
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dynamic surface chemistry of colloidal metal chalcogenide quantum dots†
Roberto
Grisorio
 ab,
Danila
Quarta
ac,
Angela
Fiore
ac,
Luigi
Carbone
a,
Gian Paolo
Suranna
ab and
Carlo
Giansante
*a
ab,
Danila
Quarta
ac,
Angela
Fiore
ac,
Luigi
Carbone
a,
Gian Paolo
Suranna
ab and
Carlo
Giansante
*a
aCNR NANOTEC, Istituto di Nanotecnologia, via Monteroni, 73100 Lecce, Italy. E-mail: carlo.giansante@nanotec.cnr.it
bDICATECh – Dipartimento di Ingegneria Civile, Ambientale, del Territorio, Edile e di Chimica, Politecnico di Bari, via Orabona 4, 70125 Bari, Italy
cDipartimento di Matematica e Fisica ‘E. De Giorgi’, Università del Salento, via per Arnesano, 73100 Lecce, Italy
First published on 7th August 2019
Abstract
The chemical species (ligands) at the surface of colloidal inorganic semiconductor nanocrystals (QDs) mediate their interactions with the surroundings. The solvation of the QDs reflects a subtle interplay between ligand–solvent and ligand–ligand interactions, which eventually compete with the coordination of the ligands at the QD surface. The QD surface coordination and solvation are indeed fundamental to preserve their optoelectronic properties and to foster the effective application of QD-based inks and nanocomposites. Here we investigate such ligand interactions by exploiting diffusion ordered NMR spectroscopy (DOSY), which is suggested as an essential complement to spectral line width analysis. To this end, we use colloidal metal chalcogenide (CdS, CdSe, and PbS) QDs with (metal-)oleate ligands at their surface in several solvents exhibiting different viscosities and polarities. We demonstrate that the ligand shell is dynamically bound to the metal chalcogenide QDs, and is thus in equilibrium between the QD surface and the surrounding solvent. Such dynamic equilibria depend on ligand–solvent interactions, which are more prominent in aliphatic, rather polar solvents that favor the solvation of the ligands and, as a consequence, their displacement from the QD surface. In addition, the ligand–ligand interactions, which are more relevant for larger QDs, contribute to the stabilization of the ligand bonding at the QD surface.
Introduction
Surfaces – and interfaces – are intrinsically important at the nanoscale. Colloidal inorganic semiconductor nanocrystals (QDs) are remarkable examples of materials in which surfaces may significantly affect structures and properties.1–3 Amongst inorganic semiconductors, metal chalcogenides (ME) are, probably, the most investigated nanomaterials. As-synthesized colloidal ME QDs are commonly conceived as metal-rich inorganic cores coordinated by electron donor organic amphiphilic molecules (the X species in QDs with the general chemical formula [MmEn]X2(m−n)) or, alternatively, as stoichiometric inorganic cores coordinated by electron withdrawing metal complexes (MX2 in QDs with the chemical formula [ME]n(MX2)m−n).4–6 The X (anionic) species and MX2 (neutral) metal complexes are expected to coordinate {111} metal-rich and {100} stoichiometric facets, respectively, which are the relevant termination of ME QDs with cubic crystal structure. In any case, the ligand shell guarantees overall charge neutrality and balance of the QDs and confer them colloidal stability.7–10 With this work, we contribute to the description of the subtle balance between ligand–solvent, ligand–ligand, and ligand–core interactions in ME QDs (specifically, CdS, CdSe, and PbS) with an expected cubic crystal structure and bearing (metal-)oleate ligands at their surface. To this end, we used nuclear magnetic resonance spectroscopy to infer, from spectral line widths and diffusion coefficients determined by diffusion ordered spectroscopy measurements,11–13 the ligand interactions with the solvent, with other ligands, and with the QD surface. We found that ligands at the surface of ME QDs undergo dynamic equilibria with the surroundings; this finding is valid for all the inorganic semiconductors investigated here, albeit the lability of PbS QDs appears as more pronounced than that of CdE QDs. Aliphatic solvents effectively solvate the ligand shell, although rather polar solvents, such as chloroform and dichloromethane, markedly shift the equilibria towards dissociated ligands and cores. Aromatic solvents are, instead, excluded from the ligand shell. In addition, the larger the QDs, the tighter the ligand shell, thus yielding less labile nanostructures. These findings support a conception of colloidal QDs (which can be extended to metal chalcogenides) as dynamic chemical species, with surface structure and composition that depend on the surroundings.Experimental section
