the network, as BiG-SCAPE does, has the danger that large groups of closely related gene clusters may be forced into multiple individual families. BiG-SCAPE partly addresses this by grouping GCFs into a higher-level organization, gene cluster clans, which comprise multiple related GCFs. Using these clan annotations or using GCF annotations at different cut-offs can remedy problems due to excessive ‘splitting’ behaviour, when attempting to match sets of gene clusters to metabolites or phenotypes.
Furthermore, it is important to note that most current BGC identification algorithms have not been designed to predict the exact borders of BGCs. AntiSMASH, for example, takes a greedy approach and extends the BGC region upstream and downstream of the core biosynthetic genes to ‘play it safe’ and make sure that no important functional enzyme-coding genes are missed. When the extension region is relatively large compared to the actual BGC, however, this can lead to BGCs being grouped into different GCFs (at certain cut-offs) when the BGCs lie in different genomic contexts across organisms.
Fragmented (meta)genome assemblies with partial BGCs can also be problematic. BiG-SCAPE provides a glocal alignment mode that is able to often still match partial gene clusters to their corresponding regions within full BGCs; thus, it is frequently still able to link partial to corresponding complete BGCs within the network. However, for very small contigs, even this mode may fail to do so.
The application of gene cluster networking approaches to large genomic datasets has made clear that vast numbers of GCFs of unknown function exist for which it is very difficult to estimate the structures or functions of their products. While, as mentioned above, genome mining tools have enabled prediction of the molecular structures of some BGC products, these predictions remain error-prone, especially for unusual monomers and rare modifications from less-studied organisms. This is due to the fact that limited training data is available for many enzyme families, while enzyme function can be very diverse and may evolve rapidly and dynamically. Integration with metabolome data has important potential to improve current predictive capacities, as metabolomics data could be used to error-correct genome-based predictions of chemical (sub)structures,26 and thus learn from the data. Additionally, metabolome data can be used to link BGCs and GCFs to specific molecules, and thus dereplicate and prioritize gene clusters for experimental characterization not only based on genomic features, but also based on chemical novelty that can be predicted from MS/MS data analysis. This will help shed light on questions regarding ‘silent’ or ‘cryptic’ gene clusters as well: are the products of so many BGCs not known because they are not expressed under the conditions studied, or simply because they do not have biological activities that are being screened for?
3. Metabolome mining
Specialized metabolites have high structural diversity, a result of evolutionary adaptation to a multitude of abiotic and biotic challenges. Furthermore, considerable metabolic variation can arise from individual pathways, as the production of specialized metabolites is influenced by a suite of complex processes. These may include dynamic transcriptional regulation of biosynthetic enzyme-encoding genes as well as enzymatic promiscuity in substrate acceptance or regioselectivity and substrate availability within the cell and environment. Genome sequencing has revealed that microorganisms often have a higher biosynthetic potential than the number of metabolites that has been observed in the laboratory. This suggests significant potential for discovery of chemical novelty. In order to identify new metabolites, there is a need for technological advances in metabolomic measurements, as well as for data methods (analysis, curation, storage and standardisation) that can effectively survey and compare larger sets of species, samples and conditions.Advances in method development and highly sensitive analytical instrumentation, especially Mass Spectrometry (MS), have allowed metabolite extracts of increasing complexity to be investigated. As a result, MS-based metabolomic technologies have been widely applied in the field of natural products. Development of new computational tools to highlight and identify target metabolites of interest has further aided our understanding of these complex systems. The scale of the data generated using MS-based metabolomics has fuelled the development of automated analysis methods to compare and identify metabolites. However, the chemical complexity and diversity of natural product extracts often makes assigning structural information to metabolomics signals (metabolite annotation) and structural elucidation of metabolites (metabolite identification) very challenging processes. In mass spectrometry-based metabolomics studies, multiple ways of data acquisition are possible, each with their own advantages and disadvantages. Typically, the aim is to capture the entire metabolome (using full scan or ‘MS1’ mode), which is advantageous to accurately quantify metabolites.
However, it is often difficult to reliably annotate metabolites from MS1 data for a variety of reasons, such as the fact that multiple distinct metabolites often share the same molecular formula and mass. Acquiring fragmentation spectra of metabolites (MS/MS or tandem MS mode, Fig. 2a) has distinct advantages to annotate and identify metabolites. These MS/MS spectra can be regarded as bar codes or fingerprints of metabolites, and several software tools have been developed to exploit this structural information. An initial step is usually to compare experimental MS/MS spectra to library spectra (Fig. 2b) to detect known metabolites, or analogues thereof, a process also known as dereplication. The reliability of this matching procedure is dependent on many factors that include experimental data quality and spectral database content that differs from database to database. It is therefore wise to check the results across various databases. Moreover, although spectral libraries are currently growing, their contents are far from completely covering the natural product metabolome. For example, GNPS spectral libraries currently contain MS/MS reference spectra for about 2.5% of known natural products. Thus, during such annotation efforts, one has to keep in mind that if presented with a yet unknown metabolite that is spectrometrically very similar to those of multiple metabolites or stereoisomers present in libraries, one needs to report the possible candidate structure space rather than a single candidate. A single extract can contain thousands of metabolites, with experiments routinely consisting of hundreds of samples. It is thus unsurprising that for most of those metabolites there is no reference data available, leaving many metabolomics signals unmatched. Therefore, despite the wealth of untargeted MS data generated, annotation remains a challenge. In practice, less than five percent of the total chemical entities in a sample can be reliably annotated to the structural level.27
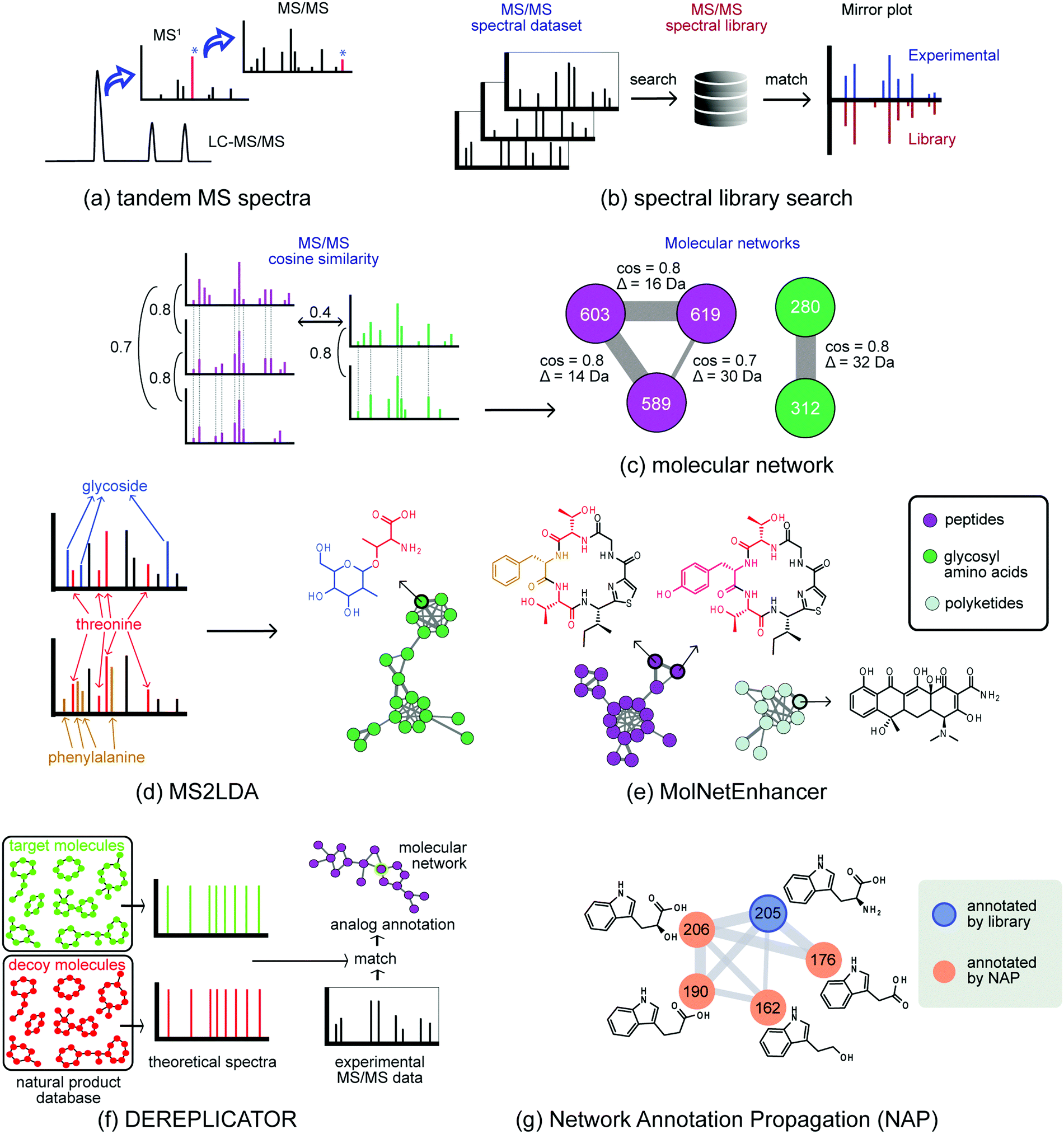 | ||
| Fig. 2 Molecular networking technologies to chart metabolic diversity. (a) In metabolomics, crude microbial extracts are analyzed by untargeted LC-MS/MS, from which the most intense ions detected in MS1 are selected and fragmented to acquire MS/MS spectral data. (b) In spectral library search, each MS/MS spectrum from the dataset is searched against a spectral library in order to find a good spectral match. (c) Molecular network organizes MS/MS datasets by spectral similarity (cosine score), in which spectra with high similarities are grouped together, forming molecular networks. (d) MS2LDA recognizes co-occurrence of ions related to structural motifs; this substructural information can be combined with annotations from other in silico tools (NAP, DEREPLICATOR, ClassyFire) (e) MolNetEnhancer integrates outputs from these tools to annotate substructures and classify the chemical classes present in the investigated dataset. (f) DEREPLICATOR is an in silico tool able to annotate known peptidic natural products. (g) NAP – Network Annotation Propagation makes use of the network topology and propagates the annotation from a spectral library match (based on in silico annotation tools) through the spectral network to improve the annotation of analogs by reranking the most plausible candidate structure based on overlapping structural fingerprints. | ||
