
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidant speciation and anionic ligand effects in the gold-catalyzed oxidative coupling of arenes and alkynes†
Manuel
Hofer
a,
Teresa
de Haro
a,
Enrique
Gómez-Bengoa
 b,
Alexandre
Genoux
a and
Cristina
Nevado
*a
b,
Alexandre
Genoux
a and
Cristina
Nevado
*a
aDepartment of Chemistry, University of Zürich, Winterthurerstrasse 190, Zürich, CH-8057, Switzerland. E-mail: cristina.nevado@chem.uzh.ch
bDepartamento de Química Orgánica I, Universidad del Pais Vasco, Apdo 1072, CP-20080 Donostia-San Sebastián, Spain
First published on 31st July 2019
Abstract
The mechanism of the gold-catalyzed oxidative cross-coupling of arenes and alkynes has been studied in detail combining stoichiometric experiments with putative reaction intermediates and DFT calculations. Our data suggest that ligand exchange between the alkyne, the Au(I)-catalyst and the hypervalent iodine reagent is responsible for the formation of both an Au(I)-acetylide complex and a more reactive “non-symmetric” I(III) oxidant responsible for the crucial Au(I)/Au(III) turnover. Further, the reactivity of the in situ generated Au(III)-acetylide complex is governed by the nature of the anionic ligands transferred by the I(III) oxidant: while halogen ligands remain unreactive, acetato ligands are efficiently displaced by the arene to yield the observed Csp2–Csp cross-coupling products through an irreversible reductive elimination step. Finally, the nature of competitive processes and catalyst deactivation pathways has also been unraveled. This detailed investigation provides insights not only on the specific features of the species involved in oxidative gold-catalyzed cross couplings but also highlights the importance of both ancillary and anionic ligands in the reactivity of the key Au(III) intermediates.
Introduction
Aryl alkynes have found widespread use as building blocks in the synthesis of numerous natural products, bioactive molecules and organic materials.1 In recent years, metal-catalyzed Csp2–H bond functionalizations have been explored as an alternative strategy to classical Pd-catalyzed cross-coupling reactions for the efficient construction of Csp2–Csp bonds.2 These approaches are attractive because they avoid the otherwise necessary pre-functionalization of the aromatic partner. However, and in contrast to the large body of metal-catalyzed Csp2–H arylation reactions,3 the direct Csp2–H alkynylation of arenes has been much less explored. Few examples though have shown the viability of this strategy.4 In 2010, the Cu-catalyzed direct alkynylation of electron deficient polyfluoroarenes with terminal alkynes using O2 as the oxidant was reported by Su et al.5 Despite its efficiency, the reaction is barely catalytic and relies on the acidity of the Csp2–H bond in the arene substrate. A different approach focused on the stoichiometric use of alkynyliodonium species as an electrophilic source of acetylenic moieties in the presence of catalytic amounts of late transition metals as demonstrated by Waser et al.6 In this context, our group reported an oxidative alkynylation of arenes via Au-catalyzed C–H functionalization of both Csp- and Csp2–H bonds (eqn (1) and Scheme 1).7 One of the most remarkable features of this protocol was the use of “deactivated” electron rich arenes and electron deficient alkynes as coupling partners. A catalytic amount of Ph3PAuCl in combination with commercially available PhI(OAc)2 as the stoichiometric oxidant was found to be effective in producing new Csp2–Csp bonds.8 A stoichiometric version of this transformation had been already described by Fuchita and co-workers back in 2001.9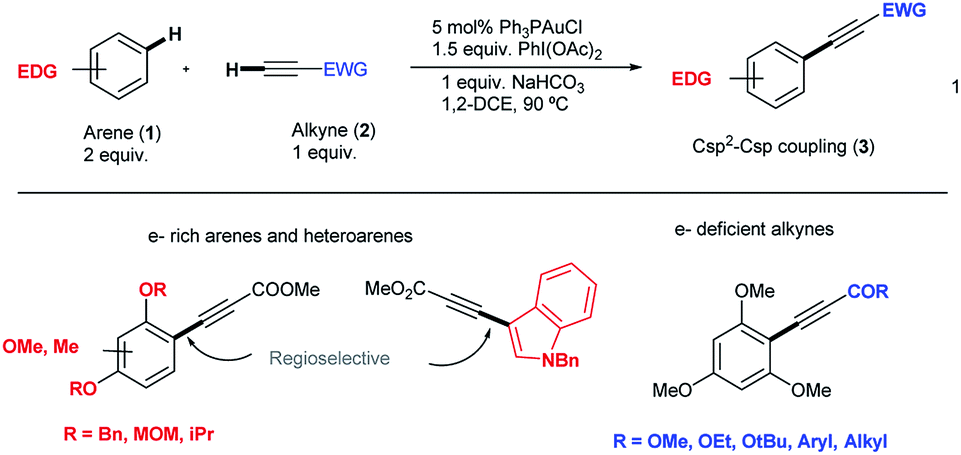 | ||
| Scheme 1 Au-catalyzed alkynylation of arenes. | ||
In the catalytic version, a hypervalent iodine reagent10 was selected as the oxidant on the basis of significant evidence that these species could promote Pd(II)/Pd(IV) catalytic cycles.11 We thus anticipated that a Au(I)/Au(III) catalytic turnover could be implemented under the reaction conditions. Furthermore, the ability of Au(III) species to trigger Csp2–H activation in electron rich arenes is also well established.9,12 Although Au(I)/Au(III)-catalyzed reactions have recently emerged as powerful tools for C–C cross couplings,13 with few notable exceptions,14 the mechanistic understanding of these processes is still limited and the characterization of putative intermediates is scarce. We report herein a detailed investigation aiming to elucidate the factors governing both reactivity and selectivity in these transformations.
In our previous work,7 the mechanistic rationale involved: (i) an equilibrium between the free alkyne and the Au catalyst with the aid of a base to form a Au(I)-acetylide complex (I); (ii) oxidation of I with PhI(OAc)2 to form Au(III) species II; (iii) arene auration to produce intermediate III which evolves via reductive elimination (iv) yielding product IV (Scheme 2, path A.1). Alternatively, in line with Waser's reports on the stoichiometric use of alkynyliodonium salts,6 a ligand exchange between PhI(OAc)2 and Au(I)-acetylide (I) to give an alkynyliodonium intermediate (V) could also be proposed. Arene addition to give VI followed by β-Au elimination would then furnish product IV as shown in path B.1 of Scheme 2. Alternatively, transmetalation between alkynyliodonium salt V and putative aryl-Au(III) species (VII) produced in situ within the oxidative reaction media12 could also deliver intermediate III, which would yield the observed products after reductive elimination as shown in Scheme 2, path B.2.
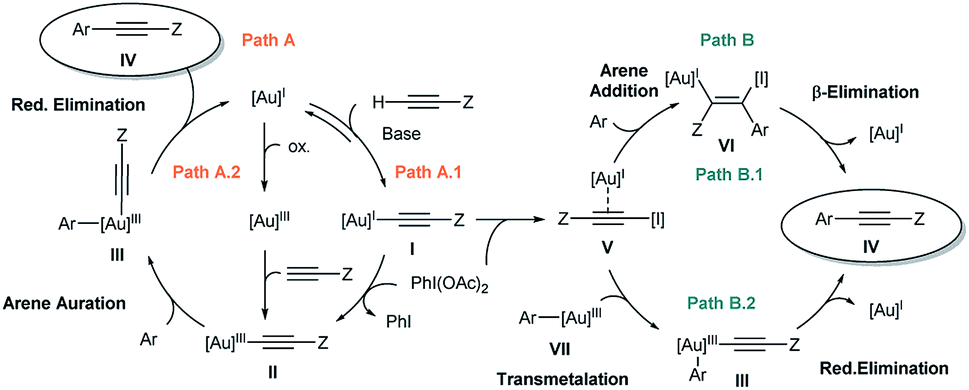 | ||
| Scheme 2 Plausible mechanisms for Au-catalyzed alkynylation of arenes. | ||
Our preliminary study left open several key questions: first and foremost, the role of PhI(OAc)2 needed to be established, whether it functioned as a stoichiometric oxidant to achieve the Au(I)/Au(III) turnover or as an electrophilic source to exchange and then cross-couple the alkyne to an electron rich arene, or both. In addition, the order of steps needed to be clarified as to whether the transfer of the alkyne to the gold(I) complex (path A.1) or a direct oxidation (path A.2) was involved in the initial step of the catalytic cycle. Furthermore, neither detailed information about the metal coordination sphere in the proposed Au(III) intermediates II and III nor about the oxidant environment was available from these initial investigations.7
Results and discussion
Initial experiments15 to investigate the feasibility of pathways B.1 and B.2 focused on alkynyliodonium salts (V) in order to reveal their potential role as intermediates in these transformations.Treatment of methyl 3-(phenyl(tosyloxy)-λ3-iodanyl)propiolate with stoichiometric amounts of 3,5-dimethoxytoluene (4) and Ph3PAuCl or Ph3PAuOAc at 90 °C did not furnish the desired Csp2–Csp cross coupling product and only decomposition of the alkynyliodonium salt was detected (Fig. 1a and b). Identical experiments in the presence of gold(III) complexes like Ph3PAuCl3 or Au(OAc)3/PPh3 showed a similar outcome (Fig. 1c and d). Interestingly, in the case of Ph3PAuCl3 formation of 2-chloro-1,5-dimethoxy-3-methylbenzene as the by-product could be observed.12a These control experiments (Section 2 in the ESI†) led us to rule out pathways B.1 and B.2 and the participation of alkynyliodonium species V as intermediates in these transformations.