Materials
All chemicals were of the highest purity commercially available unless otherwise noted and were used as received; a complete list is reported in the ESI.†Material synthesis
Colloidal ME QDs were synthesized according to slightly modified14 well-established procedures15–17 in a three-neck flask connected to a standard Schlenk line setup under oxygen- and water-free conditions; detailed synthetic and purification procedures appear in the ESI.†Material characterization
1H Nuclear Magnetic Resonance (NMR) measurements were performed with a Bruker Avance spectrometer operating at 700 MHz equipped with a Cryo-Probe optimized for 1H observation; optical absorption was recorded with a Varian Cary 5000 UV-Vis-NIR spectrophotometer; Transmission Electron Microscopy (TEM) images were recorded with a Jeol Tem 1011 microscope operated at an accelerating voltage of 100 kV. Details are reported in the ESI.†Results and discussion
Identifying species at the QD surface
NMR spectroscopy represents a powerful tool to investigate the surface of colloidal QD dispersions.10,11,18–20 The proper, albeit inherently empirical, purification of as-synthesized QDs allows NMR spectra to be acquired in which the resonances of the nuclei constituting the organic species at the QD surface are clearly recognizable. In this work, we used colloidal ME QDs, namely CdS, CdSe, and PbS QDs. These colloidal QDs were synthesized upon hot-injection of E precursors in 1-octadecene solutions of M-oleate complexes (see ESI† for details of the synthetic procedures).15–17 Therefore, oleic acid was the only amphiphilic reagent used in the syntheses as it is capable of coordinating the QD surface (with the exception of PbS QDs with a diameter below 2 nm; see ESI†). This constraint in the synthetic procedures permits to reduce the complexity of the investigated systems. We remark that, despite controversies on the presence of hydroxyl anions at the PbE QD surface,9,12,21,22 only one oleate ligand per excess Pb atom is commonly found in PbE QDs,4,7,9,12 whereas Cd-(oleate)2 is expected to be at the surface of CdE QDs.5,20 Therefore, the X species in QDs with the chemical formula [ME]n(MX2)m−n generally refers to anionic ligands. To identify species at the QD surface, the 1H-NMR spectrum of PbS QDs was compared with the components of the ligand shell (i.e., the X species, which we represented by the oleic acid reagent, and the MX2 metal complexes, represented by the Pb-oleate precursor). The NMR spectra shown in Fig. 1 were recorded in toluene-d8 and CDCl3, probably the most common solvents used to disperse the QDs, which display similar viscosities (0.560 and 0.537 mPa s at 20 °C, respectively) but different polarities (hereafter associated with the relative permittivities, which are 2.38 and 4.81, respectively).23 First of all, we observe that the NMR spectra do not show features related to 1-octadecene, i.e. the solvent used in the QD syntheses – and in the preparation of the Pb-oleate precursor. As a general consideration, the spectral features of PbS QDs in toluene-d8 are downfield shifted (by ≈0.3 ppm) and broader (by almost an order of magnitude) than those of the mere ligands (Fig. 1a). The line broadening peculiar to QD ligands is commonly attributed to the reduced rotational degrees of freedom of surface bound species, increasing the spin–spin relaxation time constant (T2) that is inversely proportional to the spectral line width (Δν = 1/πT2). In addition, it is worth noting that the methylene protons close to the carbonyl group (namely, the α and β protons) shift remarkably downfield when bound either to the Pb cation or to the QD surface, which is mainly attributed to the deprotonation of carboxylic acids and the lack of hydrogen bonding.12,24,25 Narrow resonances that are slightly upfield shifted compared to the main peaks are absent in the NMR spectra of the QDs (this is particularly evident in the vinylene region in which signals are not overlapped with other resonances). This accounts for properly purified samples in which unbound ligands had been efficiently removed upon purification (which was pursued by repeated precipitation with a non-solvent – methanol – and redispersion in a good solvent – toluene – before transfer in the relevant deuterated solvent). In CDCl3, instead, the spectral features of the oleyl moiety bound at the QD surface are similar to those of the model oleic acid and Pb-oleate species, whereas they show narrower signals compared to toluene-d8 (Fig. 1b). Hence, the role of the solvent in the NMR features of PbS QDs is clearly highlighted in Fig. 1.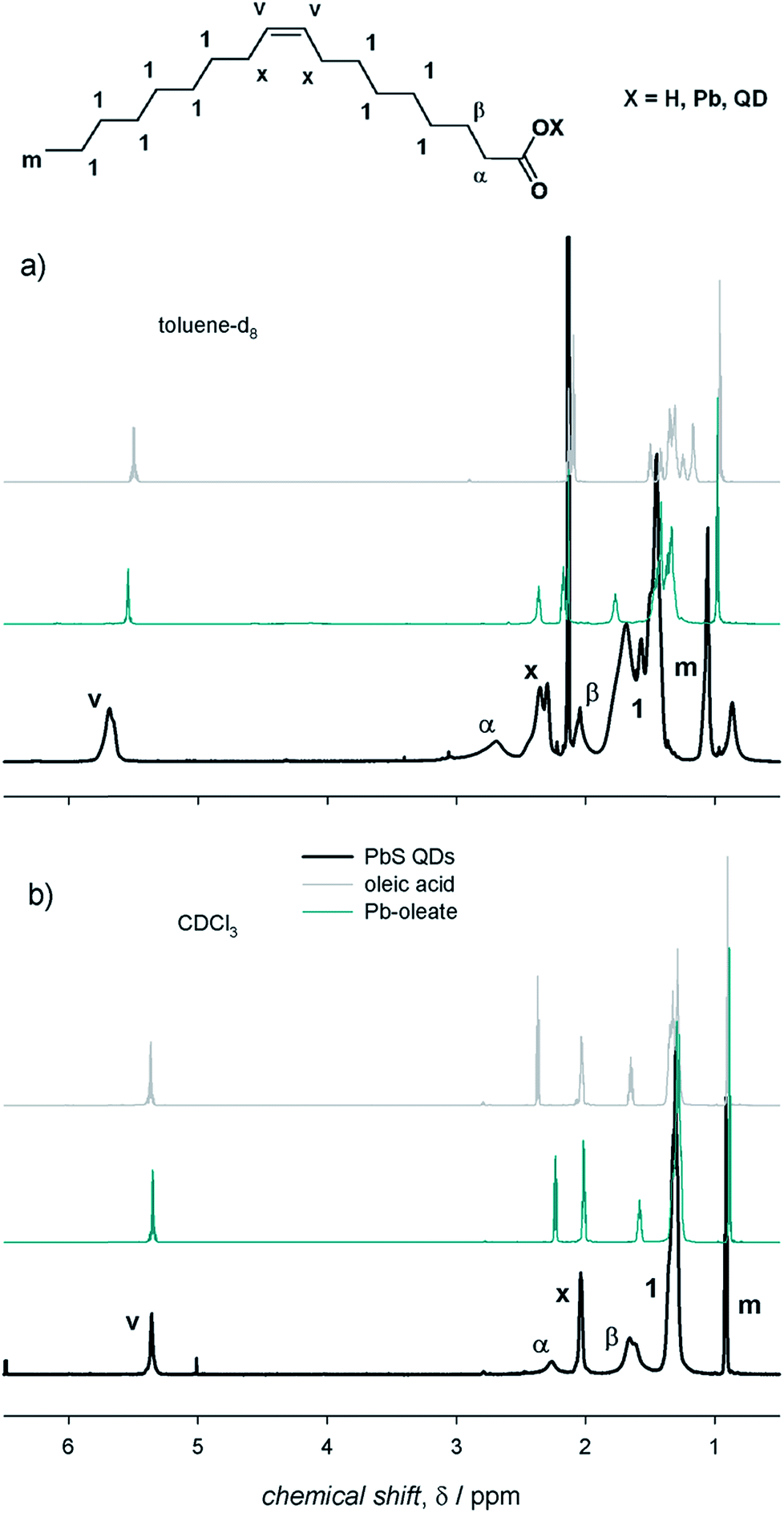 | ||