To facilitate exploratory data analysis, new tools have been developed that aim to group structurally-related metabolites together based on the spectral similarity of their fragmentation spectra (Fig. 2). Here, Global Natural Product Social (GNPS) molecular networking improved spectral comparison within and across samples.11 In general, metabolites with a similar chemical architecture yield similar fragmentation spectra. Molecular networking groups parent ions (represented by nodes) by fragmentation pattern similarity (represented by edges) to form Molecular Families (MFs) of related metabolites (Fig. 2c). This facilitates analysis of large data sets, also across multiple organisms, as molecular networks can be used to find metabolites related to a known molecule of interest, or to correlate the presence of a MF to, e.g., a biological activity. Here, it is important to take into account that approaches that are dependent on a network structure (i.e. the formation of MFs from fragmentation spectra, analogous to grouping BGCs into GCFs) have their caveats – which are similar to those discussed in the genome mining section. Briefly, choosing appropriate thresholds for networking is non-trivial, the quality of the network reflects the quality of the input data, and the parameters selected to define families influence the outcome. To connect two metabolites based on their mass fragmentation spectra, it is important to consider which distance metrics (i.e. spectral similarity scoring system) to use, as in any clustering analysis. For example, a scoring method may or may not account for differences between the two parent ions of the spectral pair. Furthermore, the threshold to enable connection of metabolites in the molecular network needs to be critically considered, as more stringent thresholds will lead to the formation of smaller MFs, whereas more lenient thresholds will lead to larger MFs.
During the last five years, GNPS11 has grown to a mass spectrometry ecosystem: a free, public, web-based platform, where it is possible to upload and store raw or processed data sets (e.g., in an open data format such as mzML, mzXML or MGF) and analyze them with several different available statistical and annotation tools. GNPS provides an online repository for public deposition/retrieval and archival called the Mass Spectrometry Interactive Virtual Environment (MassIVE) data repository. Here, researchers can search for related public datasets and either download or integrate these into a current workspace for reanalysis with different parameters or newly available tools, integration into molecular networks including their own data, or comparative analyses. If the spectral data is publicly available, it is straight-forward to re-analyze data and notifications can be set up: every time when a new dataset with a similar chemical profile is submitted, when new annotations are submitted, or when new spectral matches are found to the crowdsourced GNPS curated spectral libraries, the researcher receives a message. This interactivity facilitates a global shared data community (i.e., the “living data” concept), which can greatly enhance both the availability and number of annotated features, ultimately aiding the metabolomic identification and continuous learning about a data set that is deposited publicly.
Once the researcher has one target compound (MS/MS spectrum) of interest or relevance, it is possible to query for its presence in all the GNPS public datasets through MASST, akin to performing an NCBI BLAST search with a genomic sequence.28 In GNPS, the researcher can also compare a public dataset to their own data, thanks to the development of ReDU, which brings standardization for the metadata associated to the dataset.29 ReDU makes it possible to select a specific subset of the public data based on fixed ontology terms, thereby overcoming difficulties in matching metadata across different datasets. For instance, the same bacterial species may be deposited in the repository with different names, such as Salinispora arenicola, S. arenicola, Salinispora, marine bacteria, etc. With >2900 well-documented datasets of microbial origin, ReDU will undoubtedly contribute to natural product discovery by allowing researchers to find the same or related metabolites across all microbial data – and thereby getting an idea of a metabolite's chemical novelty and distribution over known and measured organisms.
Besides GNPS, several other metabolomics tools and platforms are available as well for data processing, analysis, and sharing (for the underlying publications, see ref. 30 and references therein). MS-DIAL (with MS-Finder) and MzMine have grown into platforms with active user base and functionalities from initial processing up to library matching and annotation of unknown signals. Another platform with such capabilities is XCMS online, from where MetLin is available as well: MetLin is currently the largest spectral library that can be searched in. Finally, next to GNPS-MassIVE, MetabolomicsWorkbench and MetaboLights each constitute an evolving metabolomics repository that promotes community sharing of both mass spectrometry-based and NMR-based metabolomics data. From its initial launch, MetaboLights has adhered to ontologies and is now also offering an increasing suite of analysis tools to make use of the public metabolomics data.31
Spectral matching and molecular networking have been shown to assist in annotating and organizing metabolomics datasets; however, for many mass features in a molecular network library, matching does not provide any structural information. In the past decade, several in silico annotation approaches have been developed to provide insights into the chemical nature of metabolites which cannot be annotated using library matching. Such in silico tools typically result in a ranked list of possible candidate structures from predicted MS/MS spectra or from public compound databases. Alternatively, they provide substructure annotations or chemical class annotations. As a result, they greatly assist in assessing chemical novelty of natural extracts, since known scaffolds or classes can be assigned to mass features to inform prioritization.
One class of methods exploits the fact that specialised metabolites usually consist of several building blocks assembled by biosynthetic machinery. Recognising these building blocks directly from metabolomics data represents an appealing strategy to elucidate the natural product. In recent years, several approaches have been developed in this area.32,33 For example, the MS2LDA tool identifies parts of metabolites (substructures) in untargeted datasets through the unsupervised detection of co-occurring molecular fragments using a text mining inspired algorithm (Fig. 2d).32 The resulting mass fragment patterns are termed Mass2Motifs, and they require annotation by researchers. MS2LDA has been applied to extracts from plants, fungi, and bacteria. From each of these datasets, tens of Mass2Motifs were annotated with substructure information culminating in several hundred annotated substructure patterns. Recently, MotifDB (http://ms2lda.org/motifdb/) was built to capture this expert knowledge, enabling easy reuse. Where some Mass2Motifs consist of specific mass fragments or neutral losses that uniquely match to MS/MS spectra in experimental data, other Mass2Motifs may contain more widely occurring mass fragments; in such cases, their presence in experimental data should be manually validated. In addition, in various sample types, Mass2Motifs could represent different isomeric substructures and care should be taken with transferring Mass2Motif annotations across very different sample types. Different chemistry will result in different sets of mass fragments and neutral losses that the user will have to structurally annotate to provide a (bio)chemical interpretation. To analyse substructures in context of metabolite diversity, annotations from MS2LDA can be used to annotate molecular networks; the MolNetEnhancer software facilitates this (Fig. 2e)34 and thereby eases manual validation of Mass2Motif presence and annotations.
