Enhanced oxygen evolution on visible light responsive TaON photocatalysts co-loaded with highly active Ru species for IO3− reduction and Co species for water oxidation†
Received
24th February 2017
, Accepted 29th March 2017
First published on 29th March 2017
Abstract
Loading an appropriate cocatalyst significantly enhances the activity of semiconductor photocatalysts in both conventional one-step water splitting and Z-scheme-type water splitting with a redox couple. In the present study, we examined the catalytic activity of Ru-based cocatalysts for IO3− reduction to improve the efficiency of O2 evolution on a TaON photocatalyst under visible light with an IO3− acceptor, which is an important half component in Z-scheme water splitting systems. X-ray photoelectron spectroscopy analyses and electrochemical measurements revealed that the calcination temperature in the loading of Ru species from aqueous RuCl3 significantly affected the catalytic activity for the reduction of IO3−. Calcination at or below 200 °C produced Ru(OH)xCly species that drastically enhanced the reduction of IO3−, whereas that at 300 °C generated conventional RuO2 that showed lower activity for IO3− reduction than Ru(OH)xCly but considerably enhanced the water oxidation. Co-loading the Ru(OH)xCly and Co cation species as water oxidation cocatalysts significantly improved O2 evolution on the TaON photocatalyst by more than a factor of two compared to that loaded with only the conventional RuO2 cocatalyst. This suggests the effectiveness of co-loading two different cocatalysts that are independently optimized for reduction and oxidation.
Introduction
Photocatalytic water splitting using semiconductor materials has received much attention because of the potential for clean H2 production from water by harvesting abundant solar light. In particular, the effective utilization of visible light is of interest for achieving practically efficient H2 production.1–3 Improving the efficiency of existing visible light responsive photocatalysts has been extensively attempted by applying various strategies, such as surface modification (e.g., cocatalyst loading),4–6 morphology control,7,8 and hetero-junction introduction.9,10 Among them, loading a suitable cocatalyst onto photocatalyst particles has been widely studied as an effective way to improve the performance of existing photocatalyst materials. The role of the cocatalyst has generally been regarded as (1) enhancing the charge separation in the photocatalyst, and/or (2) facilitating the surface chemical reactions by the carriers, i.e., water reduction and/or oxidation for one-step water splitting. For most photocatalysts used for one-step water splitting, loading of the cocatalysts that enhance water reduction (e.g., NiOx, Rh/Cr2O3) is often employed to improve the total efficiency of water splitting.11–13 Recent studies have also used an effective O2-evolving cocatalyst (e.g., Mn3O4, IrO2),11,14,15 co-loaded with a H2-evolving cocatalyst.11,15 Cocatalyst loading also influences the efficiencies of Z-scheme-type water splitting systems in which two different photocatalysts are combined by a redox couple. In some cases, loading cocatalysts that facilitate reduction or oxidation of a redox mediator, rather than water, significantly influence the total efficiency. Specifically, the reduction of IO3− to I− (IO3− + 6e− + 3H2O → I− + 6OH−) often becomes the rate-determining step in O2 evolving part, necessitating the loading of effective cocatalysts, such as PtOx or RuO2.5,16 Loading a cocatalyst with a lower overpotential on a photocatalyst for IO3− reduction increases the rate of O2 evolution from an aqueous solution containing IO3− as an electron acceptor.5,16 Additionally, co-loading a WO3 photocatalyst with PtOx and RuO2 species showed an O2 evolution rate that was two times greater than that provided by the photocatalyst loaded with only PtOx.17 Although this enhancement was explained by enhanced water oxidation on RuO2, the contribution of the RuO2 cocatalyst to the reduction of IO3− cannot be excluded. Such bi-functionality of the RuO2 cocatalyst (i.e., both for IO3− reduction and water oxidation) was also suggested for a RuO2-loaded TaON photocatalyst, whereby Ru species were mainly loaded as oxides by the impregnation of aqueous (NH4)2RuCl6 followed by calcination in air above 350 °C.18 The high activity of RuO2 for water oxidation is well known in electrochemical fields;19,20 however, the activity of RuO2 or related species for IO3− reduction, especially the relation between the chemical species of Ru and the catalytic activities, has not been examined in detailed.
In the present study, we loaded Ru species onto TaON photocatalysts and electrodes at different temperatures, evaluated their photocatalytic and catalytic activities for IO3− reduction, and performed detailed physicochemical characterization to reveal the optimum Ru chemical species for IO3− reduction. Oxynitrides, such as TaON, have recently been examined as promising materials because of their favourable band levels for water splitting under visible light,21,22i.e., more negative (higher) levels of valence band maxima (VBM) compared to those of conventional metal oxides. However, the more negative VBM decrease the overpotential for water oxidation; thus, co-loading an O2-evolving cocatalyst is expected to improve the total efficiency of such photocatalysts. Thus, Co species were co-loaded onto Ru-loaded TaON as water oxidation cocatalysts to further improve the photocatalytic activity of TaON for O2 evolution under visible light in the presence of IO3− as a reversible electron acceptor.
Experimental
Preparation of TaON particles
TaON particles were prepared according to a previously reported method21 as follows. Commercially available Ta2O5 particles (10 g, 99.9%, Kojundo Chemical Laboratory Co., Ltd.) were placed onto a quartz boat, positioned inside a quartz tube (internal diameter: 26 mm), and then kept under a stream of Ar gas (99.999%, 100 mL min−1) for more than 30 min to replace the residual air. Then, the Ar gas was replaced with a stream of NH3 (99.99%, 60 mL min−1). With the NH3 steam, the temperature was increased to 850 °C and maintained for 24 h for nitriding. After cooling to room temperature under the stream of NH3, the gas was completely replaced by an Ar stream. The X-ray diffraction (XRD) pattern of the sample could be attributed to the monoclinic phase of TaON (see Fig. S1(a)†), as reported in a previous study,21 and the photoabsorption edge was almost consistent with a reported value21 of approximately 500 nm (see Fig. S1(b)†). The particle diameters of the obtained samples were confirmed to be approximately 200–500 nm from the scanning electron microscopy (SEM) images (see Fig. S2†).