| Fig. 1 The 1H-NMR spectra of air-equilibrated 0.1 mM solutions of oleic acid, Pb-oleate, and PbS QDs in (a) toluene-d8 and (b) CDCl3. | ||
A comparison between colloidal metal chalcogenide QDs
We deemed it worthwhile to compare the behavior of different ME QDs. The 1H-NMR spectra of colloidal ME QDs having similar diameters (≈3 nm) are shown in Fig. 2 (Fig. S1–S3† show the absorption spectra and TEM micrographs of these QDs). Colloidal CdE QDs in toluene-d8 show appreciably broader resonances compared to PbS QDs (Δν ≈ 70–100 Hz for the vinylene protons of CdE QDs and Δν ≈ 50 Hz for PbS QDs). This is a common experimental observation,12,13 which may be tentatively related to the synthetic conditions: indeed, CdE QDs were synthesized at temperatures above 250 °C,15,16 whereas the syntheses of PbS QDs were conducted at 110 °C.17 Such high temperatures could have promoted the isomerization of the vinylene moiety leading to a mixture of oleic and elaidic acids (the cis and trans isomers, respectively, of 9-octadecenoic acid) that may broaden the corresponding peak (δ ≈ 5.5 ppm) and the nearby methylene protons (referred to as x protons in Fig. 1; δ ≈ 2.2 ppm).26 Another possible contribution to line broadening may also come from a heterogeneous distribution of bound ligands, which may be more pronounced in CdE QDs compared to PbS QDs. Nevertheless, homogeneous broadening, which is related to the solvation of the ligand shell, provides significant contribution to the observed spectral line widths. Indeed, the vinylene resonances, which account for the inner ligand shell, are broader by a factor of up to about 5 than the terminal methyl resonances, which account for the outer ligand shell (δ ≈ 1.0 ppm; Δν ≈ 30–40 Hz for CdE QDs and Δν ≈ 10 Hz for PbS QDs in toluene-d8). The homogeneous broadening is particularly exacerbated for α protons (δ ≈ 2.7 ppm), whose proximity with the QD surface usually leads to barely detectable resonances.27 However, α protons can be clearly observed in PbS QD spectra (Δν ≈ 100 Hz), whereas they are markedly broadened and downfield shifted in CdE QDs. In analogy with previous findings,12 much broader resonances are observed in toluene-d8 compared to CDCl3. In this regard, CdE QDs in CDCl3 are characterized by line widths (Δν ≈ 50 Hz for the vinylene protons) that are similar to those of PbS QDs in toluene-d8, as also previously reported.13 Markedly narrower resonances are, instead, commonly observed for PbE QDs in CDCl3 (Δν ≈ 10 Hz for the vinylene protons of small PbS QDs).22,28,29 Such an evident discrepancy highlights substantial differences between CdE and PbE QDs, which may be related to the inorganic core composition and to the ligand interactions with the QD surface that contribute heterogeneous broadening to the spectral line width. The reliability of measured line width values is corroborated by batch to batch reproducibility of the NMR spectra of PbS QDs in toluene-d8, as shown in Fig. S4.†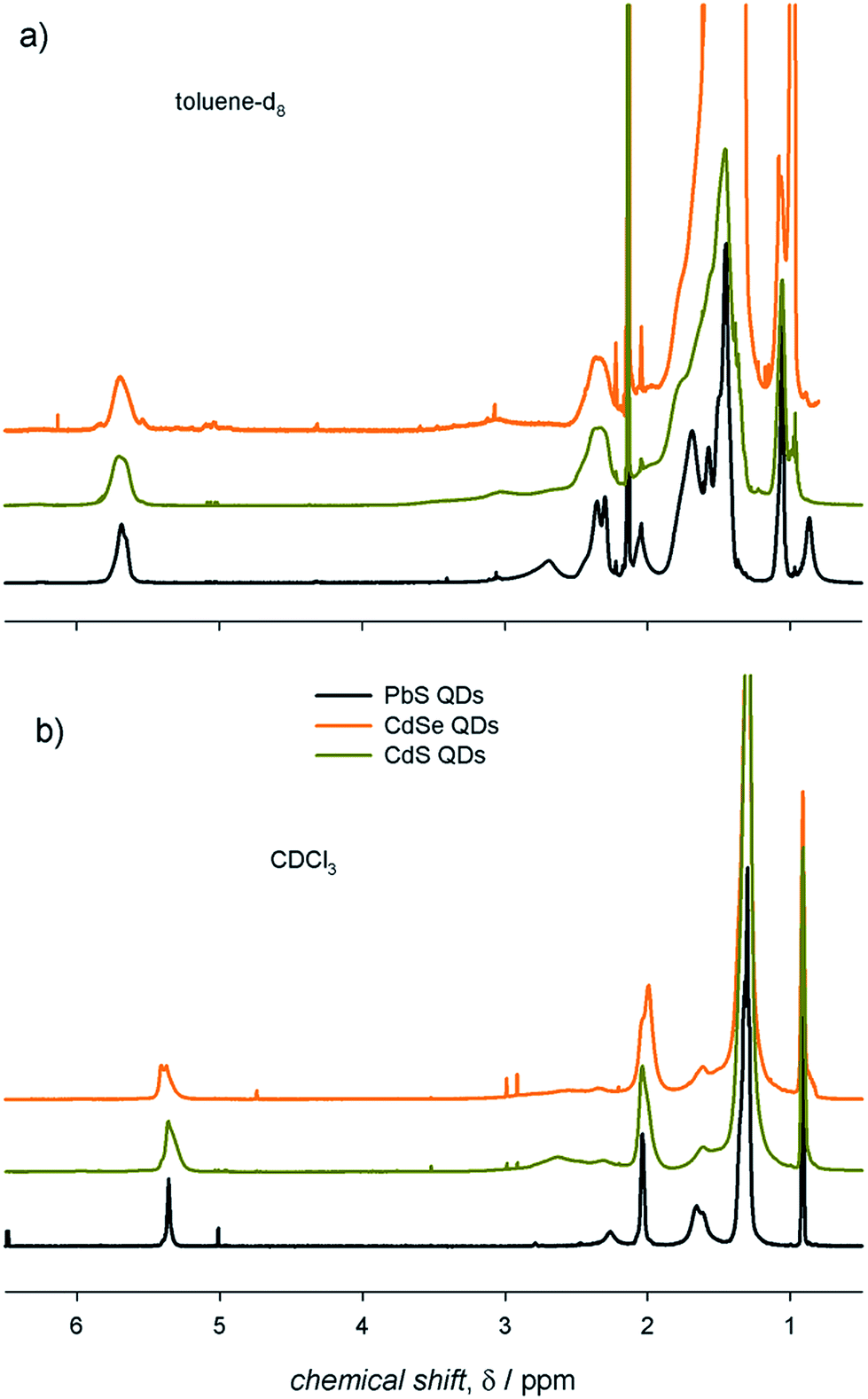 | ||
| Fig. 2 The 1H-NMR spectra of air-equilibrated 0.1 mM solutions of ME QDs with diameter ≈ 3 nm in (a) toluene-d8 and (b) CDCl3. | ||
Solvent dependence of NMR spectral features
Here, we suggest that the spectral line width indirectly accounts also for the ligand–ligand interactions, which occur simultaneously with the ligand–solvent interactions. Indeed, spectral line broadening, which is inversely proportional to the rotational degrees of freedom of the ligands themselves, may provide information on the overall ligand surroundings, i.e., both the solvent and adjacent ligands.5,10,12,13 With the aim of investigating such ligand interactions with the surroundings, we recorded 1H-NMR spectra of the colloidal ME QDs in several solvents displaying different viscosities and polarities. We used two apolar solvents such as hexane-d14 and toluene-d8 (ε/ε0 values are 1.89 and 2.38, respectively) with different viscosities (η values are 0.300 and 0.560 mPa s, respectively) and compared them with rather polar solvents such as dichloromethane-d2, chloroform-d, and dichlorobenzene-d4 (ε/ε0 values are 8.93, 4.81, and 10.1, respectively) with different viscosities (η values are 0.413, 0.537, and 1.32 mPa s, respectively).23 We note that data collected in dichloromethane-d2 should address eventual issues related to the possible acidity of chloroform-d. The corresponding 1H-NMR spectra are shown in Fig. 3. | ||
