
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective hydrogenation of CO2 into C2+ alcohols by homogeneous catalysis†
Qingli
Qian
*,
Meng
Cui
,
Zhenhong
He
,
Congyi
Wu
,
Qinggong
Zhu
,
Zhaofu
Zhang
,
Jun
Ma
,
Guanying
Yang
,
Jingjing
Zhang
and
Buxing
Han
*
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qianql@iccas.ac.cn; hanbx@iccas.ac.cn; Tel: +86-10-62562821
First published on 10th July 2015
Abstract
The hydrogenation of CO2 to produce alcohols with two or more carbons (C2+ alcohols) is of great importance, but is challenging. In this work, we found that a Ru3(CO)12/Rh2(CO)4Cl2–LiI system could catalyze the reaction effectively in 1,3-dimethyl-2-imidazolidinone (DMI) under mild conditions. Methanol, ethanol, propanol, 2-methyl propanol, butanol, and 2-methyl butanol were produced in the homogeneous catalytic reaction. The C2+ alcohols could be generated at 160 °C, which is the lowest temperature reported so far for producing C2+ alcohols via CO2 hydrogenation. The selectivity for the C2+ alcohols could be as high as 96.4% at the optimized conditions, which is higher than those reported in the literature. In addition, the catalytic system could be easily recycled. The route of the reaction for forming the C2+ alcohols was discussed on the basis of control experiments.
Introduction
Carbon dioxide (CO2) is a renewable, abundant, and cheap C1 feedstock.1 The use of CO2 as a carbon source to produce fuels and value-added chemicals is of great importance for the sustainable development of our society. The transformation of CO2 into various compounds, such as urea, salicylic acid, carbonates, polymers, alcohols, and formic acid, has been studied extensively.2The hydrogenation of CO2 to produce alcohols is one of the most important routes for converting CO2, which has received much attention, especially for producing methanol.3 In many cases, ethanol and larger alcohols (C2+ alcohols hereafter) are more desirable products, as neat fuels, fuel additives, and chemicals, than methanol.4 However, producing C2+ alcohols by CO2 hydrogenation is more difficult than producing methanol. Up to now, heterogeneous catalysts have been designed and used in the synthesis of C2+ alcohols by the catalytic hydrogenation of CO2, and some excellent results have been obtained.5–15 For example, it was found that alkali-promoted Mo/SiO2 catalysts could catalyze CO2 hydrogenation to form C2+ alcohols at 250 °C. The content of C2+ alcohols in the alcohol mixture could be 75.6%.5 Supported Rh, Fe-based, and Cu-based catalysts were combined for the synthesis of C2+ alcohols, and the multi-functional heterogeneous catalysts could promote the reaction effectively at 330–370 °C. The major C2+ alcohol was ethanol and the highest selectivity of C2+ alcohols in the alcohols was about 70%.6 Kurakata et al.7 reported that [Rh10Se]/TiO2 could promote the hydrogenation of CO2 to produce ethanol at temperatures from 250 to 450 °C, and the highest ethanol selectivity was 83%. Nieskens et al.8 fabricated a CoMoS based catalyst for synthesizing C1–C3 alcohols via CO2 hydrogenation at 340 °C, and the highest C2+ alcohol content in the alcohol mixture was 35.6%. Li et al.9 prepared a K/Cu–Zn–Fe catalyst, which was used in the reaction at 300 °C. The selectivity to C2+ alcohols reached 87.1%. Tominaga et al.16 reported CO2 hydrogenation using a Ru–Co homogeneous catalyst at 200 °C, and only methanol and ethanol were formed, with an ethanol selectivity of 26.4% in the alcohol products.
As discussed above, the synthesis of C2+ alcohols by the hydrogenation of CO2 has received considerable attention. However, in general, the heterogeneous catalysts suffer from low activity, low C2+ alcohol selectivity, and high reaction temperature. There is no doubt that exploration of the routes for highly selective CO2 hydrogenation to produce C2+ alcohols under relatively mild conditions is of great importance. In this work, we studied the hydrogenation of CO2 into C1–5 alcohols catalyzed by a Ru–Rh bimetallic homogeneous catalyst using LiI as the promoter (Scheme 1).
 | ||
| Scheme 1 Synthesis of C2+ alcohols from CO2 hydrogenation. | ||
It was found that the catalytic system could catalyze the reaction effectively under mild conditions. The liquid products were mainly methanol, ethanol, propanol, 2-methyl propanol, butanol and 2-methyl butanol, including both linear and branched alcohols. The products were distinct from those generated via homogeneous CO2 or CO hydrogenation reported in the literature. The alcohols could be generated at 160 °C. The selectivity for C2+ alcohols could be as high as 96.4% at the optimized conditions. In addition, the catalytic system could be recycled and reused.