A number of other tools, including MetFrag and MAGMa, use candidates from structure databases that are subsequently ranked based on in silico fragmentation, matching experimental mass fragments, resulting in scores for each candidate – also referred to as the combinatorial approach.35 This method has some limitations as fragment ions generated from rearrangement reactions, such as McLafferty rearrangements that occur in natural product classes such as flavonoids, cannot be assigned to a structural feature. Other approaches use machine learning. For example, CSI:FingerID, enables the generation of fragmentation trees to match MS/MS spectra to candidate structures using support vector machines. In the case of spectral prediction methods, such as CFM:ID, fragmentation spectra of known metabolites are used to computationally learn fragmentation patterns of a large set of structures. Subsequently, those in silico created spectra are matched to experimental MS/MS data. Whilst this has worked for a number of small (human) metabolites, the spectral prediction of larger natural products remains too challenging to derive high-quality annotations through spectral matching. One reason is the relatively low number of natural products with reference MS/MS data to train an appropriate spectral prediction model. Another strategy is to consider the metadata, for example, the location of the molecules or by applying taxonomically informed scoring to improve annotation. Altogether, in silico annotation tools often reduce the analysis time needed to assign structural information to metabolomics signals, while minimizing the number of possible options for validation.
For specific classes of natural products, dedicated approaches have been developed that dereplicate metabolites by using innovative strategies to match MS/MS-based fragments to fragment patterns predicted from chemical structure databases. For example, DEREPLICATOR (Fig. 2f) systematically links structures from a large peptidic natural products database to mass fragmentation spectra by comparison with theoretical spectra generated based on specific in silico fragmentation rules.36 Statistical assessment of the results is provided by also matching to a decoy database that contains non-existent peptides with similar amino acid compositions. Recently, DEREPLICATOR+ was launched, which extended this annotation strategy to polyketides, flavonoids, terpenes, and other classes of natural products.37 As even large peptidic databases are incomplete, the VarQuest tool was employed to facilitate modification-tolerant searches for peptidic structures and to predict where these modifications are located on the peptide scaffold.38 This allowed the annotation of almost 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 peptide variants in publicly available data.
000 peptide variants in publicly available data.
The combination of library matches, dereplication results and substructure predictions can be utilized to annotate molecular networks to a substantial degree. Recently developed methods to do so include Network Annotation Propagation (NAP)39 (Fig. 2g) which utilizes network topology to increase the number of relevant candidate structures for metabolomics signals, and MolNetEnhancer (Fig. 2e), which provides fine-grained molecular details by showing the presence of substructure patterns (Fig. 2d) as well as a higher-level chemical overview of the data through chemical class annotations of candidate structures in the context of their MFs (Fig. 2e).34 Such enhanced molecular networks facilitate rapid ways of exploring metabolite space across large datasets, by providing a global overview of chemical diversity.
Many annotation tools are dependent on candidate structures present in spectral databases. However, for quite a few mass signals no relevant candidates can be found – or even none at all. The above introduced CFM-ID is able to predict spectra from structures and thereby expands the search space for candidate structures. Recently, MetWork was established, which combines the network structure of molecular networks with the spectral predictions of CFM-ID, which yield informative structural information. MetWork ‘imagines’ which chemical variants could be represented by nodes based on a number of annotated molecules (using a set of appropriate sample-specific reaction rules). This is then tested by matching these variants to their theoretical spectra.40
The above tools are all dependent on measured LC-MS/MS spectral metabolomics data. These data files consist of interleaved full scan LC-MS spectra capturing the samples’ metabolic content and mass spectrometry fragmentation spectra where typically a subset of metabolites is fragmented. Whilst metabolomics aims to capture all metabolites of an organism, in practice this is impossible due to various steps during a typical (untargeted) metabolomics workflow, in particular the extraction and mass spectrometry analysis settings. For example, more polar solvents will extract the more polar metabolites from a sample, including sugars and charged molecules, but excluding most apolar metabolites such as lipids (and vice versa for apolar solvents). Furthermore, mass spectrometry analyzers cannot collect decent spectra across the entire m/z domain at the same sensitivity and a mass window needs to be provided; thereby focusing on either the smaller m/z values or the larger ones. In addition, the quality of MS/MS spectra (i.e., how many mass fragments are visible and how abundant are they?) is dependent on how well metabolites fragment under the experimental conditions. Indeed, some compound classes (e.g., polyketides) are more difficult to break than others (e.g., peptides and saccharides) due to the nature of the bonds that connect the underlying monomers. Thus, differential data acquisition settings and various amounts of background noise, for example due to interfering mass features that are co-isolated in the fragmentation cell, may hamper the correct matching of experimental spectra to library spectra. Moreover, to detect a metabolite in the mass spectrometer, the metabolite needs to be ionised. However, not all metabolites ionise that easily, and for some metabolite classes, other analytical detection techniques will need to be employed. Available alternatives to mass spectrometry are ultraviolet (UV) absorption to better detect chromophores in natural products, and nuclear magnetic resonance (NMR) spectroscopy, which can provide more detailed structural information at the cost of being less sensitive than mass spectrometry is.
Data quality plays a large role in GNPS molecular networking as well, especially as mass spectrometry fragmentation data is typically generated by selecting a window of masses rather than just one metabolite mass. Thus, often, “chimeric” MS/MS spectra are obtained that contain mass fragments from two or more fragmented metabolites. Such chimeric spectra can give rise to false connections in molecular networks. Virtually all mass spectra contain noise, and when this is substantial, it affects spectral similarity scores – typically in a negative manner, as noise is random and not shared by a spectral pair, resulting in a lower spectral score and no connection. In recent years, denoising strategies have been proposed, several of which are available in the GNPS platform.
From the perspective of metabolomics, much can be gained by attempting further integration with genomic (and transcriptomic) data. For example, linking BGCs to metabolites to understand biosynthetic pathways and integrate chemical class/structural information based on genome and metabolome data. Specifically, there is major potential in using genome-based predictions to inform chemical structure elucidation, in order to link genes to metabolites. Furthermore, linking BGCs to transcriptomics data can increase our understanding of which conditions lead to the production of different subsets of chemistry. The application of these integrated omics datasets to biology-driven questions including phylogeny, ecological function and understanding microbial (and host–microbe) interactions provides an exciting opportunity. In the next section, we will explore how these and other approaches can be leveraged for metabologenomic integration.
4. Metabologenomic integration
Linking of information across datasets is useful, as it enables structural and functional annotation. The term ‘metabologenomics’ has been coined to encompass the methodology used to integrate these complex datasets.41 Whilst functional annotations in genomics and metabolomics are increasingly present in databases to which new experimental data can be matched, it has been estimated that approximately 50% of the proteins have reliable functional annotations, whereas the total number of reference metabolomics data is such that 2–5% of observed molecules can be reliably matched to known molecules.27 Furthermore, complex samples such as fecal or soil extracts contain a multitude of microbial species and metabolites, also from other sources such as food and drugs; therefore, identifying the producing microorganism(s) is challenging. To find product–producer pairs, especially in metagenome/metabolome linking, several approaches have been demonstrated. These can be broadly defined as pattern-based, correlation-based and feature-based.42,43Pattern-based genome mining was one of the first correlation-based integration strategies to combine the analysis of BGCs across strains with molecular networking demonstrated the success of this approach for larger datasets (Fig. 3a).43 In this methodology, genome mining information (presence/absence of BGCs) was collected to form patterns across 35 Salinispora strains. The fact that a molecular network was generated for these same strains meant that correlations in BGC-metabolite pairs could be facilitated by manually exploring how the two patterns were overlapping, ultimately accelerating the linking of unknown BGC to known metabolites in addition to the prioritisation of new biosynthetic and chemical space. This was exemplified by the discovery of retimycin A, a quinomycin-type depsipeptide linked to the BGC NRPS40.43 Based on a recent study by Tobias et al.44 for a total of 22 known BGCs across 30 Photorabdus and Xenorabdus strains (totalling 660 possible combinations), the following statistics were deduced: in 119 (18%) cases, both BGC and its product were found, and in 479 (73%) of the cases, neither the BGC nor its product were found. Furthermore, in 61 (9%) cases the BGC was found in a strain, but not its product, whereas in one case (0.00151%) the product was found in a strain in which its corresponding BGC was not predicted to be present. Whereas such numbers will fluctuate43 and are highly dependent on the genome and metabolome data quality, we can infer two important things from these numbers: (i) when no BGC for a certain product is found in a genome, it is highly unlikely that this metabolite is produced by this organism, and (ii) in 66% of the cases where a BGC was found in a strain, its specialized molecule was also measured by mass spectrometry. This gives an indication of what we might expect from larger data sets as well.