Loading of cocatalysts onto TaON particles
Cocatalyst loading onto TaON particles was performed by an impregnation method from aqueous solutions containing each cation precursor followed by calcination at different temperatures in air. The Ru-species were loaded using commercially available RuCl3·nH2O powders (n = 1–3, 36–44% as Ru, Wako Pure Chemical Industries, Ltd.) as the precursors. The prepared TaON particles (2.5 g) were first suspended in an aqueous solution (∼5 mL) containing the required amount of RuCl3·nH2O (0.1, 0.5, 1.0, or 1.5 wt%, calculated as metallic Ru with respect to TaON) and stirred using a glass rod in an evaporation dish in a hot-water bath until complete dryness. The samples were calcined at different temperatures (150, 200, 250, 300, 400, and 500 °C) for 1 h in air, and the heating rate was fixed at 5 °C min−1 for all calcination temperatures. The obtained samples are referred to as Ru-x (x = calcination temperature) hereafter. The amount of the Ru cocatalyst was set at 1.0 wt% unless otherwise stated. As for the loading of Co species, commercial Co(NO3)2·6H2O powders (98.0%, Wako Pure Chemical Industries, Ltd.; 0.1, 0.5, and 1.0 wt%, calculated as metallic Co with respect to TaON) were used as the precursor. After impregnation of the precursor onto TaON according to a procedure similar to that above, the samples were calcined at different temperatures (100, 200, and 300 °C, heated to the desired temperature at 5 °C min−1) for 1 h in air. The loading amount and calcination temperature were set to 0.1 wt% and 100 °C, respectively, which were considered nearly optimum conditions based on the results for photocatalytic O2 evolution from the AgNO3 aqueous solution (see Fig. S3 and S4†). The as-prepared sample is referred to as Co-100. As for the electrochemical measurements, a greater amount (1.0 wt%) of the Co precursor was loaded onto the TaON particles to obtain higher currents for evaluating the catalytic activity of the Co species. The X-ray photoelectron spectroscopy (XPS) measurement (see Fig. S5†) strongly suggested that the main species loaded onto TaON is Co(OH)2, which has been demonstrated to enhance water oxidation on photocatalysts, such as g-C3N4.23 For co-loading of Ru and Co species, the Ru species were first loaded, and then the Co species were subsequently loaded by the procedure described above (basically set to 0.1 wt% and 100 °C). The co-loaded samples are referred to as Ru-x-Co (x = calcination temperature for the pre-loading of Ru species).
Characterization
The prepared samples were characterized by powder XRD (MiniFlex II, Rigaku Corp., Cu Kα), UV-visible diffuse reflectance spectroscopy (UV-vis, V-670, JASCO Corp., reference sample: BaSO4), Brunauer–Emmett–Teller (BET) specific surface area measurements (BEL SORP-mini II, MicrotracBEL Corp.), scanning electron microscopy (SEM, VE-9800, KEYENCE Corp.), transmission electron spectroscopy (TEM, JEM-2100F, JEOL Ltd.), energy dispersive X-ray spectroscopy (EDS, EX-24200M1G2T, JEOL Ltd.), and XPS (MT-5500, ULVAC-PHI, Inc., Mg Kα). A small amount of Au nanoparticles was sputtered onto each sample using magnetron sputtering (MSP-1S, Vacuum Device Inc.) prior to the XPS measurements. The binding energy of the Au 4f7/2 peak was adjusted to 84.0 eV to correct the chemical shifts of the other elements.
Electrochemical measurements
To evaluate the catalytic activity of Ru or Co species for IO3− reduction or water oxidation, the TaON particles loaded with these species were coated onto a conductive glass substrate (FTO, Asahi Glass Co., Ltd.) using the following squeegee method. Pastes of Ru-x (TaON loaded with 1.0 wt% Ru species at different temperatures) or Co-100 (TaON loaded with 1.0 wt% Co species at 100 °C) were prepared by mixing them thoroughly with a small amount of Milli-Q water. The particle pastes were spread (4 cm × 1.5 cm, ∼1 mg) onto an FTO substrate and then dried in air at room temperature. The electrochemical cell used for the measurements consisted of a prepared electrode as the working electrode, Ag/AgCl as the reference electrode, a Pt wire as the counter electrode, and a Na2SO4 aqueous solution (0.1 M, 70 mL, pH ∼ 6 (without adjustment)) as the electrolyte. The solutions were purged with Ar gas to remove dissolved air before the electrochemical measurements, and then the applied potential of the working electrode was controlled using a potentiostat (VersaSTAT 3, Princeton Applied Research Co., Ltd.). The onset potential of the cathodic current was investigated using linear sweep voltammetry in the Na2SO4 aqueous solution containing IO3− (1.0 or 5.0 mM) under dark conditions to verify the overpotential for the reduction of IO3− on the prepared electrodes. The anodic photocurrent was investigated by linear sweep voltammetry in the Na2SO4 aqueous solution under visible light irradiation using a 300 W Xe lamp (Cermax, LX-300F) equipped with a cutoff filter (Hoya, L-42) and cold mirror (Kenko, CM-1) that afforded irradiation within a limited wavelength range of 400–800 nm to verify the electrocatalytic activity for water oxidation of the prepared electrodes.
