
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nitride–TiO2 hybrid modified with hydrogenase for visible light driven hydrogen production†
Christine A.
Caputo
a,
Lidong
Wang
b,
Radim
Beranek
b and
Erwin
Reisner
*a
aChristian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, Cambridge University, Lensfied Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk; Web: http://www-reisner.ch.cam.ac.uk
bFaculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstraße 150, 44780 Bochum, Germany
First published on 29th June 2015
Abstract
A system consisting of a [NiFeSe]–hydrogenase (H2ase) grafted on the surface of a TiO2 nanoparticle modified with polyheptazine carbon nitride polymer, melon (CNx) is reported. This semi-biological assembly shows a turnover number (TON) of more than 5.8 × 105 mol H2 (mol H2ase)−1 after 72 h in a sacrificial electron donor solution at pH 6 during solar AM 1.5 G irradiation. An external quantum efficiency up to 4.8% for photon-to-hydrogen conversion was achieved under irradiation with monochromatic light. The CNx–TiO2–H2ase construct was also active under UV-free solar light irradiation (λ > 420 nm), where it showed a substantially higher activity than TiO2–H2ase and CNx–H2ase due, in part, to the formation of a CNx–TiO2 charge transfer complex and highly productive electron transfer to the H2ase. The CNx–TiO2–H2ase system sets a new benchmark for photocatalytic H2 production with a H2ase immobilised on a noble- and toxic-metal free light absorber in terms of visible light utilisation and stability.
Introduction
The use of efficient electrocatalysts in artificial photocatalytic schemes has been an area of recent interest for the conversion of protons to hydrogen using sunlight. Specifically, the use of redox enzymes in photocatalytic schemes highlights the importance of investigating the compatibility of biological systems with light harvesting materials and testing the stability of the resultant bio-hybrid assemblies.1 Hydrogenases (H2ases) are the most efficient noble-metal free electrocatalysts for H2 production and achieve a turnover frequency (TOF) of more than 1000 s−1 with a small overpotential.2 H2ases also show impressive H2 production rates and yields in sacrificial photocatalytic schemes in pH neutral aqueous solution.1a In these systems, a photoexcited light absorber provides electrons to the protein via an internal wire, the iron–sulfur electron relay, to the active site where proton reduction occurs. Examples are the immobilization of a H2ase on Ru-sensitised TiO2,3 on Cd-based quantum dots4 as well as homogeneous systems using the H2ase with a covalently linked photosystem I5 or in combination with an organic dye,6 and multi-component systems with a dye and a soluble redox mediator.7Polymeric carbon nitride (polyheptazine or melon, herein CNx) is a promising visible-light absorber for the photocatalytic generation of H2.8 We have recently reported the use of CNx as a light harvesting material in combination with a H2ase and a H2ase-inspired synthetic Ni catalyst for solar H2 generation.9 The CNx–H2ase system showed sustained catalysis with a turnover number (TON) of more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 after 70 h solar light irradiation. However, this hybrid system suffered from a weak interaction between the H2ase and the CNx surface, and consequently, poor electron transfer from CNx to the H2ase. Furthermore, CNx–H2ase only showed efficient H2 production up to wavelengths of approximately 420 nm and therefore only limited visible light harvesting capabilities.
000 after 70 h solar light irradiation. However, this hybrid system suffered from a weak interaction between the H2ase and the CNx surface, and consequently, poor electron transfer from CNx to the H2ase. Furthermore, CNx–H2ase only showed efficient H2 production up to wavelengths of approximately 420 nm and therefore only limited visible light harvesting capabilities.