Results and discussion
Different catalytic systems were tested for the reaction, and the results are listed in Table 1. The corresponding selectivities for the different alcohols are shown in detail in Table S1.† When LiI was used as the promoter and 1,3-dimethyl-2-imidazolidinone (DMI) as the solvent, the combined catalyst Ru3(CO)12/Rh2(CO)4Cl2 could catalyze the hydrogenation of CO2 very effectively, with methanol, ethanol, propanol, 2-methyl propanol, butanol, and 2-methyl butanol as the products, and other products being negligible in the reaction solution (Fig. S1†). The alcohol products, including both linear and branched C1–C5 alcohols, are different from those obtained via CO hydrogenation using Ru and/or Rh catalysts, since they give largely linear hydrocarbons over heterogeneous catalysts, and produce only C1 and C2 oxygenates in homogeneous catalysis.17 Very interestingly, the selectivity for C2+ alcohols could be as high as 96.4% (Entry 1), which is much higher than those reported in the literature.| Entry | Catalyst | Promoter | Solvent | STYc of alcohols | C2+OH% |
|---|---|---|---|---|---|
| a Reaction conditions: 28.2 μmol Ru catalyst and 51.5 μmol Rh catalyst (based on the metal), 2.26 mmol promoter, 2 mL solvent, 4 MPa CO2 and 4 MPa H2 (at room temperature), 200 °C and 12 h. b Precipitate was observed after the reaction. c STY stands for space time yield (C mmol L−1 h−1), which is one of the commonly used units, especially when multi-metals are utilized. | |||||
| 1 | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | DMI | 12.86 | 96.4 |
| 2b | Ru3(CO)12, Rh2(CO)4Cl2 | — | DMI | 0.36 | 2.8 |
| 3 | Ru3(CO)12, Rh2(CO)4Cl2 | KI | DMI | 14.36 | 8.3 |
| 4 | Ru3(CO)12, Rh2(CO)4Cl2 | LiCl | DMI | 16.17 | 17.7 |
| 5 | Ru3(CO)12 | LiI | DMI | 2.43 | 0.4 |
| 6b | Rh2(CO)4Cl2 | LiI | DMI | 1.07 | 2.9 |
| 7 | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | NMP | 5.11 | 72.4 |
| 8b | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | 1-Methyl piperidine | 2.07 | 0.0 |
| 9b | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | DMF | 7.64 | 0.0 |
| 10b | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | THF | 0.0 | — |
| 11b | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | Cyclohexane | 0.0 | — |
| 12b | Ru3(CO)12, Rh2(CO)4Cl2 | LiI | Water | 1.45 | 6.5 |
| 13b | RuCl3·3H2O, Rh2(CO)4Cl2 | LiI | DMI | 2.73 | 7.4 |
| 14b | Ru3(CO)12, RhCl3·xH2O | LiI | DMI | 3.38 | 5.7 |
| 15b | Ru3(CO)12, Rh6(CO)16 | LiI | DMI | 3.40 | 25.4 |
The promoter LiI played an important role in accelerating the reaction. Without the promoter, a little amount of methanol was generated, and the amount of the C2+ alcohols was negligible (Entry 2). In the presence of LiI, the reaction solution was clear after reaction, but black fine metal powder was found when LiI was not added, indicating that LiI could stabilize the catalyst. When LiI was replaced by KI, the catalyst was also stable at the reaction conditions with a high yield of methanol, but the selectivity to C2+ alcohols was very low (Entry 3). The results show that the promoter affected the activity, selectivity, and stability of the catalyst. The superiority of LiI in promoting the synthesis of C2+ alcohols may be partly attributed to the stronger Lewis acidity of the lithium cation, which could offer suitable coordination sites during the catalytic reaction. The anionic counterpart of the promoter also evidently influenced the catalytic performance. When LiCl was used, the selectivity for C2+ alcohols was much lower (Entry 4). The better performance of the iodide anion may be ascribed to its stronger nucleophilicity, which would promote the chain growth reaction.