Photocatalytic reactions
Photocatalytic O2 evolution was conducted using a Pyrex glass reaction vessel connected to a closed gas circulation system. Photocatalyst particles (50 mg) were suspended in Milli-Q water (250 mL) containing 1 mM of IO3− (NaIO3, Wako Pure Chemical Industries, Ltd., 99.5%) as an electron acceptor by continuously stirring using a stir bar and magnetic stirrer. The suspension was thoroughly degassed by repeated evacuation of the gas phase by a vacuum pump with a liquid nitrogen trap. After the introduction of Ar gas to the system (∼50 Torr), the suspension was irradiated using the Xe lamp equipped with the L-42 cutoff filter and CM-1 cold mirror (400 < λ < 800 nm). In some cases, the photocatalyst particles were suspended in Milli-Q water (100 mL) containing 20 mM of Ag+ (Wako Pure Chemical Industries, Ltd., 99.8%) as a sacrificial electron acceptor in the presence of a small amount (200 mg) of lanthanum oxide (La2O3, Wako Pure Chemical Industries, Ltd., 99.5%) to maintain the pH ≈ 8.0.21,24 The reactions in the presence of Ag+ were conducted using a flow reaction system under Ar flow (15 mL min−1). Before the reactions, the solutions were completely purged with Ar gas. Visible light was irradiated from the Xe lamp equipment. The temperature of the solution was kept at 15 °C during the reaction by a flow of external circulating cooling water. The evolved gas was analyzed using gas chromatography (GC-8A, Shimadzu; thermal conductivity detector, Molecular Sieve 5A, Ar carrier).
Results and discussion
Characterization of Ru species on TaON particles
Fig. 1 shows XPS spectra of the TaON samples loaded with Ru-species from RuCl3·nH2O at different calcination temperatures (Ru-x, x: 150, 200, 250, and 300 °C); the loading amount was set at 1.0 wt%. The XPS spectra of two commercial samples (RuCl3·nH2O and RuO2) and a lab-made Ru(OH)x sample that was prepared from RuCl3·nH2O (see ESI† for the detailed preparation) are also shown in Fig. 1 for comparison. Ru3d5/2 peaks are observed around 282.0 eV for both Ru-150 and Ru-200; this position is similar to but appreciably lower than that of the reference RuCl3·nH2O. The Cl/Ru ratios were calculated from the peak area of XPS spectra of the Ru-150 and Ru-200 samples and were lower (∼1.7) than that of the original RuCl3·nH2O (∼3). This strongly suggests that the partially hydroxylated Ru species, e.g., Ru(OH)xCly,25,26 were loaded onto TaON by the wet-impregnation process followed by calcination at relatively low temperatures. The Ru3d5/2 peak of the samples is slightly shifted to a lower binding energy after calcination at 250 °C and drastically shifted to a lower energy at 300 °C (∼281.0 eV). The Cl/Ru ratio (∼1.4) is less than the original value (∼3) after calcination at 250 °C. The Cl2p peak is negligible after calcination at 300 °C. These findings indicate that the Ru-300 sample mainly contains oxidized Ru species, i.e., conventional RuO2, as reported in a previous study.27 Ru-250 probably contains both Ru(OH)xCly and RuO2.
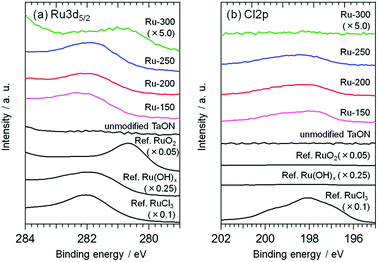 |
| | Fig. 1 XPS spectra of TaON samples loaded with Ru species prepared at different calcination temperatures and those of reference samples: (a) Ru3d5/2 region and (b) Cl2p region. The binding energy of the deposited Au was adjusted to 84.0 eV. | |
Fig. 2 shows TEM images of these Ru-x samples. Nanoparticles with diameters less than 3 nm are found on the surface of both the Ru-150 and Ru-200 samples (Fig. 2 and S6(a)–(d)†), whereas such particles are not observed on the unmodified TaON samples (see Fig. S7†). As shown in Fig. S8,† the EDS spectrum of the Ru-200 sample (analyzed within the same region shown in Fig. 2) confirms the presence of Ru species. Therefore, the observed nanoparticles on the Ru-150 and Ru-200 samples are undoubtedly Ru(OH)xCly species. However, larger particles with diameters of ∼10 nm are observed on both the Ru-250 and Ru-300 samples, which are certainly a mixture of Ru(OH)xCly and RuO2 species for Ru-250 and predominantly RuO2 species for Ru-300.