Here, we selected a hybrid material consisting of TiO2 (Hombikat UV 100, anatase, BET surface area: 300 m2 g−1, crystallite size < 10 nm) surface-modified with CNx polymer as a light absorbing hybrid material for the photocatalytic system with a H2ase for three main reasons (Fig. 1; see ESI and Fig. S1† for synthesis and characterisation). Firstly, CNx–TiO2 can be readily prepared on a gram scale by heating TiO2 nanoparticles in the presence of urea, an inexpensive and sustainable material.10
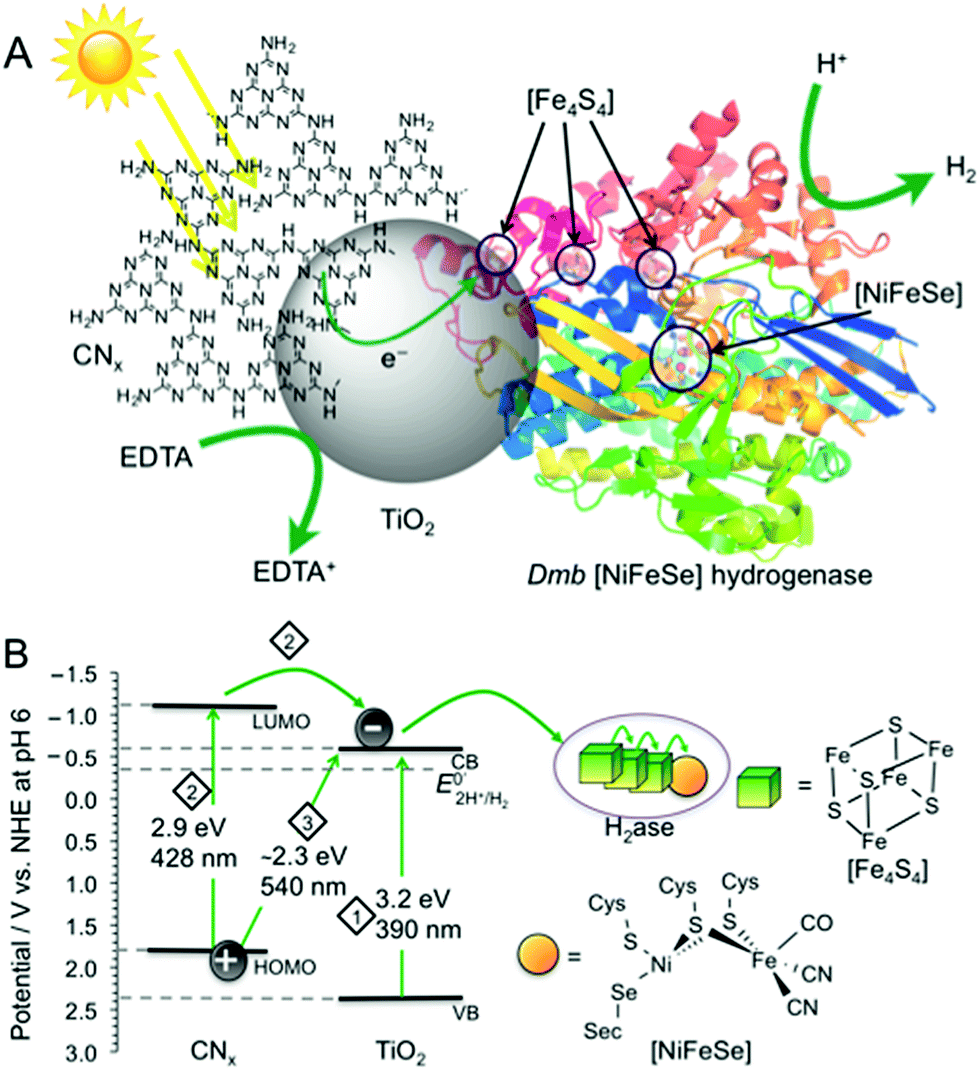 | ||
| Fig. 1 (A) Schematic representation of photo-H2 production with Dmb [NiFeSe]–H2ase (PDB ID:1CC1)14 on CNx–TiO2 suspended in water containing EDTA as a hole scavenger. (B) Irradiation of CNx–TiO2 can result in photo-induced electron transfer by three distinct pathways: (1) TiO2 band gap excitation (2) excitation of CNx (HOMOCNx–LUMOCNx), followed by electron transfer from LUMOCNx into the conduction band of TiO2 (CBTiO2). (3) Charge transfer excitation with direct optical electron transfer from HOMOCNx to CBTiO2. The CBTiO2 electrons generated through pathways 1 to 3 are then transferred via the [Fe4S4] clusters to the [NiFeSe] H2ase active site. | ||
Secondly, CNx–TiO2 provides us with substantially improved solar light harvesting performance compared to individual CNx and TiO2. Band gap excitation of TiO2 (pathway 1; Fig. 1) efficiently utilises the UV spectrum (band gap of 3.2 eV for anatase TiO2 with CBTiO2 at approximately −0.6 V vs. NHE at pH 6).11 A significant portion of the visible spectrum is utilised with CNx–TiO2 as it can, upon photo-excitation of CNx, perform photoinduced electron transfer from the LUMOCNx to CBTiO2 (pathway 2). In addition, direct optical electron transfer can occur from the HOMOCNx (with contributions of molecular orbitals formed upon interaction of CNx with TiO2)12 directly to the CBTiO2 (pathway 3), extending the absorption even further into the visible region (up to 540 nm). This absorption pathway 3 is based on strong coupling between CNx covalently grafted onto TiO2, resulting in strong charge-transfer absorption. Conclusive evidence of this charge-transfer includes previously reported spectroscopic, photoelectrochemical, and theoretical investigations.12,13 The generated CBTiO2 electrons provide the H2ase with an overpotential of approximately 0.2 V for proton reduction.
