
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen deficient α-Fe2O3 photoelectrodes: a balance between enhanced electrical properties and trap-mediated losses†
Mark
Forster
a,
Richard J.
Potter
b,
Yichuan
Ling
d,
Yi
Yang
d,
David R.
Klug
c,
Yat
Li
d and
Alexander J.
Cowan
*a
aUniversity of Liverpool, Stephenson Institute for Renewable Energy, Department of Chemistry, Liverpool, L69 7ZF, United Kingdom. E-mail: acowan@liverpool.ac.uk
bUniversity of Liverpool, School of Engineering, George Holt Building, Brownlow Hill, Liverpool, L69 3GH, United Kingdom. E-mail: rjpott@liverpool.ac.uk
cImperial College London, Department of Chemistry, Exhibition Road, London, SW7 2AZ, United Kingdom. E-mail: d.klug@imperial.ac.uk
dUniversity of California, Department of Chemistry and Biochemistry, 1156 High Street, Santa Cruz, California 95064, United States of America. E-mail: yatli@ucsc.edu
First published on 28th April 2015
Abstract
Intrinsic doping of hematite through the inclusion of oxygen vacancies (VO) is being increasingly explored as a simple, low temperature route to preparing active water splitting α-Fe2O3−x photoelectrodes. Whilst it is widely accepted that the introduction of VO leads to improved conductivities, little else is verified regarding the actual mechanism of enhancement. Here we employ transient absorption (TA) spectroscopy to build a comprehensive kinetic model for water oxidation on α-Fe2O3−x. In contrast to previous suggestions, the primary effect of introducing VO is to block very slow (ms) surface hole – bulk electron recombination pathways. In light of our mechanistic research we are also able to identify and address a cause of the high photocurrent onset potential, a common issue with this class of electrodes. Atomic layer deposition (ALD) of Al2O3 is found to be particularly effective with α-Fe2O3−x, leading to the photocurrent onset potential shifting by ca. 200 mV. Significantly TA measurements on these ALD passivated electrodes also provide important insights into the role of passivating layers, that are relevant to the wider development of α-Fe2O3 photoelectrodes.
Introduction
There is intense interest in the use of photoelectrochemical (PEC) systems for the production of solar fuels. Amongst the more promising photoanodes for PEC water oxidation is hematite (α-Fe2O3), a non-toxic, abundant, low-cost, and relatively inert material. The bandgap and band energies of α-Fe2O3 (∼2.1 eV) lead to a maximum theoretical solar-to-hydrogen (STH) efficiency of ∼15%,1 however actual achieved STH efficiencies of α-Fe2O3 photoelectrodes are substantially below this value and are typically ∼1–2%.2 This has been proposed to be due to multiple limiting factors, including; poor conductivity,3 short electron–hole lifetimes,4 slow oxygen evolution reaction kinetics5 and a low visible light absorption coefficient coupled to a short hole diffusion length (2–4 nm).6 Numerous approaches to improving the activity of α-Fe2O3 have been explored. Higher photocurrent densities have been achieved through nanostructuring which has the aim of increasing the concentration of charges generated close to the semiconductor liquid junction (SCLJ) to overcome the short hole diffusion length.7 In such nanostructured electrodes, dopants are known to be critical for photoelectrochemical activity with un-doped α-Fe2O3 electrodes being electrically insulating7 and often photoelectrochemically inactive.8 A wide variety of extrinsic dopants have now been explored including Si,8–10 Sn,11–13 Ti,13–15 Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16,17 and these lead to both enhanced long range charge transport and the formation of a sufficient electric field for charge separation at the SCLJ within the nanostructured domains.8
16,17 and these lead to both enhanced long range charge transport and the formation of a sufficient electric field for charge separation at the SCLJ within the nanostructured domains.8
The introduction of oxygen vacancies (VO) has also recently been shown to be an effective approach to improve the activity of α-Fe2O3 photoelectrodes.18–20 A report by Li et al. on the decomposition of β-FeOOH nanowires in an oxygen deficient atmosphere demonstrated the highly active nature of oxygen deficient hematite (α-Fe2O3−x) photoelectrodes for water oxidation, with a photocurrent of 3.4 mA cm−2 being achieved under 100 mW cm−2, and in this paper we examine the factors behind the high activity of these same electrodes.19 The enhanced activity, ease of inclusion of VO, ability to process at temperatures as low as 350 °C21 and reports of a synergistic effect of extrinsic dopants with intrinsic VO sites20,22 has led to a surge of interest in the controlled inclusion of VO in hematite over the last 2 years. Given the potential utility of this approach it is important that the fundamental processes associated with VO inclusion are elucidated.23–26
The higher incident photon to current efficiency (IPCE) of α-Fe2O3−x (64% at 1.50 VRHE) compared to the air annealed sample (α-Fe2O3, 0.57% at 1.50 VRHE) in the report of Li et al. correlated with the presence of Fe2+ sites as measured by XPS and a large increase in the measured donor density (Nd).19 It has been known for over 25 years that the inclusions of VO/Fe2+ sites within α-Fe2O3 leads to formation of a donor band ∼80 meV below the conduction band.27 The inclusion of VO's is suggested to improve activity through a number of mechanisms including; improved charge transport, higher charge separation yields and decreased contact resistances at the semiconductor/transparent conducting oxide interface, with various studies citing differing contributions from each effect.13,21,23–26 However, to date research has concentrated on material development and few direct measurements of the actual mechanism of enhanced activity for α-Fe2O3−x are reported.