 |
| | Fig. 2 TEM images of TaON samples loaded with Ru species prepared at different calcination temperatures. | |
The role of Ru species loaded onto TaON particles at different temperatures
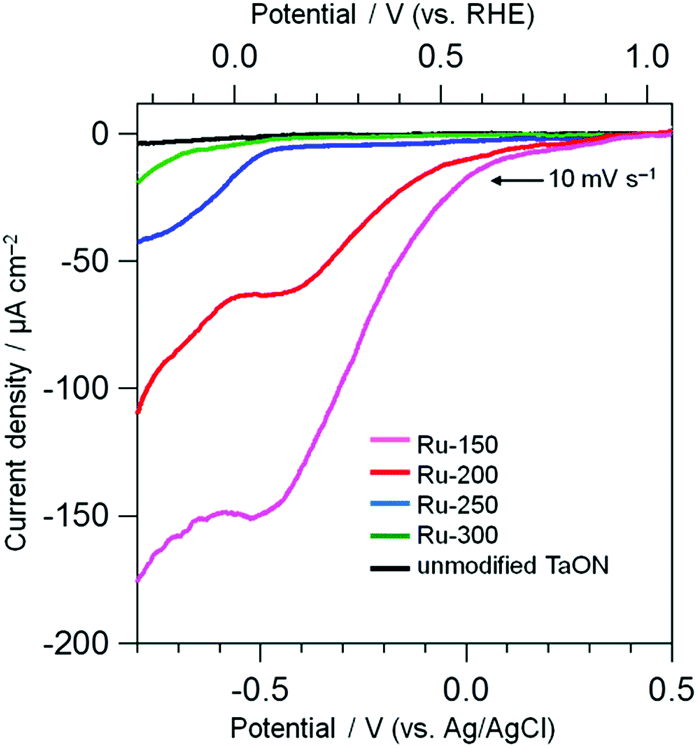
Electrochemical measurements were performed to evaluate the catalytic activity of Ru species loaded onto TaON for the reduction of IO3− and oxidation of water. The electrodes were prepared by spreading the Ru-x samples (TaON particles loaded with Ru-species at different temperatures) or unmodified TaON particles onto an FTO substrate using the squeegee method without any calcination (just drying). Fig. 3 shows current–potential curves for these electrodes in a Na2SO4 aqueous solution containing IO3− (1.0 mM) under dark conditions. Each scan was measured in the negative direction at a rate of 10 mV s−1. No clear cathodic current onset is observed on the unmodified TaON, even at much more negative potentials compared to the standard redox potential of IO3−/I− at this pH (+0.47 V vs. Ag/AgCl), indicating the negligibly low catalytic activity of the unmodified TaON surface for the reduction of IO3−, as suggested in the previous report.18 However, the Ru-300 sample, which contains mainly RuO2 species, clearly exhibits the onset corresponding to the reduction of IO3− (see Fig. S9† for the current with different concentrations of IO3−) at around −0.6 V vs. Ag/AgCl, confirming the reported catalytic activity of RuO2 for the reduction of IO3−.18 Surprisingly, the onset potentials on the Ru-150 and Ru-200 electrodes, both of which mainly contain Ru(OH)xCly species, are observed around much more positive potentials of +0.1 V vs. Ag/AgCl, along with astonishingly higher current densities. As shown in Fig. S9,† the cathodic currents on both Ru-150 and Ru-200 samples increase with increasing concentrations of IO3− but with almost the same onset potentials at around +0.1 V vs. Ag/AgCl, strongly suggesting that the cathodic current is derived from IO3− reduction not from the reduction of Ru species themselves. These findings strongly suggest that the Ru(OH)xCly species possess much higher catalytic activities for IO3− reduction than the conventional RuO2 species. Such catalytic activities of Ru(OH)xCly for IO3− reduction have not been reported.
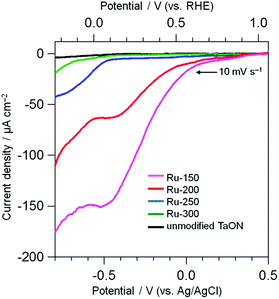 |
| | Fig. 3 Current–potential curves of TaON electrodes loaded with Ru species and an unmodified TaON electrode in a Na2SO4 solution (0.1 M) containing NaIO3 (1.0 mM) under dark conditions. | |
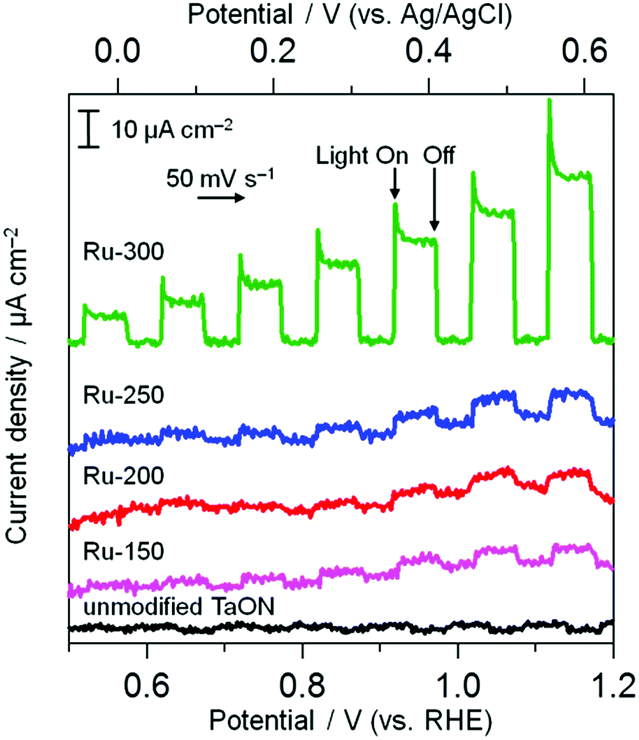
The electrocatalytic activities of these Ru species for water oxidation were investigated by monitoring the anodic response in a Na2SO4 aqueous solution under visible light irradiation because of the n-type nature of the TaON semiconductor (shown in Fig. 4). Unmodified TaON shows a negligible photoresponse within the potential range from +0.5 to +1.2 V vs. RHE. However, Ru-300 exhibits a clear anodic photocurrent, indicating that RuO2 species effectively catalyze water oxidation, as reported.18,28 As for other samples (Ru-150, Ru-200, and Ru-250), the anodic photocurrents appreciably increase compared to the unmodified sample, but the extent of the increase is much less than that for Ru-300. This slight increase in anodic photocurrent is probably due to the moderate catalytic activity of the Ru(OH)xCly species for water oxidation and/or the oxidation of such Ru(OH)xCly species themselves by photogenerated holes without catalyzing water oxidation. The Ru-250 sample exhibits a mixed property between Ru-300 and Ru-200 in both cathodic and anodic process, supporting the XPS results indicating that the Ru-250 sample contains both RuO2 and Ru(OH)xCly species. These results strongly indicate that Ru(OH)xCly species prepared at or below 200 °C exhibit much higher activities for IO3− reduction than the conventional RuO2 species that are prepared by calcination at or above 300 °C, whereas such RuO2 exhibits a higher activity for the oxidation of water than the Ru(OH)xCly species. As shown above, the Ru(OH)xCly species show much higher catalytic activities for IO3− reduction than conventional RuO2.