Thirdly, the H2 evolution catalyst employed in this study, Desulfomicrobium baculatum (Dmb) [NiFeSe]–hydrogenase is not only known for its high H2 evolution activity, lack of H2 inhibition and O2-tolerance,6,14b,14c,15 but also for its titaniaphilicity.3a This high affinity of the enzyme to adsorb strongly to TiO2 stems presumably from a protein surface rich in glutamatic and aspartic acid residues close to the distal [Fe4S4] cluster, which act as anchor sites to TiO2 and allow for stable binding and efficient electron flow into the hydrogenase active site (Fig. 1A).1a,3a Thus, the CNx–TiO2 hybrid is expected to support a more robust H2ase-particle interaction than with CNx alone, which would result in improved charge transfer and ultimately increased catalytic turnover for H2 production.
Results and Discussion
Photocatalytic systems were assembled by dispersing CNx–TiO2 particles in an aqueous electron donor solution (0.1 M; 2.98 mL) in a photoreactor vessel (headspace volume: 4.74 mL; see ESI† for experimental details). The vessel was sonicated under air (15 min) before sealing and purging with an inert gas (2% CH4 in N2). The H2ase (16.5 μL, 3 μM) was then added and the photo-reactor purged again to ensure anaerobic conditions. The stirred suspension was irradiated at 25 °C with a solar light simulator (air mass 1.5 global filter, I = 100 mW cm−2) and the headspace H2 was quantified at regular time intervals by gas chromatography against the internal CH4 standard. The conditions were optimised for maximum turnover frequency (TOFH2ase) by varying the electron donor and pH of the solution (Table S1; Fig. S2 and S3†). Optimised conditions consisted of ethylenediamine tetraacetic acid (EDTA; 0.1 M) as the electron donor at pH 6. A ratio of semiconductor (5 mg unless otherwise noted) to H2ase (50 pmol) was used for ease of comparison to previously reported photosystems with Dmb [NiFeSe]–H2ase.3,6,9Solar (UV-visible) irradiation (λ > 300 nm) of CNx–TiO2–H2ase under standard conditions generated an initial TOFH2ase of (2.8 ± 0.3) × 104 h−1 or 8 s−1 with the production of 5.85 ± 0.59 μmol H2 after 4 h and 28 ± 3 μmol H2 with an overall TONH2ase > (5.8 ± 0.6) × 105 after 72 h (Fig. 2 and S4†). Negligible amounts of H2 were detected in the absence of H2ase, CNx–TiO2 or EDTA. UV band gap excitation of TiO2 did not result in the accumulation of O2, which suggests that holes generated upon UV band gap excitation of TiO2 are either efficiently quenched by EDTA directly or scavenged after being trapped by CNx.