| Fig. 3 The (a, c, and e) 1H-NMR spectra and (b, d, and f) Stejskal–Tanner plots of air-equilibrated 0.1 mM solutions of (a and b) PbS, (c and d) CdSe, and (e and f) CdS QDs with diameter ≈ 3 nm in hexane-d14, toluene-d8, chloroform-d, dichloromethane-d2, and dichlorobenzene-d4 as indicated in the corresponding panels. | ||
With the expectation that an effective ligand solvation may lead to the eventual displacement of such ligands from the QD surface, we also used diffusion ordered NMR spectroscopy (DOSY) to probe the diffusion of the ligands in the aforementioned solvents.30 On this basis, such measurements provide insights on the ligand interactions with the QD surface, thus complementing information obtained by spectral line width analysis. The Stejskal–Tanner plots31 obtained from DOSY measurements for the colloidal ME QD dispersions in the five different solvents are also shown in Fig. 3. Such plots yielded the diffusion coefficients of the ligands in the colloidal QD dispersions, thus permitting to estimate, by means of the Stokes–Einstein equation, the QD solvodynamic diameters (dsolv).32 We note that the vinylene resonances (the inner ligand shell) and the terminal methyl resonances (the outer ligand shell), although showing drastically different line widths, diffuse with the same coefficients with the pulsed field gradient strength (decays of both vinylene and methyl intensities in toluene-d8 and chloroform-d are shown in Fig. S5†). At first glance, it is evident that both toluene-d8 and dichlorobenzene-d4, the only aromatic solvents employed in this work, yielded broader, more downfield shifted resonances and slower diffusion than the other solvents (Fig. 3). As opposed, the aliphatic solvents, namely dichloromethane-d2, chloroform-d, and hexane-d14, yielded rather narrow resonances. In addition, only the latter, apolar solvent, presented a monoexponential decay of the vinylene intensity with the pulsed field gradient strength. Both dichloromethane-d2 and chloroform-d instead showed biexponential decays (Fig. 3). However, the measured diffusion coefficients and, consequently, the apparent solvodynamic diameters show values (dsolv = 1.8 and 4.0 nm, respectively, for PbS QDs in dichloromethane-d2) smaller than those expected for tightly bound ligands (i.e., close to the sum of the QD core and the ligand shell; we indeed measured in toluene-d8, dsolv ≈ 5.1 nm). We suggest that such values are affected by ligand desorption dynamics, i.e. ligands at the QD surface undergo dynamic equilibrium with the surroundings. Such dynamic equilibrium may occur on time scales comparable with those of the DOSY experiments (100 ms), thus resulting in ligand desorption kinetics faster than 10 s−1. The 1H-NMR spectra and the Stejskal–Tanner plots for large PbS QDs (i.e., with diameter of ≈5 nm, thus larger than the QDs shown in Fig. 3) in different solvents are shown in Fig. S6.† These lines of experimental evidence qualitatively support the general character of the solvent dependence of both line widths and diffusion coefficients discussed above. Moreover, DOSY measurements performed on these large PbS QDs show that a fast diffusing component observed in dichloromethane-d2 at 22 °C is, instead, not observed at low temperature (i.e., at 0 °C), yielding a single diffusing species with a coefficient that approaches the value measured in toluene-d8 at 22 °C (Fig. S7†). The monoexponential decay observed in dichloromethane-d2 at 0 °C corroborates the notion of dynamic equilibria involving ligands at the QD surface. We speculate that the labile surface species are mainly the M-oleate complexes, which coordinate the stoichiometric facets of the colloidal ME QDs. Indeed, oleate displacement from metal-rich facets would require protonation of such ligands leading to charge neutral species; in addition, previous experimental and theoretical studies concur in attributing a lower thermodynamic stability to M-oleate ligands,12,20,33,34 which is related to the ratio of adsorption/desorption kinetics. Given the very similar diffusion observed for all ME QDs in toluene-d8, the ligand dynamic equilibria appear to be more prominent for PbS QDs compared to CdE QDs, as evinced from the Stejskal–Tanner plots of ME QDs in dichloromethane-d2 at 22 °C (Fig. S8†).
Therefore, we suggest that NMR measurements that combine both spectral line widths and diffusion coefficients, performed on colloidal ME QD solutions in different solvents, can be exploited to infer the ligand interactions with the surroundings: i.e., the ligand interactions with (i) the solvent and the concomitant ligand interactions with (ii) adjacent ligands and with (iii) the QD surface.
The information contained in the 1H-NMR spectra and the Stejskal–Tanner plots shown in Fig. 3 can be resumed in the plots displayed in Fig. 4, in which the vinylene peak line width and the apparent ligand shell thickness (determined from the solvodynamic diameter of the QDs) are reported as a function of the solvent viscosities and relative permittivities. We remark that the apparent ligand shell thickness values determined in CDCl3 and CD2Cl2 were obtained by considering the ME QD solvodynamic diameter as the weighted mean of the two values (based on the pre-exponential factors) found in the biexponential Stejskal–Tanner plots (which are reported, along with the other values, in Table S1†).
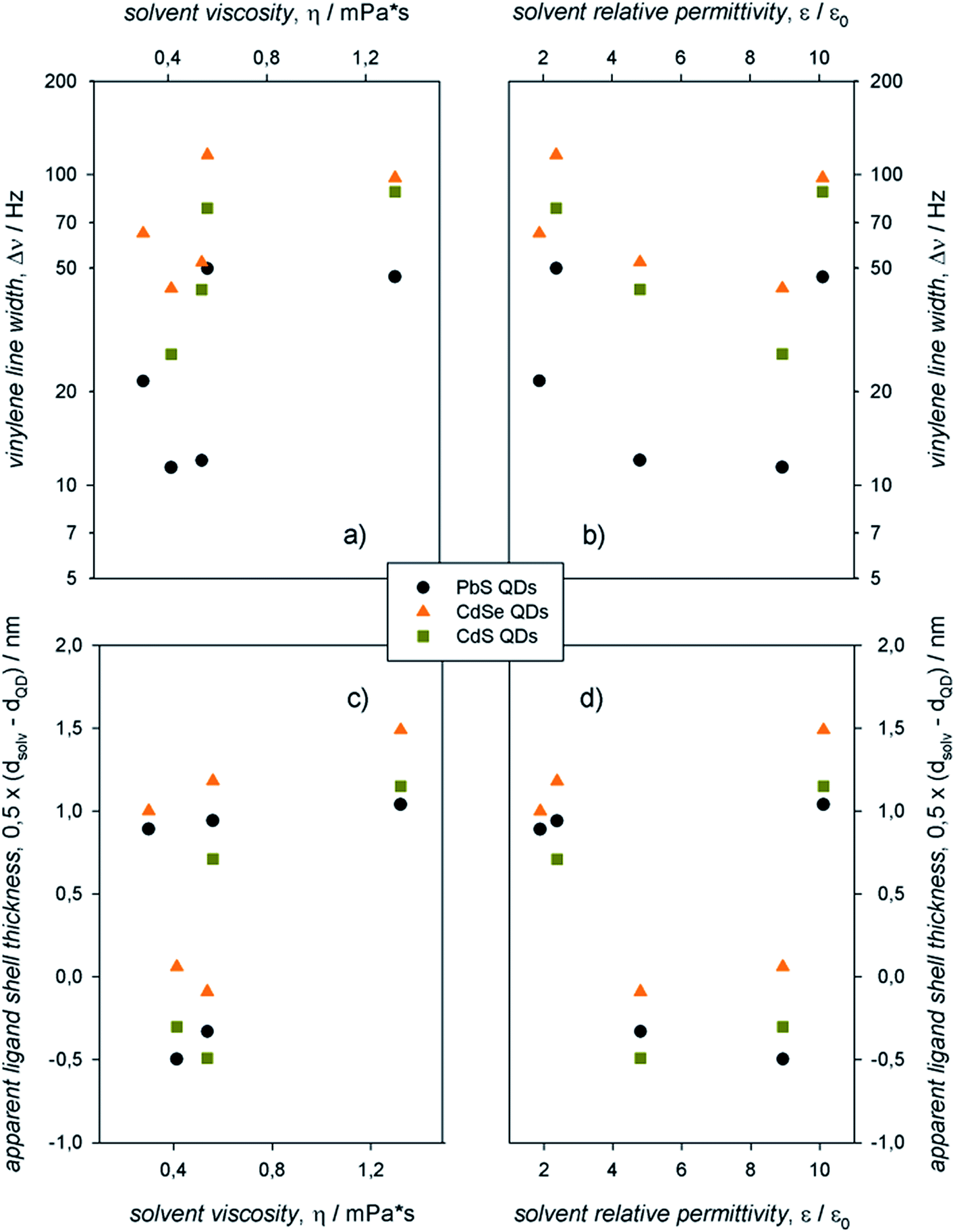 | ||
| Fig. 4 Plots of the (a and b) vinylene linewidth and of the (c and d) apparent ligand shell thickness as a function of the solvent (a and c) viscosity and (b and d) dielectric constant for air-equilibrated 0.1 mM solutions of CdS, CdSe, and PbS QDs with diameter ≈ 3 nm. | ||
In the same solvent, colloidal PbS QDs showed markedly narrower resonances compared to CdE QDs. For QDs with the same core composition, aliphatic solvents (C6D14, CDCl3, and CD2Cl2) yielded narrower resonances compared to the aromatic solvents (C6D5CD3 and C6D4Cl2). Data on the apparent ligand shell thickness, instead, provide information that cannot be gathered from line width measurements.13 In this case, no remarkable dependence on QD composition was observed. Indeed, negligible values of the apparent ligand shell thickness were obtained for all the colloidal ME QDs in aliphatic, rather polar solvents (such as CDCl3 and CD2Cl2). In addition, the apparent ligand shell thickness for ME QDs in either aromatic or apolar solvents (C6D14, C6D5CD3, and C6D4Cl2) showed similar values, which are rather comparable with, or slightly lower than, the length of the ligand oleyl moiety.
Size dependence of NMR spectral features
To assess an eventual dependence on QD diameter for the ligand interactions with the surroundings, we measured the 1H-NMR spectra of PbS QDs of different sizes ranging between ≈2 and 7 nm (shown in Fig. 5). For any given 1H-NMR spectrum, the inner vinylene resonances were broader than the terminal methyl resonances, thus reflecting the penetration in the ligand shell by solvent molecules. As discussed above, much broader resonances were observed in toluene-d8 compared to CDCl3 for QDs of the same batches. In addition, it can be clearly observed that the line broadening increased with the QD diameter, which was also accompanied by a downfield shift that was particularly evident in toluene-d8.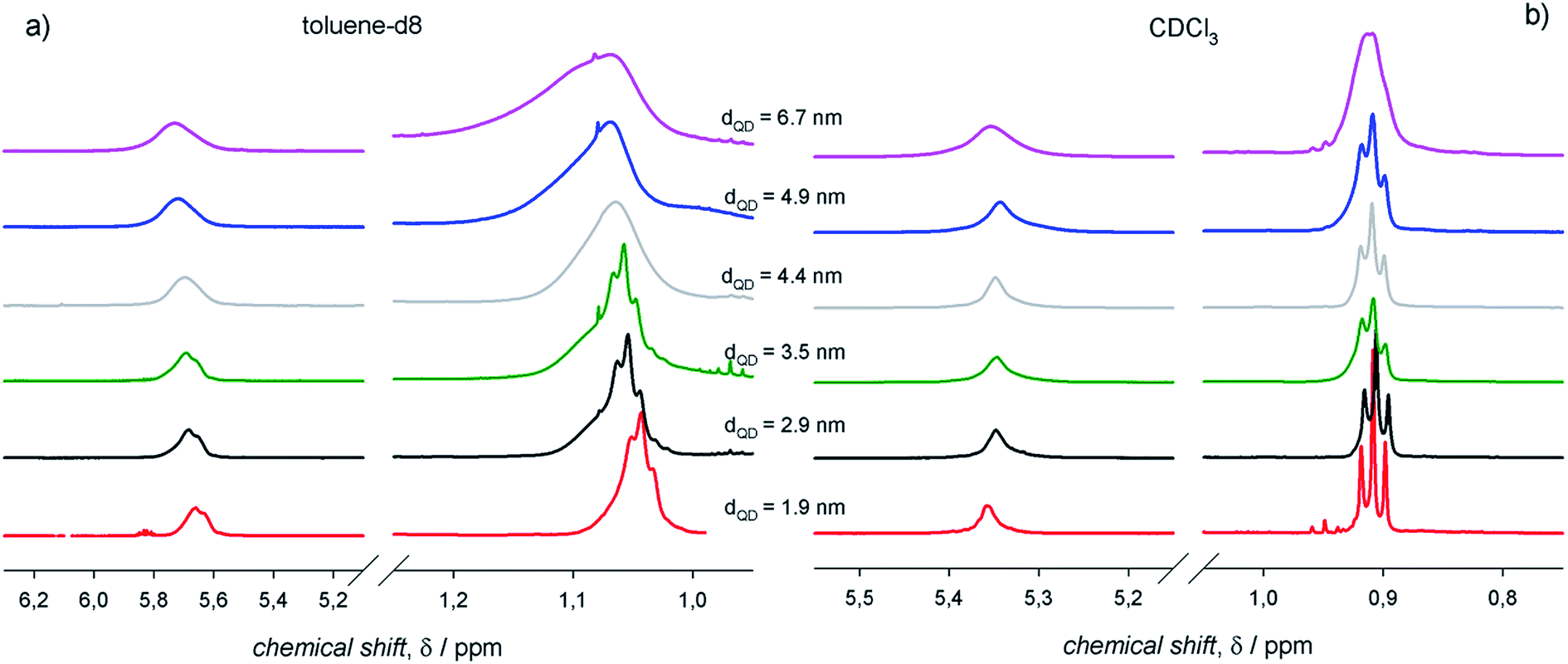 | ||
| Fig. 5 The 1H-NMR spectra of air-equilibrated 0.1 mM solutions of PbS QDs with different diameters in (a) toluene-d8 and (b) CDCl3. | ||
The information gathered from line width and diffusion coefficient measurements on PbS QDs of different diameters can be resumed in the plots shown in Fig. 6. In agreement with previous studies determining that the heterogeneous line broadening, which is related to the intrinsic heterogeneity of (ligands at) the QD surface, can be considered almost size independent,13 the observed increase of the total line width with the QD size (Fig. 6a) can be ascribed to reduced ligand–solvent interactions in large QDs. This suggests that larger QDs bear a ligand shell that is more compact than that of smaller QDs, consequently hindering the penetration of solvent molecules. A tight ligand shell may imply significant ligand–ligand interactions. In analogy with the findings on the line width, also the apparent ligand shell thickness displayed an appreciable increase with the QD size (Fig. 6b). Such an increase of the apparent ligand shell thickness with the QD size was observed in different solvents (Fig. 6b; again, the apparent ligand shell thickness values measured in CDCl3 and CD2Cl2 were obtained by considering the ME QD solvodynamic diameter as the weighted mean of the two values found by biexponential fitting; corresponding values are listed in Table S2†). These lines of experimental evidence further support the attribution of a tight ligand shell to large QDs in either apolar or aromatic solvents. Such an attribution is based on the evidence that larger QDs show larger facets bearing more ligands per facet compared to smaller QDs, with a proportionally less relevant contribution from edges and vertexes. This may lead to a consequent increase of the number of adjacent ligands fostering ligand–ligand