We also used Ru3(CO)12 (Entry 5) and Rh2(CO)4Cl2 (Entry 6) separately, but the yield of the product and the selectivity to the C2+ alcohols was very low, indicating the synergistic effect of the two catalysts in accelerating the reaction. Thus we choose the space time yield (STY) to express the catalytic activity, which may give an integrated evaluation of the bimetallic catalytic system. The reaction was also carried out in other solvents, and it was demonstrated that DMI was the best solvent for the reaction (Entries 1 and 7–12). One of the main reasons is that the catalyst was stable in DMI, but it was not stable in most of the other solvents used. In N-methyl-2-pyrrolidone (NMP), the catalyst was also stable, but the efficiency of the reaction was lower than that in DMI. This indicates that the solvent effect is also important for the reaction. Using LiI as the promoter and DMI as the solvent, the performance of other mixed catalysts, such as RuCl3·3H2O/Rh2(CO)4Cl2, Ru3(CO)12/RhCl3·xH2O, and Ru3(CO)12/Rh6(CO)16, were also studied (Entries 13–15), but the efficiencies were lower than that of Ru3(CO)12/Rh2(CO)4Cl2 because of their poor stability. The results above indicate that the catalytic system composed of Ru3(CO)12/Rh2(CO)4Cl2, LiI, and DMI had good activity, selectivity, and stability for the hydrogenation of CO2 to generate C2+ alcohols. Therefore, the effects of the reaction conditions were further studied using this catalytic system.
Fig. 1 depicts the results of the CO2 hydrogenation conducted at different temperatures. At 150 °C, only methanol and ethanol were formed, and methanol was the major product. When the temperature reached 160 °C, the yields of methanol and ethanol increased, and C3+ alcohols emerged. Thus, 160 °C was the initial temperature for the obvious formation of C3+ alcohols. So far this is the lowest temperature reported for the formation of these alcohols. The yields of all the alcohols increased as the temperature rose. From 180 to 200 °C, the methanol yield underwent a dramatic drop, accompanied with an evident increase in the yields of the target C2+ alcohols. In the range of 200–220 °C, the yields of methanol and ethanol were nearly unchanged with increasing temperature, but the yields of the other alcohols increased continuously with increasing temperature. The main reason is that the methanol formed can be further transformed into ethanol, and the ethanol can be converted into larger alcohols, which will be discussed in more detail in the following paragraphs. The yield of methanol is much lower than that of ethanol in this temperature range because methanol is more reactive than ethanol. Therefore, the methanol generated was converted into ethanol quickly.
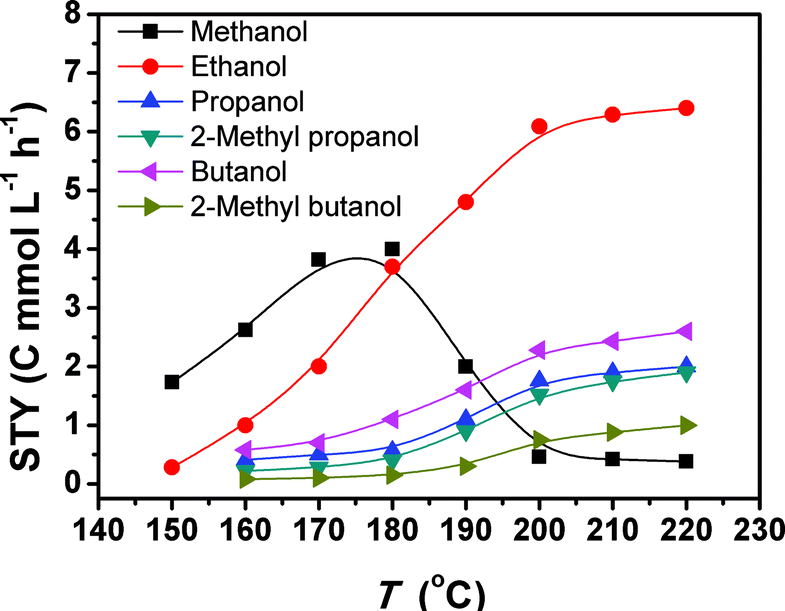 | ||
| Fig. 1 The space time yields (STY) of the alcohols at different temperatures. Reaction conditions: 28.2 μmol Ru3(CO)12 and 51.5 μmol Rh2(CO)4Cl2 (based on the metal), 2.26 mmol LiI, 2 mL DMI, 4 MPa CO2 and 4 MPa H2 (at room temperature), and 12 h. | ||