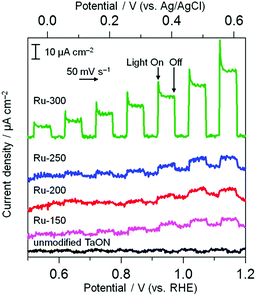 |
| | Fig. 4 Current–potential curves of TaON electrodes loaded with Ru species and an unmodified TaON electrode in a Na2SO4 solution (0.1 M) under visible light (λ > 400 nm). | |
Photocatalytic O2 evolution on TaON loaded Ru species at different temperatures in an aqueous solution containing IO3− as an electron acceptor
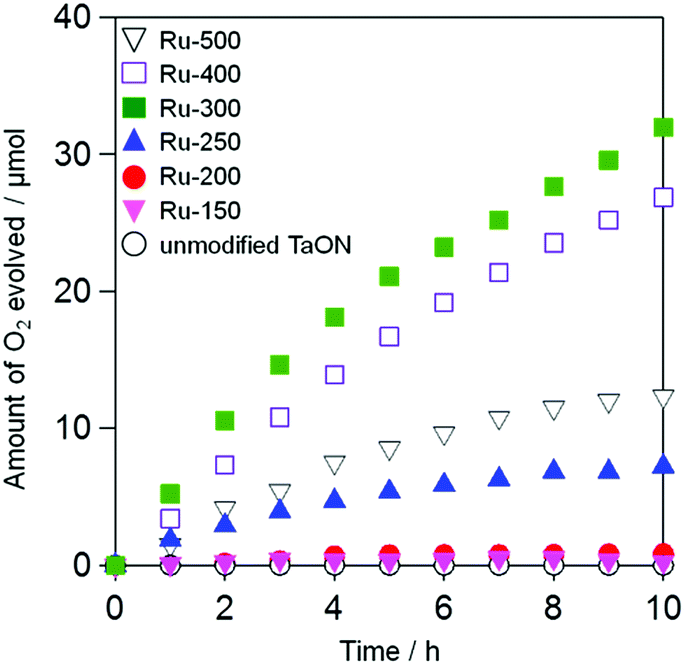
Fig. 5 shows the time courses of photocatalytic O2 evolution on Ru-loaded TaON samples (i.e., Ru-x) that were prepared at different temperatures in an aqueous solution containing IO3− (1.0 mM) as an electron acceptor under visible light irradiation (400 < λ < 800 nm). No O2 evolution was observed over the unmodified TaON particles (see open white circles). Although the rates of O2 evolution on the TaON samples loaded with Ru species at or below 200 °C are quite small, the samples calcined at or above 250 °C exhibit obvious O2 evolution at steady rates. The rate of O2 evolution increased with increasing calcination temperatures from 250 to 300 °C, whereas it slightly and drastically decreased at 400 and 500 °C, respectively. The influence of the amount of Ru (0.1, 0.5, 1.0, and 1.5 wt%) on the O2 evolution rate was examined at a fixed temperature of 300 °C, which is an optimum temperature for the sole loading of Ru species. As shown in Fig. S10,† the rates are rarely affected by the Ru amounts.
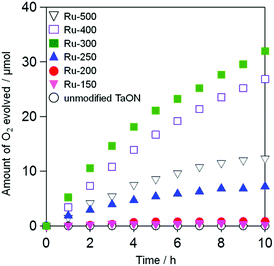 |
| | Fig. 5 Time courses for the photocatalytic oxidation of water on the TaON photocatalysts loaded with Ru species prepared at different temperatures and an unmodified TaON photocatalyst in an aqueous solution (250 mL) containing NaIO3 (1.0 mM) under visible light (amount of photocatalyst: 50 mg, amount of Ru: 1.0 wt% as metal, λ > 400 nm). | |
The O2 evolution in the present system with IO3− should proceed via the following equations:29,30
| | | IO3− + 6e− + 3H2O → I− + 6OH− | (1) |
The unmodified TaON particles showed appreciably high rates of O2 evolution from aqueous AgNO3 (initial rate of O2 evolution: ∼40 μmol h−1, see Fig. S11†) with a reasonable value compared to that previously reported.21,24 This indicates again that the low activity of the unmodified TaON sample for O2 evolution from aqueous NaIO3 is due to the negligible activity of the TaON surface for the multi-electron reduction of IO3− (eqn (1)), which causes a significant recombination of electrons and holes. The loading of RuO2 species, which were impregnated from RuCl3·nH2O at 300 °C or from (NH4)2RuCl6 at 350 °C, on TaON effectively triggered the O2 evolution from aqueous NaIO3.18 However, neither the photocatalytic nor the catalytic activity of TaON loaded with Ru species at temperatures below 250 °C was examined in previous studies.18,28 The electrochemical and photocatalytic activity results for the Ru-300 sample in the present study support the arguments in the previous study that the loaded RuO2 species promote both reduction of IO3− and oxidation of water.18 However, both the Ru-150 and Ru-200 samples showed negligibly low rates of O2 evolution despite the existence of Ru(OH)xCly species that exhibit much higher activities for IO3− reduction. These results evoke the possibility that the low activity of Ru-150 and Ru-200 samples for O2 evolution from aqueous NaIO3 are derived from the insufficient efficiency for water oxidation on the unmodified TaON and/or the Ru species by photogenerated holes.