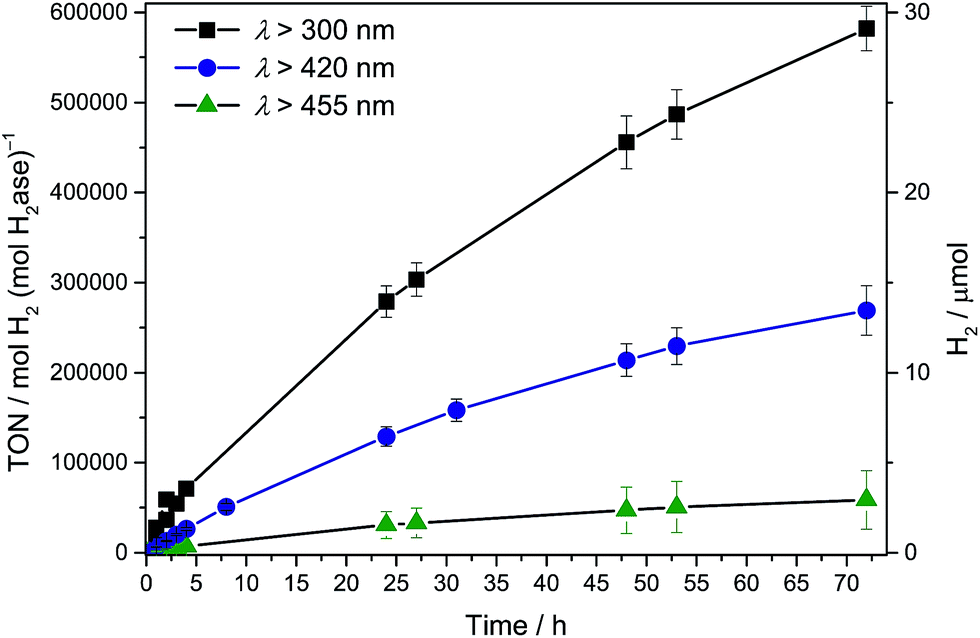 | ||
| Fig. 2 Photocatalytic H2 production with Dmb [NiFeSe]–H2ase (50 pmol) with CNx–TiO2 (5 mg) in EDTA (pH 6, 0.1 M, 3 mL) under AM 1.5G irradiation at an intensity of 1 Sun at λ > 300, 420 and 455 nm. | ||
To qualitatively determine the contributions from the three excitation pathways in Fig. 1B, irradiation was also performed with different long-pass filters. The CNx–TiO2–H2ase system was studied under visible light irradiation at λ > 420 nm to study the contribution of CNx to light absorption (pathways 2 & 3) without the contribution of intrinsic absorption by TiO2 (pathway 1). A photoactivity with an initial TOFH2ase of 6353 ± 635 h−1 was observed, which results in the generation of 1.31 ± 0.13 μmol H2 after 4 h. After 72 h, 13 ± 1 μmol of H2 were generated with a TONH2ase of more than (2.6 ± 0.3) × 105 (Fig. 2).
Subsequently, irradiation was carried out at λ > 455 nm to investigate the contribution of the direct charge-transfer from the HOMOCNx to CBTiO2 to the photoactivity. A TOFH2ase of 1096 ± 175 h−1 with the evolution of 0.26 ± 0.06 μmol H2 after 4 h and 2.9 ± 1.6 μmol H2 after 72 h was observed, which corresponds to 17% of the visible light activity. This suggests that all three pathways in Fig. 1B contribute to the UV-vis photoactivity, whereas pathways 2 and 3 are responsible for the visible-light response of CNx–TiO2–H2ase. Previous investigations of CNx–TiO2 hybrids have shown that their activity is limited by the strong electronic coupling between CNx and TiO2 leading not only to intense visible light absorption but also to fast back electron transfer (primary recombination).13,16
In order to study the role of TiO2 as heterogeneous electron relay in CNx–TiO2–H2ase in more detail, a sample of CNx–ZrO2 (15 mg) was also tested with the H2ase. The negative CBZrO2 at approximately −1.35 V vs. NHE at pH 6, prevents electron injection from LUMOCNx (approximately −1.25 V vs. NHE at pH 6).17 This band level mismatch allowed us to demonstrate that spatial proximity of surface-bound H2ase to CNx alone cannot promote productive electron transfer as no H2 was observed with CNx–ZrO2–H2ase (λ > 300 nm; Fig. S4†). Thus, charge transfer from the LUMOCNx into CBZrO2 (pathway 2) is not possible, nor is the direct electron transfer from HOMOCNx to CBZrO2 (pathway 3), which are crucial to the formation of H2 with the hybrid material.