The results in Fig. 1 suggest that 200 °C is a suitable temperature. We further studied the effects of other parameters on the reaction at this temperature, and the results are given in Table 2. The corresponding selectivities to different alcohols are shown in Table S2.† The C2–C5 alcohols were generated at all the conditions. At a fixed pressure ratio of CO2 and H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the total yield of the alcohols and the selectivity to the C2+ alcohols increased remarkably as the total pressure was raised from 2 MPa to 10 MPa (Entries 1–5). At the same pressure, the total yield of the alcohols increased with the partial pressure of H2 (Entries 4, 6, 7), but the selectivity to C2+ alcohols was highest at a CO2:H2 pressure ratio of 1:1. The dosage of LiI was crucial for the alcohols generation (Entry 2 of Table 1, Entries 4, 8, and 9 of Table 2). When the LiI dosage was in the range of 0–1.13 mmol, both the total yield of the alcohols and the selectivity to C2+ alcohols increased significantly with the increase in the dosage. As the LiI dosage was increased to 2.26 mmol, the amount of the alcohols generated decreased slightly but the C2+ alcohols selectivity increased greatly. However, as the dosage further increased to 3.39 mmol, the selectivity to C2+ alcohols remained high, but the total yield of the alcohols reduced considerably. The main reason may be that more active sites were occupied by iodide anions as an excess amount of LiI was used, inhibiting the hydrogenation reaction. The atomic ratio of Ru and Rh also affected the yield of the reaction. At the same total amount of Ru and Rh (79.7 μmol), 28.2 μmol Ru and 51.5 μmol Rh gave the highest total yield of the alcohols and the selectivity for C2+ alcohols (Entries 5 and 6 of Table 1, Entries 4, 10, 11, 12 of Table 2). As expected, the total yield of the alcohols increased with an increasing amount of the catalyst (Entries 4, 13, 14, 15), but the yield was less sensitive to the amount of catalyst as the amount was large enough.
:1), the total yield of the alcohols and the selectivity to the C2+ alcohols increased remarkably as the total pressure was raised from 2 MPa to 10 MPa (Entries 1–5). At the same pressure, the total yield of the alcohols increased with the partial pressure of H2 (Entries 4, 6, 7), but the selectivity to C2+ alcohols was highest at a CO2:H2 pressure ratio of 1:1. The dosage of LiI was crucial for the alcohols generation (Entry 2 of Table 1, Entries 4, 8, and 9 of Table 2). When the LiI dosage was in the range of 0–1.13 mmol, both the total yield of the alcohols and the selectivity to C2+ alcohols increased significantly with the increase in the dosage. As the LiI dosage was increased to 2.26 mmol, the amount of the alcohols generated decreased slightly but the C2+ alcohols selectivity increased greatly. However, as the dosage further increased to 3.39 mmol, the selectivity to C2+ alcohols remained high, but the total yield of the alcohols reduced considerably. The main reason may be that more active sites were occupied by iodide anions as an excess amount of LiI was used, inhibiting the hydrogenation reaction. The atomic ratio of Ru and Rh also affected the yield of the reaction. At the same total amount of Ru and Rh (79.7 μmol), 28.2 μmol Ru and 51.5 μmol Rh gave the highest total yield of the alcohols and the selectivity for C2+ alcohols (Entries 5 and 6 of Table 1, Entries 4, 10, 11, 12 of Table 2). As expected, the total yield of the alcohols increased with an increasing amount of the catalyst (Entries 4, 13, 14, 15), but the yield was less sensitive to the amount of catalyst as the amount was large enough.