Photocatalytic O2 evolution on TaON co-loaded Ru and Co species in an aqueous solution containing IO3− as an electron acceptor
The co-loading of effective cocatalysts, such as Co species, for water oxidation was investigated to enhance the rate of water oxidation of TaON samples loaded with Ru(OH)xCly. Additional cocatalysts were loaded below 200 °C to avoid changing the chemical state of the pre-loaded Ru species. Co species, such as Co(OH)2, formed at lower temperatures have been demonstrated to enhance water oxidation on various photoanodes and photocatalysts, such as g-C3N4.23 Although loading CoOx or Co-Pi particles onto TaON photoanodes enhances the water oxidation and thereby improves the stability,31,32 no study has reported the improved performance of TaON photoanodes and photocatalysts with the simple loading of such Co(OH)2 species. As described in the experimental section, the loading of Co species by impregnation of aqueous Co(NO3)2 and subsequent calcination at 100 °C in air considerably enhance the O2 evolution in the presence of Ag+ as sacrificial electron acceptor under visible light, even with a small amount of loading of 0.1 wt% (see Fig. S3 and S4†). However, no obvious cathodic current was observed on the TaON electrode loaded with the Co species at 100 °C, even in the solution containing IO3− (see Fig. S9(f)†), indicating that the Co species on the TaON samples possess quite low activity for IO3− reduction. Therefore, we attempted to deposit the Co species at 100 °C on TaON particles sequentially after loading Ru species at various temperatures and evaluated their photocatalytic activities for O2 evolution from aqueous NaIO3.
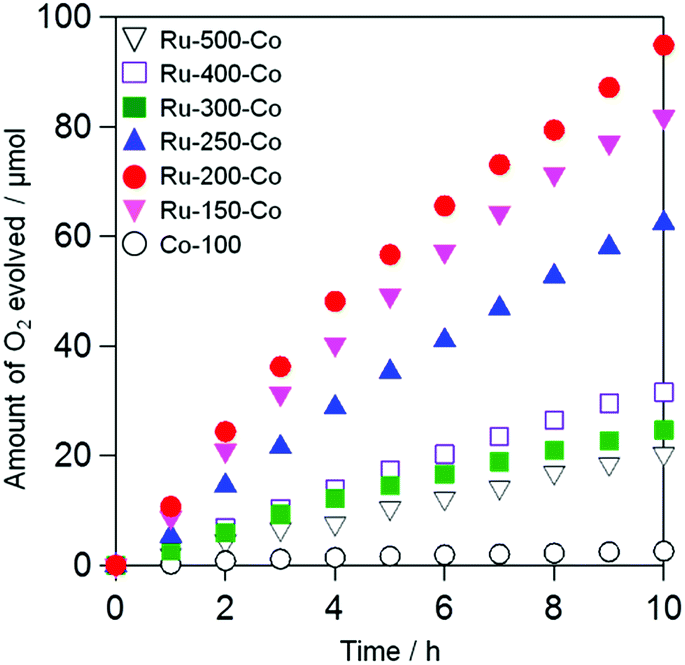
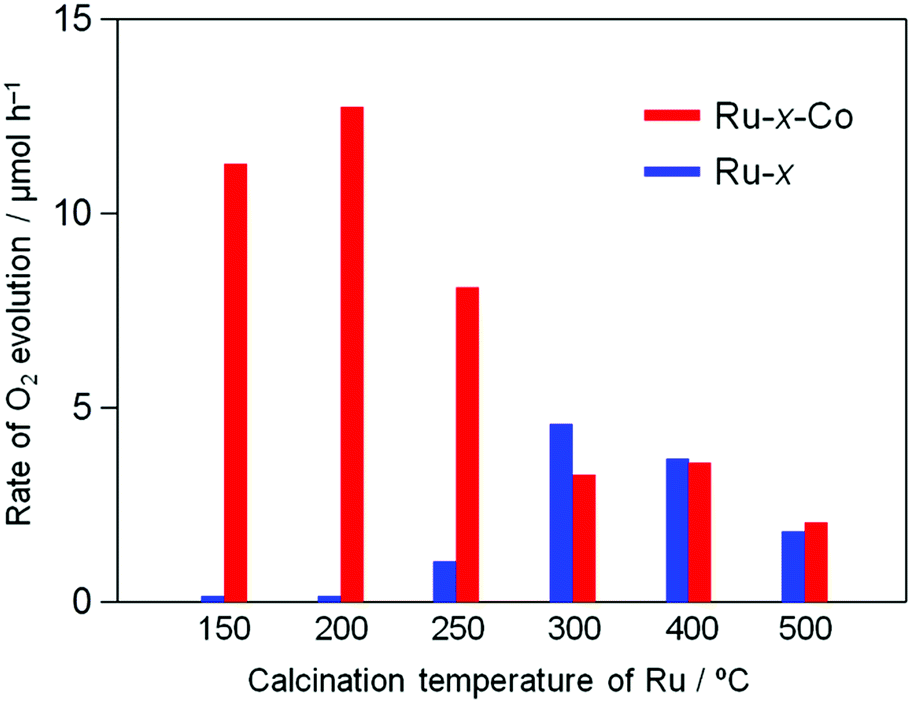
Fig. 6 shows the time courses of photocatalytic O2 evolution on TaON samples co-loaded with Ru and Co species (i.e., Ru-x-Co) in an aqueous solution containing IO3− as an electron acceptor under visible light irradiation. Almost no O2 evolution was observed on TaON loaded with the Co species alone because of the negligible activity of the Co species for IO3− reduction, as clearly demonstrated in an electrochemical measurement (Fig. S9(f)†). In contrast, all of the Ru-x-Co samples exhibited obvious O2 evolution with steady rates; among them, the Ru-200-Co sample exhibited the highest rate (∼12.5 μmol h−1). Even after the reaction, the presence of Ru species was clearly confirmed by means of XPS analysis (see Fig. S12†), while the peak position of Ru species slightly shifted to lower energy at which the main peak of Ru(OH)x is observed. Additionally, the ratio of Cl to Ru significantly decreased from ∼1.7 to 0.2 after the reaction. These findings indicate that the majority of Ru(OH)xCly species were transformed to Ru(OH)x species during reaction. However, the rate of O2 evolution was almost unchanged within 10 h of irradiation, strongly suggesting that both the Ru(OH)x and Ru(OH)xCly species work as reduction site of IO3− on the present TaON photocatalysts. The comparable catalytic activity of Ru(OH)x-loaded TaON electrode to Ru-200 one for electrochemical IO3− reduction (see Fig. S13†) supports this assumption. As for Co species, the peak position of the Co2p XPS spectrum of the Ru-200-Co sample after reaction was slightly shifted to lower energy and the intensity of satellite peak (at ∼786.6 eV) attributed to Co2+ species decreased (see Fig. S14†). Although we cannot identify the precise composition at current stage, it appears that Co species loaded were partially oxidized to form CoO and/or CoO(OH) species that also function as water oxidation catalyst.33