For comparison, H2 production was also tested with CNx (5 mg) and H2ase (50 pmol) in the absence of metal oxide under standard conditions. A TONH2ase of 14852 ± 1485 was obtained after 4 h with an initial TOF of 6288 ± 649 h−1 when irradiated with UV-visible light (λ > 300 nm, Table S1†). Under visible light irradiation (λ > 420 nm), a TONH2ase of 2375 ± 267 was observed after 4 h and no H2 was produced at λ > 455 nm, demonstrating the substantially enhanced activity with CNx–TiO2–H2ase compared to CNx–H2ase at all wavelengths (Fig. S4†).
Experiments were also performed with TiO2–H2ase. While the system showed comparable activity under UV-visible irradiation due to efficient band gap excitation of TiO2 (pathway 1), it showed significantly reduced activity under visible only irradiation at λ > 420 nm and displayed negligible H2 yields at λ > 455 nm compared to CNx–TiO2–H2ase (Fig. S4†).9 Thus, UV-band gap excitation of TiO2 dominates the absorption of the CNx–TiO2–H2ase hybrid material under UV-light irradiation, which becomes less significant under visible irradiation.
The effect of light intensity on the photocatalytic activity (λ > 300 nm) was studied by employing neutral density filters. A photoactivity of approximately 90% remained when employing a 50% absorbance filter (50 mW cm−2) and 44% of activity remained with an 80% filter (20 mW cm−2; Fig. S5†). The initial non-linear decrease in activity implies that the system is not limited by light at 1 Sun intensity as has been observed previously with synthetic H2 evolution catalyst-modified Ru dye-sensitised TiO2 systems.18
The CNx–TiO2–H2ase system sets a new benchmark for visible light driven and prolonged H2 production with a heterogenised H2ase without the need for expensive or toxic materials.3,4,9 A part of this improvement can be attributed to the direct optical electron transfer (pathway 3) within CNx–TiO2, which draws the absorption of solar light significantly into the visible spectrum.