| Entry | Ru/Rh [μmol] | LiI [mmol] | CO2/H2 [MPa] | STY of alcohols | C2+ [%] |
|---|---|---|---|---|---|
| a Reaction conditions: Ru3(CO)12/Rh2(CO)4Cl2 were used as the catalysts and their dosage was based on the metal, LiI was used as the promoter, 2 mL DMI, 200 °C, and 12 h. | |||||
| 1 | 28.2/51.5 | 2.26 | 1/1 | 1.13 | 77.0 |
| 2 | 28.2/51.5 | 2.26 | 2/2 | 3.39 | 90.6 |
| 3 | 28.2/51.5 | 2.26 | 3/3 | 5.37 | 92.6 |
| 4 | 28.2/51.5 | 2.26 | 4/4 | 12.86 | 96.4 |
| 5 | 28.2/51.5 | 2.26 | 5/5 | 14.10 | 96.1 |
| 6 | 28.2/51.5 | 2.26 | 2/6 | 20.66 | 39.0 |
| 7 | 28.2/51.5 | 2.26 | 6/2 | 3.17 | 84.2 |
| 8 | 28.2/51.5 | 1.13 | 4/4 | 14.25 | 40.6 |
| 9 | 28.2/51.5 | 3.39 | 4/4 | 5.88 | 97.1 |
| 10 | 8.0/71.7 | 2.26 | 4/4 | 3.32 | 84.0 |
| 11 | 39.9/39.9 | 2.26 | 4/4 | 12.07 | 76.9 |
| 12 | 55.8/23.9 | 2.26 | 4/4 | 8.57 | 80.4 |
| 13 | 0/0 | 2.26 | 4/4 | 0 | — |
| 14 | 14.1/25.8 | 2.26 | 4/4 | 4.48 | 47.8 |
| 15 | 42.3/77.3 | 2.26 | 4/4 | 16.31 | 93.9 |
We carried out experiments on the reuse of the catalytic system. After reaction, the alcohols generated in the reaction were removed under vacuum, which was confirmed by GC analysis. Then the catalyst, solvent (DMI), and the LiI were used directly for the next run. The results of the reuse experiments are given in Table S3.† The yield of the total alcohols and the selectivity to C2+ alcohols did not change obviously after five cycles (12 h each cycle), indicating that the catalyst was stable for at least 60 h at this temperature.
Fig. 2 presents the time course for the formation of the alcohols. Methanol, ethanol and propanol were generated within 1 h and their yields increased with time. After 6 h, a considerable amount of 2-methyl propanol, butanol, and 2-methyl butanol could be detected and their amounts increased with time. The methanol content began to decrease quickly and the amounts of the higher alcohols increased continuously with the reaction proceeding. After 12 h, the methanol content was low and did not change considerably with time. At the same time, the ethanol content began to decrease slowly, and the content of the C3+ alcohols continued to increase with reaction time. The yield of methanol passed through a maximum with increasing reaction time. With the increase of reaction time, some of the methanol is transformed into ethanol and the ethanol can be further converted, and so on.
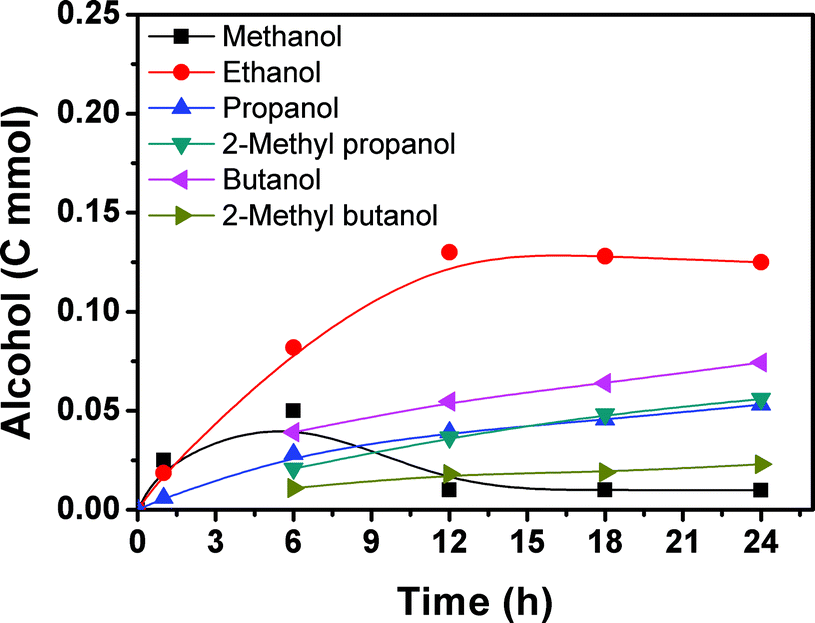 | ||
| Fig. 2 Time course of the alcohol formation. Reaction conditions: 28.2 μmol Ru3(CO)12 and 51.5 μmol Rh2(CO)4Cl2 (based on the metal), 2.26 mmol LiI, 2 mL DMI, 4 MPa CO2 and 4 MPa H2 (at room temperature), and 200 °C. | ||
The results above suggest that methanol was formed from CO2 and H2 in the reaction. The methanol acts as the intermediate for the generation of ethanol, and the ethanol can be converted into larger alcohols in the reaction process. In order to obtain more evidence to support this argument, we carried out tracer experiments by adding small amounts of 13CH3OH or 13C2H5OH in the reaction system at 200 °C with a reaction time of 12 h. The GC-MS results with 13CH3OH and 13C2H5OH are shown in Fig. S2 and S3,† respectively. When 13CH3OH was used as the tracer, C2+ alcohols containing 13C were yielded. Similarly, when the 13C2H5OH tracer was added in the reaction system, 13C was present in some C3+ alcohols. Therefore, it can be concluded that in the hydrogenation of CO2 for obtaining the alcohols, the methanol and ethanol formed act as intermediates for forming the larger alcohols.