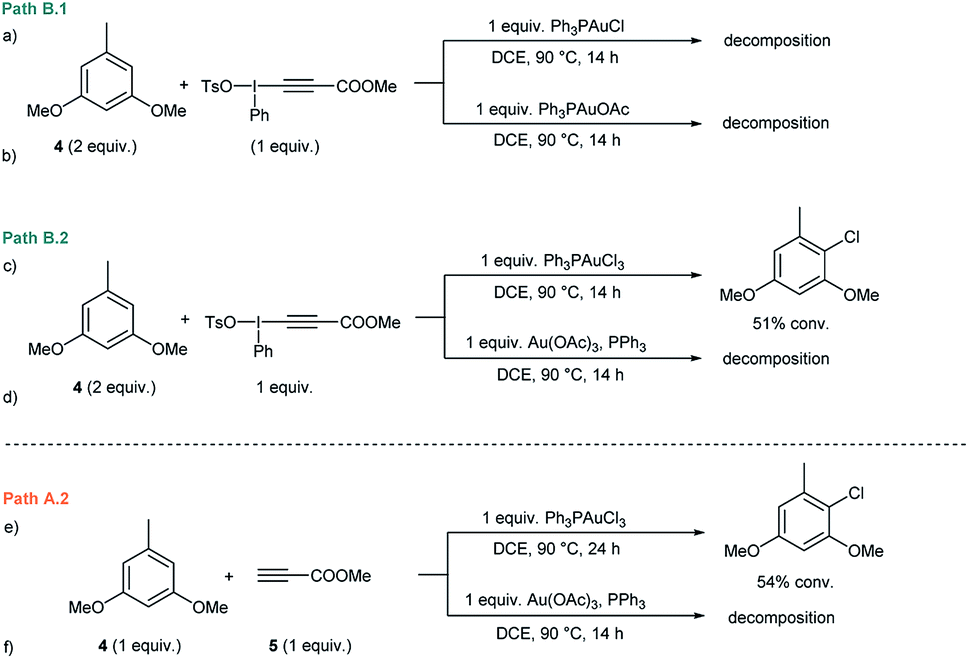 | ||
| Fig. 1 (a–d) Stoichiometric experiments involving alkynyliodonium species (paths B.1 and B.2 in Scheme 2). (e and f) Stoichiometric experiments regarding path A.2. | ||
Experiments to investigate pathway A.2 involved the participation of the alkyne in the presence of gold(III) species. However, stoichiometric experiments with 3,5-dimethoxytoluene (4), methyl propiolate (5) and Ph3PAuCl3 or Au(OAc)3/PPh3 at 90 °C showed the formation of the arylchloride in the case of Ph3PAuCl3 but no participation of the alkyne (Fig. 1e and f). To explore the direct oxidation of the initial catalyst, Ph3PAuCl was treated with an excess of PhI(OAc)2 at 90 °C. However, no reaction was observed even after prolonged heating and just Ph3PO could be detected in trace amounts (Fig. S2 and S3 in the ESI†). These experiments suggest that the neutral Ph3PAuCl complex used as the catalyst is scarcely oxidized by PhI(OAc)2 under the reaction conditions, in contrast to previous results obtained for PhICl2 which furnished Ph3PAuCl3 in 96% yield even at room temperature.16In situ oxidation of the Au(I) catalyst also seems to be at the outset of the Au-catalyzed oxidative oxo- and aminoarylation of alkenes with boronic acids.14a,b However, the results described herein clearly indicate that the present alkynylation reaction proceeds, at least at the outset, through an alternative reaction mechanism.
Formation and reactivity of Au(I)-acetylide (8)
To investigate path A.1, a careful spectroscopic analysis (1H and 31P NMR) of the reaction mixture stemming from the reaction between 3,5-dimethoxytoluene (4) and methyl propiolate (5) under the standard conditions (5 mol% Ph3PAuCl, 1.5 equiv. PhI(OAc)2, 1 equiv. NaHCO3) was performed. The reaction showed the presence of three species: the initial catalyst Ph3PAuCl, [(Ph3P)2Au]Cl (7) and a Ph3PAu(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CO2Me) complex (8) (Fig. 2a, S4 and S5 in the ESI†).
C–CO2Me) complex (8) (Fig. 2a, S4 and S5 in the ESI†).
Complex 8 appears already after the first minutes of the reaction and it disappears towards the end whereas Ph3PAuCl and 7 are present after the starting materials have been completely consumed. Ph3PO could not be detected in the reaction mixture. These results indicate that the phosphine ligand remains bound to the metal center and thus does not get oxidized by PhI(OAc)2 in appreciable quantities. In situ generated phosphine-free Au(III) species have been proved to be the productive intermediates in the recently reported Au-catalyzed cross coupling reaction of aryl silanes with arenes.14c Interestingly, a catalytic reaction in the presence of IPrAuCl gave no product conversion, thus highlighting the importance of the ancillary ligand in these transformations.
Once the species detected during the reaction had been identified, we decided to interrogate in detail both the mechanism for the formation, as well as the reactivity of the Au(I)-acetylide complex 8. We monitored the formation of 8 from Ph3PAuCl, methyl propiolate (5) and NaHCO3 by both 1H and 31P NMR performing the reaction in CD2Cl2. Experimentally, the formation of complex 8 is not a favorable process, and even after prolonged heating, it could only be detected in marginal amounts (Fig. 2b, S6 and S7 in the ESI†). In contrast, the same reaction in the presence of PhI(OAc)2 revealed the presence of 8 after only 5 minutes (Fig. 2c, S8 and S9 in the ESI†), in line with the spectroscopic analysis of a catalytic reaction (Fig. 2a). Interestingly, the reaction of Ph3PAuOAc with 5 proceeded quantitatively at room temperature in the absence of oxidant producing 8 and AcOH in only 10 minutes (Fig. 2d). On the other hand, the reverse reaction, although not unfeasible, is not a favorable process. These observations suggested an additional and unexpected new role of the oxidant in the initial steps of the reaction: PhI(OAc)2 favors the formation of the observed complex 8 (Fig. S10–S13 in the ESI†).
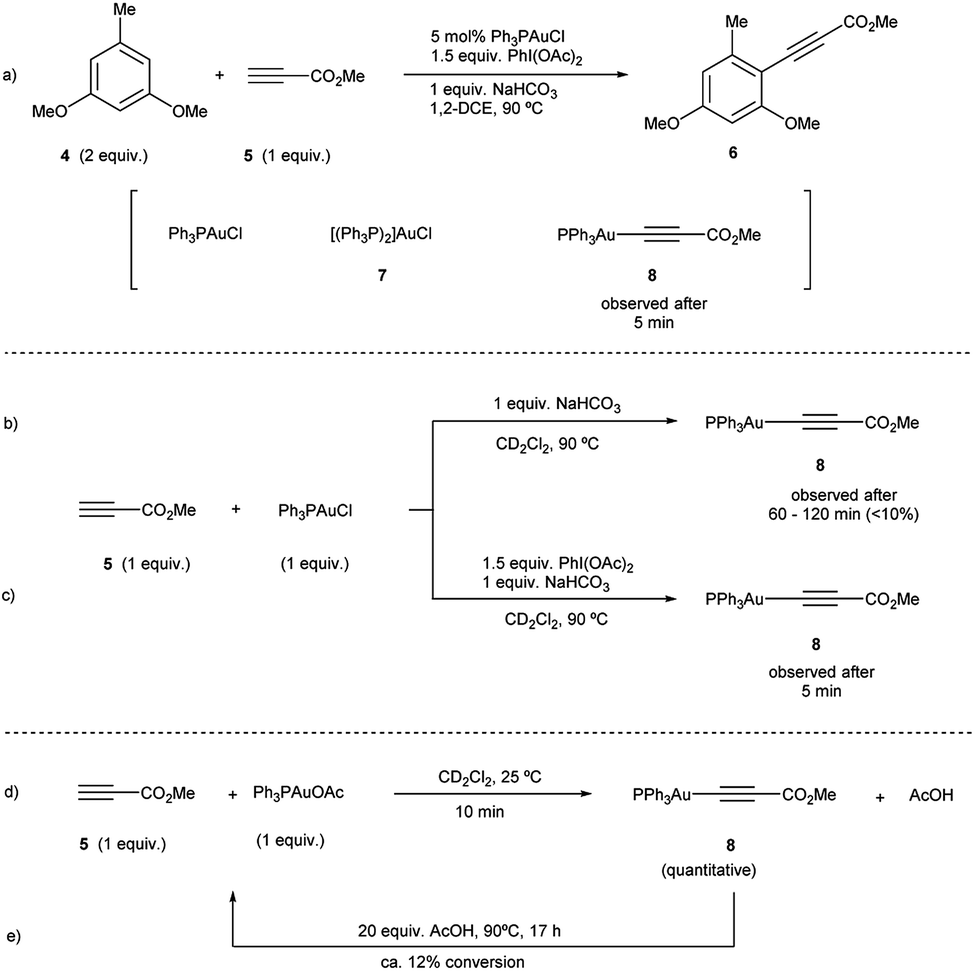 | ||
| Fig. 2 (a) Standard catalytic reaction. (b and c) Experiments towards the formation of Au(I)-acetylide 8 from Ph3PAuCl and alkyne 5 in the absence or presence of PhI(OAc)2, respectively. (d and e) Reactivity of Ph3PAuOAc towards methyl propiolate (5). | ||
The reactivity of the Au(I)-acetylide 8 was studied next. Gold acetylides have been proposed as productive reaction intermediates in different transformations including the formation of Au vinylidenes17 or the Au-catalyzed homo-18 and heterocoupling19 of alkynes. The reaction of 8 with PhI(OAc)2 in CD2Cl2 was monitored by 1H and 31P NMR. Indeed, no conversion was observed up to 60 °C while only very low conversion into Ph3PAuOAc and Ph3PAuCl was detected even after prolonged heating at 90 °C (eqn (2), Fig. S14 and S15 in the ESI†).20 These results indicate that 8 is hardly oxidized with PhI(OAc)2 and also that the putative oxidation product Ph3PAu(CC–CO2Me)(OAc)29 is rather unstable under the reaction conditions undergoing rapid reductive elimination to give Ph3PAuOAc and 3-(acetyloxy)-methyl propiolate (which decomposes in situ due to its highly labile nature).