The enzyme loading onto CNx–TiO2 was calculated based on the BET surface area of 111 m2 g−1, a crystallite surface area of ∼314 nm2 per particle and an estimation that approximately one-quarter of the surface area of TiO2 is accessible for the enzyme to adsorb. This equates to ∼0.1 H2ase per particle of CNx–TiO2. The approximate 1:10 enzyme:particle ratio allows the H2ase to function at the maximum rate (i.e., TOF) as the maximum electron flux of conduction band electrons is directed towards a single enzyme. To qualitatively determine the amounts of surface-bound and solubilised H2ase in the optimised system, H2ase (50 pmol) was loaded onto CNx–TiO2 (5 mg) in aqueous EDTA solution by stirring under N2 for 15 min. The suspension was centrifuged and the supernatant decanted (see ESI† for experimental details). The CNx–TiO2–H2ase pellet was re-dispersed in fresh EDTA solution (3 mL, 0.1 M, pH 6) and the photocatalytic vessel purged with 2% CH4 in N2. The suspension was then irradiated (λ > 420 nm) and H2 production monitored (Fig. 3). The H2 production activity was nearly identical to a sample that was not centrifuged, both in the presence and absence of methyl viologen (MV2+, see below), indicating that attachment of H2ase to CNx–TiO2 is essentially quantitative. The substantially improved adsorption of the enzyme on the TiO2 surface compared to the inert CNx polymer therefore also contributes to the increased activity of CNx–TiO2–H2ase compared to CNx–H2ase. Previously an 88% decrease in photoactivity was observed with the poorly interacting CNx–H2ase after centrifugation and re-dispersion in fresh electron donor buffer.9
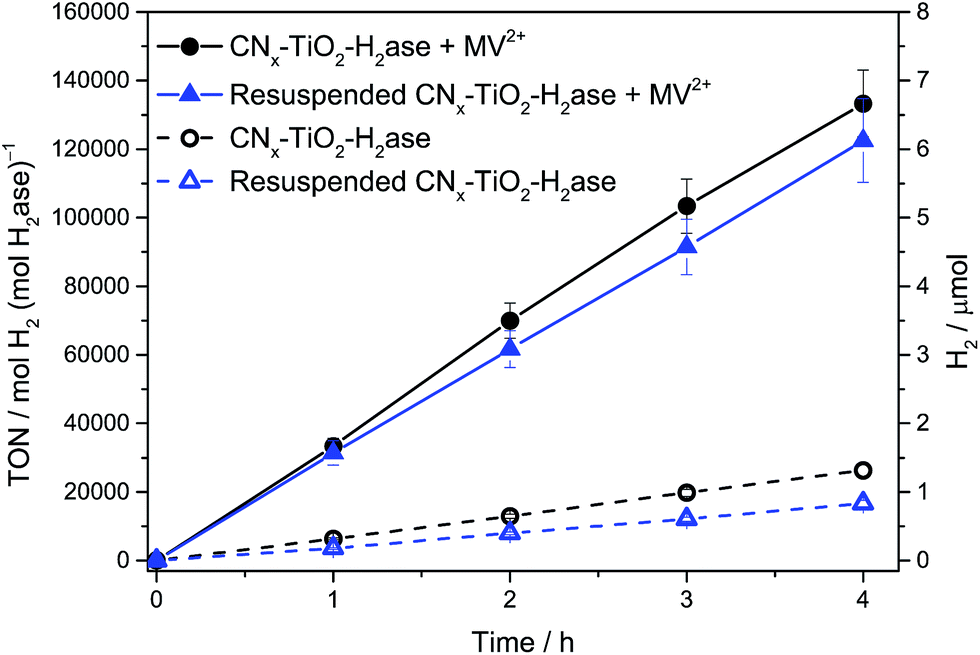 | ||
| Fig. 3 Photocatalytic H2 production using Dmb [NiFeSe]–H2ase (50 pmol) in EDTA (pH 6, 0.1 M, 3 mL) with CNx–TiO2 (5 mg) under optimised conditions before and after centrifugation and re-suspension in fresh EDTA buffer solution followed by 1 Sun irradiation (λ > 420 nm). Results are also shown in the presence and absence of redox mediator, methyl viologen (MV2+). | ||
The external quantum efficiency (EQE) of the CNx–TiO2–H2ase system was measured by applying narrow band pass filters (λ = 360 ± 10 nm; I = 2.49 mW cm−2 and 400 ± 10 nm; I = 4.34 mW cm−2; see ESI† for experimental details). UV-irradiation gave an EQE of approximately 4.8% and under visible irradiation an EQE of 0.51% was obtained. These values are more than a 10-fold improvement over the UV and visible EQE for the CNx–H2ase system,9 which can be attributed to the improved light absorption (Fig. S6†) and increased electron transfer rate due to adsorption of the H2ase onto the particle surface.