interactions at the expense of the ligand–solvent interactions. This argument holds regardless of the controversial size dependent ligand coverage esteems: we previously reported on a ligand coverage that increases with QD diameter,12 whereas the opposite dependence was also reported.13 Nevertheless, a tight ligand shell results in more thermodynamically stable and, most likely, less kinetically labile coordination of ligands at the QD surface. In addition, it may result that the reduced ligand–solvent interactions may favor the ligand–core interactions. This is supported by the slight size dependent increase of both the line width and the apparent ligand shell thickness in aliphatic, polar solvents, such as chloroform-d.
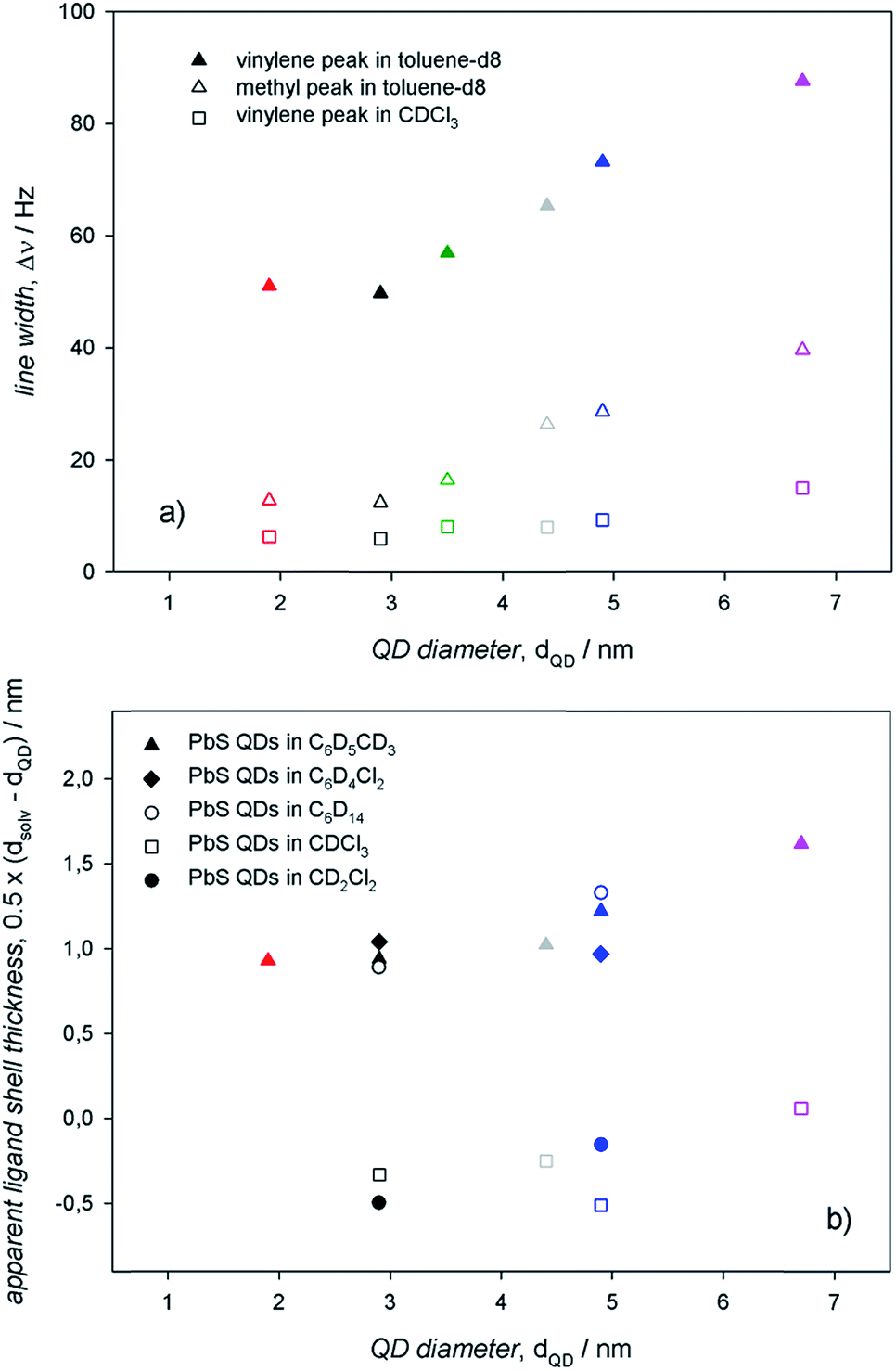 | ||
| Fig. 6 (a) Plots of the vinylene and methyl linewidths of air-equilibrated 0.1 mM solutions of PbS QDs in toluene-d8 as a function of the QD diameter. (b) Plots of the apparent ligand shell thickness of air-equilibrated 0.1 mM solutions of PbS QDs in different solvents (indicated in the legend) as a function of the QD diameter. | ||
Conclusion
Here we have demonstrated that the (metal-)organic chemical species at the surface of colloidal metal chalcogenide QDs undergo dynamic equilibrium with the surroundings. This observation relies on the analysis of 1H-NMR spectral line widths and diffusion coefficients of the oleyl moieties that constitute the ligand shell of CdS, CdSe, and PbS QDs. Such an analysis permits to infer the interactions of the ligands with the surrounding species, i.e., the solvent, the adjacent ligands at the QD surface, and the inorganic core to which the ligands are bound. We suggest the use of DOSY as a powerful tool to complement the information gathered from spectral line width analysis. Indeed, the line width can barely be related to ligands that are highly dynamically bound at the QD surface, whereas the measurement of the apparent diffusion coefficients may uncover the presence of chemical species in fast exchange between the QD surface and the solvent. Our study thus fosters the notion of colloidal QDs as nanostructures whose components, the ligands and the core, are not statically bound, but in dynamic equilibrium with the surroundings.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
C. G. thanks Progetto di ricerca MIUR PON 2014–2020, Energia per l'Ambiente TARANTO (Project Number: ARS01_00637).References
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- C. M. Fang, M. A. van Huis, D. Vanmaekelbergh and H. W. Zandbergen, ACS Nano, 2010, 4, 211–218 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS.
- I. Moreels, K. Lambert, D. De Muynck, F. Vanhaecke, D. Poelman, J. C. Martins, G. Allan and Z. Hens, Chem. Mater., 2007, 19, 6101–6106 CrossRef CAS.
- N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 18536–18548 CrossRef CAS PubMed.
- C. Giansante and I. Infante, J. Phys. Chem. Lett., 2017, 8, 5209–5215 CrossRef CAS PubMed.
- I. Moreels, B. Fritzinger, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2008, 130, 15081–15086 CrossRef CAS PubMed.
- B. Fritzinger, R. K. Capek, K. Lambert, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2010, 132, 10195–10201 CrossRef CAS PubMed.
- D. Zherebetskyy, M. Scheele, Y. Zhang, N. Bronstein, C. Thompson, D. Britt, M. Salmeron, P. Alivisatos and L.-W. Wang, Science, 2014, 344, 1380–1384 CrossRef CAS PubMed.
- K. De Nolf, S. M. Cosseddu, J. J. Jasieniak, E. Drijvers, J. C. Martins, I. Infante and Z. Hens, J. Am. Chem. Soc., 2017, 139, 3456–3464 CrossRef CAS PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- R. Grisorio, D. Debellis, G. P. Suranna, G. Gigli and C. Giansante, Angew. Chem., Int. Ed., 2016, 55, 6628–6633 CrossRef CAS PubMed.
- J. De Roo, N. Yazdani, E. Drijvers, A. Lauria, J. Maes, J. S. Owen, I. Van Driessche, M. Niederberger, V. Wood, J. C. Martins, I. Infante and Z. Hens, Chem. Mater., 2018, 30, 5485–5492 CrossRef CAS.
- C. Giansante, Nanoscale, 2019, 11, 9478–9487 RSC.
- W. W. Yu and X. G. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS PubMed.
- H. Shen, H. Wang, Z. Tang, J. Z. Niu, S. Lou, Z. Du and L. S. Li, CrystEngComm, 2009, 11, 1733–1738 RSC.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS PubMed.
- A. J. Morris-Cohen, M. Malicki, M. D. Peterson, J. W. J. Slavin and E. A. Weiss, Chem. Mater., 2013, 25, 1155–1165 CrossRef CAS.
- E. Drijvers, J. De Roo, J. C. Martins, I. Infante and Z. Hens, Chem. Mater., 2018, 30, 1178–1186 CrossRef CAS.
- H. Beygi, S. A. Sajjadi, A. Babakhani, J. F. Young and F. van Veggel, Appl. Surf. Sci., 2018, 457, 1–10 CrossRef CAS.
- J. L. Peters, J. de Wit and D. Vanmaekelbergh, Chem. Mater., 2019, 31, 1672–1680 CrossRef CAS PubMed.
- R. C. Weast, M. J. Astle, W. H. Beyer and C. Chemical Rubber, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, CRC Press, Boca Raton, Fla, 1984 Search PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- R. Grisorio, M. E. Di Clemente, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, M. Irimia-Vladu and G. P. Suranna, Nanoscale, 2019, 11, 986–999 RSC.
- H. Najbjerg, J. F. Young and H. C. Bertram, J. Agric. Food Chem., 2011, 59, 8994–9000 CrossRef CAS PubMed.
- J. De Roo, S. Coucke, H. Rijckaert, K. De Keukeleere, D. Sinnaeve, Z. Hens, J. C. Martins and I. Van Driessche, Langmuir, 2016, 32, 1962–1970 CrossRef CAS PubMed.
- C. Giansante, I. Infante, E. Fabiano, R. Grisorio, G. P. Suranna and G. Gigli, J. Am. Chem. Soc., 2015, 137, 1875–1886 CrossRef CAS PubMed.
- D. M. Kroupa, N. C. Anderson, C. V. Castaneda, A. J. Nozik and M. C. Beard, Chem. Commun., 2016, 52, 13893–13896 RSC.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- R. R. Knauf, J. C. Lennox and J. L. Dempsey, Chem. Mater., 2016, 28, 4762–4770 CrossRef CAS.
- M. Saniepay, C. Mi, Z. Liu, E. P. Abel and R. Beaulac, J. Am. Chem. Soc., 2018, 140, 1725–1736 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis of colloidal ME QDs; spectroscopic data; TEM images. See DOI: 10.1039/c9na00452a |
| This journal is © The Royal Society of Chemistry 2019 |