In sharp contrast, the reaction of 8 in the presence of PhICl2 cleanly proceeded at room temperature to give cis-Ph3PAu(CC–CO2Me)(Cl)2 keep "cis-Ph3PAu(CC–CO2Me)(Cl)2 (10)" in single line(10), whose structure could be confirmed by X-ray diffraction analysis (eqn (3)). These results not only showcase the different oxidizing abilities of PhI(OAc)2vs. PhICl2 but also the influence of the ligand transferred by the hypervalent iodine reagent on the stability of the corresponding Au(III) intermediates produced in the reaction mixture. When 8 and PhICl2 were stirred at higher temperature, reductive elimination on 10 occurred, furnishing Ph3PAuCl, which is oxidized in the presence of the remaining oxidant to Ph3PAuCl3. In this case, the by-product stemming from reductive elimination (i.e. 3-chloro-methyl propiolate 1121) could be clearly observed (eqn (4), Fig. S20 and S21 in the ESI†).
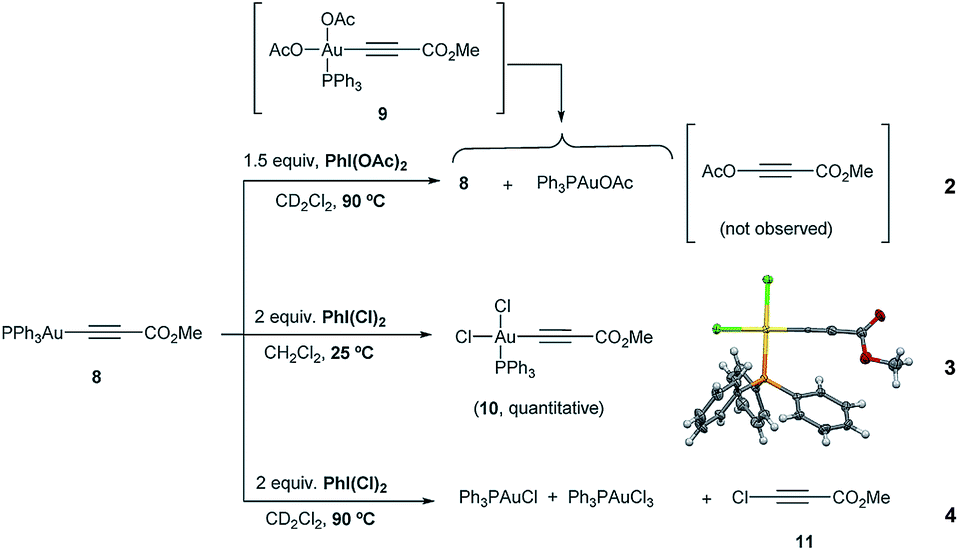
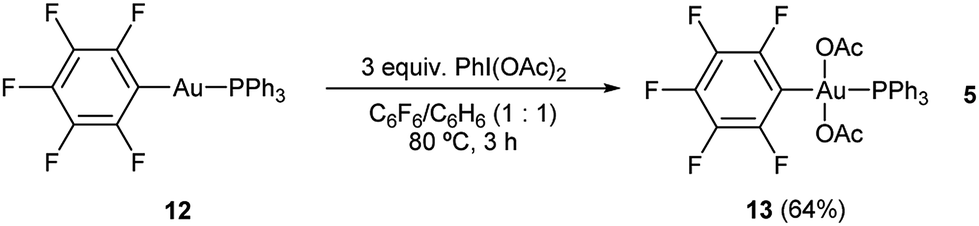
Due to the labile nature of complex 9, we decided to seek an alternative model system to study the reactivity of the putative Au(III) intermediates produced during the aryl alkynylation reaction. Ph3PAuC6F5 (12) was selected expecting that the electron deficient nature of the pentafluorophenyl ligand could mimic that of the propiolate unit while offering a more stable platform for the isolation of gold(III) species. Reaction of Ph3PAuC6F5 (12) with PhI(OAc)2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of hexafluorobenzene/benzene at 80 °C delivered trans-Ph3PAu(C6F5)(OAc)2 (13) in 64% yield according to our previously reported procedure (eqn (5)).22
:1 mixture of hexafluorobenzene/benzene at 80 °C delivered trans-Ph3PAu(C6F5)(OAc)2 (13) in 64% yield according to our previously reported procedure (eqn (5)).22
Reactivity of putative Au(III)-intermediates
We set out to examine the reactivity of complexes 10 and 13 towards the species present in the media during the standard aryl alkynylation reaction, namely: methyl propiolate (5), Au(I)-acetylide complex (8) and electron-rich arenes in a stoichiometric fashion. The results of this study have been summarized in Table 1. Interestingly, trans-Ph3PAu(C6F5)(OAc)2 (13) reacted with methyl propiolate (5) at 25 °C to give methyl 3-(pentafluorophenyl)-prop-2-ynoate (14) in 68% yield together with Au(I)-acetylide complex 8 as a result of the double replacement of both acetato ligands with free alkyne followed by reductive elimination (see Fig. S22 and S23 in the ESI†). The reaction of 13 with complex 8 was also illustrative, providing 14 in 62% yield together with Ph3PAuOAc. Since no Au(I)-acetylide complex 8 was detected at the end of the reaction, we have to assume that upon a first Au(I)/Au(III) transmetalation (which could also be described as a Au(I)/Au(III) ligand exchange reaction), Csp2–Csp reductive elimination occurs fast, preventing a second ligand transfer between the different gold species (see Fig. S24 and S25 in the ESI†). Finally, the reactions of 13 with 1,3,5-trimethoxybenzene, 1,3-dimethoxytoluene 4 and N-methyl indole were also enlightening as they proceeded efficiently towards the corresponding cross-coupling products 15, 16 and 17 in 85, 74 and 85% yield, respectively.16b,23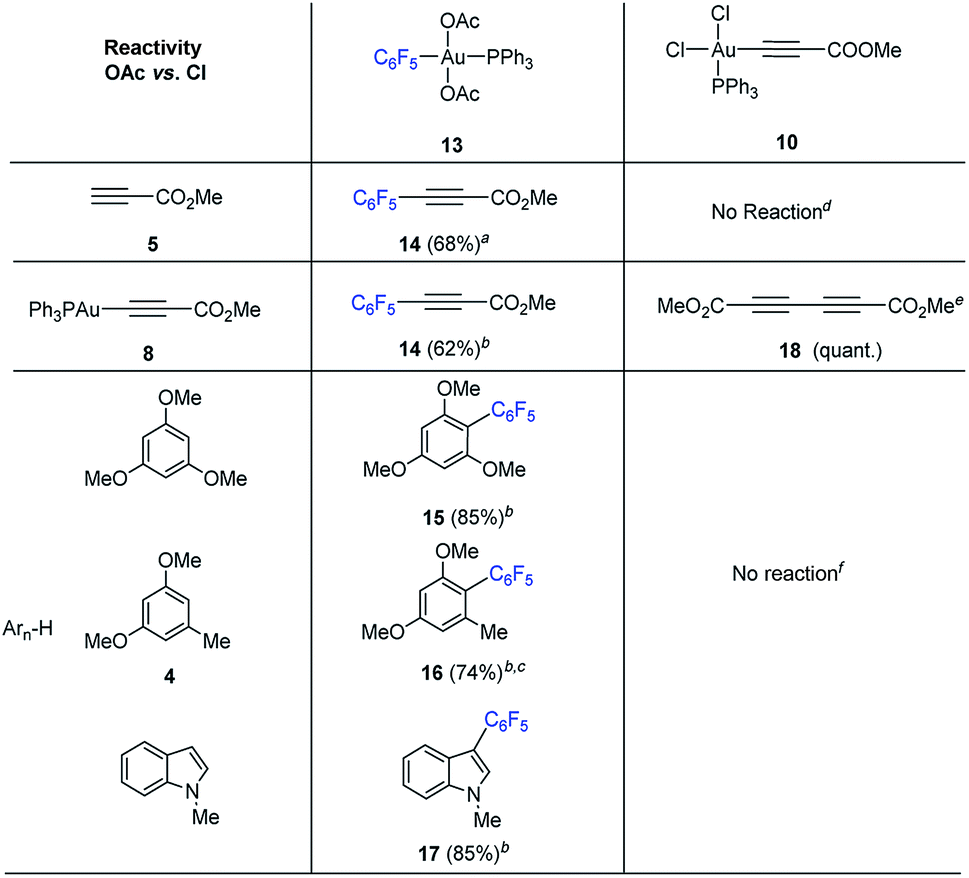