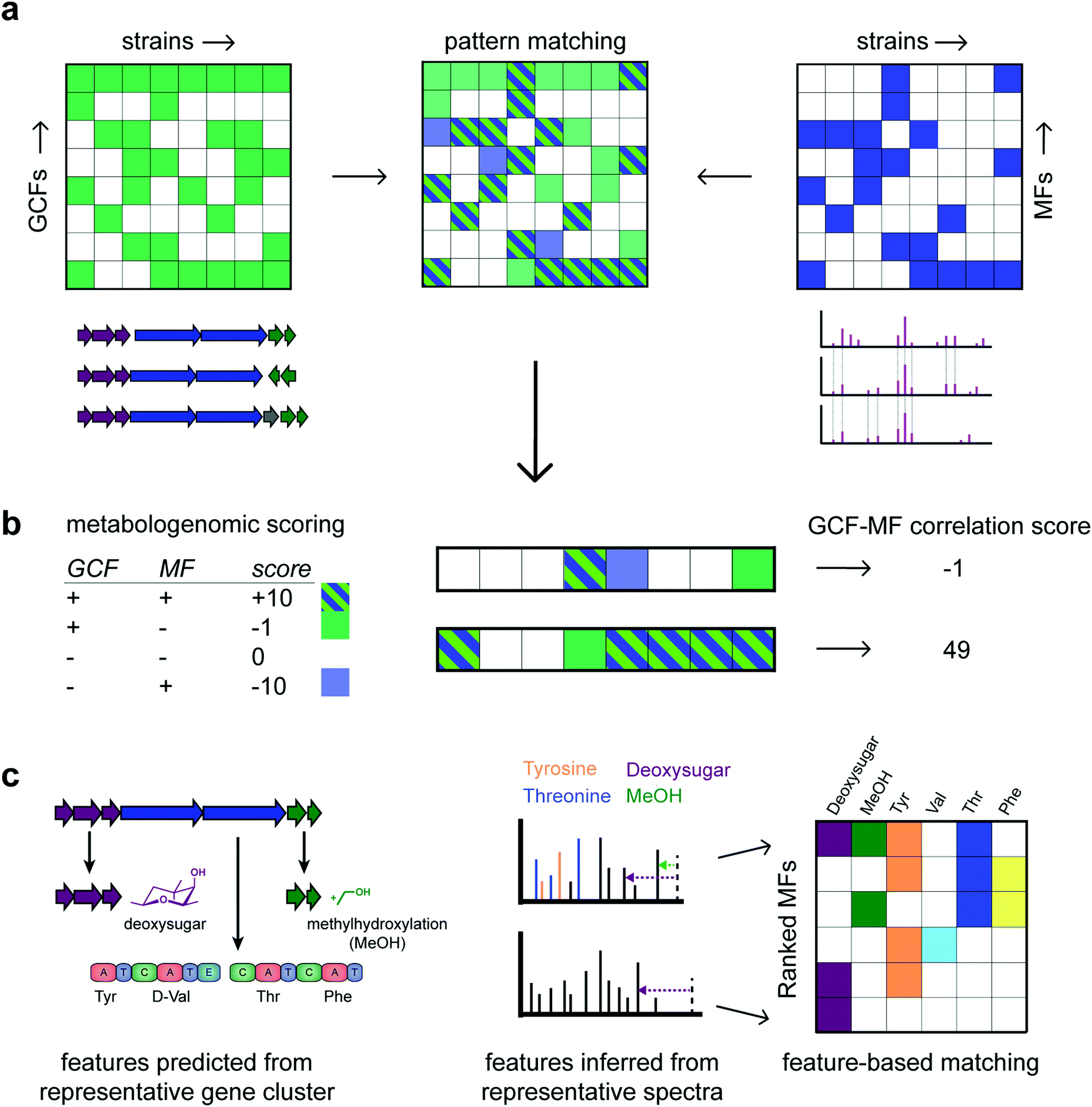 | ||
| Fig. 3 Various types of matching gene cluster families (GCFs) to molecular families (MFs) have been proposed. Panel (a) describes “pattern based matching” where the two presence/absence matrices for GCFs and MCFs across all the strains are combined in one table where manually promising candidate links are identified. (b) In “correlation based matching”, a correlation based score is calculated such as the one proposed by Goering et al.41 Using such a scoring, GCF-MF links can be ranked to focus follow-up studies on a relevant subset. Here, the lower of the two GCF-MF links is clearly more promising than the upper link with scores of 49 and −1, respectively. Finally, panel (c) highlights the concept of “feature based matching” where structural features learnt from the genome are matched to those inferred from the metabolome. In the example, 6 structural features could be predicted from the GCF of which a representative gene cluster is depicted. In MFs, the presence/absence of these structural features is then determined. In the example, the top row MF has 4 of the 6 structural features present as highlighted in a representative spectrum. In contrast, the lowest ranking MF has only the deoxysugar loss present, making it the least likely candidate to match with the GCF. It is of note that the genome provides unique structural information on the most likely stereochemistry of the amino acids following the presence/absence of epimerization domains. In the example, the valine moiety is most likely D-valine, something that is typically impossible to tell from metabolomics data. Hence, a match is made based on the presence/absence of the valine moiety – independent of its stereochemistry. | ||
Historically, these correlation-based approaches have been largely manual and often targeted based on BGC information to direct the prioritization of chemistry. However, in recent years, automated methods have emerged that consider correlation metrics and statistical frameworks to rank promising links between gene cluster and molecular families or genes and mass spectra. A metabologenomic score was introduced to link gene clusters to mass spectra of molecules based on their presence/absence patterns across strains45 (Fig. 3b). The score takes into account similar assumptions as learnt from the Photorabdus and Xenorabdus study described above: the presence of a molecule in the absence of a likely gene cluster producing it is heavily penalized whereas cryptic BGCs are penalized less. Thus, such scores formalize the quality of a match and it enables to rank matches at the large-scale that is required nowadays. Correlation scores could be calculated between gene cluster families and individual spectra as well. Future scoring metrics could explore different boost and penalty values, take into consideration the size of GCFs and MFs, as well as the inclusion or exclusion of fragmented or incomplete BGCs.
There have also been advances in feature-based integration strategies, although more so for ‘modular’ natural product classes due to their relatively well-defined building blocks (Fig. 3c). From the genome, monomers such as amino acid moieties and glycosyl moieties, as well as enzymatic modifications such as methylation and hydroxylation, can be predicted from gene cluster sequences. Similarly, substructures can be predicted from mass spectra. GCFs and MFs can thus be ranked based on the number of corresponding structural features (Fig. 3c). For example, sub-clusters (represented by sets of genes that co-evolve and cooperate to synthesize a certain chemical moiety)24 could in the future be automatically matched to chemical substructures (represented by co-occurring mass fragments, neutral losses), and/or (groups of) mass differences as discovered by metabolome mining (e.g., Mass2Motifs from MS2LDA).24,32 Feature-based matching approaches have been successfully pioneered for glycosylated and peptidic metabolites targeting glycosyl groups and amino acids, termed glycogenomics and peptidogenomics.46,47 We expect that the correlative and feature based matching approaches can be jointly used to refine genome–metabolome links; for example, by first using a correlative approach followed by a reranking based on the presence/absence of structural features. Once confident links have been identified, metabolites can be connected to their producers. In addition, complementary structural information from the genome that is hard to observe or infer from metabolomics (e.g., the stereochemistry of amino acids in nonribosomal peptides) can be exploited for structural elucidation of specialised metabolites.
Some fully automated methods exist for linking mass spectra to molecular structures, by matching structural features predicted from genomics to those inferred from metabolomics. Specifically, for non-ribosomal peptides (NRPs) there is NRPquest,26 and for ribosomally synthesized and post-translationally modified peptides (RiPPs) there are MetaMiner48 and DeepRiPP.50 While these pattern-feature based methods are distinct in detail and the type of natural products they target, they share similar principles (Fig. 4). Starting from metabolomics data and BGCs, these methods consist of (some of) the following steps: (a) predicting hypothetical small molecule products from BGCs, (b) predicting the fragmentation patterns and theoretical spectra of these hypothetical molecules, (c) matching mass spectra against the theoretical spectra, allowing for a specific number of modifications, (d) computing the statistical significance of the matches, (e) calculating the false discovery rate of matches, and (f) forming a molecular network of significant identifications.