We previously showed that a significantly increased photoactivity was observed under standard conditions using CNx–H2ase upon addition of an excess of the redox mediator MV2+, producing up to 77 μmol H2 after 69 h of UV-visible irradiation.9 A long-term experiment with H2ase (50 pmol), CNx–TiO2 (5 mg) and added MV2+ (5 μmol) in aqueous EDTA (0.1 M) at pH 6 was performed with both λ > 300 nm light and with visible light only (λ > 420 nm). Under UV-visible irradiation after 72 h, the CNx–TiO2–MV–H2ase system produced 193 μmol H2 with a TONH2ase of > 3.8 × 106 and an initial TOFH2ase of 35 s−1 (Fig. S7†). Under visible-light only, 66 μmol H2 was produced with a TONH2ase of 1.3 × 106 and an initial TOFH2ase of 9 s−1 (Fig. S8†). The ratio of the amount of hydrogen produced in the presence and absence of MV2+ can be used to estimate the relative efficiency of the charge transfer from material to H2ase. Under full spectrum irradiation (λ > 300 nm) with CNx–H2ase the ratio was found to be 22, whereas for both TiO2–H2ase and CNx–TiO2–H2ase systems the ratio was 5. This strongly supports the fact that there is a significant improvement in the charge transfer from a TiO2-based material to H2ase. In addition, this ratio remains constant when the wavelength of light used is restricted to the visible region (λ > 420 nm).
The H2 production rates in the presence of MV2+ are significantly higher than those obtained in the absence of MV2+. The blue colour of the vials containing MV2+ is indicative of the formation of reduced MV+˙ in solution (Fig. S9†). By comparison, addition of MV2+ to the previously reported Ru-dye-sensitised TiO2–H2ase system caused a slight decrease in activity, which was attributed to the decreased availability of electrons for the H2ase and the absorption of incident photons by MV+˙.3a Here, solubilised MV+˙ does not limit light absorption by CNx–TiO2 significantly and is able to efficiently donate electrons to surface-bound H2ase, resulting in increased H2 production. This result implies that interfacial electron transfer from CNx–TiO2 to H2ase is still not fully optimised in this system, where the orientation of the H2ase is not fully ‘directed’. Ideally, the distance from the CNx–TiO2 surface to the [Fe4S4] electron transport chain should be minimised and an improved orientation of the enzyme would allow trapping of CBTiO2 electrons more efficiently for maximised turnover.19
Favourable electron transfer kinetics at the CNx–TiO2–H2ase interface can be assumed based on previous reports. Electron transfer in the order of 107 s−1 was reported from CdS nanorods to an [FeFe]–H2ase isolated from Clostridium acetobutylicum.4c In addition, a long lived photo-excited state lifetime of τ1/2 ∼ 0.8 s was previously reported for TiO2 conduction band electrons in a photocatalytic system with Ru dye-sensitised TiO2 and electron transfer to co-immobilised molecular cobaloxime catalysts occurred with τ1/2 ∼ 5 to 50 μs.20 Based on these reports, we can assume that a reasonably long-lived TiO2 conduction band electron is generated and that H2ase is capable of readily collecting these electrons.
Conclusions
In summary, solar light driven H2 production with a semi-biological system consisting of TiO2 modified with polymeric CNx and immobilised H2ase has been demonstrated. We have shown that by improving the surface interaction of the enzyme with the light harvesting CNx material, specifically by adsorption of the enzyme onto the TiO2 surface, H2 generation is drastically improved. Another important factor is the improved visible light absorption by direct CNx excitation (pathway 2) and CNx–TiO2 charge transfer (pathway 3), which enables high photoactivity. The CNx–TiO2–H2ase assembly achieved a TOF of 8 s−1 and TON of > 5.8 × 105 after 72 h in the absence of an external soluble redox mediator, thereby setting a new benchmark for photochemical architectures based on abundant and non-toxic materials and a heterogenised H2ase. The additional use of the redox mediator MV2+ allowed for the photo-generation of H2 with a TOF of 35 s−1 and a TON of > 3.8 × 106. This work advances the use of hybrid photocatalytic schemes by integrating highly active electrocatalysts with advanced light absorbing materials such as CNx–TiO2, which is shown to be compatible with H2ases in aqueous solution.Acknowledgements
We acknowledge support by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), the OMV Group and a Marie Curie fellowship to C.C. (GAN 624997624997). R.B. and L.W. acknowledge financial support by the MIWFT-NRW within the project “Anorganische Nanomaterialien für Anwendungen in der Photokatalyse”. We thank Dr J. C. Fontecilla-Camps and Dr C. Cavazza (CNRS Grenoble, France) for providing us with Dmb [NiFeSe] hydrogenase, Ms Marielle Bauzan (CNRS Marseilles, France) for growing the bacteria, and Dr Michal Bledowski for assistance with CNx–TiO2 synthesis.Notes and references
- (a) E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS; (b) P. W. King, Biochim. Biophys. Acta, 2013, 1827, 949–957 CrossRef CAS PubMed.