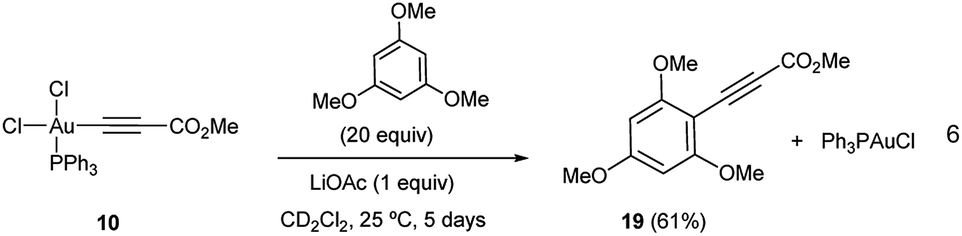
The reactivity pattern observed for cis-dichloro(methoxy-carbonylethynyl) (triphenylphosphine)-gold(III) (10) turned out to be completely different as shown on the right column of Table 1. In contrast to 13, complex 10 did not react with methyl propiolate (5) (Fig. S26 and S27 in the ESI†) although it underwent transmetalation with Au(I)-acetylide complex 8 even at −25 °C to give alkyne homocoupling product 18 (ref. 24) and Ph3PAuCl. As in the previous case, only one Cl/alkyne ligand exchange took place (Fig. S28 and S29 in the ESI†). Also in contrast to 13, cis-Ph3PAu(CC–CO2Me)(Cl)2 (10) proved to be completely unreactive towards electron-rich aromatic nucleophiles even after prolonged heating at 130 °C (Fig. S30 and S31 in the ESI†).16b,25 The experiments summarized in Table 1 showcase the strong differences in reactivity for diacetato-Au(III) vs. dichloro-Au(III) complexes26 and highlight the importance of the oxidant of choice, i.e. the ligand that ultimately the oxidant transfers onto the metal center, for a productive reaction outcome. In line with this hypothesis, replacement of the chloro ligands in 10 by reaction with 1 equivalent of LiOAc in the presence of 1,3,5-trimethoxybenzene in excess resulted in the clean formation of Ph3PAuCl and alkynylation product 19 in 61% yield (eqn (6)).
Additional stoichiometric experiments with Au(I)-acetylide complex 8 were designed. When the reaction of 3,5-dimethoxytoluene (4) was run using Au(I)-acetylide 8 as the stoichiometric alkynylating agent in the presence of PhI(OAc)2 and NaHCO3 only traces of the desired cross-coupling product 6 were detected (eqn (7), Fig. S32 and S33 in the ESI†). In contrast, when methyl propiolate (5) was incorporated into the reaction, arylalkyne product 6 was clearly observed after only one hour even if in low conversion (eqn (8), Fig. S34 and S35 in the ESI†). A catalytic version of this reaction using 5 mol% of 8 or Ph3PAuOAc also afforded 6 although again, in a much less efficient manner compared to the standard conditions (eqn (9), Fig. S36 and S37 in the ESI†).
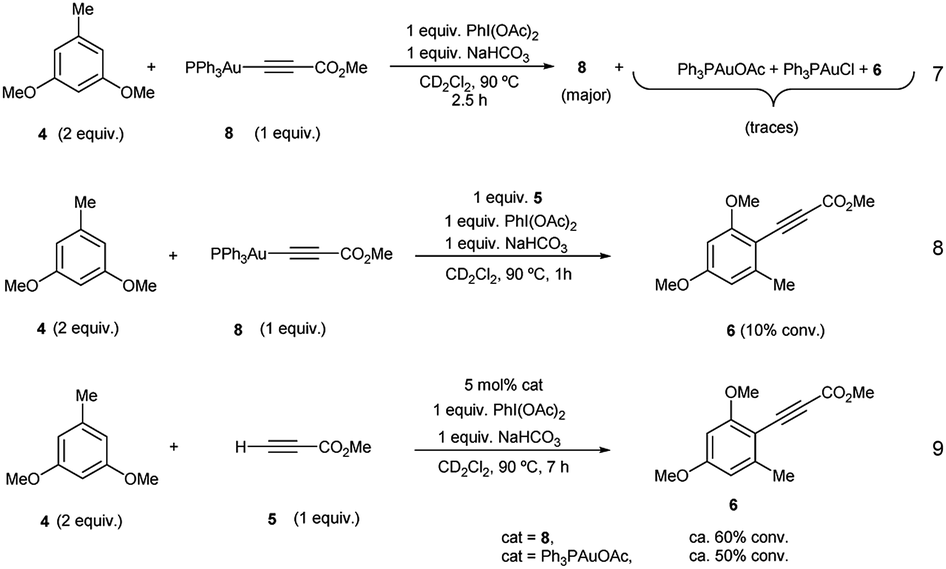
These experiments clearly suggest that the presence of free alkyne in the reaction mixture favors a productive reaction outcome and together with eqn (2) highlight that PhI(OAc)2is not an efficient oxidant for8and the putative Ph3PAu(CC–CO2Me)(OAc)2 (9) complex is not a highly competent reaction intermediate. Additionally, the reactions shown in Fig. 2b–d indicated that the oxidant is involved in the activation of the alkyne. We hypothesized that the formation of 8 could occur by ligand exchange on Ph3PAuCl in the presence of PhI(OAc)2 to form Ph3PAuOAc which rapidly activates the alkyne 5 to form 8 and AcOH, which is then quenched by NaHCO3 present in the reaction media (Fig. 2d). This proposal is supported by recent experiments of Shi et al., showing the formation of R3PAuOAc in the presence of R3PAuCl and PhI(OAc)2 by MALDI-MS analysis.27 Thus, to gain a deeper insight into the specific nature of the individual steps involved in these transformations, DFT calculations and additional control experiments were carried out.