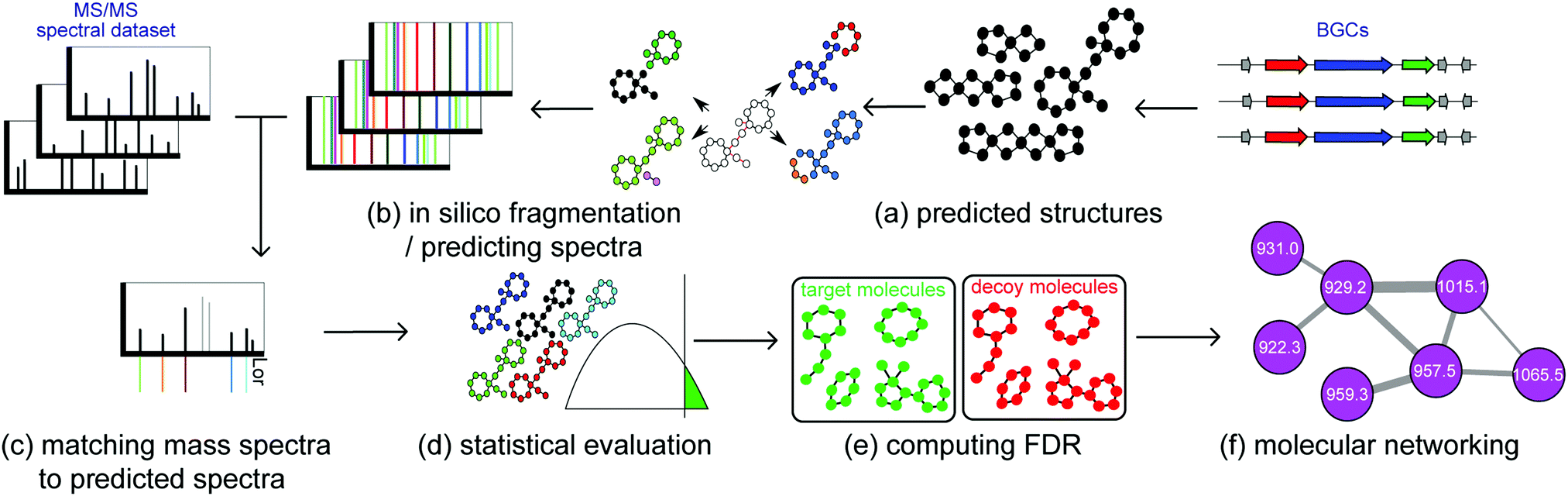 | ||
| Fig. 4 Starting from metabolomics data and biosynthetic gene clusters, substructure-based approaches for integrating metabolomics and genomics data consist of the following steps. (a) Predicting hypothetical small molecule products of the biosynthetic gene clusters (here each node represents a monomer, e.g. an amino acid or a ketide, and each edge represents a bond between monomers, e.g. amide bond), (b) predicting the fragmentation pattern and theoretical spectra of these hypothetical molecules during mass spectrometry, (c) matching mass spectra against the theoretical spectra, allowing for a specific number of modifications, (d) computing statistical significance of the matches, (e) calculating the false discovery rate of matches, (f) forming molecular network of significant identifications. | ||
(a) Predicting hypothetical small molecule products of BGCs. In case of NRPs, multiple algorithms are used for predicting amino acid specificities of A-domains.22 In case of RiPPs, BGCs are predicted based on modification enzymes found in different RiPP classes: open reading frames (ORFs) within the BGCs are extracted as precursor RiPPs, and based on enzymes present in the BGC, modifications are incorporated in the precursor RiPPs to form mature RiPP structures.
(b) Predicting the fragmentation pattern and theoretical spectra of these hypothetical molecules during mass spectrometry. In the case of peptides, fragmentation patterns are formed by disconnecting amide bonds between nitrogen and carbon. In case of more general small molecules, fragmentation patterns are formed by disconnecting nitrogen–carbon, oxygen–carbon, and carbon–carbon bonds.
(c) Matching mass spectra against the theoretical spectra, allowing for a specific number of modifications. Usually prediction of hypothetical small molecules based on genome mining is erroneous, due to the difficulties involved in predicting post-translational and post-assembly modifications. These modifications can be discovered using modification-tolerant searches of mass spectra against hypothetical small molecules.38
(d) Computing the statistical significance of the matches. Raw scores between hypothetical small molecules and spectra are defined as the number of peaks shared between the two. These scores are usually biased toward molecules with higher molecular weights. Therefore, it is necessary to convert raw scores into P-values, which are defined as the ratio of randomly generated molecular structures with scores higher than the target small molecules against the mass spectra.
(e) Calculating the false discovery rate of matches. In order to compute the false discovery rate, hypothetical small molecules are randomly shuffled to form a decoy database. Then, the false discovery rate is computed as the ratio of the number of molecules identified in the decoy database, over the number of molecules identified in the target database.36,49
(f) Forming a molecular network of significant identifications. The chemical identities of the metabolites are further expanded and contextualized using molecular networking.
Despite these common approaches to integrated data analysis, several challenges have motivated the development of solutions to improve integration both within and across datasets. Key challenges include the comparability of the data due to, for example, different experimental protocols, data processing protocols, data formats, lack of structured reference or knowledge bases. There is also a lack of development of tools to check and curate data and metadata quality, as well as tools that can use or reuse this paired data and its accompanying metadata.
The first challenge discussed here is the availability of consistent, well-curated, standardised data. There has been an increasing degree of availability of whole genome sequencing and metabolomics data from the same strains, enabling complementary structural information obtained or inferred from genome as well as metabolome predictions. Different complementary sets of omics data related to the same origin are termed “paired data sets”. Over the last few years, multiple papers have published paired data sets and shown how pattern-based mining assists in coupling genomic information to molecular spectra. The more paired data sets become available, the more we can start to exploit the complementary structural information from the genome and metabolome and link gene clusters to their products and thereby molecules to their producers. The latter is especially useful in metagenomics and meta-metabolomics experiments, where a molecule can potentially be produced by many different bacterial strains. Efforts are ongoing to create a platform called the Paired omics Data Platform (https://pairedomicsdata.bioinformatics.nl), where existing and novel paired data sets can be recorded to provide an overview of existing paired data sets and thereby stimulate reuse for natural product discovery. Furthermore, additional omics data such as transcriptomics and peptidogenomics, as well as metadata, can be added to genomic and metabolomic data. Transcriptome data, for example, can guide researchers to BGCs that are actively expressed under the same conditions where metabolites of interest are being observed.50 A comprehensive set of linked chemical and genomic features in the paired data sets will benefit the entire natural product research community and beyond. Validated links can be exploited by experimentalists to quickly assess whether specialized metabolite products are known for predicted BGCs. Moreover, computational biologists can use the validated links as anchor points to train machine learning models to computationally link genome and metabolome data.
The second challenge is the importance of data and metadata quality, quality filters and quality vs. quantity of paired data. Relatively poor quality data is likely to generate inaccurate annotations and can consequently lead to erroneous hypotheses about the biological system under investigation. The quality of public data is frequently questioned and it is therefore important to develop standardized workflows for generating quality control reports. As noted earlier, there are several variables to be considered that influence the data quality, including how the sample was handled and pre-processed, extraction procedure, analytical methods employed, data processing and many others. Some authors suggest that protocol standardization facilitates a better integration of omics data; however, such standard operating protocols may not always result in the best quality data for individual use cases. In addition, the presence of uninformative features in both metabolomics and genomics data, which may come from baseline or poor quality spectra or gene reads, can complicate integrative analysis workflows. Therefore, filtering steps are essential for statistical analyses; however, the same filters may also remove relevant features from the dataset. The use of some quality controls can help to overcome this issue and largely improve the quality of the final data. Altogether, the choice of which datasets to include in paired data analysis is a compromise between selecting higher-quality sample data and datasets and the total number of samples. Since more paired datasets will typically result in a higher chance of finding relevant patterns, weighed choices about data quality have to be made.
Comprehensive, curated, and standard-adhering sample information is crucial, and this includes metadata and the validity of the links between omics data sets. To achieve this, the use of domain-specific ontologies and linking these ontologies across domains not only helps to standardize sample information; its application also facilitates the linking of information: for example, smart use of ontologies can ensure that not only direct (exact) terms will match, but also indirect (related) terms. On both the genomics and metabolomics sides, several initiatives have spearheaded developments in creating uniform metadata with extensive sample information.
