
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible disassembly of metallasupramolecular structures mediated by a metastable-state photoacid†
Suzanne M.
Jansze
 ,
Giacomo
Cecot
and
Kay
Severin
*
,
Giacomo
Cecot
and
Kay
Severin
*
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch
First published on 20th April 2018
Abstract
The addition of a metastable-state photoacid to solutions containing metal–ligand assemblies renders the systems light responsive. Upon irradiation, proton transfer from the photoacid to the ligand is observed, resulting in disassembly of the metallasupramolecular structure. In the dark, the process is fully reversed. Light-induced switching was demonstrated for six different metal–ligand assemblies containing PdII, PtII or RuII complexes and bridging polypyridyl ligands. The methodology allows liberating guest molecules with light.
Introduction
The controlled manipulation of supramolecular structures with light represents an interesting option to create functional nanoscale devices.1 A common strategy to implement light-responsiveness in metal–ligand assemblies is the utilization of photochromic ligands. Light allows altering the geometry of the ligand, which in turn may result in a structural reconfiguration of the metallasupramolecular assembly.1,2 For example, bipyridyl ligands containing dithienylethene units have been used for the light-induced conversion of a Pd24L48 capsule into small metallamacrocycles,3 to create Pd2L4 cages with light-dependent host–guest chemistry,4 and for the photo-induced sol–gel switching of a metallogel.5 Ligands containing the azobenzene motif were employed to modulate the host–guest chemistry of a spherical Pd12L24 complex,6 to alter the solubility of a coordination cage,7,8 and to control the catenation of a palladium metallacycle.9The construction of light-responsive metal–ligand assemblies with photochromic ligands is not straightforward. The design of suited ligands can be challenging, and the preparation of the ligands may involve substantial synthetic efforts. In this study, we describe an alternative and more general approach to render metal–ligand assemblies light-responsive: the addition of a metastable-state photoacid. We demonstrate that the acidification by the photoacid upon irradiation can be used for the disassembly of metallasupramolecular structures.10 The process is reversible, and the metallasupramolecular structures form again in the dark at room temperature (Scheme 1).
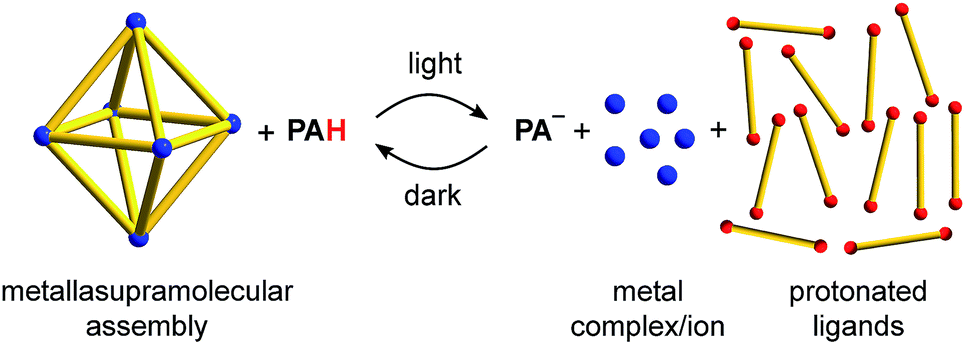 | ||
| Scheme 1 Reversible disassembly of metallasupramolecular structures by means of a metastable-state photoacid (PAH). The M6L12 coordination cage is used as a representative example of a metal–ligand assembly. | ||