DFT studies and characterization of the oxidizing species
In line with the experimental observations summarized in Fig. 2, calculations confirmed that formation of Au(I)-acetylide 8 from Ph3PAuCl in the absence of oxidant is a highly unfavorable process even in the presence of base (+19.1 kcal mol−1, Fig. 3a). The lack of reactivity observed for Ph3PAuCl in the presence of PhI(OAc)2 could also be confirmed. A potential Au(Cl)/I(OAc) exchange is also disfavored (+12.8 kcal mol−1), and thus such an equilibrium would be strongly shifted towards the starting materials (Fig. 3b). When alkyne is added into the system, the energies of these two equilibria remain unchanged. However, the trace amounts of Ph3PAuOAc that could be produced rapidly react with the free alkyne present in the media to give Au(I)-acetylide complexes and acetic acid, which will be quenched with the base present in the reaction (Fig. 3c and 2d). The energy for this process decreases to +6.3 kcal mol−1. Thus, the second equilibria will drive the first one towards the right, influenced by the presence of free alkyne. Furthermore, the in situ generated PhI(OAc)(Cl) intermediate presents a much lower activation energy towards the oxidation of Au(I) acetylide viaTS3 (+20.1 kcal mol−1) compared to PhI(OAc)2viaTS3′ (+29.2 kcal mol−1) (Fig. 3d).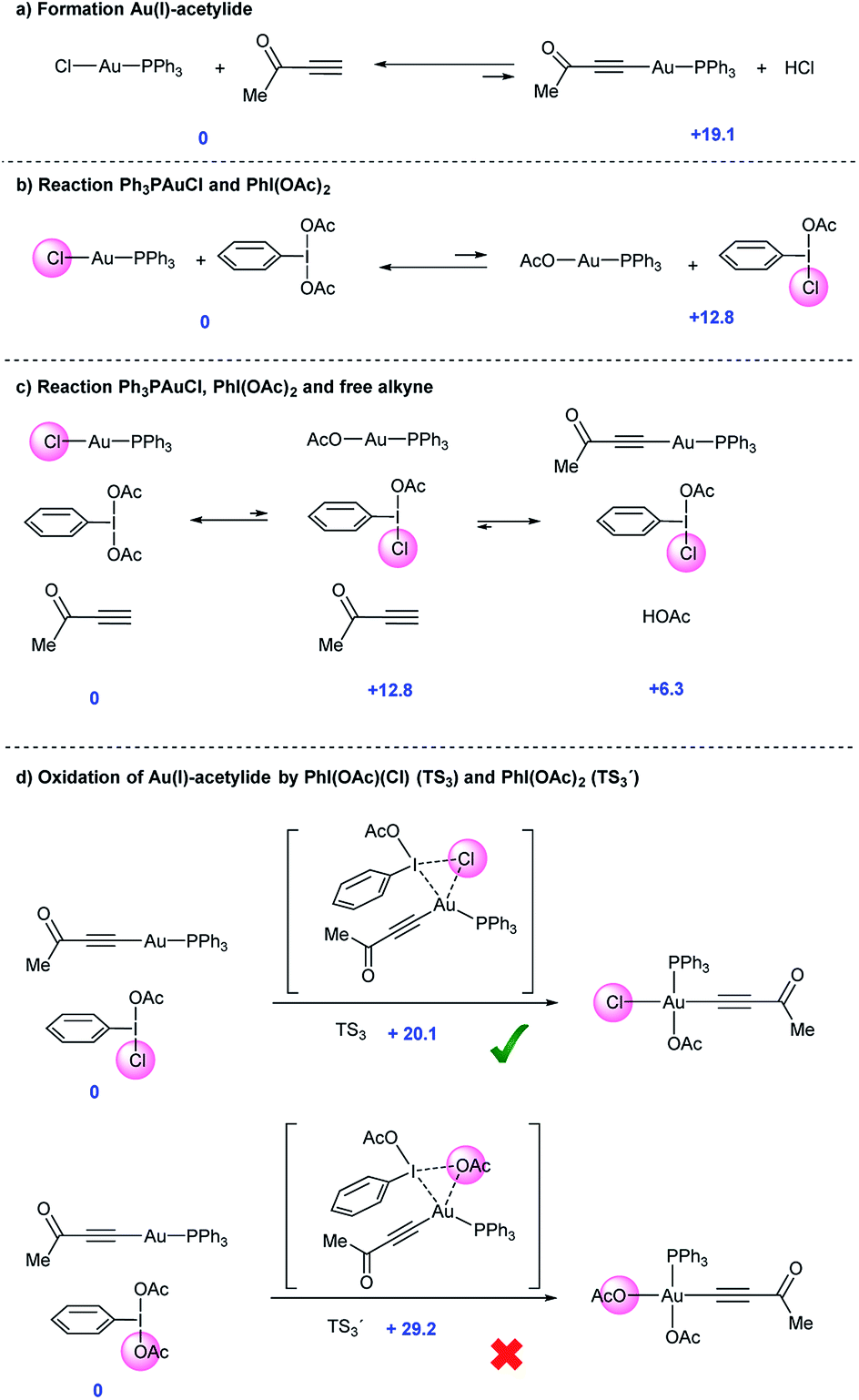 | ||
| Fig. 3 (a–d) Computed Gibbs free energy values (kcal mol−1) for the potential reaction between Ph3PAuCl, alkyne (methyl alkynyl ketone) and hypervalent iodine reagents calculated with the M06 functional. | ||
Stoichiometric experiments were subsequently designed to support the hypothesis of a Au(Cl)/I(OAc) ligand exchange triggered by the presence of free alkyne and the formation of a more reactive “non-symmetrical” oxidant. In an attempt to detect such species by highly sensitive 19F NMR spectroscopy, a fluorine-containing iodonium diacetate, namely m-F-C6H4I(OAc)2 (20), was synthesized.28
The in situ kinetic studies of a reaction between 20, methyl propiolate (5) and Ph3PAuCl revealed the consumption of 20 and the simultaneous formation of a new product with a characteristic 19F signal at 107.5 ppm which was assigned to m-F-C6H4I(OAc)(Cl) (21) (Fig. 4a and b). The oxidizing potential of 21 is higher than that of 20 as already revealed by the DFT calculations (Fig. 3d) and thus the Au(I)-acetylide complex 8 which has been generated in situ can be slowly oxidized even at room temperature, thus preventing the accumulation of 21 in the reaction media. In the absence of other species, an OAc-alkyne ligand exchange reaction on the Au(III)-acetylide intermediate (red path) or a transmetalation between the Au(I) and Au(III)-acetylide species coexisting in the reaction media (blue path) could explain the formation of homocoupling product 18, which is produced in a comparable ratio to that in which 20 is consumed (Fig. 4c). Additional experiments were carried out to support the proposed structure of compound 21: the reaction of m-F-C6H4I(Cl)2 with 1 equivalent of AgOAc delivered, after only 5 min, the same species observed in the 19F NMR spectrum, thus confirming the proposed composition of the “non-symmetric” oxidant (Fig. 4d) (for these and additional control experiments, see Section 3.8 in the ESI†).
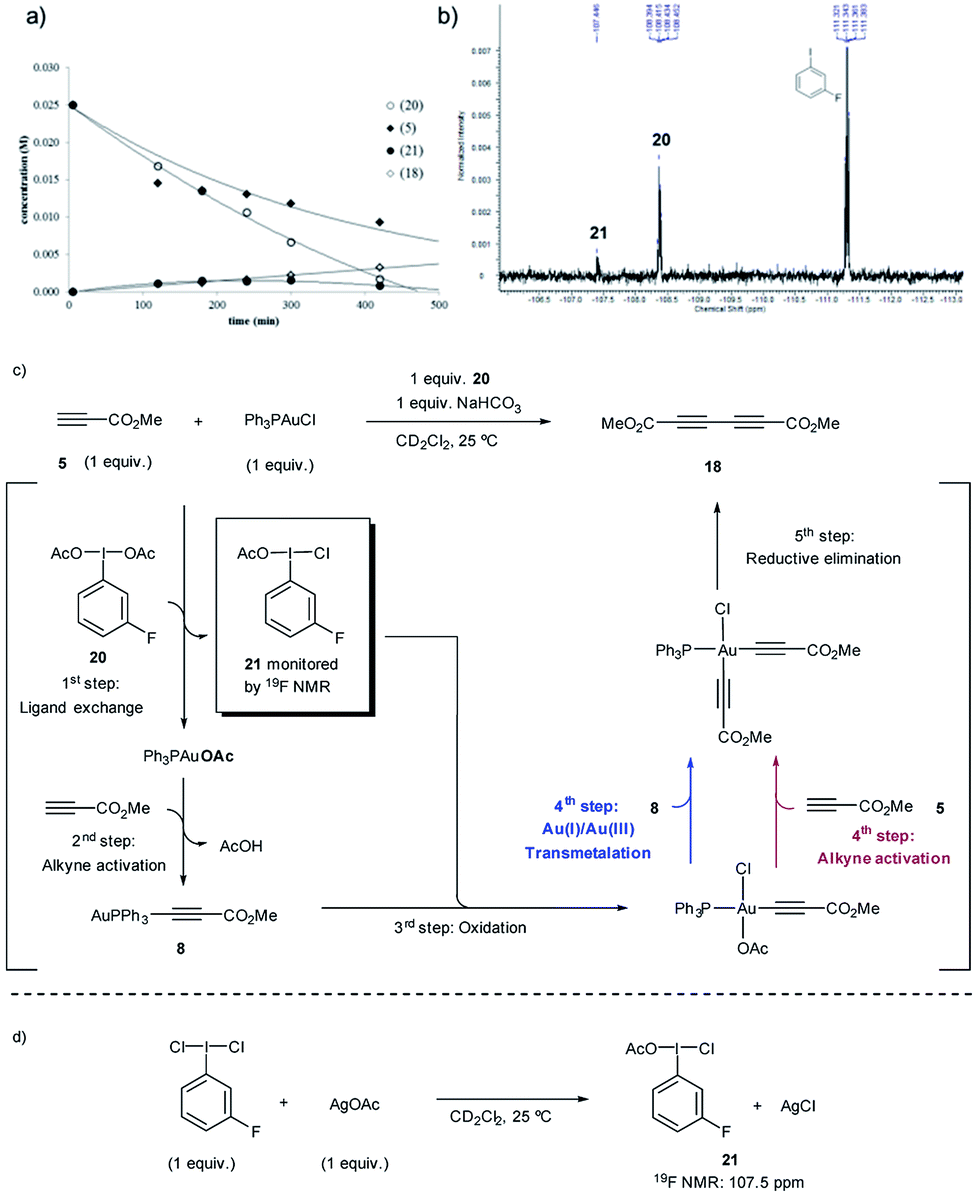 | ||
| Fig. 4 Formation of “non-symmetric” I(III) species by reaction of m-F-C6H4I(OAc)220 with Ph3PAuCl and methyl propiolate (5): (a) Evolution of the temporal concentration of reactants, intermediates and product. (b) 19F NMR traces of the reaction mixture. (c) Rationalization of the observed species. (d) Alternative synthesis and characterization of m-F-C6H4I(OAc)(Cl) (21). | ||
To confirm the ability of chloride transfer from Ph3PAuCl to PhI(OAc)2, the standard cross-coupling reaction was performed in the presence of 1 equivalent of (n-Bu)4NCl (see Section 3.9 in the ESI†). As expected, the initial excess of chloride in the reaction mixture inhibited the formation of the desired cross-coupling product. In turn, 2-chloro-3,5-dimethoxytoluene could be detected, pointing towards in situ generated 21, which in this case is produced in abundant quantities in the reaction media, as the chlorinating agent. In line with these results, in the absence of chloride available for ligand exchange, the performance of 8 or Ph3PAuOAc as catalysts (eqn (9)) delivered the cross-coupling product in lower yield compared to the standard conditions.