Recent publications show exciting opportunities regarding new ways in which paired genomics and metabolomics data sets can yield novel microbial–metabolite relationships. Morton and co-workers introduced mmvec, which uses metabolome data and taxonomic profiles as input and then applies a single-layer neural network to learn the co-occurrence probabilities between measured metabolites and microbes.51 Mmvec does not use correlation analyses to link microbes to metabolites, but uses probability-based metrics to identify the most likely microbe–metabolite co-occurrences. Such a method holds great promise as it overcomes some of the limitations of correlation-based approaches when applied to non-absolute quantitative data (as microbial taxonomy and metabolite information typically are) whilst still presenting a ranked list of interactions. Another recent tool linking taxonomic information to metabolite abundance is MelonnPan, which predicts community metabolomes from microbial community profiles.52 Mallick and coworkers show how their computational framework successfully recovers metabolic trends for more than 50% of microbiome-associated molecules – thereby providing insights in the metabolic capacity of communities for which only metagenomics data is available. Similarly, Cao et al. developed a method for detecting microbiota-associated small molecules based on the patterns of co-occurrence of molecular and microbial features across multiple microbiomes, and further mapping each molecule to the phylogenetic clade responsible for its production/transformation.53 These approaches aid in linking bacterial taxa or gene clusters to metabolite products using correlation-based or neural networking (machine learning) methods. Ultimately, the success of linking genome and metabolome mining workflows for NP discovery will depend on platform and infrastructure development. This is always a chicken and egg conundrum. The tools need to be developed to analyze the data but then we also need appropriate data to develop the tools. Successful infrastructure needs access to relevant training data. These data are well-curated data, often assembled via community-knowledge akin to the way MIBiG and GNPS are capturing the knowledge by the community in computer-readable formats to enable integrated analysis across data types.
5. Opportunities
Over the last decade and a half, the cost of sequencing has dropped by nine orders of magnitude, and during the same time, the costs to generate mass spectrometric data have dropped by two orders of magnitude. This has resulted in increasing numbers of laboratories that can collect both these data types. For example, the Qiita platform54 has public genome data on hundreds of thousands of microbiome samples, and GNPS has public mass spectrometry data for thousands of microbial samples including cultures and microbiome samples (tens of thousands), as well as metabolomics on samples from the American Gut Project (http://humanfoodproject.com/americangut/), the Global FoodOmics (https://globalfoodomics.org), Tiny Earth (https://tinyearth.wisc.edu/) and the (integrative) Human Microbiome Project (https://hmpdacc.org/ihmp/). Thus, Qiita and GNPS currently contain thousands of samples for which both metabolomics and sequencing (predominantly 16S-rRNA sequences, but increasingly whole genome sequences as well) have been collected. These data include isolates (around 1700 – predominantly Streptomyces spp., Salinispora spp., Cyanobacteria spp., and human microbiome related bacterial species) and, predominantly, human gut metagenomes (more than 500) and 16S-rRNA amplicon sequences (over 2000). However, due to the complexity of linking the data sets in practice, the utility of this data has not yet been fully realized nor exploited.Starting with the pairing of such data sets, we are learning how to begin navigating this opportunity. To facilitate the integration of multiple omics methods, standardization is critical. Although this certainly allows more overarching research questions to be asked, no universal method will ever exist for all data types and questions. However, we see opportunities to use more standardized data formats, including metadata, using controlled vocabularies when possible, and to report data in a reusable format. Here, journals could play their role to define clear requirements of what needs to be deposited in the public domain and how the data is linked in an easily accessible format. For example, with sequence data, most data is deposited in the public domain file due to journal requirements. With mass spectrometry data there are currently tens of different formats and no requirement by the scientific community to deposit data with an accession number. In our opinion, data should be publicly shared as soon as possible, preferably prior to paper publication. One way, aside from a community-enforced sharing requirement, could be to incentivise sharing, if more knowledge or data is gained by the user when shared publicly. In this regard, GNPS and the work at MetaboLights and the Metabolomics Workbench are major initiatives.29,31 Overall, we expect that the amount of public data will increase fuelled by many open data initiatives funded by the general public.
Genomics and metabolomics datasets are increasingly correlated to each other, in particular for the modular biosynthetic pathways for polyketides and nonribosomal peptides, and often by manual approaches. However, the use of more complex statistical methods to integrate these data are urgently needed. For example, metabologenomic scoring could be improved by taking into account prior probabilities for observing certain links, or by correcting for structure in the data, such as phylogenetic relationships between species. Such future improvements will also expand the successful linking of gene clusters to molecules for any compound class going beyond the modular metabolites. Also in general, additional sophisticated tools have to be developed in order to create the connection between these two areas of expertise. This is not a simple task, as interdisciplinary knowledge at the interface of chemistry, biology and informatics is required. Making these combined omics tools also openly accessible to the community will be pivotal to our understanding of chemical biology. In this regard, the recent efforts by Qiime2, originally designed and focused on genomics data, to also enable metabolomics analyses are very promising.55
The computationally-driven correlation- and feature-based matching (Fig. 3b and c) rely on gene cluster and spectral similarity scoring across data sets and accurate presence/absence patterns across strains. We foresee possible improvements in similarity scoring through machine learning developments and increasingly large sets of reference library MS/MS spectra becoming available that will boost genome-metabolome matching. Recent exciting developments of new tools allow for more reliable annotation of compound classes and substructures. We think that such additional structural features as chemical compound class predictions (i.e., from antiSmash, MolNetEnhancer or CSI:FingerID, etc.) could assist in reranking and refinement of linking results when matched to compound class predictions inferred from the genome, i.e., a terpene biosynthesis cluster is more likely to match to a predicted terpene molecular family than a family of NRPS. Furthermore, on the genomics side, the number of validated BGCs to molecular structure links are increasingly fuelled by initiatives such as MiBIG;21 such datasets facilitate algorithmic development to predict structural features from genomic sequences. In addition, new strategies to improve structural predictions, such as computational predictions of substrate specificities and regioselectivities of core as well as tailoring biosynthetic enzymes, will also contribute to improved linking through feature-based matching. Synthetic biology can also aid in this regard, by synthesizing and assaying targeted sets of enzymes or enzymatic domains that would fill key gaps in current training sets.
To conclude, we foresee that major computational advances will be needed to exploit the full potential of paired omics datasets already available. With the increasingly higher-throughput genomics and metabolomics pipelines available, we expect more paired data sets to become available. Ultimately, both developments will fuel each other, as computational advances to link data will stimulate the generation of paired data sets. As showcased by the many community initiatives and tools that have appeared over the last five years, it is clear that the era of integrated omics analysis has well and truly started. The question will be what these integrated capabilities will look like – perhaps it will be a facebook-like network infrastructure that, instead of connecting data to people, will be used to connect different types of information about molecules. We look forward to all the new and exciting developments in the years ahead.
Conflicts of interest
MHM is a co-founder of Design Pharmaceuticals and a member of the scientific advisory board of Hexagon Bio. PCD is a member of the scientific advisory boards of Sirenas and Cybele.References
- R. Mendes, M. Kruijt, I. de Bruijn, E. Dekkers, M. van der Voort, J. H. M. Schneider, Y. M. Piceno, T. Z. DeSantis, G. L. Andersen, P. A. H. M. Bakker and J. M. Raaijmakers, Science, 2011, 332, 1097–1100 CrossRef CAS PubMed.
- V. J. Carrión, J. Perez-Jaramillo, V. Cordovez, V. Tracanna, M. de Hollander, D. Ruiz-Buck, L. W. Mendes, W. F. J. van Ijcken, R. Gomez-Exposito, S. S. Elsayed, P. Mohanraju, A. Arifah, J. van der Oost, J. N. Paulson, R. Mendes, G. P. van Wezel, M. H. Medema and J. M. Raaijmakers, Science, 2019, 366, 606–612 CrossRef PubMed.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS PubMed.
- L. J. Cohen, D. Esterhazy, S.-H. Kim, C. Lemetre, R. R. Aguilar, E. A. Gordon, A. J. Pickard, J. R. Cross, A. B. Emiliano, S. M. Han, J. Chu, X. Vila-Farres, J. Kaplitt, A. Rogoz, P. Y. Calle, C. Hunter, J. K. Bitok and S. F. Brady, Nature, 2017, 549, 48–53 CrossRef CAS PubMed.
- M. H. Medema and M. A. Fischbach, Nat. Chem. Biol., 2015, 11, 639–648 CrossRef CAS PubMed.
- R. D. Kersten and P. C. Dorrestein, ACS Chem. Biol., 2009, 4, 599–601 CrossRef CAS PubMed.