Only methanol and ethanol were produced as alcohol products in the homogeneous CO2 hydrogenation.16 It was also reported that only C1 and C2 oxygenates were yielded via CO hydrogenation using homogeneous Ru and/or Rh catalysts.17,18 Whereas the alcohols generated by our catalytic system included C1–C5 alcohols of both linear and branched structures. This suggests that the reaction pathway of CO2 hydrogenation using our catalytic system is obviously different from those of the CO2 or CO hydrogenation reported in the literature.16–18
Conclusions
In summary, we have studied the performance of different catalysts, promoters, and solvents for the synthesis of C2+ alcohols by the hydrogenation of CO2. It is discovered that Ru3(CO)12, Rh2(CO)4Cl2, and LiI exhibit an excellent synergistic effect in catalyzing the reaction using DMI as the solvent. The Ru3(CO)12/Rh2(CO)4Cl2/LiI-DMI homogeneous catalytic system can catalyze the reaction effectively and selectively at relatively mild conditions. The target C2+ alcohols start to form at 160 °C. The selectivity to the C2+ alcohols can reach 96.4%, and the catalytic system can be reused. In the reaction, methanol is first formed, and the small alcohol can act as the intermediate for generating the larger ones. The C2+ alcohols include both linear and branched alcohols, which is distinct from those produced via homogeneous CO2 or CO hydrogenation reported in the literature. We believe that many other catalytic systems can be explored for the hydrogenation of CO2 by combination of various homogeneous catalysts, co-catalysts, and solvents.Experimental
Chemicals
Ruthenium carbonyl (Ru3(CO)12, purity > 98%) was purchased from Adamas Reagent, Ltd. Tetracarbonyl-di-μ-chlorodirhodium(I) (Rh2(CO)4Cl2, Rh 50.1–52.9%), rhodium(III) chloride hydrate (RhCl3·xH2O, Rh 38.5–45.5%), anhydrous lithium iodide (LiI, 99.95%), potassium iodide, (KI, 99.9%), and 1-methyl piperidine (99%) were obtained from Alfa Aesar China Co., Ltd. Hexarhodiumhexadecacarbonyl (Rh6(CO)16, 98%) was provided by J&K Chemical Ltd. (Shanghai). Ruthenium(III) chloride hydrate (RuCl3·3H2O, Ru 36.7%) was provided by Shenyang Jinke Reagent Co., Ltd. 1,3-Dimethyl-2-imidazolidinone (DMI, 99%) was purchased from TCI Shanghai Co., Ltd. N-Methyl-2-pyrrolidone (NMP, 99.5%), N,N-dimethylformamide (DMF, 99.5%) and cyclohexane (99.5%) were provided by Sinopharm Chemical Reagent Co., Ltd. Tetrahydrofuran (THF, A.R. Grade) was obtained from Beijing Chemical Company. Toluene (99.8%, HPLC) was obtained from Xilong Chemical Co., Ltd. Methanol-13C (99 atom% 13C) and ethanol-13C2 (99 atom% 13C) were purchased from Sigma-Aldrich Co. LLC. The CO2 (99.99%) and H2 (99.99%) were provided by Beijing Analytical Instrument Company.Hydrogenation of CO2
All the reactions were conducted in a 16 mL Teflon-lined stainless steel reactor equipped with a magnetic stirrer. In a typical experiment, known amounts of the Ru and/or Rh catalysts, LiI or another promoter, tracer (methanol-13C or ethanol-13C2 if used), and 2 mL solvent were loaded into the reactor. The reactor was sealed and purged three times with CO2 of 3 MPa, and then CO2 and hydrogen were charged to the desired pressure at room temperature, respectively. The reactor was placed in an air bath of constant temperature, and the magnetic stirrer was started at 800 rpm. After reaction, the reactor was cooled in an ice-water bath for 1 h, the residual gas was released carefully in a hood. The liquid mixture was analyzed by GC (Agilent 7890B) equipped with a flame ionization detector and a HP-5 capillary column (0.32 mm in diameter and 30 m in length) using toluene as the internal standard. Identification of the liquid products was done using a GC-MS (SHIMADZU-QP2010) as well as by comparing the retention times of the standards in the GC traces (Fig. S4†). The yields of the products were calculated from the GC data.To test the reusability of the catalytic system, the alcohols formed in the reaction were removed at 80 °C under vacuum for 1.5 h, and the catalytic system (Ru3(CO)12–Rh2(CO)4Cl2–LiI/DMI) was used directly for the next run.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21373234, 21173239, 21303224, 21321063) and the Chinese Academy of Sciences (KJCX2.YW.H30). We are grateful for the help of Dr Jinchao Wei in the GC-MS analysis.Notes and references
- (a) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley–VCH, Weinheim, 2010 Search PubMed; (b) M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- (a) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (b) T. Yu, R. Cristiano and R. G. Weiss, Chem. Soc. Rev., 2010, 39, 1435–1447 RSC; (c) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (d) P. G. Jessop, F. Joó and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS PubMed; (e) Y. H. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS PubMed; (f) K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS PubMed; (g) O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC; (h) X. J. Cui, X. C. Dai, Y. Zhang, Y. Q. Deng and F. Shi, Chem. Sci., 2014, 5, 649–655 RSC; (i) C. Liu, J. H. Xie, G. L. Tian, W. Li and Q. L. Zhou, Chem. Sci., 2015, 6, 2928–2931 RSC; (j) Y. Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- (a) W. Wang, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC; (b) C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed; (c) S. Wesselbaum, T. Vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed; (d) J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed; (e) C. H. Lim, A. M. Holder, J. T. Hynes and C. B. Musgrave, J. Am. Chem. Soc., 2014, 136, 16081–16095 CrossRef CAS PubMed; (f) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. V. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (g) N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- (a) G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS PubMed; (b) J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- T. Tatsumi, A. Muramatsu and H. Tominaga, Chem. Lett., 1985, 593–594 CrossRef CAS.
- (a) T. Inui and T. Yamamoto, Catal. Today, 1998, 45, 209–214 CrossRef CAS; (b) T. Inui, T. Yamamoto, M. Inoue, H. Hara, T. Takeguchi and J. B. Kim, Appl. Catal., A, 1999, 186, 395–406 CrossRef CAS.
- H. Kurakata, Y. Izumi and K. Aika, Chem. Commun., 1996, 389–390 RSC.
- L. S. Davy, D. Nieskens, Y. Ferrari, R. Liu and J. Kolonko, Catal. Commun., 2011, 14, 111–113 CrossRef PubMed.
- S. G. Li, H. J. Guo, C. R. Luo, H. R. Zhang, L. Xiong, X. D. Chen and L. L. Ma, Chem. Lett., 2013, 143, 345–355 CAS.
- R. Kieffer, M. Fujiwara, L. Udron and Y. Souma, Catal. Today, 1997, 36, 15–24 CrossRef CAS.
- M. Kishida, K. Yamada, H. Nagata and K. Wakabayashi, Chem. Lett., 1994, 555–556 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef CAS.
- K. K. Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67–81 CrossRef.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Energy, 1997, 22, 343–348 CrossRef CAS.
- K. Okabe, H. Yamada, T. Hanaoka, T. Matsuzaki, H. Arakawa and Y. Abe, Chem. Lett., 2001, 904–905 CrossRef CAS.
- K. Tominaga, Y. SaSaki, M. Saito, K. Hagihara and T. Watanabe, J. Mol. Catal., 1994, 89, 51–56 CrossRef CAS.
- P. M. Maitlis, J. Mol. Catal. A: Chem., 2003, 204–205, 55–62 CAS.
- R. W. Wegman and K. G. Moloy, US Pat., 4 727 200, 1988.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables. See DOI: 10.1039/c5sc02000j |
| This journal is © The Royal Society of Chemistry 2015 |