Results and discussion
For our investigations, we decided to use the merocyanine photoacid PAH, the structure of which is depicted in Scheme 2. Upon exposure to violet light, PAH undergoes a ring-closing reaction to give the spiropyran PA−, along with liberation of a proton (Scheme 2).11 The spiropyran form is metastable and accumulates under irradiation. As a result, a pH drop of more than 2 units can be achieved. When light irradiation is stopped, the merocyanine form PAH is regenerated.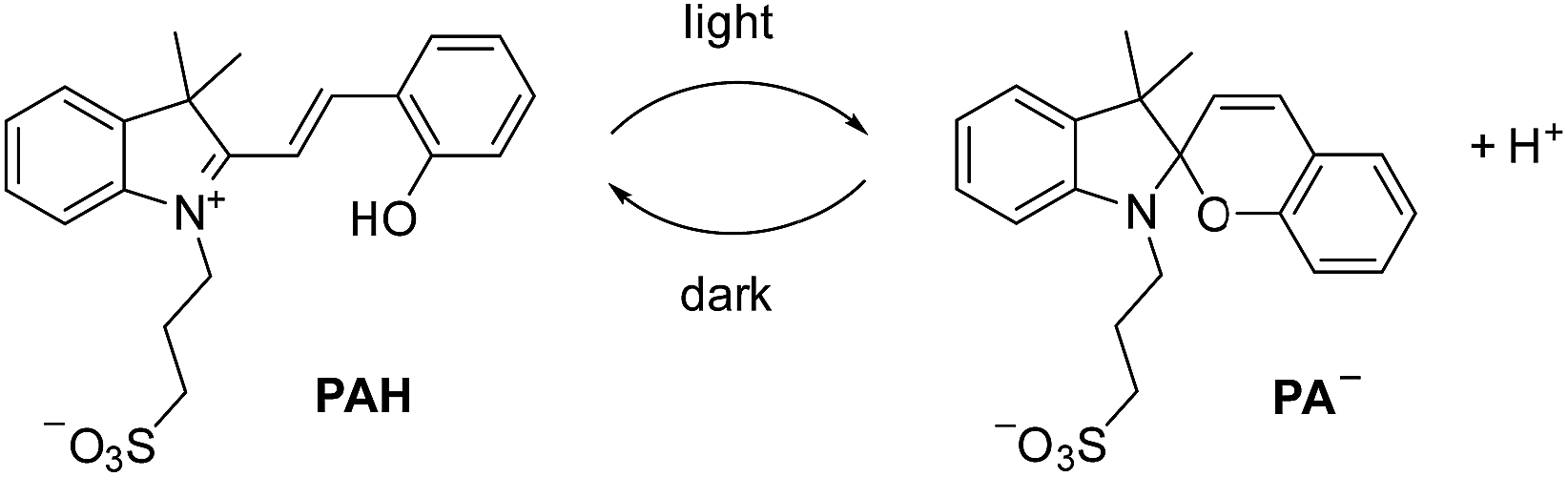 | ||
| Scheme 2 Irradiation with light converts the merocyanine PAH into the spiropyran PA−, with concomitant liberation of a proton. | ||
The metastable-state photoacid PAH was introduced by Liao and co-workers in 2011.12 Since then, PAH and structurally related compounds have been used in the context of supramolecular chemistry. Applications include the light-induced complexation of a pyridinium salt by a macrocyclic receptor,13 the switching of a rotaxane-based molecular shuttle,14 the light-controlled reversible self-assembly of nanorods,15 the reversible chromism of guests in coordination cages,16 the modulation of a hydrazine switch,17 the reversible photo-induced gel–sol transition of a dipeptide gel,18 and the controlled aggregation of nanoparticles,19 among others.11,20 We wanted to explore if PAH is also suited for controlling the structures of metal–ligand assemblies by light.
First, we performed test reactions with the PdII complex [(dppp)Pd(py)2](OTf)2 (1). Complex 1 features a strongly bond 1,3-bis(diphenylphosphino)propane (dppp) ligand and two labile pyridine (py) ligands. We chose 1, because PdII complexes with chelating diphosphine ligands are popular building blocks in metallasupramolecular chemistry.2,21 As connecting units, polypyridyl ligands are often employed, and the pyridyl ligands in 1 are surrogates for more intricate ligands. A solution of complex 1 (125 μM) and PAH (8 equiv.) was prepared in the dark using a mixture of CD3CN and D2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as the solvent. The solution was then irradiated for 20 minutes with violet light using a commercial LED panel (λ = 425 nm). After irradiation, the sample was immediately analysed by 1H and 31P NMR spectroscopy, and further NMR spectra were recorded after 8 hours in the dark.
:2) as the solvent. The solution was then irradiated for 20 minutes with violet light using a commercial LED panel (λ = 425 nm). After irradiation, the sample was immediately analysed by 1H and 31P NMR spectroscopy, and further NMR spectra were recorded after 8 hours in the dark.