- D. D. Nguyen, A. V. Melnik, N. Koyama, X. Lu, M. Schorn, J. Fang, K. Aguinaldo, T. L. Lincecum Jr, M. G. K. Ghequire, V. J. Carrion, T. L. Cheng, B. M. Duggan, J. G. Malone, T. H. Mauchline, L. M. Sanchez, A. Marm Kilpatrick, J. M. Raaijmakers, R. De Mot, B. S. Moore, M. H. Medema and P. C. Dorrestein, Nat. Microbiol., 2016, 2, 16197 CrossRef CAS PubMed.
- K. B. Kang, M. Ernst, J. J. J. van der Hooft, R. R. da Silva, J. Park, M. H. Medema, S. H. Sung and P. C. Dorrestein, Plant J., 2019, 98, 1134–1144 CAS.
- Y. Zou, W. Xue, G. Luo, Z. Deng, P. Qin, R. Guo, H. Sun, Y. Xia, S. Liang, Y. Dai, D. Wan, R. Jiang, L. Su, Q. Feng, Z. Jie, T. Guo, Z. Xia, C. Liu, J. Yu, Y. Lin, S. Tang, G. Huo, X. Xu, Y. Hou, X. Liu, J. Wang, H. Yang, K. Kristiansen, J. Li, H. Jia and L. Xiao, Nat. Biotechnol., 2019, 37, 179–185 CrossRef CAS PubMed.
- A. Almeida, S. Nayfach, M. Boland, F. Strozzi, M. Beracochea, Z. J. Shi, K. S. Pollard, D. H. Parks, P. Hugenholtz, N. Segata, N. C. Kyrpides and R. D. Finn, bioRxiv, 2019 DOI:10.1101/762682.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. B. P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O’Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- J. C. Navarro-Muñoz, N. Selem-Mojica, M. W. Mullowney, S. A. Kautsar, J. H. Tryon, E. I. Parkinson, E. L. C. De Los Santos, M. Yeong, P. Cruz-Morales, S. Abubucker, A. Roeters, W. Lokhorst, A. Fernandez-Guerra, L. T. D. Cappelini, A. W. Goering, R. J. Thomson, W. W. Metcalf, N. L. Kelleher, F. Barona-Gomez and M. H. Medema, Nat. Chem. Biol., 2020, 16, 60–68 CrossRef PubMed.
- D. Meleshko, H. Mohimani, V. Tracanna, I. Hajirasouliha, M. H. Medema, A. Korobeynikov and P. A. Pevzner, Genome Res., 2019, 29, 1352–1362 CrossRef CAS PubMed.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS PubMed.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed.
- S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS PubMed.
- K. Blin, V. P. Andreu, E. L. C. de los Santos, F. Del Carratore, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, D625–D630 CrossRef CAS PubMed.
- K. Palaniappan, I.-M. A. Chen, K. Chu, A. Ratner, R. Seshadri, N. C. Kyrpides, N. N. Ivanova and N. J. Mouncey, Nucleic Acids Res., 2020, 48, D422–D430 Search PubMed.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. de Bruijn, Y. H. Chooi, J. Claesen, R. Cameron Coates, P. Cruz-Morales, S. Duddela, S. Düsterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. N. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kötter, D. Krug, J. Masschelein, A. V. Melnik, S. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H.-W. Nützmann, G. Pan, A. Pati, D. Petras, F. Jerry Reen, F. Rosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gómez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y.-Q. Cheng, J. Clardy, R. J. Cox, R. De Mot, S. Donadio, M. S. Donia, W. A. van der Donk, P. C. Dorrestein, S. Doyle, A. J. M. Driessen, M. Ehling-Schulz, K.-D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Höfte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H.-J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Méndez, M. Metsä-Ketelä, J. Micklefield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Müller, B. A. Neilan, M. Nett, J. Nielsen, F. O’Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J.-L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Süssmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glöckner, Nat. Chem. Biol., 2015, 11, 625 CrossRef CAS PubMed.
- J. A. van Santen, G. Jacob, A. L. Singh, V. Aniebok, M. J. Balunas, D. Bunsko, F. C. Neto, L. Castaño-Espriu, C. Chang, T. N. Clark, J. L. Cleary Little, D. A. Delgadillo, P. C. Dorrestein, K. R. Duncan, J. M. Egan, M. M. Galey, F. P. J. Haeckl, A. Hua, A. H. Hughes, D. Iskakova, A. Khadilkar, J.-H. Lee, S. Lee, N. LeGrow, D. Y. Liu, J. M. Macho, C. S. McCaughey, M. H. Medema, R. P. Neupane, T. J. O’Donnell, J. S. Paula, L. M. Sanchez, A. F. Shaikh, S. Soldatou, B. R. Terlouw, T. A. Tran, M. Valentine, J. J. J. van der Hooft, D. A. Vo, M. Wang, D. Wilson, K. E. Zink and R. G. Linington, ACS Cent. Sci., 2019, 5, 1824–1833 CrossRef CAS PubMed.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Muñoz, B. R. Terlouw, J. J. J. van der Hooft, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2020, 48, D454–D458 Search PubMed.
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 CrossRef CAS PubMed.
- M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS PubMed.
- F. Del Carratore, K. Zych, M. Cummings, E. Takano, M. H. Medema and R. Breitling, Commun. Biol., 2019, 2, 83 CrossRef PubMed.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
- H. Mohimani, W.-T. Liu, R. D. Kersten, B. S. Moore, P. C. Dorrestein and P. A. Pevzner, J. Nat. Prod., 2014, 77, 1902–1909 CrossRef CAS PubMed.
- R. R. da Silva, P. C. Dorrestein and R. A. Quinn, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12549–12550 CrossRef CAS PubMed.
- M. Wang, A. K. Jarmusch, F. Vargas, A. A. Aksenov, J. M. Gauglitz, K. Weldon, D. Petras, R. da Silva, R. Quinn, A. V. Melnik, J. J. J. van der Hooft, A. M. Caraballo-Rodríguez, L. F. Nothias, C. M. Aceves, M. Panitchpakdi, E. Brown, F. Di Ottavio, N. Sikora, E. O. Elijah, L. Labarta-Bajo, E. C. Gentry, S. Shalapour, K. E. Kyle, S. P. Puckett, J. D. Watrous, C. S. Carpenter, A. Bouslimani, M. Ernst, A. D. Swafford, E. I. Zúñiga, M. J. Balunas, J. L. Klassen, R. Loomba, R. Knight, N. Bandeira and P. C. Dorrestein, Nat. Biotechnol., 2020, 38, 23–26 CrossRef CAS PubMed.
- A. K. Jarmusch, M. Wang, C. M. Aceves, R. S. Advani, S. Aguire, A. A. Aksenov, G. Aleti, A. T. Aron, A. Bauermeister, S. Bolleddu, A. Bouslimani, A. M. C. Rodriguez, R. Chaar, R. Coras, E. O. Elijah, M. Ernst, J. M. Gauglitz, E. C. Gentry, M. Husband, S. A. Jarmusch, K. L. Jones, Z. Kamenik, A. Le Gouellec, A. Lu, L.-I. McCall, K. L. McPhail, M. J. Meehan, A. V. Melnik, R. C. Menezes, Y. A. M. Giraldo, N. H. Nguyen, L. F. Nothias, M. Nothias-Esposito, M. Panitchpakdi, D. Petras, R. Quinn, N. Sikora, J. J. J. van der Hooft, F. Vargas, A. Vrbanac, K. Weldon, R. Knight, N. Bandeira and P. C. Dorrestein, bioRxiv, 2019 DOI:10.1101/750471.
- R. Spicer, R. M. Salek, P. Moreno, D. Cañueto and C. Steinbeck, Metabolomics, 2017, 13, 106 CrossRef PubMed.
- K. Haug, K. Cochrane, V. C. Nainala, M. Williams, J. Chang, K. V. Jayaseelan and C. O’Donovan, Nucleic Acids Res., 2020, 48, D440–D444 Search PubMed.
- J. J. J. van der Hooft, J. J. J. van der Hooft, J. Wandy, M. P. Barrett, K. E. V. Burgess and S. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13738–13743 CrossRef PubMed.
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299–302 CrossRef PubMed.
- M. Ernst, K. B. Kang, A. M. Caraballo-Rodríguez, L.-F. Nothias, J. Wandy, C. Chen, M. Wang, S. Rogers, M. H. Medema, P. C. Dorrestein and J. J. J. van der Hooft, Metabolites, 2019, 9, E144 CrossRef PubMed.