Finally, DFT calculations were carried out to map the entire energy potential surface (Fig. 5). The mixture of free alkyne, PhI(OAc)2 and Ph3PAuCl was taken as the ground state of energy (G = 0 kcal mol−1), mimicking the initial experimental conditions. As detailed in Fig. 5, the Cl/OAc anion exchange between the gold and the iodine center through TS1 (with Au–O1 and I–Cl bond distances of 2.35 and 2.90 Å respectively) leads to a first high energy mixture, INT1 (+12.8 kcal mol−1), which is readily transformed into INT2 (+6.3 kcal mol−1) by deprotonation of the alkyne, alkynyl-gold complex formation and HOAc release viaTS2 (with bond distances: Au–C1 = 2.34 Å, Au–O1 = 2.46 Å and C1–H = 1.23 Å).29 The oxidation of the alkynyl-gold complex by the active oxidant species PhI(OAc)(Cl) presents an affordable activation energy (+20.1 kcal mol−1 from INT2 to TS3), involving the rupture of the I–Cl bond (I–Cl = 3.10 Å, Au–Cl = 2.67 Å, Au–I = 3.32 Å and Au–C1 = 2.0 Å). The alternative Au(I) to Au(III) oxidation involving the I–OAc bond of PhI(OAc)(Cl) is disfavoured by more than 8 kcal mol−1 with respect to TS3 (Fig. 3d). After the oxidation viaINT3, iodobenzene is released to form a highly stable neutral intermediate INT5A (−17.9 kcal mol−1).
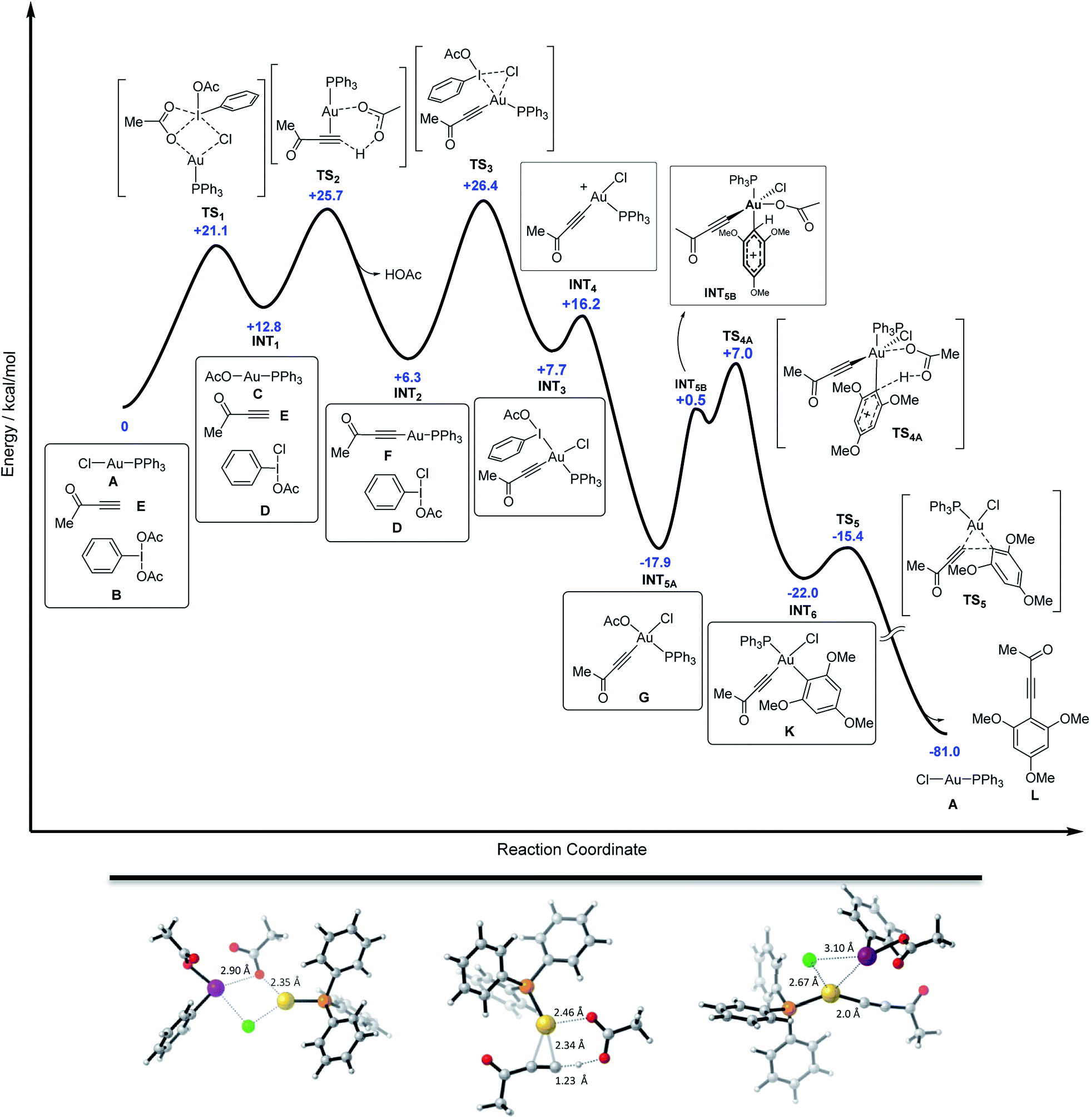 | ||
| Fig. 5 Top: complete energy profile for the reaction between Ph3PAuCl, alkyne (methyl alkynyl ketone), PhI(OAc)2 and arene (1,3,5-trimethoxybenzene). Gibbs free energy values in blue calculated with the M06 functional. Bottom: structures of transition states TS1, TS2 and TS3 (left to right) highlighting the relevant distances. | ||
The arene reacts then with INT5A and the acetate ligand abstracts the proton to restore the aromaticity viaTS4A in an overall highly exergonic process to give INT6 (−29.7 kcal mol−1 from INT3), which is followed by a fast reductive elimination (the energy profile calculated for a dissociative interaction of the arene with INT3 can be found in Fig. S69 in the ESI†).15 Deuterium labelling experiments on the arene carried out in our seminal study7 showed no primary KIE, in line with the present DFT results in which arene auration is not turnover limiting. Ph3PAuCl is formed in the final stage, re-entering the cycle, which shows an overall reaction energy of −81 kcal mol−1. Thus, the DFT calculations support the hypothesis of the transformation of gold(I)-chloride into gold(I)-acetate (INT1), and this into Au(I)-acetylide (INT2) through two up-hill equilibria. The activation energies for these processes are comparable to that of the subsequent oxidation step by the in situ generated PhI(OAc)(Cl) via transition state TS3 and also to that of the attack of the arene onto the alkynyl-gold(III) intermediate viaTS4A. DFT calculations also confirmed the lability of the aryl-aurate intermediate (INT6), which rapidly evolves via reductive elimination towards the cross-coupling product regenerating the Ph3PAuCl catalyst.23,30
Proposed catalytic cycle
The data presented in previous sections enabled a more detailed mechanism for the Au-catalyzed alkynylation of arenes to be proposed based on a better understanding of both oxidant and catalyst speciation for a productive reaction outcome (Scheme 3).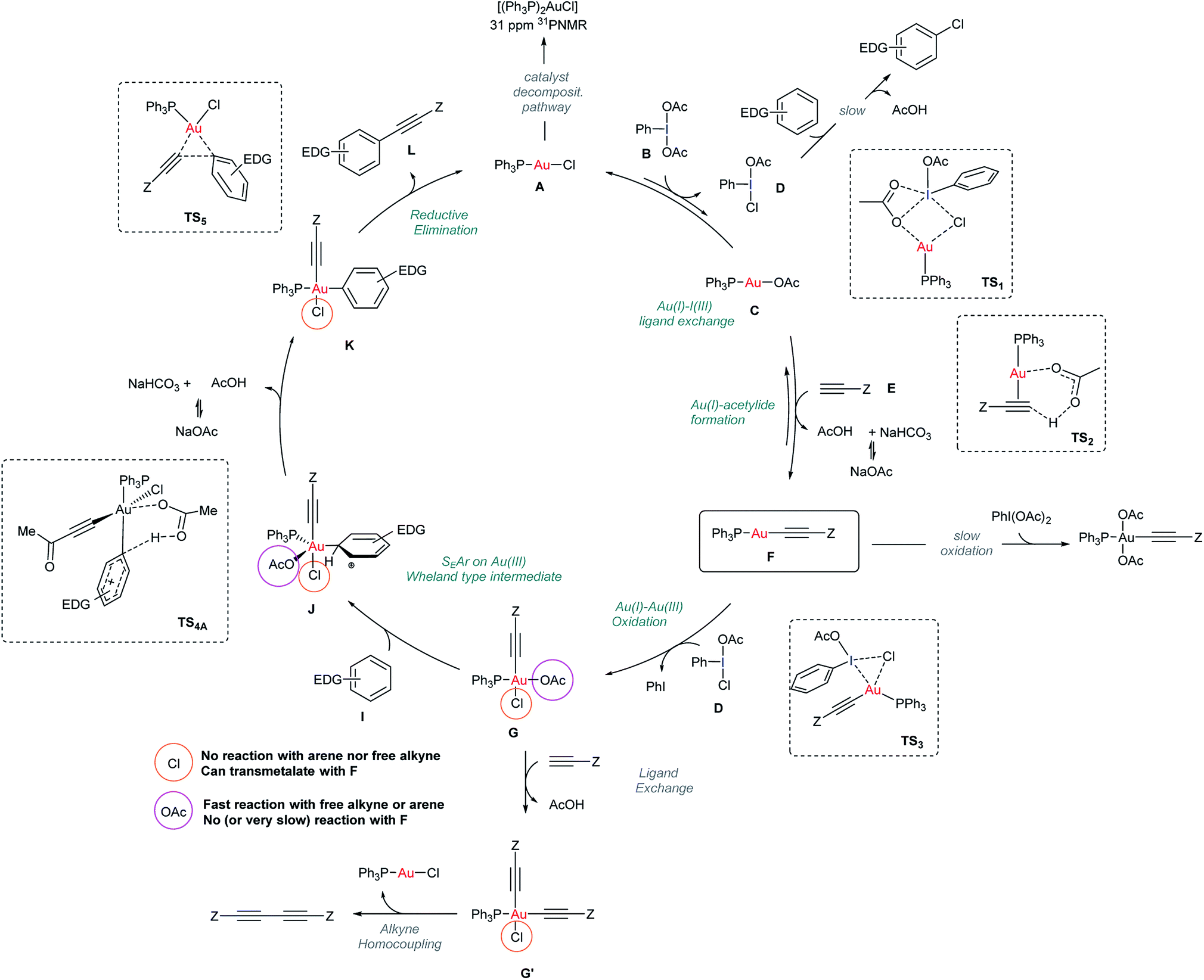 | ||
| Scheme 3 Mechanistic proposal for the Au-catalyzed alkynylation of arenes. Structures of TS1, TS2, TS3 and TS5 stem from DFT calculations reported in Fig. 5. | ||
At the outset of the reaction, the formation of an Au(I)-acetylide complex F takes place. However, the reaction of methyl propiolate (E) and Ph3PAuCl in the presence of a base to give acetylide complex F is not a favorable process (Fig. 2b). In contrast, the same reaction in the presence of PhI(OAc)2 revealed the formation of F after only 5 minutes (Fig. 2c), in line with the spectroscopic analysis of a catalytic reaction (Fig. 2a). These results led us to discard a facile equilibrium between the Au pre-catalyst and the alkyne while suggesting a new role for the oxidant in the initial steps of this transformation. Studies, including F-labeling experiments and DFT calculations, support a mechanistic scenario involving multiple equilibria between the alkyne, oxidant and gold. Initially, a ligand exchange between Ph3PAuCl and PhI(OAc)2 delivers Ph3PAuOAc and a non-symmetric oxidant, PhI(OAc)(Cl). As shown in Fig. 2d, 3a and b, free alkyne reacts with the trace amounts of Ph3PAuOAc to give Au(I)-acetylide complex F and acetic acid, which is quenched in the presence of NaHCO3. Experimentally, the formation of a non-symmetric hypervalent iodine m-F-PhI(OAc)(Cl) (21) could also be monitored by 19F NMR (Fig. 4). This new oxidant formed in situ presents a lower activation energy towards the oxidation of F into Au(III)-acetylide complex G compared to PhI(OAc)2 (ΔΔG†ca. 9 kcal mol−1, Fig. 3d). Experiments summarized in eqn (2) and (7)–(9) clearly suggest that free alkyne favors a productive reaction outcome and also that PhI(OAc)2 is not an efficient oxidant for 8 nor is the putative Ph3PAu(CC–CO2Me)(OAc)2 (9) complex a highly competent reaction intermediate. Still, alternative reaction pathways operating with PhI(OAc)2 as the oxidant cannot be completely ruled out (eqn (9)). Putative analogues of Au(III) complexes G (10 and 13 in Table 1) were used as mechanistic probes in stoichiometric experiments which revealed the crucial role of anionic ligands in the reaction outcome. Thus, acetato ligands on the Au(III) center can be rapidly exchanged in the presence of arenes whereas the corresponding chlorides remain unreacted.
A competitive OAc/alkyne exchange in G to give G′ can occur although in a sufficiently slower rate to enable a productive cross coupling reaction rather than the undesired homocoupling of alkyne, which is sometimes observed as a minor by-product in these transformations. Although a Au(I)/Au(III) transmetalation involving the chloride ligands towards the formation of a bis-alkynyl Au(III) intermediate cannot be completely ruled out,18,31 control experiments indicate that this process might be slow under the present reaction conditions (see Fig. 4c and S51–S57 in the ESI†).
The proposed Au(Cl)–I(OAc) exchange in the first steps of the reaction produces a “non-symmetric” ArI(Cl)(OAc) oxidant responsible for reaction by-products stemming from the direct oxidation (i.e. chlorination) of the arene (<5%) (Section 4.2 in the ESI†).32 Finally, the mechanism for catalyst decomposition has also been studied. Slow de-coordination of Ph3P from the neutral starting complex Ph3PAuCl or other phosphine-Au species involved in the reaction results in the formation of trace amounts of (Ph3P)2AuX (7) visible in the 31P NMR of the standard catalytic reaction media (Fig. 2a). Control experiments revealed that these species are catalytically inactive and do not interfere with a productive reaction outcome (see Section 4.3 in the ESI†).33
Conclusions
A detailed investigation of the gold-catalyzed alkynylation of arenes including kinetic and stoichiometric experiments together with DFT calculations has provided an insightful perspective on the mechanism of this transformation. A ligand exchange involving the alkyne, Au(I)-catalyst and oxidant is needed to form both a Au(I)-acetylide complex and a more reactive “non-symmetric” oxidant PhI(OAc)(Cl) responsible for the crucial Au(I)/Au(III) turnover. Both processes, i.e. the formation of Au(I)-acetylide and its oxidation, are connected through an equilibrium which evolves along the reaction progress. Reaction of the electron rich arenes with the in situ generated Au(III)-alkynyl intermediate occurs to produce a short-lived aryl-alkynyl-Au(III) complex, which evolves by reductive elimination to produce the observed cross-coupling products and the Au(I) catalyst. The mechanisms of both catalyst decomposition and competing side reactions have also been unraveled.A few of the lessons learned in this study may also be applicable to other gold-catalyzed oxidative cross-couplings employing I(III) oxidants. Unexpectedly, a ligand exchange between the gold(I) pre-catalyst and the initial hypervalent iodine might be the key to produce the suitable gold(I)-species to enable activation of the alkyne in the first place. Furthermore, the same process provides the appropriate oxidizing species, capable of producing reactive Au(III)-intermediates. This process is influenced by both the nature of the ancillary ligand on gold and by the presence of other reaction partners which can shift this up-hill equilibria. Oxidation is also an energetically demanding process which translates into a Au(III)-intermediate, whose reactivity will be fine-tuned by the nature of the anionic ligands transferred by the oxidant: while acetato ligands favor activation of the arene and are easily displaced to give the cross-coupling products, chlorides are much less reactive and thus stabilize these species favoring transmetalation processes. We believe that this mechanistic study supporting Au(I)/Au(III) redox catalytic cycles provides novel insights, useful not only for the development of new gold catalyzed oxidative transformations but also for the improvement and fine tuning of already available ones.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC Starting grant agreement no. 307948) and the Swiss National Science Foundation (SNF 200020_146853) for financial support and the SGI/IZO-SGIker UPV/EHU and Schrödinger (UZH) for allocation of computational resources. We thank Dr A. Linden for the X-ray crystal structure determination of 10 (CCDC-1008765).†Notes and references
- (a) P. J. Garatt, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, p. 271 Search PubMed; (b) Acetylene Chemistry: Chemistry, Biology and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1, p. 2 Search PubMed; (b) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- For recent reviews in C–H arylation, see: (a) Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH, Weinheim, Germany, 2009, p. 311 Search PubMed; (b) R. Giri, B.-F. Shi, K.-M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (d) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS.
- (a) F. Besselievre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553–9556 CrossRef CAS PubMed; (b) Z. Shao and F. Peng, Angew. Chem., Int. Ed., 2010, 49, 9566–9568 CrossRef CAS PubMed; (c) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096–2098 CrossRef CAS PubMed; (d) N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358–2361 CrossRef CAS PubMed; (e) B. Pacheco Berciano, S. Lebrequier, F. Besselievre and S. Piguel, Org. Lett., 2010, 12, 4038–4041 CrossRef CAS PubMed; (f) C. Theunissen and G. Evano, Org. Lett., 2014, 16, 4488–4491 CrossRef CAS PubMed.
- Y. Wei, H. Zhao, J. Kan, W. Su and M. Hong, J. Am. Chem. Soc., 2010, 132, 2522–2523 CrossRef CAS PubMed.
- (a) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346–9349 CrossRef CAS PubMed; (b) J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304–7307 CrossRef CAS PubMed; (c) J. P. Brand, C. Chevalley and J. Waser, Beilstein J. Org. Chem., 2011, 7, 565–569 CrossRef CAS PubMed; (d) J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem.–Eur. J., 2012, 18, 5655–5666 CrossRef CAS PubMed; (e) Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743–6747 CrossRef CAS PubMed; (f) Y. Li and J. Waser, Beilstein J. Org. Chem., 2013, 9, 1763 CrossRef PubMed; (g) J. P. Brand, Y. Li and J. Waser, Isr. J. Chem., 2013, 53, 901–910 CrossRef CAS.
- T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512–1513 CrossRef CAS PubMed.
- For a Au-catalyzed tandem allenoate cyclization/alkynylation protocol with Selectfluor as the oxidant, see: M. N. Hopkinson, J. E. Ross, G. T. Giuffredi, A. D. Gee and V. Governeur, Org. Lett., 2010, 12, 4904–4907 CrossRef CAS PubMed.
- Y. Fuchita, Y. Utsonomiya and M. Yasutake, J. Chem. Soc., Dalton Trans., 2001, 2330–2334 RSC.
- (a) V. V. Zhdankin, ARKIVOC, 2009, 1 Search PubMed; (b) R. M. Moriarty, Org. Chem., 2005, 70, 2893–2903 CrossRef CAS PubMed; (c) J. P. Brand, D. Fernandez Gonzalez, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102–115 RSC For a seminal example of Au(I)/Au(III) involving hypervalent iodine reagents, see: A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 386–388 RSC.
- (a) N. R. Deprez and M. Sanford, Inorg. Chem., 2007, 46, 1924–1935 CrossRef CAS PubMed; (b) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147–1149 CrossRef CAS PubMed; (c) T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tang, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793–3801 CrossRef CAS PubMed; (d) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824–889 CrossRef CAS PubMed.
- (a) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053–3059 CrossRef CAS; (b) K. S. Liddle and C. Parkin, J. Chem. Soc., Chem. Commun., 1972, 26 RSC; (c) P. W. J. deGraaf, J. Boersma and G. J. M. Van der Kerk, J. Organomet. Chem., 1976, 105, 399–406 CrossRef CAS; (d) K. A. Porter, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 4922–4927 CrossRef CAS.
- For seminal contributions on Au(I)/Au(III) manifolds, see: (a) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592–597 CrossRef CAS; (b) L. Cui, G. Zhang and L. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 3884–3887 CrossRef CAS PubMed; (c) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112–3115 CrossRef CAS; (d) A. Iglesias and K. Muñiz, Chem.–Eur. J., 2009, 15, 10563–10569 CrossRef CAS PubMed; (e) M. N. Hopkinson, A. Tessier, A. Salisbury, G. T. Giufredi, L. E. Combettes, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2010, 16, 4739–4744 CrossRef CAS PubMed; (f) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS PubMed; (g) W. E. Brenzovich, J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728–4731 CrossRef CAS PubMed; (h) L. T. Ball, M. Green, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724–4727 CrossRef CAS PubMed; (i) A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS PubMed; (j) T. de Haro and C. Nevado, Angew. Chem., Int. Ed., 2011, 50, 906–910 CrossRef CAS PubMed; (k) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed; (l) C. X. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef PubMed. For selected reviews on this specific topic see: (m) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493–497 CrossRef CAS; (n) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS PubMed; (o) M. Hopkinson, A. D. Gee and V. Governeur, Chem.–Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed; (p) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (q) T. de Haro and C. Nevado, Synthesis, 2011, 16, 2530–2539 Search PubMed; (r) C. Nevado and T. de Haro, in New Strategies in Chemical Synthesis and Catalysis, ed. B. Pignataro, Wiley-VCH, Weinheim, 2012, p. 247 Search PubMed.
- (a) E. Tkatchouck, N. P. Mankad, D. Benitez, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef PubMed; (b) W. E. Brenzovich, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard III and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5922 CrossRef PubMed; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254–264 CrossRef CAS PubMed; (d) J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778–1781 CrossRef CAS PubMed; (e) M. Joost, A. Zeineddine, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed; (f) D. A. Rosca, J. A. Wright, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167–2175 CrossRef PubMed; (g) D. A. Rosca, J. Fernández-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015, 1, e1500761 CrossRef PubMed; (h) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245–254 CrossRef CAS PubMed.
- Additional control experiments, experimental procedures, and full characterization of new compounds can be found in the accompanying ESI.†.
- (a) M. Hofer and C. Nevado, Eur. J. Inorg. Chem., 2012, 9, 1338–1341 CrossRef; (b) M. Hofer and C. Nevado, Tetrahedron, 2013, 69, 5751–5757 CrossRef CAS.
- (a) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS; (b) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed; (c) A. Simmoneau, F. Jaroschik, D. Lesage, M. Karanik, R. Guillot, M. Malacria, J.-C. Tabet, J.-P. Goddard, L. Fensterbank, V. Gandon and Y. Gimbert, Chem. Sci., 2011, 2, 2417–2422 RSC; (d) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolf and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4469 CrossRef CAS PubMed; (e) M. M. Hansmann, M. Rudolf, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2012, 52, 2593–2598 CrossRef PubMed.
- For a Au-catalyzed oxidative homocoupling of alkynes using Selectfluor, see: A. Leyva-Pérez, A. Doménech, S. I. Al-Resayes and A. Corma, ACS Catal., 2012, 2, 121–126 CrossRef . The authors propose the transmetalation between two alkynyl-Au species in different oxidation states (Au(I)/Au(III)) as the key step for the formation of the new Csp–Csp bond.
- For an Au-catalyzed heterocoupling of alkynes, see: H. Peng, Y. Xi, N. Ronaghi, B. Dong, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2014, 136, 13174–13177 CrossRef CAS PubMed . The gold activation of the triple bond on an alkynyl-Au(III) intermediate is proposed as the key step for this transformation. Control experiments in our system ruled out the π-activation of complex 8 under the standard reaction conditions.
- Ph3PAuCl, which is observed in minimal quantities, is produced by the slow activation of the chlorinated solvent as demonstrated by additional control experiments (see Fig. S16–S19 in the ESI†).
- B. B. Snider, D. M. Roush, D. J. Rodini, D. Gonzalez and D. Spindell, J. Org. Chem., 1980, 45, 2773–2785 CrossRef CAS.
- M. Hofer, A. Genoux, R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1021 CrossRef CAS PubMed.
- W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159–164 CrossRef CAS PubMed.
- The homocoupling production of methyl propiolate 18 presents a characteristic signal at 3.81 ppm in the 1H NMR: J. A. Varela, L. Castedo, M. Maestro, J. Mahía and C. Saá, Chem.–Eur. J., 2001, 7, 5203–5213 CrossRef CAS.
- The reactivity observed for complex 10 could also be reproduced with its pentafluorophenyl analogue Ph3PAu(C6F5)(Cl)2. See ref. 16.
- (a) A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618–6621 CrossRef CAS PubMed; (b) K. J. Stowers and M. S. Sanford, Org. Lett., 2009, 11, 4584–4587 CrossRef CAS PubMed; (c) D.-A. Rosca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247–7249 RSC.
- D.-H. Zhang, L.-Z. Dai and M. Shi, Eur. J. Org. Chem., 2010, 28, 5454–5459 CrossRef.
- J. G. Sharefkin and H. Saltzman, Org. Synth., 1963, 43, 62 CrossRef CAS.
- AcOH reacts with the NaHCO3 present in the reaction media to form NaOAc thus bringing the effective energy of INT2 to an even lower level.
- (a) J. Vicente, M. D. Bermudez, J. Escribano, M. P. Carrillo and P. G. Jones, J. Chem. Soc., Dalton Trans., 1990, 3083–3089 RSC; (b) J. Vicente, M. D. Bermudez and J. Escribano, Organometallics, 1991, 10, 3380–3384 CrossRef CAS; (c) T. Lauterbach, M. Livendahl, A. Rosellon, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed; (d) M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328–1332 CrossRef CAS.
- O. Schuster and H. Schmidbaur, Organometallics, 2005, 24, 2289–2296 CrossRef CAS.
- Products of propiolate polymerization, which have been observed in the presence of gold nanoparticles ( A. Leyva-Perez, J. Oliver-Meseguer, J. R. Cabrero-Antonino, P. S. Rubio-Marques, S. I. Al-Resayes and A. Corma, ACS Catal., 2013, 3, 1865–1873 CrossRef CAS ) were not detected in the reaction mixtures.
- (a) T. J. Harrison, J. A. Kozak, M. Corbella-Pané and G. R. Dake, J. Org. Chem., 2006, 71, 4525–4529 CrossRef CAS PubMed; (b) G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed; (c) A. Zhdanko, M. Ströbele and M. E. Maier, Chem.–Eur. J., 2012, 18, 14732–14744 CrossRef CAS PubMed; (d) M. Kumar, J. Jasinski, G. B. Hammond and B. Xu, Chem.–Eur. J., 2014, 20, 3113–3119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1008765. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02372k |
| This journal is © The Royal Society of Chemistry 2019 |