The 1H NMR spectrum of the mixture before irradiation shows two doublets in the region between 7.5 and 9 ppm, which can be assigned to the photoacid PAH (Scheme 3, left side). In addition, one can observe a doublet at ∼8.5 ppm, resulting from the pyridine NCH protons. Irradiation leads to the nearly complete disappearance of the two PAH signals. The NCH signal of the bound pyridine ligands is no longer observable. Instead, three new signals appear. These peaks can be attributed to the formation of protonated pyridine (1b), as evidenced by comparison with a reference sample of pyridinium chloride (Scheme 3, spectrum on the top left side). After 8 hours in the dark, the NMR spectrum of the sample is indistinguishable from that of the sample before irradiation. The 31P NMR spectrum of the starting mixture shows a broad peak at ∼9 ppm, which is converted to a sharper signal at ∼12 ppm after irradiation (Scheme 3, right side). A similar signal is observed when complex [(dppp)Pd](OTf)2 is dissolved in CD3CN/D2O. We attribute this signal to a complex [(dppp)PdL2] (1a), with L being either solvent and/or triflate. Again, the spectrum converts back to that of the starting mixture when the sample is kept in the dark. Taken together, the data are strong evidence for a light-induced displacement of the pyridine ligands, which is fully reversed in the dark.
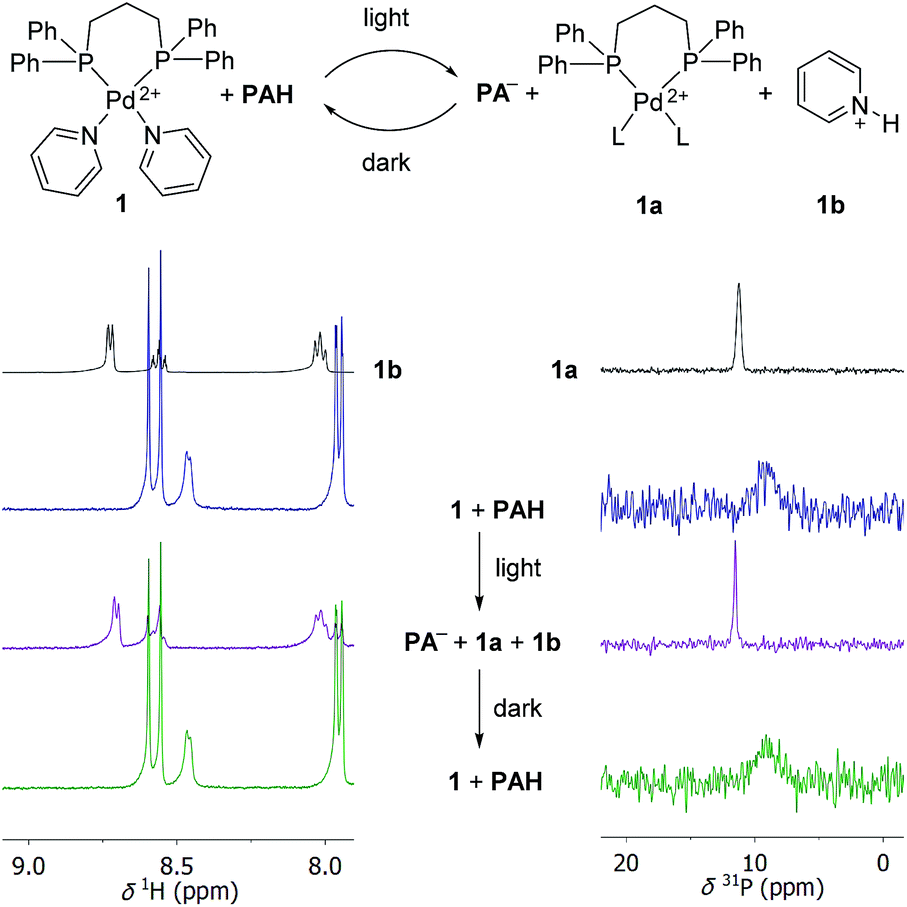 | ||
| Scheme 3 Irradiation of a mixture of complex 1 (125 μM) and the photoacid PAH (8 equiv.) in CD3CN/D2O (8:2) by violet light results in the displacement of the pyridyl ligands as evidenced by the 1H NMR (left side) and 31P NMR spectra (right side). The triflate anions of 1 are not shown for clarity. L indicates solvent or OTf−. | ||
Similar test reactions were performed with the model complexes [(py)4Pd](BF4)2 (2), [(bipy)3Fe](BF4)2 (3), and [(bipy)3Zn](BF4)2 (4) (bipy = 2,2′-bipyridyl). These kind of coordination motifs are also found in numerous metallasupramolecular assemblies.22 As it was observed for 1, it was possible to dissociate the N-donor ligands by irradiation with light, and re-complexation was observed in the dark (for details see the ESI†). For the Pd complex 2 and the Zn complex 4, the photo-induced conversion was very high (>80%), whereas for the Fe complex 3, only partial disassembly was observed (∼46% of complex 3 remained intact). We have also attempted to break complex 3 with a Brønsted acid, namely trifluoroacetic acid (TFA, 24 equiv. with respect to Fe). As with PAH, only partial disassembly was observed, indicating that the Fe complex is less susceptible to an acid-induced ligand displacement.
Encouraged by the results with the model complexes 1–4, we next examined reactions with larger and structurally more complex metal–ligand assemblies. Six different metallasupramolecular structures were prepared using the pyridyl ligands depicted in Fig. 1. More specifically, we have prepared an octahedral M6L12 cage with [Pd(py*)4]2+ (py* = substituted pyridine) complexes at the vertices, bridging ligands A, and a [B(p-C6H4F)4]− guest molecule (5),23 two M6L4 cages with gyrobifastigium-like geometry and [(dppp)Pd(py*)]2+ or [(dcpm)Pt(py*)]2+ complexes connecting the tetratopic ligand B (6 and 7, dcpm = bis(dicyclohexylphosphino)methane),24 a coordination barrel containing [(dcpm)Pt(py*)]2+ complexes and ligand C (8),24a a trigonal prismatic Ru complex with trispyridyl ligand D (9),25 and an M2L4 cage based on ligand E with [Pd(py*)4]2+ vertices (10)26 (Fig. 2).
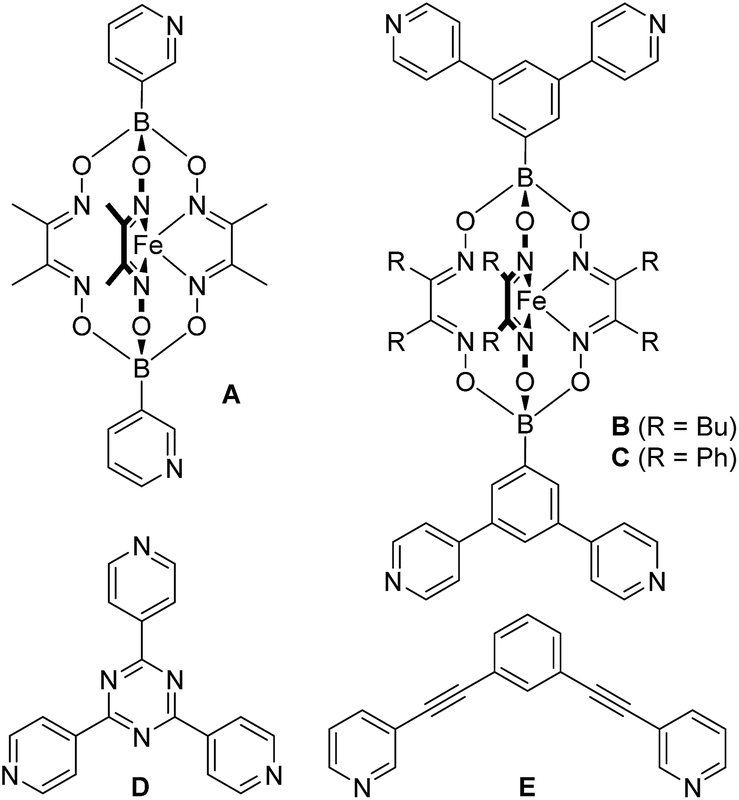 | ||
| Fig. 1 Structures of the pyridyl ligands A–E. | ||
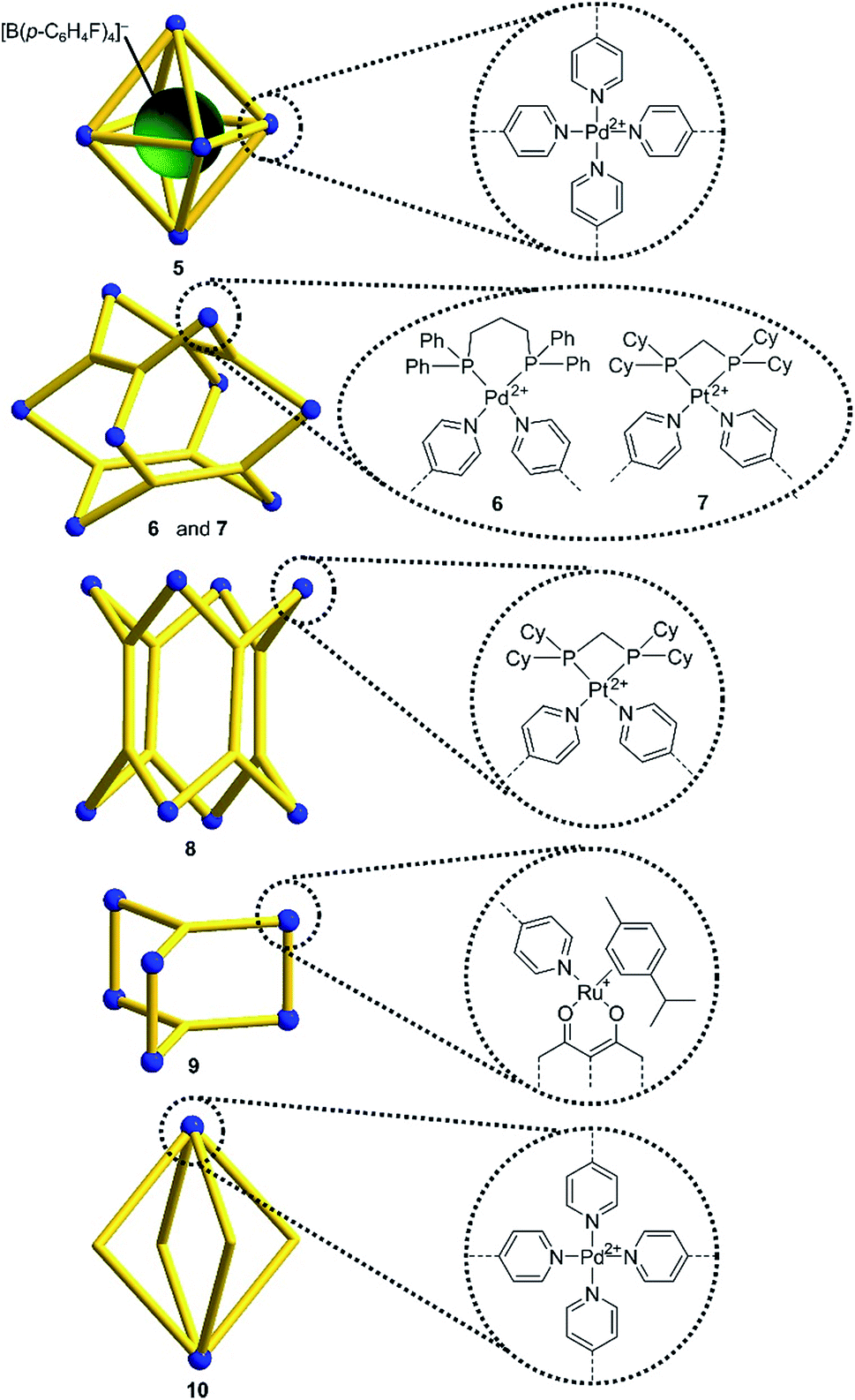 | ||
| Fig. 2 Schematic representation of the metallasupramolecular structures used in this study. Details about the bridging ligands (yellow) are given in ESI.† | ||
First test reactions with complex 5 and TFA (24 equiv.) showed that the complex can be fully disassembled by acid. We then set up an experiment similar to what has been described for the model complexes. Assembly 5 was dissolved together with an excess of the photoacid PAH in a mixture of CD3CN and D2O (8:2) in the dark. After irradiation with violet light, we immediately