- (a) F. A. Armstrong, N. A. Belsey, J. A. Cracknell, G. Goldet, A. Parkin, E. Reisner, K. A. Vincent and A. F. Wait, Chem. Soc. Rev., 2009, 38, 36–51 RSC; (b) A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC; (c) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- (a) E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed; (b) E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- (a) B. L. Greene, C. A. Joseph, M. J. Maroney and R. B. Dyer, J. Am. Chem. Soc., 2012, 134, 11108–11111 CrossRef CAS PubMed; (b) A. Bachmeier, V. C. C. Wang, T. W. Woolerton, S. Bell, J. C. Fontecilla-Camps, M. Can, S. W. Ragsdale, Y. S. Chaudhary and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 15026–15032 CrossRef CAS PubMed; (c) M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef CAS PubMed.
- (a) C. E. Lubner, R. Grimme, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 404–414 CrossRef CAS PubMed; (b) H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed; (c) M. Ihara, H. Nishihara, K.-S. Yoon, O. Lenz, B. Friedrich, H. Nakamoto, K. Kojima, D. Honma, T. Kamachi and I. Okura, Photochem. Photobiol., 2006, 82, 676–682 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- (a) I. Okura, Coord. Chem. Rev., 1985, 68, 53–99 CrossRef CAS; (b) O. A. Zadvornyy, J. E. Lucon, R. Gerlach, N. A. Zorin, T. Douglas, T. E. Elgren and J. W. Peters, J. Inorg. Biochem., 2012, 106, 151–155 CrossRef CAS PubMed.
- (a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed; (b) D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed; (c) M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- (a) R. Beranek and H. Kisch, Photochem. Photobiol. Sci., 2008, 7, 40–48 RSC; (b) D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 9975–9978 CrossRef CAS PubMed.
- (a) A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed; (b) M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed; (c) Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- M. Bledowski, L. Wang, A. Ramakrishnan, O. V. Khavryuchenko, V. D. Khavryuchenko, P. C. Ricci, J. Strunk, T. Cremer, C. Kolbeck and R. Beranek, Phys. Chem. Chem. Phys., 2011, 13, 21511–21519 RSC.
- M. Bledowski, L. Wang, A. Ramakrishnan, A. Bétard, O. V. Khavryuchenko and R. Beranek, ChemPhysChem, 2012, 13, 3018–3024 CrossRef CAS PubMed.
- (a) E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS PubMed; (b) A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS PubMed; (c) C. S. A. Baltazar, M. C. Marques, C. M. Soares, A. M. DeLacey, I. A. C. Pereira and P. M. Matias, Eur. J. Inorg. Chem., 2011, 948–962 CrossRef CAS.
- (a) M. C. Marques, R. Coelho, A. L. De Lacey, I. A. C. Pereira and P. M. Matias, J. Mol. Biol., 2010, 396, 893–907 CrossRef CAS PubMed; (b) A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061–7063 RSC.
- (a) L. Wang, M. Bledowski, A. Ramakrishnan, D. König, A. Ludwig and R. Beranek, J. Electrochem. Soc., 2012, 159, H616–H622 CrossRef CAS; (b) M. Bledowski, L. Wang, S. Neubert, D. Mitoraj and R. Beranek, J. Phys. Chem. C, 2014, 118, 18951–18961 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533 CrossRef CAS.
- F. Lakadamyali, M. Kato and E. Reisner, Faraday Discuss., 2012, 155, 191–205 RSC.
- A. Bachmeier and F. Armstrong, Curr. Opin. Chem. Biol., 2015, 25, 141–151 CrossRef CAS PubMed.
- (a) F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed; (b) A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02017d |
| This journal is © The Royal Society of Chemistry 2015 |