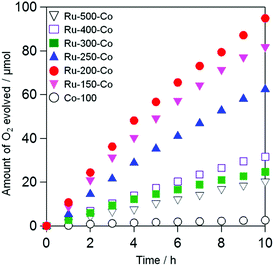 |
| | Fig. 6 Time courses for the photocatalytic oxidation of water on the TaON photocatalysts loaded with Ru and Co species prepared at different temperatures and a TaON photocatalyst loaded with Co species in an aqueous solution (250 mL) containing NaIO3 (1.0 mM) under visible light (amount of photocatalyst: 50 mg, amount of Ru: 1.0 wt% as metal; Co: 0.1 wt% as metal, λ > 400 nm). | |
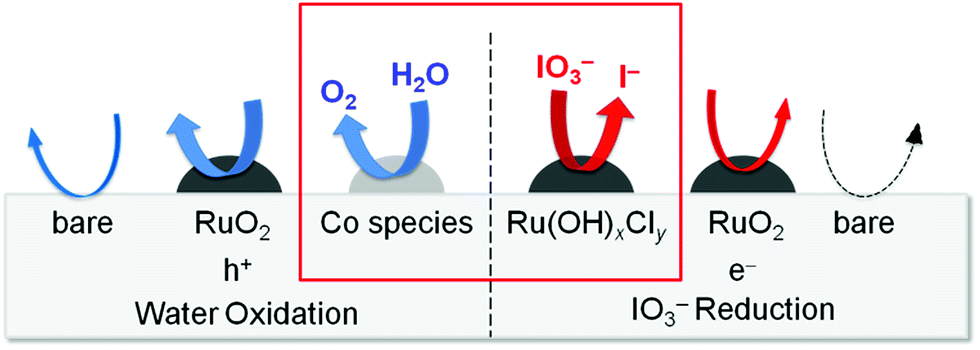
The Ru-200 sample showed a negligibly low activity without loading of any Co species (see Fig. 5). Fig. 7 summarizes the rates of O2 evolution on Ru-x-Co and Ru-x for comparison; the rates were estimated from the amount of O2 evolution at 1–3 h. The Ru-200-Co sample exhibited the highest O2 evolution rate, which was more than twice that on the Ru-300 sample, (the optimum photocatalyst for O2 evolution in the presence of IO3− was the species with only Ru loading). This significant improvement in the O2 evolution activity verifies our hypothesis that the co-loading of the reduction and oxidation sites, which are independently optimized for each reaction, will provide a much higher total efficiency. As seen in Fig. 7, co-loading Co species onto the samples loaded with Ru species at or below 250 °C drastically increased the rate of O2 evolution. In stark contrast, the rates of O2 evolution on the TaON samples loaded with RuO2 (or similar) at or above 300 °C were almost unchanged by the co-loading of the Co species. Fig. 8 summarizes the speculated role and activities of cocatalysts loaded on TaON. The unmodified TaON particles rarely catalyzed the reduction of IO3−, whereas they possess moderate activity for the oxidation of water, which is sufficient for the O2 evolution with sacrificial Ag+ but insufficient for that with the reversible electron acceptor, IO3−. The Ru(OH)xCly species, which were loaded onto TaON by calcination at or below 200 °C possess a significantly higher activity for the reduction of IO3− than conventional RuO2. However, the activity of Ru(OH)xCly species for water oxidation is negligibly low, which is quite different from that of conventional RuO2. Consequently, co-loading of the Ru(OH)xCly species and the Co species promotes IO3− reduction and water oxidation, respectively, which greatly enhances O2 evolution with the IO3− electron acceptor. The reduction of IO3−, which requires six electrons, often becomes the rate-determining step for O2 evolution in Z-scheme water splitting systems with IO3−/I− redox, and thereby necessitates development and loading of effective cocatalyst, such as PtOx and RuO2.5,16 We discovered the high activity of the Ru(OH)xCly (or Ru(OH)x) species, which will provide the opportunity to improve the O2 evolution efficiency on O2-evolving photocatalysts employed in Z-scheme water splitting systems.