recorded 1H and 19F NMR spectra, followed by additional spectra after a waiting period of 8 hours in the dark. Preliminary studies revealed that the addition of small amounts of NaCl facilitated the disassembly process, and all subsequent experiments with 5 were performed with six equivalents of NaCl. It is worth noting that chloride ions are known for their ability to decompose PdII cages.27 For complex 5, six equivalents of NaCl were not sufficient to induce detectable decomposition, even though we were able to disrupt the assembly at higher NaCl concentrations. The beneficial effect of small amounts of NaCl for the photoswitching process might be due to stabilization of the PdII ions after ligand displacement.
The 1H NMR spectrum after irradiation indicates the acid-induced de-complexation of the pyridyl ligands (Scheme 4, left side). Additional evidence for the disassembly of the octahedron was obtained by analysing the 19F NMR spectra (Scheme 4, right side). The initial spectrum shows two signals, one for the encapsulated [B(p-C6H4F)4]− guest molecule, and one for non-bound [B(p-C6H4F)4]−. After irradiation, only ‘free’ [B(p-C6H4F)4]− (5c) is observed. In the dark, the M6L12 cage is re-formed, as corroborated by the 1H NMR spectrum, and the 19F NMR signal of the encapsulated borate anion. Overall, it is evident that light irradiation leads to a destruction of the metallasupramolecular structure and liberation of the guest. The process is reversible: in the dark, the cage with the encapsulated guest is formed again.