- K. Scheubert, F. Hufsky and S. Böcker, J. Cheminf., 2013, 5, 12 CAS.
- H. Mohimani, A. Gurevich, A. Mikheenko, N. Garg, L.-F. Nothias, A. Ninomiya, K. Takada, P. C. Dorrestein and P. A. Pevzner, Nat. Chem. Biol., 2017, 13, 30–37 CrossRef CAS PubMed.
- H. Mohimani, A. Gurevich, A. Shlemov, A. Mikheenko, A. Korobeynikov, L. Cao, E. Shcherbin, L.-F. Nothias, P. C. Dorrestein and P. A. Pevzner, Nat. Commun., 2018, 9, 4035 CrossRef PubMed.
- A. Gurevich, A. Mikheenko, A. Shlemov, A. Korobeynikov, H. Mohimani and P. A. Pevzner, Nat. Microbiol., 2018, 3, 319–327 CrossRef CAS PubMed.
- R. R. da Silva, M. Wang, L.-F. Nothias, J. J. J. van der Hooft, A. M. Caraballo-Rodríguez, E. Fox, M. J. Balunas, J. L. Klassen, N. P. Lopes and P. C. Dorrestein, PLoS Comput. Biol., 2018, 14, e1006089 CrossRef PubMed.
- Y. Beauxis and G. Genta-Jouve, Bioinformatics, 2019, 35, 1795–1796 CrossRef CAS PubMed.
- A. W. Goering, R. A. McClure, J. R. Doroghazi, J. C. Albright, N. A. Haverland, Y. Zhang, K.-S. Ju, R. J. Thomson, W. W. Metcalf and N. L. Kelleher, ACS Cent. Sci., 2016, 2, 99–108 CrossRef CAS PubMed.
- S. Soldatou, G. H. Eldjarn, A. Huerta-Uribe, S. Rogers and K. R. Duncan, FEMS Microbiol. Lett., 2019, 366, fnz142 CrossRef CAS PubMed.
- K. R. Duncan, M. Crüsemann, A. Lechner, A. Sarkar, J. Li, N. Ziemert, M. Wang, N. Bandeira, B. S. Moore, P. C. Dorrestein and P. R. Jensen, Chem. Biol., 2015, 22, 460–471 CrossRef CAS PubMed.
- N. J. Tobias, H. Wolff, B. Djahanschiri, F. Grundmann, M. Kronenwerth, Y.-M. Shi, S. Simonyi, P. Grün, D. Shapiro-Ilan, S. J. Pidot, T. P. Stinear, I. Ebersberger and H. B. Bode, Nat. Microbiol., 2017, 2, 1676–1685 CrossRef CAS PubMed.
- J. R. Doroghazi, J. C. Albright, A. W. Goering, K.-S. Ju, R. R. Haines, K. A. Tchalukov, D. P. Labeda, N. L. Kelleher and W. W. Metcalf, Nat. Chem. Biol., 2014, 10, 963–968 CrossRef CAS PubMed.
- R. D. Kersten, Y.-L. Yang, Y. Xu, P. Cimermancic, S.-J. Nam, W. Fenical, M. A. Fischbach, B. S. Moore and P. C. Dorrestein, Nat. Chem. Biol., 2011, 7, 794 CrossRef CAS PubMed.
- R. D. Kersten, N. Ziemert, D. J. Gonzalez, B. M. Duggan, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4407–E4416 CrossRef CAS PubMed.
- L. Cao, A. Gurevich, K. L. Alexander, C. B. Naman, T. Leão, E. Glukhov, T. Luzzatto-Knaan, F. Vargas, R. Quinn, A. Bouslimani, L. F. Nothias, N. K. Singh, J. G. Sanders, R. A. S. Benitez, L. R. Thompson, M.-N. Hamid, J. T. Morton, A. Mikheenko, A. Shlemov, A. Korobeynikov, I. Friedberg, R. Knight, K. Venkateswaran, W. H. Gerwick, L. Gerwick, P. C. Dorrestein, P. A. Pevzner and H. Mohimani, Cell Syst., 2019, 9, 600–608 CrossRef CAS PubMed.
- K. Scheubert, F. Hufsky, D. Petras, M. Wang, L.-F. Nothias, K. Dührkop, N. Bandeira, P. C. Dorrestein and S. Böcker, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed.
- G. C. A. Amos, T. Awakawa, R. N. Tuttle, A.-C. Letzel, M. C. Kim, Y. Kudo, W. Fenical, B. S. Moore and P. R. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11121 CrossRef CAS PubMed.
- J. T. Morton, A. A. Aksenov, L. F. Nothias, J. R. Foulds, R. A. Quinn, M. H. Badri, T. L. Swenson, M. W. Van Goethem, T. R. Northen, Y. Vazquez-Baeza, M. Wang, N. A. Bokulich, A. Watters, S. J. Song, R. Bonneau, P. C. Dorrestein and R. Knight, Nat. Methods, 2019, 16, 1306–1314 CrossRef CAS PubMed.
- H. Mallick, E. A. Franzosa, L. J. Mclver, S. Banerjee, A. Sirota-Madi, A. D. Kostic, C. B. Clish, H. Vlamakis, R. J. Xavier and C. Huttenhower, Nat. Commun., 2019, 10, 3136 CrossRef PubMed.
- L. Cao, E. Shcherbin and H. Mohimani, mSystems, 2019, 4, e00387 CrossRef PubMed.
- A. Gonzalez, J. A. Navas-Molina, T. Kosciolek, D. McDonald, Y. Vázquez-Baeza, G. Ackermann, J. DeReus, S. Janssen, A. D. Swafford, S. B. Orchanian, J. G. Sanders, J. Shorenstein, H. Holste, S. Petrus, A. Robbins-Pianka, C. J. Brislawn, M. Wang, J. R. Rideout, E. Bolyen, M. Dillon, J. Gregory Caporaso, P. C. Dorrestein and R. Knight, Nat. Methods, 2018, 15, 796–798 CrossRef CAS PubMed.
- E. Bolyen, J. R. Rideout, M. R. Dillon, N. A. Bokulich, C. C. Abnet, G. A. Al-Ghalith, H. Alexander, E. J. Alm, M. Arumugam, F. Asnicar, Y. Bai, J. E. Bisanz, K. Bittinger, A. Brejnrod, C. J. Brislawn, C. T. Brown, B. J. Callahan, A. M. Caraballo-Rodríguez, J. Chase, E. K. Cope, R. Da Silva, C. Diener, P. C. Dorrestein, G. M. Douglas, D. M. Durall, C. Duvallet, C. F. Edwardson, M. Ernst, M. Estaki, J. Fouquier, J. M. Gauglitz, S. M. Gibbons, D. L. Gibson, A. Gonzalez, K. Gorlick, J. Guo, B. Hillmann, S. Holmes, H. Holste, C. Huttenhower, G. A. Huttley, S. Janssen, A. K. Jarmusch, L. Jiang, B. D. Kaehler, K. B. Kang, C. R. Keefe, P. Keim, S. T. Kelley, D. Knights, I. Koester, T. Kosciolek, J. Kreps, M. G. I. Langille, J. Lee, R. Ley, Y.-X. Liu, E. Loftfield, C. Lozupone, M. Maher, C. Marotz, B. D. Martin, D. McDonald, L. J. McIver, A. V. Melnik, J. L. Metcalf, S. C. Morgan, J. T. Morton, A. T. Naimey, J. A. Navas-Molina, L. F. Nothias, S. B. Orchanian, T. Pearson, S. L. Peoples, D. Petras, M. L. Preuss, E. Pruesse, L. B. Rasmussen, A. Rivers, M. S. Robeson 2nd, P. Rosenthal, N. Segata, M. Shaffer, A. Shiffer, R. Sinha, S. J. Song, J. R. Spear, A. D. Swafford, L. R. Thompson, P. J. Torres, P. Trinh, A. Tripathi, P. J. Turnbaugh, S. Ul-Hasan, J. J. J. van der Hooft, F. Vargas, Y. Vázquez-Baeza, E. Vogtmann, M. von Hippel, W. Walters, Y. Wan, M. Wang, J. Warren, K. C. Weber, C. H. D. Williamson, A. D. Willis, Z. Z. Xu, J. R. Zaneveld, Y. Zhang, Q. Zhu, R. Knight and J. G. Caporaso, Nat. Biotechnol., 2019, 37, 852–857 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |