
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the Biginelli reaction mechanism by the ionic liquid effect: the combined role of supported heteropolyacid derivatives and acidic strength†
Elon F. Freitasa,
Roberto Y. Souzab,
Saulo T. A. Passos b,
José A. Diasa,
Silvia C. L. Dias*a and
Brenno A. D. Neto*b
b,
José A. Diasa,
Silvia C. L. Dias*a and
Brenno A. D. Neto*b
aLaboratory of Catalysis, Chemistry Institute, (IQ-UnB), University of Brasília, Campus Universitário Darcy Ribeiro – Asa Norte, 70910-900, Brasília-DF, Brazil. E-mail: scdias@unb.br
bLaboratory of Medicinal and Technological Chemistry, University of Brasília, Chemistry Institute (IQ-UnB), Campus Universitário Darcy Ribeiro, 70910-900, Brasília-DF, Brazil. E-mail: brenno.ipi@gmail.com
First published on 29th August 2019
Abstract
Herein, a combination of heteropolyacids and ionic liquids as a catalytic system was studied for the Biginelli multicomponent reaction; the positive ionic liquid effect associated with the acidic strength of zeolite-supported heteropolyacids made this combination an efficient catalytic system for the multicomponent synthesis of 3,4-dihydropyrimidin-2(1H)-one/thione derivatives. The acidic strength effect was evaluated, and a range was determined in which the reaction provided better results. The mechanism of the reaction was also investigated in the presence and absence of ionic liquids, and two features of paramount importance were revealed: the mechanism could be tuned to proceed through only one reaction path among three possibilities and the kinetics of the reaction was significantly faster in the presence of an ionic liquid.
Introduction
The search for more sustainable catalytic processes is of paramount importance in modern chemistry. The possibility of merging multicomponent reactions (MCRs)1–3 and ionic liquids (ILs)4–6 under catalytic conditions is therefore an attractive way to avoid the waste typically observed in linear syntheses and the use of volatile organic solvents. The combination of MCRs and ILs has indeed been regarded as “a perfect synergy for eco-compatible heterocyclic synthesis”.7ILs are widely used in the chemical industry,8 catalysis,9 biocatalysis,10 extraction processes,11 the obtention and purification of biological components,12 and other applications.13 For a long time, these ionic fluids have actually been claimed to be the solvents of the future14–16 because they represent a plausible alternative towards environmental acceptability.17 The use of ILs has proved to be essential to improve yields and/or selectivities in several reactions.18–20 The toolbox of organic methodologies has greatly expanded ever since MCRs have been incorporated as green and sustainable tools.21 For example, MCRs are being currently used in polymer modifications,22 for fluorescent nanoprobe syntheses,23 as AIE luminogens,24 as metal sensors,25 and in straight syntheses of new libraries of biologically active molecules.26
The idea of joining MCRs and ILs is in this context promising, and the expectation of achieving better results for MCRs performed in ILs compared to the use of classical organic solvents is more than reasonable. It is known that Brønsted acid catalysts can behave as superacids27,28 when supported in ILs. Heteropolyacids (HPAs) naturally display superacid behavior and are currently applied as catalysts in several organic transformations, as reviewed elsewhere.29–31 HPAs, both supported and unsupported, have also been tested as catalysts for MCRs.32–37
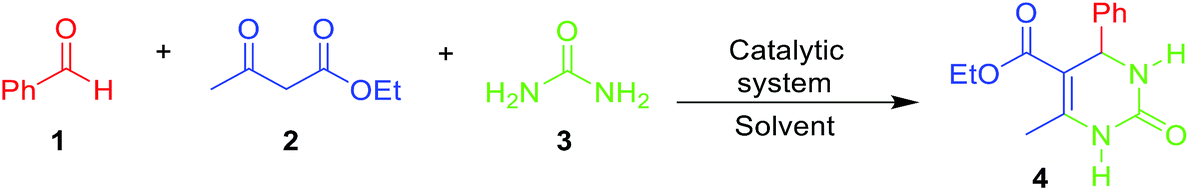
HPA derivatives embedded in IL media have been successfully applied to catalyze some MCRs.36–38 For example, a polymeric heteropolyacid-containing pyridinium IL catalyst proved to be a promising system for the multicomponent Biginelli reaction.39 The Biginelli reaction is believed by some to be the most important MCR.40 This MCR (Scheme 1) allows the direct synthesis of bioactive DHMPs (3,4-dihydropyrimidin-2(1H)-ones or -thiones) such as monastrol, piperastrol, and enastron.41–45
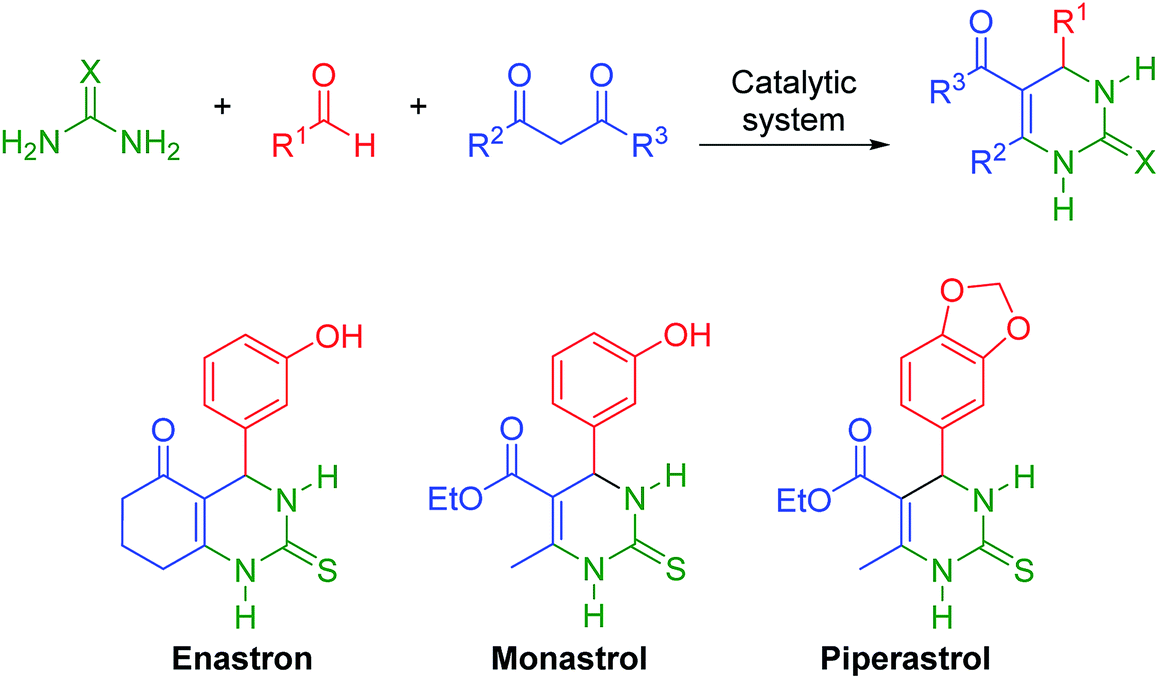 | ||
| Scheme 1 The general Biginelli reaction and examples of bioactive compounds directly obtained using this multicomponent reaction. | ||
HPAs and their derivatives have been previously used to promote the Biginelli reaction; however, in general, only moderate yields were achieved using organic solvents and no mechanistic elucidation was described.46 The need for excess reagents and other drawbacks, such as organic solvent contamination, low yields, long reaction times and no mechanistic evaluation, were also noted.46–49 Some of the authors have investigated different features of the multicomponent Biginelli reaction mechanism50–54 and other MCRs55–58 as well as heteropolyacid-catalyzed reactions.59–62 In the current work, we disclose the beneficial effect of ILs using HPA derivatives on the Biginelli reaction and the mechanistic implications for this MCR. For the first time, the reaction pathway of the Biginelli reaction has been tuned by adding an IL to the catalytic system, as will be demonstrated in due course.
Results and discussion
A comprehensive physicochemical study of the supported HPA catalysts on zeolite Y tested herein has been published by some of the authors elsewhere.59 However, some fundamental aspects of those characterizations must be briefly mentioned herein to guide the readers as to the most relevant factors and to associate the structural aspects of the materials with their activity in the tested MCR. Elemental analysis of the HPA supported on zeolite Y was performed by EDX/XRF. The values obtained were very similar to the nominal ones and were reported as the real values. The XRD results showed increased loss in the crystallinity of the supported catalysts, which was higher for the HPW-supported samples than for the HSiW-supported samples. This is a result of the stronger acidity of HPW, which destroys the zeolite Y structure in greater proportion during the preparation of the composites.59 Dispersion of either HPW or HSiW on the surface of zeolite Y leads to nanocrystals lower than 20 nm in size. The fingerprint bands of HPW and HSiW as well as the zeolite Y main bands were verified by FT-IR. The definitive presence of Keggin anions and their interactions with zeolite Y were confirmed by 31P, 29Si and 27Al MAS NMR spectroscopy, which revealed a decrease in tetrahedral Al as the amount of HPA increased. Gaseous pyridine adsorption experiments indicated that Lewis acid sites from zeolite Y interacted with the Keggin anions; as a result, the catalysts are heterogeneous in nature.59An initial model Biginelli reaction (Scheme 2 and Fig. 1) using benzaldehyde, urea and ethyl acetoacetate (equimolar quantities) without solvent and in the presence of the catalysts showed that all the HSiW-supported catalysts presented higher yields due to their higher amounts of Brønsted sites than of HPW sites, although they have slightly weaker acid strength. It was observed that pure zeolite Y contains Brønsted acid sites that are not accessible by all reagents (18% yield); therefore, the catalysis occurred on the external surface. On the other hand, the impregnation of HPA caused partial destruction of the crystalline structure of zeolite Y; once deposited on the surface of the zeolite, the strong Brønsted acidic sites (protons) of HPA were exposed, and thus the reaction yield increased. The relative numbers of HPA protons are presented in Table S1 in the ESI.†
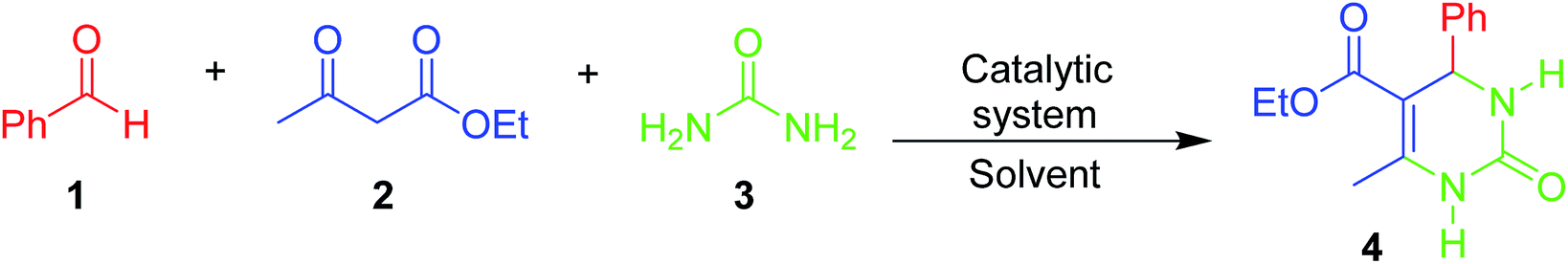 | ||
| Scheme 2 The model Biginelli reaction used to optimize the catalytic conditions in this study. All reactions were performed using equimolar quantities of the reagents. | ||
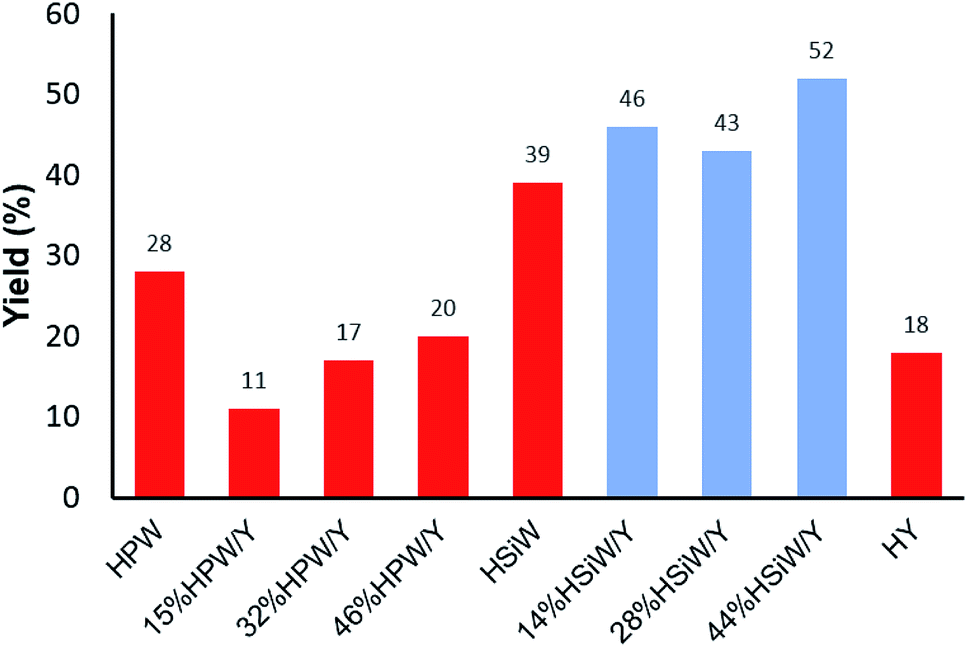 | ||
| Fig. 1 Effects of the catalyst composition on the reaction yield of the solvent-free model Biginelli reaction (1 mmol of each reagent) with a catalyst load of 30 mg after 60 min at 90 °C. | ||
One parameter that required further evaluation was the reaction temperature. The three best catalysts were chosen, and a temperature range from 50 °C to 100 °C was considered (Fig. 2). The catalyst 14% HSiW/Y generally showed lower conversion than the other catalysts. In addition, as the temperature increased, the yields also increased; thus, 100 °C was chosen as the ideal temperature to perform the MCR.
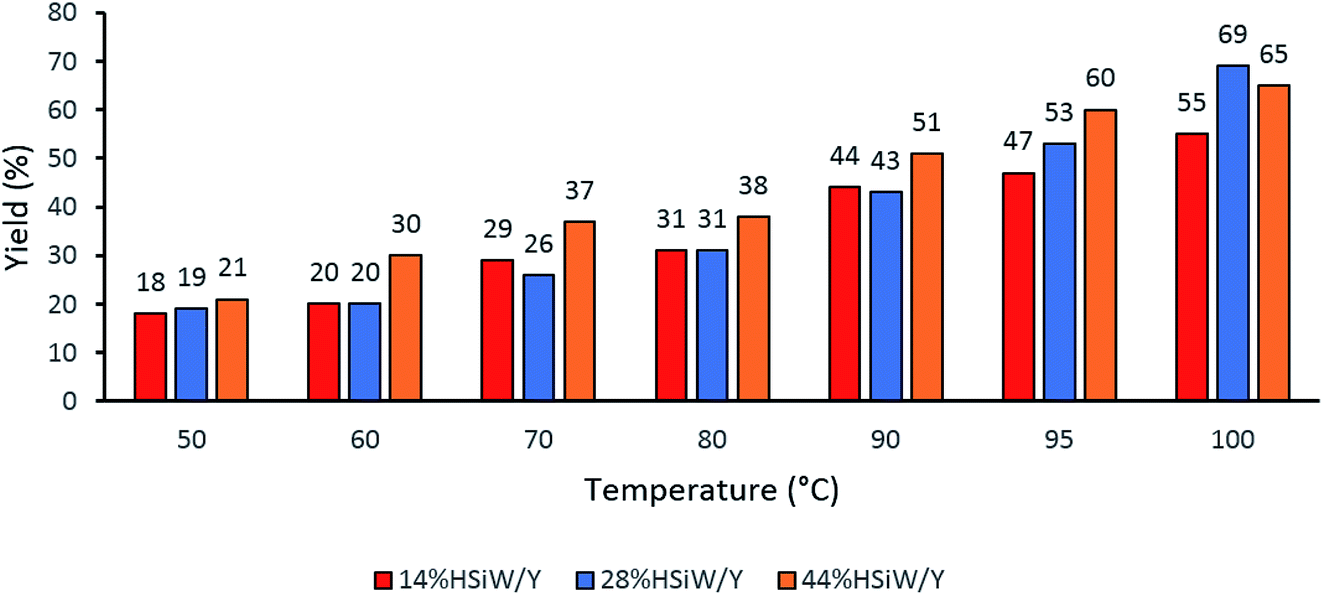 | ||
| Fig. 2 Effects of the reaction temperature on the solvent-free model Biginelli reaction yields. Reaction time of 60 min with 30 mg of catalyst, 1 mmol of each reagent and no solvent. | ||
Next, twelve solvents with different polarities were studied (Fig. 3). In a preliminary run, 95% yield was achieved for 28% HSiW/Y at 100 °C using BMI·PF6 as solvent. When using the imidazolium-based IL BMI·PF6 in a catalyst-free version of the model reaction, the DHPM yield was 35%. In the solvent-free version, but with 28% HSiW/Y as the catalyst, the highest yield was 69%. These results already indicate a positive ionic liquid effect by promoting stabilization of the charged intermediates by the formation of both ion pairs and larger supramolecular aggregates, in accordance with the literature.19,63 The presence of the IL increases the reaction rate by decreasing the activation barrier and alters the keto–enol tautomerization equilibrium of ethyl acetate.50 The formation of the enol and its stabilization by interactions with the solvent are paramount for the Biginelli reaction because the enolic form is more reactive; literature reports indicate that BMI·PF6 promotes the tautomeric equilibrium shift to enol formation.50
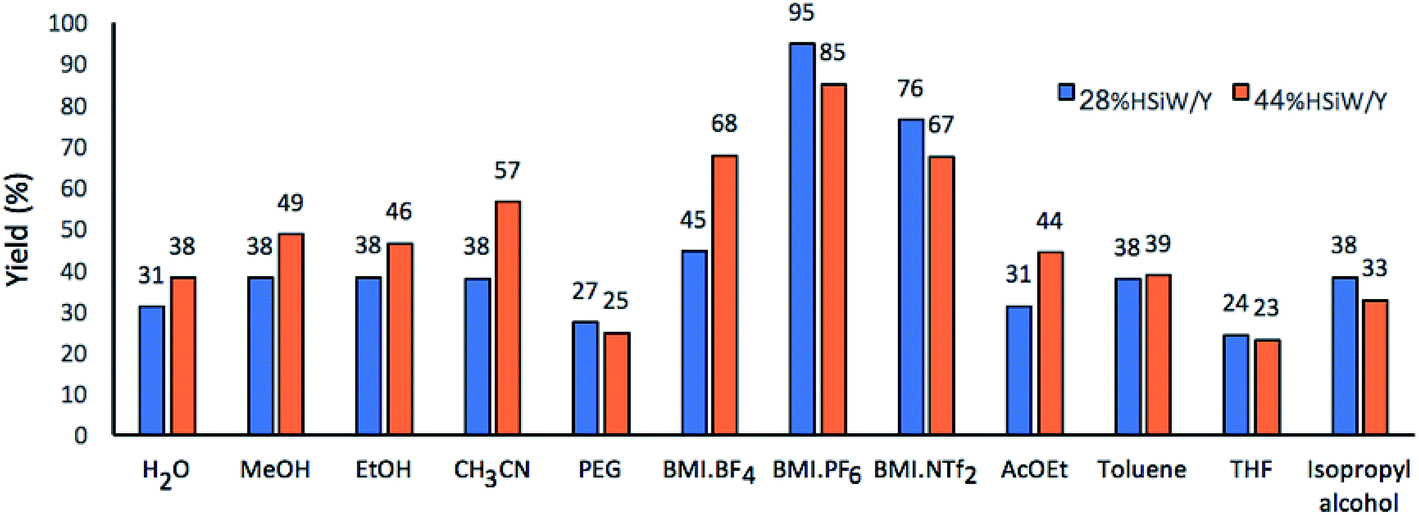 | ||
| Fig. 3 Effects of the solvent on the yield of the model Biginelli reaction. Reaction time of 60 min, 30 mg of catalyst, temperature of 100 °C, 1 mmol of each reagent and 0.5 mL of solvent. All reactions were performed in sealed Schlenk tubes to avoid solvent loss. | ||
A reaction profile was depicted (Fig. 4). The yields ranged from 16% to 83%. For both catalysts, the yields were very close within experimental error. After 60 min, the yield became steady, presenting little variation. An approximately sigmoid profile could be observed because the product precipitates from the reaction medium, thus shifting the equilibrium towards MCR adduct formation; this is in accordance with the expected profile as indicated in another study.50
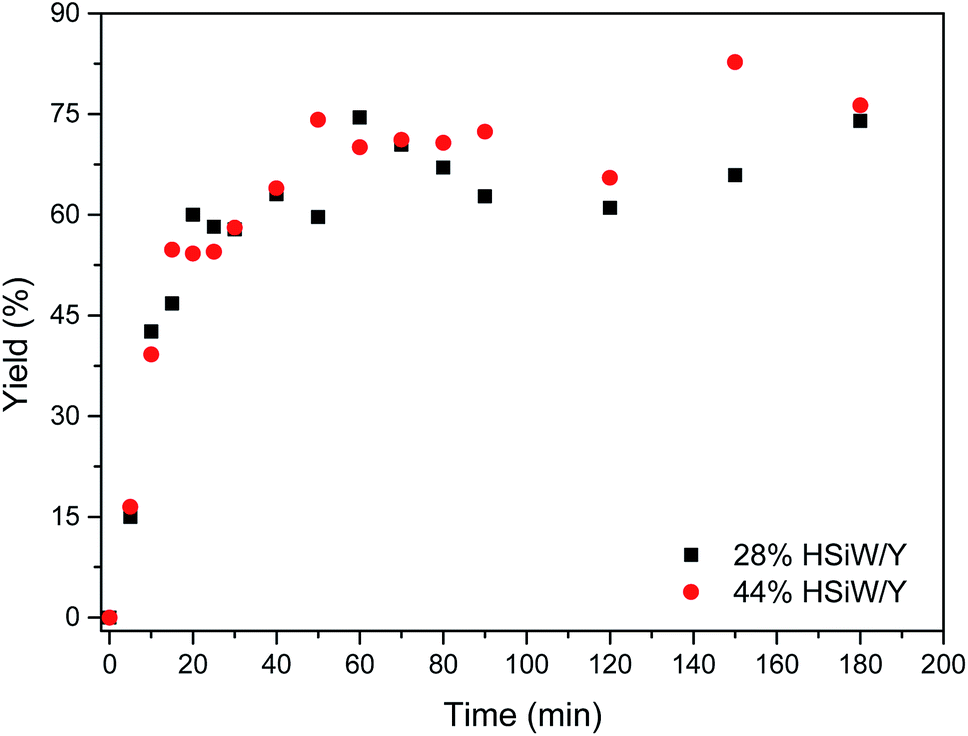 | ||
| Fig. 4 Kinetic profile for the catalytic model Biginelli reaction under the following conditions: temperature of 100 °C, 1 mmol of each reagent, 30 mg of catalyst and 0.5 mL of the solvent BMI·PF6. All yields refer to the isolated DHPM derivative. | ||
Further evaluation of the catalyst amount revealed that 50 mg of catalyst presented the best yields for both 28% HSiW/Y and 44% HSiW/Y (Fig. 5).
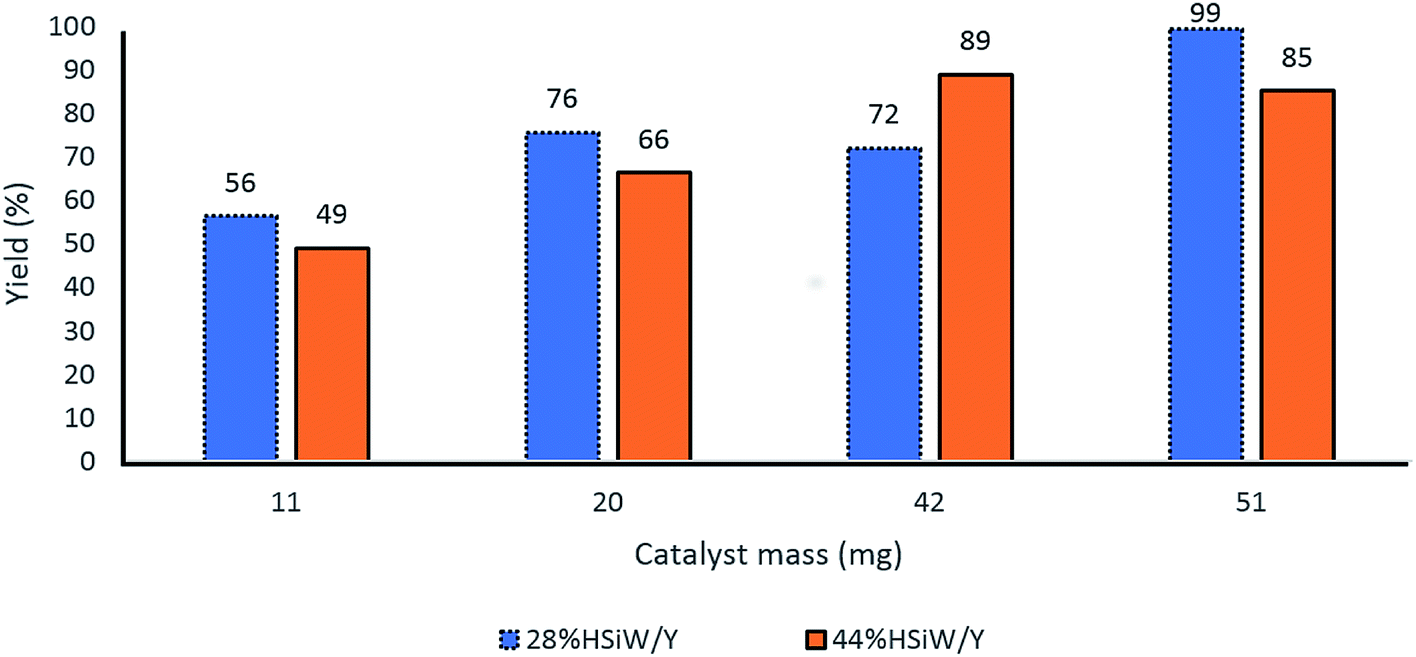 | ||
| Fig. 5 Effect of the catalyst amount on the yields of the model Biginelli reaction. Reaction time of 60 min, temperature of 100 °C, 1 mmol of each reagent, 0.5 mL of BMI·PF6. | ||
Thus, based on the optimization results for the Biginelli reaction, we noted that the best catalysts were 28% HSiW/Y and 44% HSiW/Y. Previous analyses59 of the acidities of these catalysts showed that 28% HSiW/Y had sites as strong as 123 kJ mol−1 in an amount of 0.06 mmol g−1 of catalyst, whereas 44% HSiW/Y had stronger sites (137 kJ mol−1) with 0.05 mmol g−1 of acid sites. At this point, for good performance of this reaction, acidic sites within 120 to 140 kJ mol−1 were necessary. Otherwise, the yield for these reactions decreased, which indicates the occurrence of possible parallel reactions that lead to the decomposition of the main product and reagents. These results are in accordance with the literature,52 where superacidic systems led to decomposition of the reagents and the adduct of the Biginelli reaction. Considering the nature of the acidic sites of the catalysts used herein, it is shown that the Lewis sites of the original zeolite Y were practically eliminated by the introduction of HSiW on its surface. Because HSiW is located mainly at the pore mouths of zeolite Y, the acid sites accessed by the substrates were those on the deposited nanocrystals. The Brønsted sites were therefore responsible for the catalytic Biginelli reaction.59 An in-depth analytical characterization of the best catalytic zeolite-supported heteropolyacid is shown in Fig. S1–S8 in the ESI.†
The best catalytic conditions could therefore be compared with other catalytic systems already described in the literature for the multicomponent Biginelli reaction, as shown in Table 1. It should be noted that the other procedures using zeolites or heteropolyacids (or both) require longer times and excess reagents and return lower yields in typically longer reaction times. Solvent has been proved to play a vital role to further the Biginelli reaction under catalyzed conditions;64 thus, solventless conditions are not the best option for this MCR. Some examples noted in Table 1 require toxic solvents, such as toluene or acetonitrile.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reagent proportions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3) :2:3) |
Solvent | Temp. (°C) | Time (h) | Catalyst | Yield (%) | Ref. |
| a 4-NO2PhCHO instead of PhCHO. | |||||||
| 1 | 1.00:1.00:1.00 |
Solventless | 100 | 1 | PTA@MIL-101 | 90 | 65 |
| 2 | 1.00:1.00:1.00 |
Toluene | 110 | 6 | H-MOR | 85 | 66 |
| 3 | 1.00:1.00:1.20 |
Methanol | 80 | 4 | MCM-41-HClO4 | 76 | 67 |
| 4 | 1.00:1.00:1.20 |
Ethanol | 80 | 8 | E4a | 90 | 68 |
| 5 | 1.00:1.00:1.20 |
CH3COOH | 100 | 5 | HTMA | 75 | 69 |
| 6 | 1.00:1.00:1.20 |
Solventless | 80 | 1 | H3PW12O40@SiO2 | 91 | 70 |
| 7 | 1.00:1.00:1.20 |
MeCN | 80 | 1 | PW@SiO2 | 95 | 71 |
| 8 | 1.00:1.00:1.50 |
Solventless | 80 | 1 | PMoBiV | 92 | 72 |
| 9a | 1.00:1.00:1.50 |
MeCN | 80 | 5 | 8 wt% NaY + 0.5 mM TPA | 80 | 73 |
| 10 | 1.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1.00:1.00 :1.00:1.00 |
BMI·PF6 | 100 | 1 | 28% HSiW/Y | 99 | This work |
The current methodology allowed the DHPM derivative to be obtained in almost quantitative yield using the ionic fluid BMI·PF6 as the reaction medium. Equimolar quantities of reagents were required, and no excess reagents were needed to obtain the DHPM in nearly quantitative yield. The reaction time of only 60 min is among the shortest times reported for the reaction. The catalytic system could also be recovered and reused at least three times. The following yields were obtained: 99%, 70% and 75%, respectively. The washing procedure using ethanol to purify the Biginelli adduct (see the Experimental section for details) is likely responsible for the catalyst mass loss throughout the purification; after the third cycle, only 40 mg of the solid catalyst was recovered, indicating 20% mass loss during recycling.
To gain insight into the generality of the methodology, the best system (28% HSiW/Y) was used to perform the synthesis of other DHPM derivatives. The results are summarized in Table 2.
|
|||
|---|---|---|---|
| DHPM | R1 | X | Yield (%) |
| a | Ph | O | 99 |
| b | 3-OH-Ph | S | 89 |
| c | 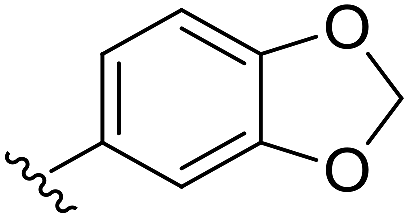 |
S | 83 |
| d | 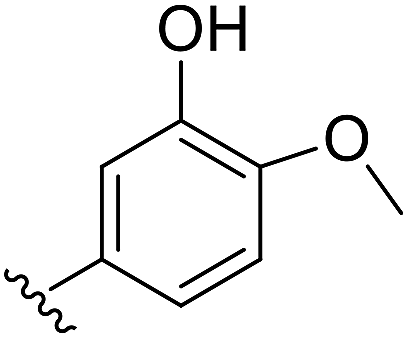 |
S | 90 |
| e | Ph | S | 96 |
| f | 2-OH-Ph | S | 89 |
| g | 3-OH-Ph | O | 91 |
| h | 4-Cl-Ph | O | 70 |
| i | 4-Br-Ph | S | 84 |
| j | 4-Br-Ph | O | 70 |
Good to excellent yields were generally obtained for the isolated DHPMs, indicating the good performance of the best catalytic system under the optimized reaction conditions. Compounds with known antitumoral activities74 (Table 2, entries b, c, d and g) could be directly obtained using the developed methodology.
The mechanism of the reaction was also investigated by electrospray (tandem) mass spectrometry75 (ESI-MS(/MS)). ESI has been successfully used to disclose several mechanistic aspects of a plethora of catalyzed and noncatalyzed reactions, as discussed in several reviews.76–83 The Brønsted acid-catalyzed Biginelli reaction mechanism was first disclosed by ESI monitoring by Eberlin and co-workers.84 Later, we demonstrated the Lewis acid-catalyzed Biginelli reaction mechanism53 and some variations of Brønsted acid-catalyzed reactions.50–52 Basically, three mechanisms may occur at the same time;56 that is, the iminium, Knoevenagel and enamine pathways (Scheme 3). The formation of the first key intermediate determines the reaction pathway, although the reactions may take place in a complex equilibrium.85 Typically, the iminium mechanism is favored, as shown in many available studies.64,86 Some catalytic conditions, however, favor the Knoevenagel mechanism87,88 or the enamine pathway.89,90 In some cases, more than one mechanism may be operating at the same time, even under catalyzed conditions.91 Due to the high complexity of the mechanism, its evaluation is an enormous challenge.
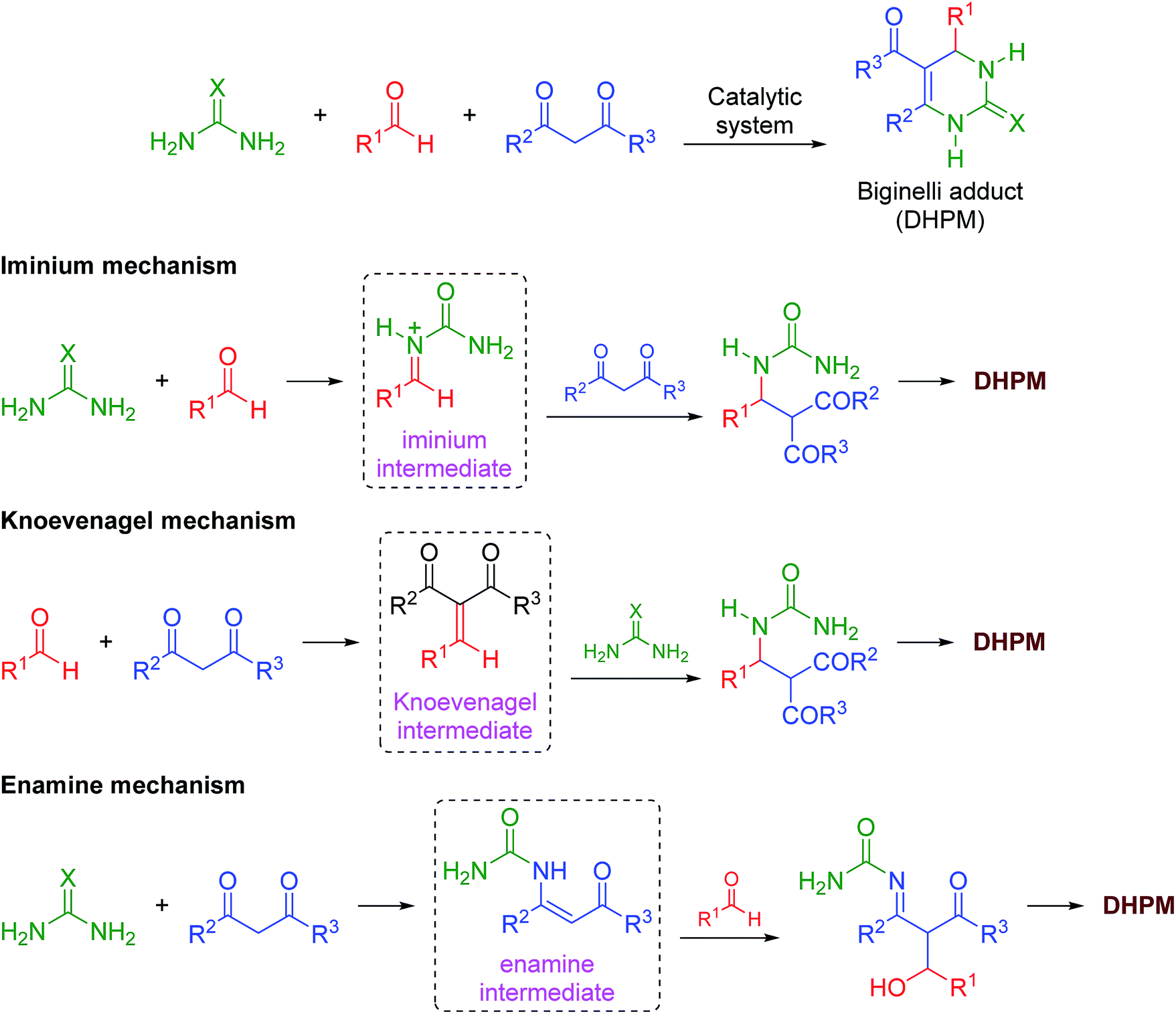 | ||
| Scheme 3 The catalyzed Biginelli reaction and the three simplified possible reaction pathways for DHPM formation. | ||
To evaluate the influence of the IL on the reaction, the analyses were monitored online during 5, 30, 60 and 90 min in the absence and in the presence of the ionic medium (Fig. 6). The ESI(+)-MS/MS characterization of all detected intermediates is presented in Fig. S9–S15 in the ESI.†
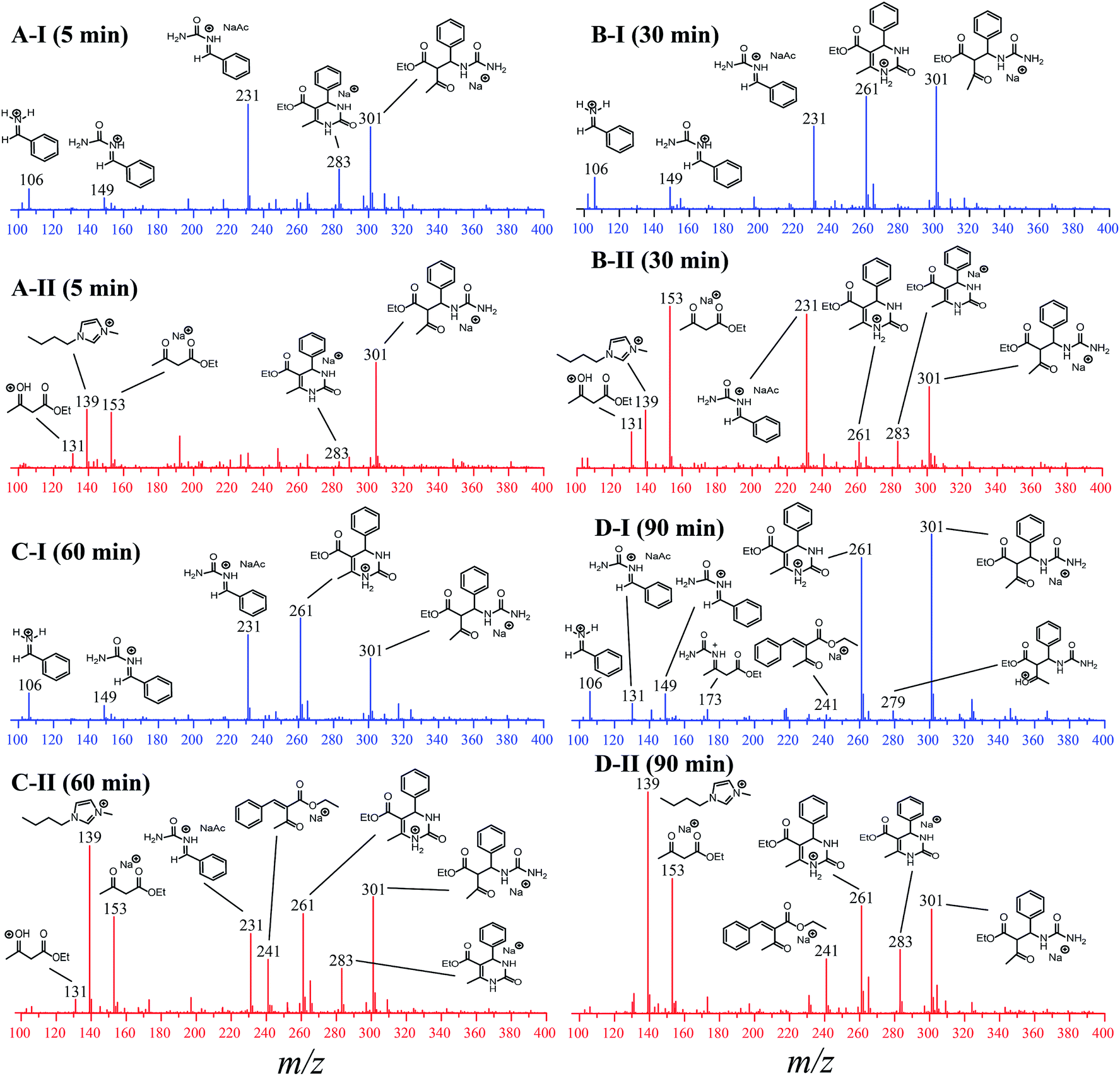 | ||
| Fig. 6 ESI-MS(+) monitoring of the Biginelli reaction in the absence (I, blue spectra) and presence (II, red spectra) of the ionic liquid BMI·PF6. (A) 5 min of reaction, (B) 30 min of reaction, (C) 60 min of reaction and (D) 90 min of reaction. Reaction conditions: catalyst (0.5 mol%), BMI·PF6 (0 or 0.5 μM), benzaldehyde (5.0 μM), ethyl acetoacetate (5.0 μM), urea (5.0 μM). The analyses were performed by dissolving the reaction mixture in methanol to afford the described concentrations. | ||
For the reaction monitored online in the absence of the IL (Fig. 6-I), the presence of a signal attributed to the iminium mechanism was noted (intermediate of m/z 149), although a Knoevenagel intermediate (m/z 241) and the enamine intermediate (m/z 173) could also be detected (Fig. 6D-I) and were further characterized by tandem MS/MS (Fig. S3 and S5†). This indicates that at least three mechanisms may be occurring at the same time, and it is not possible to identify any preference of the reaction pathway. It was also noted that the intermediate of m/z 301 (detected as a sodiated adduct) formed immediately after the addition of 1,3-dicarbonyl to the iminium ion (m/z 149); this in turn undergoes cyclization, affording the Biginelli adduct (m/z 261 or m/z 283 as the sodiated adduct). After 90 min of reaction (Fig. 6D-I) in the absence of the IL, the intermediates are noted at high intensity, and the base peak is an intermediate of m/z 301. In the initial period of the reaction (5 min, Fig. 6A-I), the base peak is the iminium intermediate (seen as the adduct of m/z 231).
For the reaction conducted in the presence of the IL BMI·PF6 (0.5 μM), a completely different reaction profile is depicted in Fig. 6-II. The Knoevenagel intermediate (m/z 241) appears to be the key intermediate during the whole transformation. In the beginning of the reaction (Fig. 6B-II), the iminium intermediate is also noted; however, the unsuccessful detection during the time course of the reaction clearly indicates that the iminium is reversibly formed and returns to the reagents (equilibrium), therefore proceeding preferentially through the Knoevenagel pathway (Fig. 6D-II). Another important feature is that the reaction is apparently faster in the presence of the ionic salt. After 5 min of reaction (Fig. 6A-II), the base peak is the last intermediate (m/z 301) prior to the Biginelli adduct formation (m/z 261 or 283), and at the end of the reaction (Fig. 6D-II), the imidazolium cation (m/z 139) is the base peak. To confirm this supposition, the monitoring was repeated using 5 μM of the IL concentration (Fig. 7); after 5 min, only the imidazolium and the Biginelli adduct were detected, and almost no intermediate could be noted except for the last intermediate (m/z 301) formed prior to the cyclization step, affording the expected DHPM (m/z 261). This result strongly indicates the positive effect of the IL during the catalyzed reaction.
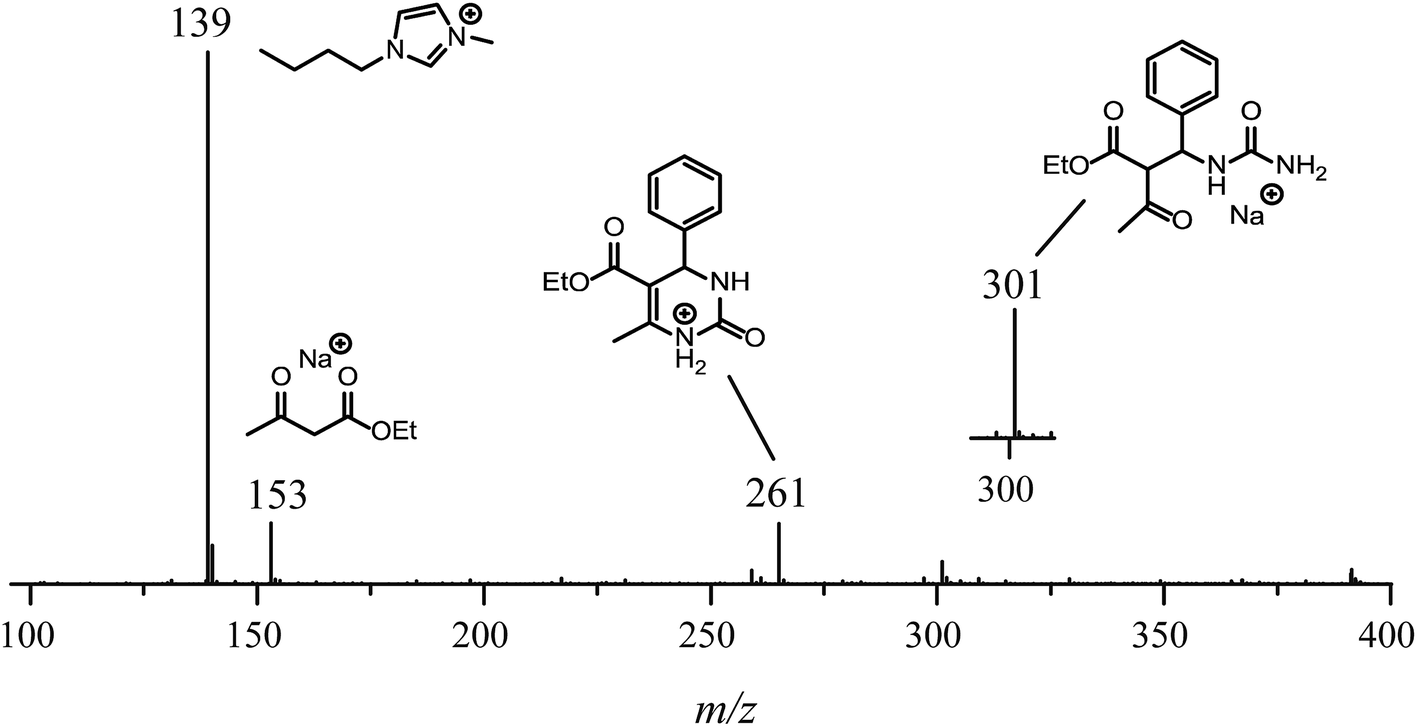 | ||
| Fig. 7 ESI-MS(+) monitoring of the Biginelli reaction in the presence of the ionic liquid BMI·PF6 after 5 min of reaction. Reaction conditions: catalyst (0.5 mol%), BMI·PF6 (5.0 μM), benzaldehyde (5.0 μM), ethyl acetoacetate (5.0 μM), and urea (5.0 μM). The analyses were performed by dissolving the reaction mixture in methanol to afford the described concentrations. The inset is a 12-fold expansion to show the intermediate of m/z 301 prior to the final cyclization step affording the Biginelli adduct. | ||
We also decided to monitor the reaction using 4-chloro benzaldehyde as the reagent (see Table 2, Entry h) because it returned the lowest yield among all the synthesized DHPMs. In principle, this experiment should allow us to verify both the mechanism and the possibility of intermediate (and/or byproduct) formation and should increase our comprehension of the transformation along with the catalytic behavior of the developed system, as shown in Fig. S16.†
The time monitoring of the reaction returned interesting results and showed no detectable enamine intermediate (m/z 173). Starting reagent signals (see Fig. S16†) were also noted during the reaction monitoring time; however, no characterizable intense byproduct signal was detected, indicating that no side reactions were taking place. These results helped to explain the lower observed yields during the synthesis of this DHPM derivative because the reagents were not fully consumed but no notable byproduct formed. The Knoevenagel intermediate (m/z 253 using the 4-chloro benzaldehyde instead of benzaldehyde), although noted in the spectra after 30 min of reaction monitoring, had a very low intensity, thus preventing its MS/MS characterization. However, the iminium intermediate (m/z 183) could be noted during the whole time course of the reaction, and its signal intensity could be monitored (Fig. 8).
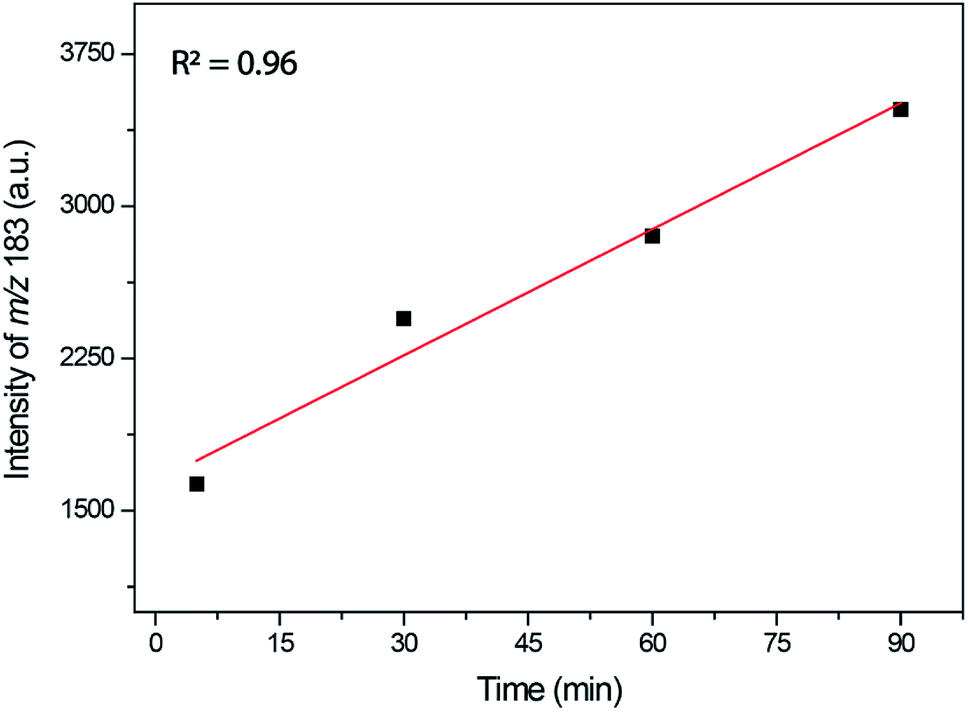 | ||
| Fig. 8 Monitoring of the iminium intermediate (m/z 183) by ESI(+)-MS during the reaction time. Reaction conditions: catalyst (0.5 mol%), BMI·PF6 (5.0 μM), 4-chloro benzaldehyde (5.0 μM) instead of benzaldehyde, ethyl acetoacetate (5.0 μM), and urea (5.0 μM). The analyses were performed by dissolving the reaction mixture in methanol to afford the described concentrations. | ||
As depicted in Fig. 8, the iminium intermediate concentration increased during the reaction course. This result has two important implications: (i) the yield is expected to be lower because the intermediate is increasing in concentration instead of being consumed, as expected during the course of a MCR. Therefore, this intermediate shows the behavior of a so-called dead-end intermediate.57,92 (ii) The results point once more to the Knoevenagel pathway because this intermediate was detected and readily consumed, as expected for the preferentially operating Biginelli reaction pathway.
Based on all the obtained results, a catalytic cycle can be proposed (Scheme 4) in the presence and in the absence of the imidazolium-based IL BMI·PF6. In the presence of BMI·PF6, the Knoevenagel pathway seems to be highly favored. The cationic intermediates are also prone to afford ion pairs and larger supramolecular aggregates when both the anion and the cation are present in the reaction. In the absence of the IL, the three reaction pathways are likely to be involved in the DHPM synthesis; this indicates that the positive IL effect not only improves the yield and shortens the reaction time, but also affects the selection of the reaction pathway. Although the iminium mechanism seems to be preferentially acting in the catalysis without ILs, the presence of intermediates from the other two possible mechanisms indicates that the catalytic system is not capable of selecting only one pathway and suppressing the possibilities of the other two; this is a key feature that is observed when the catalysis is performed in the ionic medium.
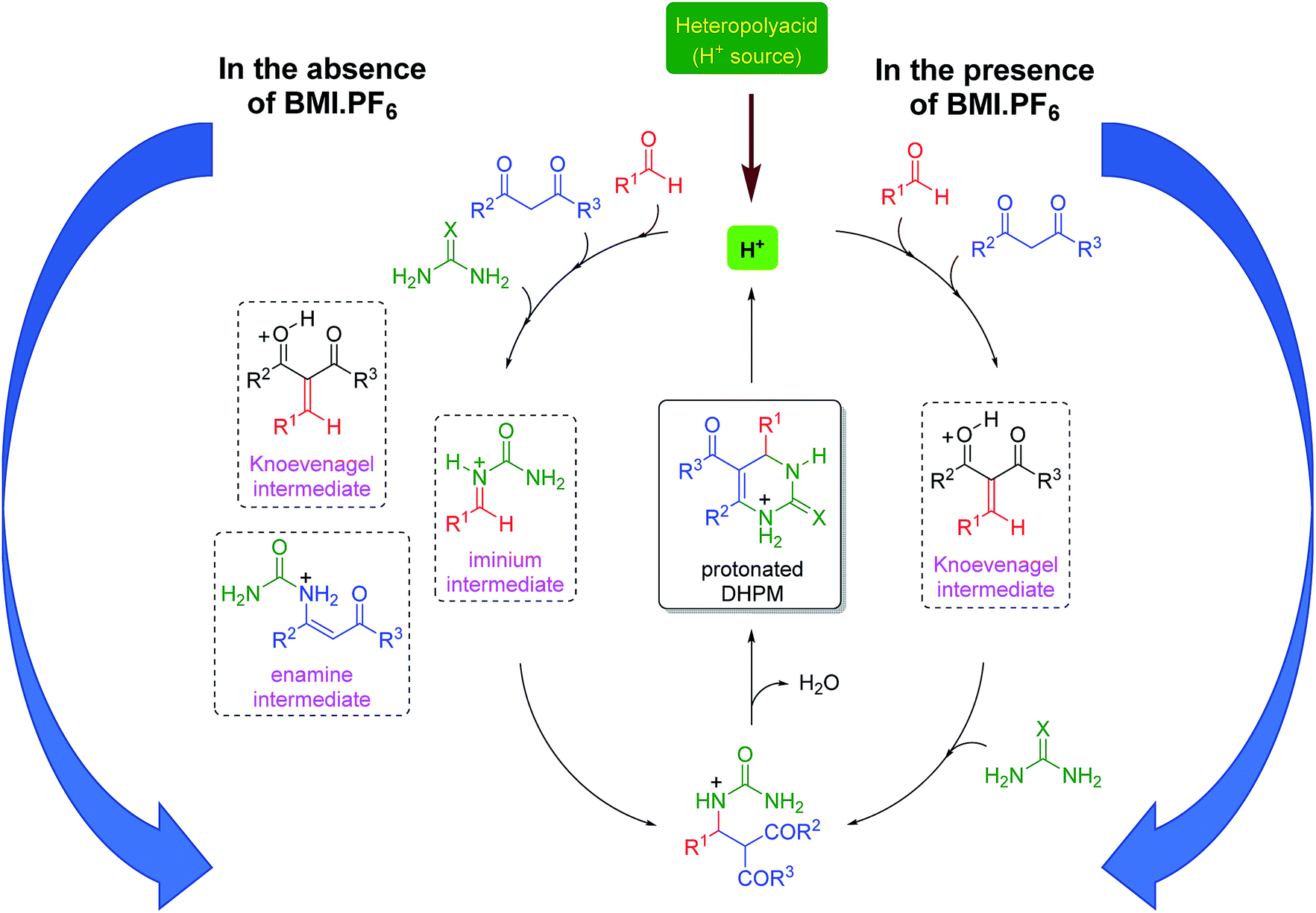 | ||
| Scheme 4 Catalytic cycle in the presence (right) and absence (left) of BMI·PF6. Note that in the presence of the ionic fluid, only one pathway is favored, whereas in its absence, three pathways are likely to be involved. | ||
In summary, a method has been developed to perform the Biginelli reaction in combination with HPAs and the limits of the acidic strength have been demonstrated. The use of zeolites as supports for HPA derivatives is an effective strategy for the development of new catalytic systems to be applied in several acid-catalyzed MCRs. The best catalytic system developed herein (28% HSiW/Y) could be applied under optimized conditions to the synthesis of several DHPMs, including some with known antitumoral activities.
The mechanism investigation by means of ESI-MS(/MS) indicated that the three possible mechanisms occur at the same time when the reaction is performed in the absence of the IL. In the presence of the IL, however, the reaction pathway can be tuned, and only one mechanism operates during the catalytic DHPM synthesis. For the first time, the mechanism pathway selection was successfully tuned simply by carrying out the reaction in an IL; this is in accordance with the positive IL effects noted for several previously described catalyzed reactions (see the cited reviews). The results described herein open an avenue of possibilities toward more efficient catalyzed MCRs and show the advantages of merging heteropolyacids and ILs not only for improving yields and shortening reaction times, but also for possibly improving the selectivity and tuning the reaction pathways during a chemical transformation.
Experimental
Preparation of the supported HPAs
NH4-Y zeolite from Zeolyst (CBV 300, SiO2/Al2O3 molar ratio = 5.1) was calcined at 550 °C for 8 h to obtain the protonic form (HY). HPA impregnation was performed in aqueous solution using 1 g of HY added to 10 mL of a 0.1 mol L−1 HCl solution containing the required amount of HPA (15, 30, and 45 wt% H3PW12O40, HPW or H4SiW12O40, HSiW, Aldrich, >99.9%). The slurry was maintained under stirring at 150 °C until solvent evaporation. Then, the catalysts were treated in a furnace at 300 °C for 4 h (denoted as X% HPA/Y).Characterization of the catalysts
Detailed conditions of the characterization have been published elsewhere.59 A general description is provided here for reference. The real amounts of the supported HPAs on the HY samples were obtained by elemental analysis using an EDX-XRF spectrometer from Shimadzu (model EDX 720). The quantity of HPA was determined by tungsten elemental analysis using analytical curves. XRD patterns were obtained with a Bruker powder diffractometer (model D8 Focus) using a copper tube (Kα = 1.5406 Å) in the 2θ region from 2 to 50°. FT-IR spectra were obtained on a Thermo Scientific (model Nicolet 6700) spectrometer. Samples were prepared by the KBr pellet method (1:100 – sample:KBr). The nature of the acid sites was determined by FT-IR spectroscopy of pyridine adsorbed on the catalysts. All acquisitions were obtained at 25 °C, with 256 scans and 4 cm−1 resolution. Magic angle spinning solid state nuclear magnetic resonance (MAS NMR) spectroscopy was performed on a Bruker Avance III HD, Ascend (600 MHz for 1H) at 14.1 T, using a 4 mm CP/MAS probe in zirconia rotors. 31P (243.1 MHz), 29Si (119.3 MHz) and 27Al (156.4 MHz) nuclei at a spin rate of 10 kHz were used to characterize the catalysts. The strengths (enthalpy) of the catalysts were measured by liquid phase incremental microcalorimetry of pyridine in cyclohexane slurries. For the calcined samples (300 °C) of HPA supported on zeolite Y, a diluted pyridine solution in cyclohexane was added to the solid slurry in anhydrous cyclohexane, and the amount of evolved heat was determined.
Catalytic reactions
The reactants benzaldehyde and ethyl acetoacetate were purified by distillation, and no treatment was performed on commercial urea. All catalysts were activated by heating treatment at 300 °C for 1 h before being used in the reaction. The standard experiment was run in a sealed Schlenk tube, in which 50 mg of catalyst was weighed and then 1 mmol of each reagent was added. 0.5 mL of BMI·PF6 was added to the system. The tube was then sealed and maintained under magnetic stirring for 60 min at 100 °C. The Biginelli adducts were purified by crystallization using hot ethanol. If no precipitation was noted, the crude mixture was purified by a chromatographic column using mixtures of hexane and ethyl acetate. All product characterizations are shown in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS.
- B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
- L. Levi and T. J. J. Mueller, Chem. Soc. Rev., 2016, 45, 2825–2846 RSC.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef CAS PubMed.
- N. Isambert, M. D. S. Duque, J. C. Plaquevent, Y. Genisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347–1357 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. M. Balbino, D. Peral, J. Carles Bayon and J. Dupont, ChemCatChem, 2015, 7, 972–977 CrossRef CAS.
- F. vanRantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- F. Pena-Pereira and J. Namiesnik, ChemSusChem, 2014, 7, 1784–1800 CrossRef CAS PubMed.
- H. Tateishi-Karimata and N. Sugimoto, Nucleic Acids Res., 2014, 42, 8831–8844 CrossRef CAS PubMed.
- O. Acevedo, J. Phys. Chem. A, 2014, 118, 11653–11666 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- K. R. Seddon, Nat. Mater., 2003, 2, 363–365 CrossRef CAS PubMed.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CAS.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- J. W. Lee, J. Y. Shin, Y. S. Chun, H. Bin Jang, C. E. Song and S.-G. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef CAS PubMed.
- H. K. Stassen, R. Ludwig, A. Wulf and J. Dupont, Chem.–Eur. J., 2015, 21, 8324–8335 CrossRef CAS PubMed.
- F. Fuenzalida, D. Aravena, C. Garcia, M. Garcia, R. Arce, G. Ramirez, C. Diaz, M. Isaacs, M. del Carmen Arevalo and M. J. Aguirre, Electrochim. Acta, 2017, 258, 959–969 CrossRef CAS.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS.
- Z. Long, L. C. Mao, M. Y. Liu, Q. Wan, Y. Q. Wan, X. Y. Zhang and Y. Wei, Polym. Chem., 2017, 8, 5644–5654 RSC.
- T. T. Hao, H. R. Liang, Y. H. Ou-Yang, C. Z. Yin, X. L. Zheng, M. L. Yuan, R. X. Li, H. Y. Fu and H. Chen, J. Org. Chem., 2018, 83, 4441–4454 CrossRef CAS PubMed.
- J. J. Yang, J. R. Li, P. F. Hao, F. D. Qiu, M. X. Liu, Q. Zhang and D. X. Shi, Dyes Pigm., 2015, 116, 97–105 CrossRef CAS.
- P. Slobbe, E. Ruijter and R. V. A. Orru, MedChemComm, 2012, 3, 1189–1218 RSC.
- C. Chiappe and S. Rajamani, Eur. J. Org. Chem., 2011, 5517–5539 CrossRef CAS.
- K. E. Johnson, R. M. Pagni and J. Bartmess, Monatsh. Chem., 2007, 138, 1077–1101 CrossRef CAS.
- L. M. Sanchez, H. J. Thomas and G. P. Romanelli, Mini-Rev. Org. Chem., 2015, 12, 115–126 CrossRef CAS.
- F. Su and Y. H. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- G. P. Romanelli and J. C. Autino, Mini-Rev. Org. Chem., 2009, 6, 359–366 CrossRef CAS.
- P. I. Villabrille, V. Palermo, A. G. Sathicq, P. G. Vazquez and G. P. Romanelli, Curr. Org. Chem., 2018, 22, 94–100 CrossRef CAS.
- S. Sadjadi, M. M. Heravi, V. Zadsirjan and V. Farzaneh, Appl. Surf. Sci., 2017, 426, 881–889 CrossRef CAS.
- V. Palermo, A. G. Sathicq, T. Constantieux, J. Rodriguez, P. G. Vazquez and G. P. Romanelli, Catal. Lett., 2016, 146, 1634–1647 CrossRef CAS.
- V. Palermo, A. Sathicq, T. Constantieux, J. Rodriguez, P. Vazquez and G. Romanelli, Catal. Lett., 2015, 145, 1022–1032 CrossRef CAS.
- S. M. Sadeghzadeh, RSC Adv., 2016, 6, 75973–75980 RSC.
- R. Tayebee, M. F. Abdizadeh, B. Maleki and E. Shahri, J. Mol. Liq., 2017, 241, 447–455 CrossRef CAS.
- M. M. Heravi, M. Mirzaei, S. Y. S. Beheshtiha, V. Zadsirjan, F. M. Ameli and M. Bazargan, Appl. Organomet. Chem., 2018, 32, e4479 CrossRef.
- J. K. Wang, Y. X. Zong, R. G. Fu, Y. Y. Niu, G. R. Yue, Z. J. Quan, X. C. Wang and Y. Pan, Ultrason. Sonochem., 2014, 21, 29–34 CrossRef CAS PubMed.
- F. S. De Oliveira, P. M. De Oliveira, L. M. Farias, R. C. Brinkerhoff, R. C. M. A. Sobrinho, T. M. Treptow, C. R. Montes D'Oca, M. A. G. Marinho, M. A. Hort, A. P. Horn, D. Russowsky and M. G. Montes D'Oca, MedChemComm, 2018, 9, 1282–1288 RSC.
- D. Russowsky, R. F. S. Canto, S. A. A. Sanches, M. G. M. D'Oca, A. de Fatima, R. A. Pilli, L. K. Kohn, M. A. Antonio and J. E. de Carvalho, Bioorg. Chem., 2006, 34, 173–182 CrossRef CAS PubMed.
- H. Y. K. Kaan, V. Ulaganathan, O. Rath, H. Prokopcova, D. Dallinger, C. O. Kappe and F. Kozielski, J. Med. Chem., 2010, 53, 5676–5683 CrossRef CAS PubMed.
- M. Oliverio, P. Costanzo, M. Nardi, I. Rivalta and A. Procopio, ACS Sustainable Chem. Eng., 2014, 2, 1228–1233 CrossRef CAS.
- D. Russowsky, F. A. Lopes, V. S. S. da Silva, K. F. S. Canto, M. G. M. D'Oca and M. N. Godoi, J. Braz. Chem. Soc., 2004, 15, 165–169 CrossRef CAS.
- M. N. Godoi, H. S. Costenaro, E. Kramer, P. S. Machado, M. G. Montes D'Oca and D. Russowsky, Quim. Nova, 2005, 28, 1010–1013 CrossRef CAS.
- L. Saher, M. Makhloufi-Chebli, L. Dermeche, B. Boutemeur-Khedis, C. Rabia, A. M. S. Silva and M. Hamdi, Tetrahedron Lett., 2016, 57, 1492–1496 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, P. Sridhar, J. S. S. Reddy, K. Nagaiah, N. Lingaiah and P. S. Saiprasad, Eur. J. Org. Chem., 2004, 552–557 CrossRef CAS.
- M. M. Heravi, F. Derikvand and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2005, 242, 173–175 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari and F. F. Bamoharram, Catal. Commun., 2006, 7, 373–376 CrossRef CAS.
- H. G. O. Alvim, T. B. Lima, A. L. de Oliveira, H. C. B. de Oliveira, F. M. Silva, F. C. Gozzo, R. Y. Souza, W. A. da Silva and B. A. D. Neto, J. Org. Chem., 2014, 79, 3383–3397 CrossRef CAS PubMed.
- L. M. Ramos, B. C. Guido, C. C. Nobrega, J. R. Corrêa, R. G. Silva, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo and B. A. D. Neto, Chem.–Eur. J., 2013, 19, 4156–4168 CrossRef CAS PubMed.
- H. G. O. Alvim, T. B. de Lima, H. C. B. de Oliveira, F. C. Gozzo, J. L. de Macedo, P. V. Abdelnur, W. A. Silva and B. A. D. Neto, ACS Catal., 2013, 3, 1420–1430 CrossRef CAS.
- L. M. Ramos, A. Tobio, M. R. dos Santos, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo, A. L. de Oliveira and B. A. D. Neto, J. Org. Chem., 2012, 77, 10184–10193 CrossRef CAS PubMed.
- G. C. O. Silva, J. R. Correa, M. O. Rodrigues, H. G. O. Alvim, B. C. Guido, C. C. Gatto, K. A. Wanderley, M. Fioramonte, F. C. Gozzo, R. de Souza and B. A. D. Neto, RSC Adv., 2015, 5, 48506–48515 RSC.
- G. A. Medeiros, W. A. da Silva, G. A. Bataglion, D. A. C. Ferreira, H. C. B. de Oliveira, M. N. Eberlin and B. A. D. Neto, Chem. Commun., 2014, 50, 338–340 RSC.
- H. G. O. Alvim, E. N. da Silva Junior and B. A. D. Neto, RSC Adv., 2014, 4, 54282–54299 RSC.
- R. Y. Souza, G. A. Bataglion, D. A. C. Ferreira, C. C. Gatto, M. N. Eberlin and B. A. D. Neto, RSC Adv., 2015, 5, 76337–76341 RSC.
- H. G. O. Alvim, G. A. Bataglion, L. M. Ramos, A. L. de Oliveira, H. C. B. de Oliveira, M. N. Eberlin, J. L. de Macedo, W. A. da Silva and B. A. D. Neto, Tetrahedron, 2014, 70, 3306–3313 CrossRef CAS.
- E. F. Freitas, M. F. Paiva, S. C. L. Dias and J. A. Dias, Catal. Today, 2017, 289, 70–77 CrossRef CAS.
- F. C. G. de Mattos, E. N. C. B. de Carvalho, E. F. de Freitas, M. F. Paiva, G. F. Ghesti, J. L. de Macedo, S. C. L. Dias and J. A. Dias, J. Braz. Chem. Soc., 2017, 28, 336–347 Search PubMed.
- P. R. S. Braga, A. A. Costa, E. F. de Freitas, R. O. Rocha, J. L. de Macedo, A. S. Araujo, J. A. Dias and S. C. L. Dias, J. Mol. Catal. A: Chem., 2012, 358, 99–105 CrossRef CAS.
- C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, J. L. de Macedo, J. A. Dias, S. C. L. Dias and K. S. P. Alvim, Appl. Catal., A, 2010, 372, 153–161 CrossRef CAS.
- T. S. Rodrigues, V. H. C. Silva, P. M. Lalli, H. C. B. de Oliveira, W. A. da Silva, F. Coelho, M. N. Eberlin and B. A. D. Neto, J. Org. Chem., 2014, 79, 5239–5248 CrossRef CAS.
- J. H. Clark, D. J. Macquarrie and J. Sherwood, Chem.–Eur. J., 2013, 19, 5174–5182 CrossRef CAS PubMed.
- M. Saikia, D. Bhuyan and L. Saikia, Appl. Catal., A, 2015, 505, 501–506 CrossRef CAS.
- S. R. Mistry, R. S. Joshi, S. K. Sahoo and K. C. Maheria, Catal. Lett., 2011, 141, 1541–1547 CrossRef CAS.
- K. Khan and Z. N. Siddiqui, Monatsh. Chem., 2015, 146, 2097–2105 CrossRef CAS.
- A. Hegedus, Z. Hell and I. Vigh, Synth. Commun., 2006, 36, 129–136 CrossRef.
- M. Tajbakhsh, B. Mohajerani, M. M. Heravi and A. N. Ahmadi, J. Mol. Catal. A: Chem., 2005, 236, 216–219 CrossRef CAS.
- M. M. Amini, A. Shaabani and A. Bazgir, Catal. Commun., 2006, 7, 843–847 CrossRef CAS.
- E. Rafiee and F. Shahbazi, J. Mol. Catal. A: Chem., 2006, 250, 57–61 CrossRef CAS.
- O. D'alessandro, A. G. Sathicq, V. Palermo, L. M. Sanchez, H. Thomas, P. Vázquez, T. Constantieux and G. Romanelli, Curr. Org. Chem., 2012, 16, 2763–2769 CrossRef.
- M. Moosavifar, C. R. Chim., 2012, 15, 444–447 CrossRef CAS.
- A. de Fatima, T. C. Braga, L. d. S. Neto, B. S. Terra, B. G. F. Oliveira, D. L. da Silva and L. V. Modolo, J. Adv. Res., 2015, 6, 363–373 CrossRef CAS PubMed.
- F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2011, 50, 5261–5263 CrossRef CAS PubMed.
- L. S. Santos, J. Braz. Chem. Soc., 2011, 22, 1827–1840 CrossRef CAS.
- R. Theron, Y. Wu, L. P. E. Yunker, A. V. Hesketh, I. Pernik, A. S. Weller and J. S. McIndoe, ACS Catal., 2016, 6, 6911–6917 CrossRef CAS.
- C. Iacobucci, S. Reale and F. De Angelis, Angew. Chem., Int. Ed., 2016, 55, 2980–2993 CrossRef CAS PubMed.
- J. Limberger, B. C. Leal, A. L. Monteiro and J. Dupont, Chem. Sci., 2015, 6, 77–94 RSC.
- K. L. Vikse, Z. Ahmadi and J. S. McIndoe, Coord. Chem. Rev., 2014, 279, 96–114 CrossRef CAS.
- A. Ray, T. Bristow, C. Whitmore and J. Mosely, Mass Spectrom. Rev., 2018, 37, 565–579 CrossRef CAS.
- L. P. E. Yunker, R. L. Stoddard and J. S. McIndoe, J. Mass Spectrom., 2014, 49, 1–8 CrossRef CAS PubMed.
- C.-C. Chen and P.-C. Lin, Anal. Methods, 2015, 7, 6947–6959 RSC.
- R. De Souza, E. T. da Penha, H. M. S. Milagre, S. J. Garden, P. M. Esteves, M. N. Eberlin and O. A. C. Antunes, Chem.–Eur. J., 2009, 15, 9799–9804 CrossRef CAS.
- B. A. D. Neto, H. G. O. Alvim and A. A. M. Lapis, in Noncovalent Interactions in Catalysis, The Royal Society of Chemistry, 2019, ch. 17, pp. 377–392 Search PubMed.
- C. O. Kappe, J. Org. Chem., 1997, 62, 7201–7204 CrossRef CAS PubMed.
- C. G. S. Lima, S. Silva, R. H. Goncalves, E. R. Leite, R. S. Schwab, A. G. Correa and M. W. Paixao, ChemCatChem, 2014, 6, 3455–3463 CrossRef CAS.
- K. Folkers and T. B. Johnson, J. Am. Chem. Soc., 1933, 55, 3784–3791 CrossRef CAS.
- I. Cepanec, M. Litvic, M. Filipan-Litvic and I. Grungold, Tetrahedron, 2007, 63, 11822–11827 CrossRef CAS.
- M. Litvic, I. Vecenaj, Z. M. Ladisic, M. Lovric, V. Vinkovic and M. Filipan-Litvic, Tetrahedron, 2010, 66, 3463–3471 CrossRef CAS.
- S. H. Zheng, Y. J. Jian, S. Xu, Y. Wu, H. M. Sun, G. F. Zhang, W. Q. Zhang and Z. W. Gao, RSC Adv., 2018, 8, 8657–8661 RSC.
- V. G. Santos, M. N. Godoi, T. Regiani, F. H. S. Gama, M. B. Coelho, R. O. M. A. de Souza, M. N. Eberlin and S. J. Garden, Chem.–Eur. J., 2014, 20, 12808–12816 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS/MS, NMR spectra and descriptions are provided. See DOI: 10.1039/c9ra03336j |
| This journal is © The Royal Society of Chemistry 2019 |