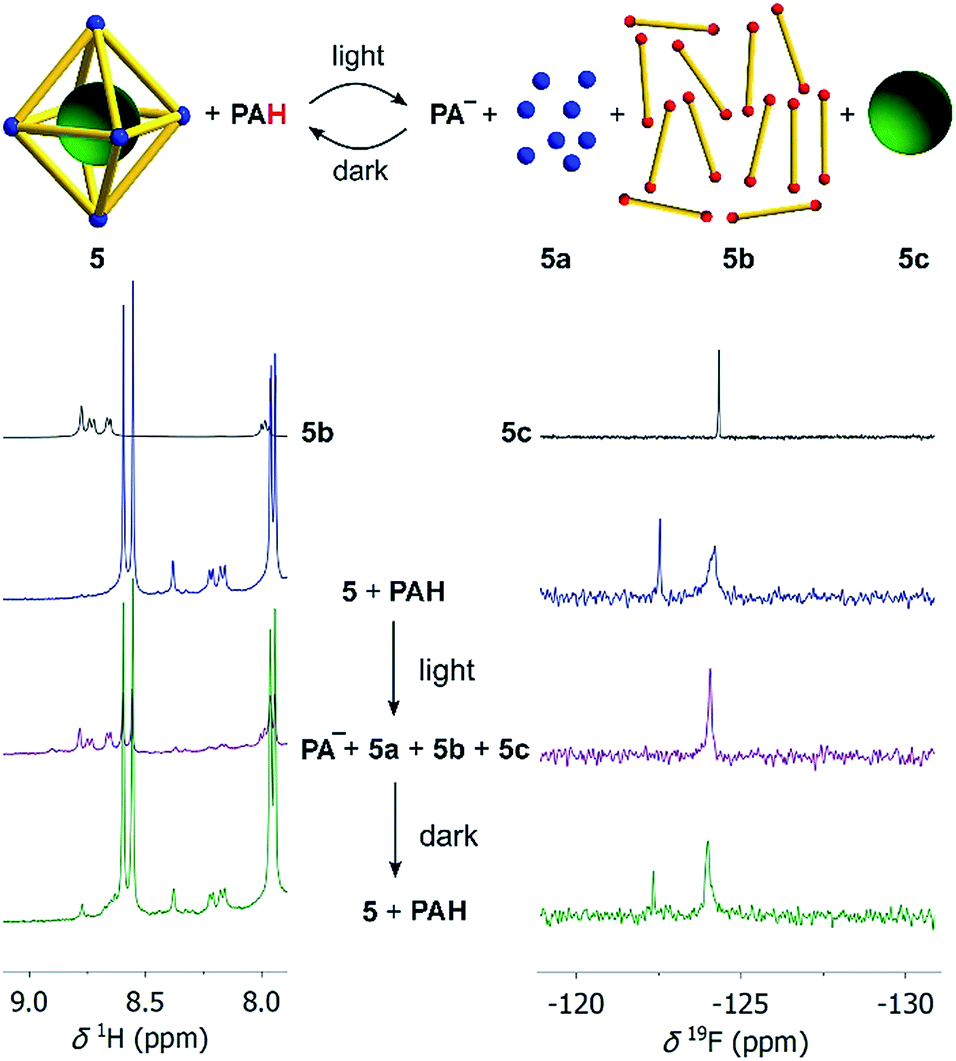 | ||
| Scheme 4 Irradiation of a mixture of cage 5 (10 μM), NaCl (60 μM), and the photoacid PAH (8 equiv. with respect to each bipyridyl ligand) in CD3CN/D2O (8:2) by violet light results in disassembly of the cage and liberation of the [B(p-C6H4F)4]− guest (green ball) as evidenced by the 1H NMR (left) and 19F NMR spectra (right). Details about the bridging ligand (yellow) are given in the ESI.† | ||
Experiments with the assemblies 6–10 gave similar results: a reversible, light-induced disassembly of the cage structures was observed by NMR spectroscopy in all cases (for details see the ESI†). For 6, 7, and 8, the switching efficiency was found to be very high, with at least 80% of the cages being disassembled by the photoacid. For 9 and 10, the process was less efficient, with only 16% (9) and 45% (10) of the cage structures being disrupted.
The difference in switching efficiency between cage 5 (>80%) and cage 10 (45%) is intriguing, given that both assemblies are based on [Pd(py*)4]2+ complexes. We were able to identify two factors which contribute to the reduced switching efficiency observed for 10: first, the photoacid binds weakly to cage 10 (Fig. S31†),28 and this interaction is expected to stabilize the assembly. Such an interaction was not observed for cage 5. Second, the pyridyl ligand A used for cage 5 is more basic than the pyridyl ligand E used for cage 10 (Fig. S32†). Consequently, acid-induced disassembly is favoured for cage 5.
The disassembly process was fully reversible in all cases. Kinetic investigations at 298 K showed that all assemblies are re-formed with half-lifes between 20 and 70 minutes. For comparison: the photoacid alone switches back with t1/2 of 2.4 minutes under these conditions (CH3CN/H2O, 8:2), indicating that the re-formation of the coordination complex is the rate-limiting step. The re-assembly of the complexes 5 and 8 was also followed at 50 °C. As expected, the reactions were faster, and complete re-assembly was observed after 30 minutes (5) or 10 minutes (8), respectively.
The robustness of the photoswitching process was demonstrated by repeated disassembly–assembly cycles using the coordination barrel 8 (Fig. 3). A mixture of the barrel and PAH was irradiated for 20 minutes, and then 1H NMR and 31P NMR spectra were recorded. After 2.5 hours in the dark, additional spectra were measured. This procedure was repeated four times. The 1H and 31P NMR spectra show barrel destruction and re-assembly over all cycles. The relative amount of the protonated ligand 8b, as determined by 1H NMR spectroscopy, was used as an indication of the switching efficiency. The process displayed good stability over the five cycles (Fig. 3). Ultimately, the sensitivity of PAH towards hydrolytic degradation represents a limit,12,13 but within the timeframe of our experiment, hydrolysis was not observed.
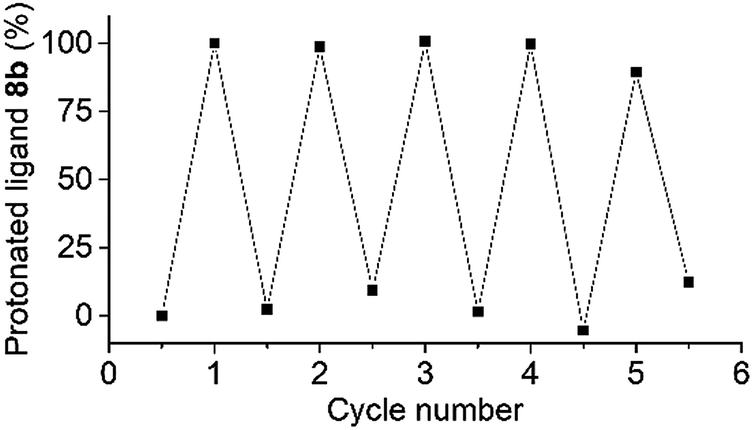 | ||
| Fig. 3 Repeated photoswitching between the coordination barrel 8 and its disassembled state. The relative amount of the protonated ligand 8b was used as an indication of the switching efficiency. For the first cycle, the amount of 8b was normalized to 0 and 100%, respectively. | ||
Conclusions
We have demonstrated that the addition of a metastable-state photoacid to solutions of metallasupramolecular assemblies renders the systems photo-responsive. Irradiation with violet light leads to proton transfer from the photoacid to the ligand, resulting in partial or full disassembly of the metal–ligand structure. The process is in all cases reversed in the dark. In principle, similar changes could be induced by addition of acids and bases. However, the utilization of light is not effecting the overall composition of the system (e.g. no accumulation of salts), and light can be used with good spatial and temporal control. Compared to the utilization of photochromic ligands, our approach offers the advantage of being more general and easier to implement. For the present study, we have focused on metallasupramolecular structures based on polypyridyl ligands, but it is expected that the methodology can be extended to assemblies with other ligands, given that the basicity of the system is within the pH range of the photoacid. We have already shown that the approach is suited for the light-induced release of guest molecules, but other applications can be envisioned as well. For example, it could be possible to control catalytic reactions with light, since some coordination cages are able to act as catalytic nanoreactors.29 Furthermore, it should be possible to use a related approach for photoswitching of metallasupramolecular polymers and metallogels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Swiss National Science Foundation, by the Marie Curie Initial Training Network “ResMoSys”, and by the Ecole Polytechnique Fédérale de Lausanne (EPFL).References
- (a) A. Díaz-Moscoco and P. Ballester, Chem. Commun., 2017, 53, 4635–4652 RSC; (b) M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC; (c) X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS; (d) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed; (e) S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- (a) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (b) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- S.-C. Wei, M. Pan, Y.-Z. Fan, H. Liu, J. Zhang and C.-Y. Su, Chem.–Eur. J., 2015, 21, 7418–7427 CrossRef CAS PubMed.
- T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133–5136 CrossRef CAS PubMed.
- J. Park, L.-B. Sun, Y.-P. Chen, Z. Perry and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 5842–5846 CrossRef CAS PubMed.
- The solubility of a coordination cage can also be modified with a photoswitchable guest. See: G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS PubMed.
- D. Zhang, Y. Nie, M. L. Saha, Z. He, L. Jiang, Z. Zhou and P. J. Stang, Inorg. Chem., 2015, 54, 11807–11812 CrossRef CAS PubMed.
- For examples of acid/base-switchable supramolecular assemblies see: (a) J. Altmann and A. Pöthing, Angew. Chem., Int. Ed., 2017, 56, 15733–15736 CrossRef PubMed; (b) S. Yi, V. Brega, B. Captain and A. E. Kaifer, Chem. Commun., 2012, 48, 10295–10297 RSC; (c) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058–10059 CrossRef CAS PubMed; (d) P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed; (e) T. Gottschalk, B. Jaun and F. Diederich, Angew. Chem., Int. Ed., 2006, 46, 260–264 CrossRef PubMed; (f) Z. Grote, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2004, 126, 16959–16972 CrossRef CAS PubMed; (g) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561–8856 CrossRef CAS; (h) N. Branda, R. M. Grotzfeld, C. Valdes and J. Rebek, J. Am. Chem. Soc., 1995, 117, 85–88 CrossRef CAS.
- Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef CAS PubMed.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- Q. Shi and C.-F. Chen, Org. Lett., 2017, 19, 3175–3178 CrossRef CAS PubMed.
- L.-P. Yang, F. Jia, J.-S. Cui, S.-B. Lu and W. Jiang, Org. Lett., 2017, 19, 2945–2948 CrossRef CAS PubMed.
- J. Guo, H. Zhang, Y. Zhoua and Y. Liu, Chem. Commun., 2017, 53, 6089–6092 RSC.
- D. Samanta, D. Galaktionova, J. Gemen, L. J. W. Shimon, Y. Diskin-Posner, L. Avram, P. Král and R. Klajn, Nat. Commun., 2018, 9, 641 CrossRef PubMed.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef CAS PubMed.
- X. Li, J. Fei, Y. Xu, D. Li, T. Yuan, G. Li, C. Wang and J. Li, Angew. Chem., Int. Ed., 2018, 130, 1921–1925 CrossRef.
- P. K. Kundo, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef PubMed.
- For an early study on this topic see: S. Silvi, A. Arduini, A. Pochini, A. Secci, M. Tamasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378–13379 CrossRef CAS PubMed.
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- (a) Z. Wu, K. Zhou, A. V. Ivanov, M. Yusobov and F. Verpoort, Coord. Chem. Rev., 2017, 353, 180–200 CrossRef CAS; (b) L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC; (c) W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC; (d) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1880 RSC; (e) S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239–2248 RSC.
- M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004–1010 RSC.
- (a) G. Cecot, M. Marmier, S. Geremia, R. De Zorzi, A. V. Vologzhanina, P. Pattison, E. Solari, F. F. Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2017, 139, 8371–8381 CrossRef CAS PubMed; (b) G. Cecot, B. Alameddine, S. Prior, R. De Zorzi, S. Geremia, R. Scopelliti, F. T. Fadaei, E. Solari and K. Severin, Chem. Commun., 2016, 52, 11243–11246 RSC.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 4695–4700 CrossRef CAS.
- D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed.
- (a) D. Preston, A. Fox-Charles, W. K. C. Loa and J. D. Crowley, Chem. Commun., 2015, 51, 9042–9045 RSC; (b) R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed; (c) J. E. M. Lewis, E. L. Gavey, S. A. Camerona and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- For examples of PdII cages which bind guest molecules with sulfonate groups see: (a) S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC; (b) G. H. Clever and M. Shionoya, Chem.–Eur. J., 2010, 16, 11792–11796 CrossRef CAS PubMed; (c) S. Hiraoka, M. Kiyokawa, S. Hashida and M. Shionoya, Angew. Chem., Int. Ed., 2010, 49, 138–143 CrossRef CAS PubMed.
- (a) M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS; (b) L. Catti, Q. Zhang and K. Tiefenbacher, Chem.–Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing synthetic procedures and experimental details. See DOI: 10.1039/c8sc01108g |
| This journal is © The Royal Society of Chemistry 2018 |