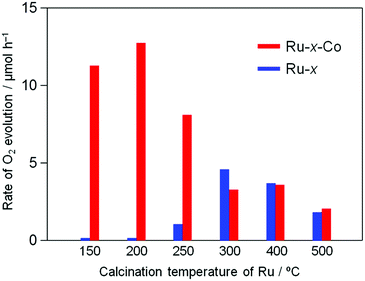 |
| | Fig. 7 Initial rate of O2 evolution on Ru-x and Ru-x-Co photocatalysts (50 mg) prepared at different temperatures in an aqueous solution (250 mL) containing NaIO3 (1.0 mM) under visible light (λ > 400 nm). | |
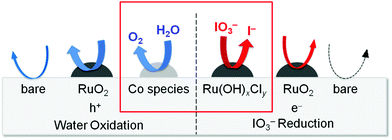 |
| | Fig. 8 Summary of the speculated role and activities of the cocatalysts loaded onto TaON. | |
Conclusions
This study focused on elucidating the catalytic activity of a Ru-based cocatalyst on a TaON photocatalyst for IO3− reduction and enhancing the O2 evolution by co-loading a Co-based cocatalyst that effectively catalyzes water oxidation. The Ru(OH)xCly species obtained by the calcination of RuCl3·nH2O at or below 200 °C showed a much higher activity for IO3− reduction than conventional RuO2, while the Ru(OH)xCly species rarely catalyzed water oxidation. However, even with loading of highly active Ru(OH)xCly (or Ru(OH)x) species, the rate of O2 evolution on TaON was scarcely enhanced, indicating that water oxidation on an unmodified TaON surface was the rate-determining step. Therefore, co-loading Co species that effectively catalyze water oxidation drastically increased the rate of O2 evolution on TaON loaded with Ru(OH)xCly (or Ru(OH)x) species. The optimum combination of each cocatalyst provided almost twice the rate of O2 evolution as that on conventional RuO2-loaded TaON, on which the RuO2 species work as bi-functional cocatalyst. These findings strongly suggest that the co-loading of two different cocatalysts independently optimized for reduction and oxidation can be an effective strategy to enhance the efficiency of water splitting systems. However, the loading site of each cocatalyst has not been controlled in the present case, i.e., some Ru(OH)xCly species are undoubtedly loaded onto the reduction sites of the photocatalyst but others might be loaded onto oxidation sites where photogenerated holes tend to accumulate. Thus, controlled loading of such a cocatalyst onto favourable reaction sites will further improve the overall efficiency.
Acknowledgements
This work was financially supported by the JSPS Grant-in-Aid for Scientific Research (b) (Grant Number 15H03849) and the Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Agency (JST). The authors are indebted to the technical division of Institute for Catalysis, Hokkaido University for their help in building the experimental equipment.
References
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- K. Maeda, T. Takata, M. Hara, N. Saito, N. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS PubMed.
- K. Maeda, H. Terashima, K. Kase, M. Higashi, M. Tabata and K. Domen, Bull. Chem. Soc. Jpn., 2008, 81, 927 CrossRef CAS.
- R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228 CrossRef CAS PubMed.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS PubMed.
- J. S. Jang, H. G. Kim and J. S. Lee, Catal. Today, 2012, 185, 270 CrossRef CAS.
- A. J. Nozik, Appl. Phys. Lett., 1977, 30, 567 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554 CAS.
- A. Iwase, H. Kato and A. Kudo, Chem. Lett., 2005, 34, 946 CrossRef CAS.
- A. Xiong, T. Yoshinaga, T. Ikeda, M. Takashima, T. Hisatomi, K. Maeda, T. Setoyama, T. Teranishi and K. Domen, Eur. J. Inorg. Chem., 2014, 767 CrossRef CAS.
- H. Suzuki, O. Tomita, M. Higashi and R. Abe, Catal. Sci. Technol., 2015, 5, 2640 CAS.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- K. Maeda, R. Abe and K. Domen, J. Phys. Chem. C, 2011, 115, 3057 CAS.
- S. Trasatti and G. Buzzanca, J. Electroanal. Chem., 1971, 29, 1 CrossRef CAS.
- J. Kiwi and M. Grätzel, Angew. Chem., Int. Ed. Engl., 1979, 18, 624 CrossRef.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698 RSC.
- G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 31, 736 CrossRef.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941 CrossRef CAS.
- M. Hara, G. Hitoki, T. Takata, J. N. Kondo, H. Kobayashi and K. Domen, Catal. Today, 2003, 78, 555 CrossRef CAS.
- V. I. Pârvulescu, S. Coman, P. Palade, D. Macovei, C. M. Teodorescu, G. Filoti, R. Molina, G. Poncelet and F. E. Wagner, Appl. Surf. Sci., 1999, 141, 164 CrossRef.
-
V. I. Pârvulescu, V. Pârvulescu, S. Coman, C. Radu, D. Macovei, E. Angelescu and R. Russu, Preparation of Catalysts VI, 1995, p. 561 Search PubMed.
- T. Arikawa, Y. Takasu, Y. Murakami, K. Asakura and Y. Iwasawa, J. Phys. Chem. B, 1998, 102, 3736 CrossRef CAS.
- M. Higashi, R. Abe, A. Ishikawa, T. Takata, B. Ohtani and K. Domen, Chem. Lett., 2008, 37, 138 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416 RSC.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, J. Photochem. Photobiol., A, 2002, 148, 71 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968 CrossRef CAS PubMed.
- M. Higashi, O. Tomita and R. Abe, Top. Catal., 2016, 59, 740 CrossRef CAS.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of electrochemical measurements using TaON loaded with Ru species prepared at different calcination temperatures, the activity of TaON loaded with Co species for O2 evolution from aqueous AgNO3, XPS spectrum of TaON loaded with Co species etc. are shown. See DOI: 10.1039/c7se00110j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Osamu
Tomita
a,
Osamu
Tomita