A further complication is that although large increases in photocurrent have been achieved through the inclusion of VO, the onset potentials remain relatively high for α-Fe2O3−x, typically 1.0 VRHE or greater19,21 compared to as low as ∼0.6 VRHE for an ALD α-Fe2O3 electrode following a high temperature (800 °C) treatment, leading to typical solar to fuels efficiencies for oxygen deficient hematite that are significantly lower than other state of the art hematites.28,29 It has been proposed by several authors that a possible cause of the high onset potentials in α-Fe2O3−x is the increased concentration of surface defect states.30,31 Surface defects in hematite are known to lead to Fermi level pinning and increased levels of trap-mediated recombination, and the selective passivation of such states in stoichiometric α-Fe2O3 has proven to be a highly effective approach to improving photoelectrode activity.30 Surface passivation treatments on extrinsically doped photoelectrodes have included high temperature annealing steps and overlayer depositions.29,32,33 The hypothesised deleterious role of surface states in oxygen deficient hematite is apparently contradicted by a recent study on hydrogen treated α-Fe2O3 which reported improved linear sweep photocurrents (recorded at 50 mV s−1) for samples only containing Fe2+ defect states at the surface, compared to photoelectrodes containing both bulk and surface VO.24 It is therefore apparent that currently no consensus exists regarding the mechanism of operation of oxygen deficient hematite. In order to systematically address the high onset potential of α-Fe2O3−x samples an improved understanding of the role of both bulk and surface VO sites and their effects on the charge carrier kinetics is required.
Transient absorption (TA) spectroscopy and transient photocurrent (TPC) measurements offer a route to directly measure the effect of material modifications and treatments on the yield and dynamics34,35 of photogenerated charges within a PEC cell.36 Previous TA spectroscopic studies of α-Fe2O3 photoelectrodes have examined numerous aspects of the photophysics and chemistry of extrinsically doped α-Fe2O3, including the role of co-catalysts on hole kinetics,31,32,37,38 and the effect of bias on charge trapping and recombination, from the fs–ms timescale.39,40 Of particular significance has been the realization that a key requirement for water splitting is the ability to accumulate very long-lived holes at the SCLJ, with apparent rate constants for water splitting ranging from 0.1–6 s−1,41,42 correlating with a measured significant thermal barrier to hole transfer.43 The slow hole transfer enables recombination between bulk electrons and surface accumulated holes on the millisecond timescale in Si–Fe2O3 at <1.2 VRHE which significantly lowers photoelectrochemical activity. Here we report the first TA study of a α-Fe2O3−x photoelectrode in a PEC cell, with the aim of identifying the key design rules required to develop more efficient defect rich photoanodes. We have chosen to study samples prepared as originally reported by some of us19 as they remain amongst the most active oxygen deficient electrodes reported, with current densities reaching ca. 4 mA cm−2 under 1 sun illumination. Our detailed kinetic analysis allows to (i) elucidate the observed bias dependent activity of α-Fe2O3−x, yielding a complete mechanistic model of the operation of oxygen deficient electrodes and (ii) to provide critical mechanistic insights into the role of ALD Al2O3 overlayers. ALD Al2O3 is found to be particularly effective in shifting the photocurrent onset potential of α-Fe2O3−x (∼0.2 V), representing a significant enhancement in photoelectrochemical water splitting efficiency.
Results and discussion
For this study fresh α-Fe2O3 (air annealed) and α-Fe2O3−x (oxygen deficient) films were prepared as previously described, details can be found in the ESI.†19 The photoelectrochemical activities of both α-Fe2O3 and α-Fe2O3−x were assessed through linear sweep voltammograms measured in 1 M NaOH using a portion of the output of a 75 W Xe lamp (ca. 0.1 sun) scanning at 5 mV s−1. We employ this low power light source in our mechanistic study as it allows us to measure the photoelectrochemical response of the sample inside the transient spectrometer using the same cell as in the TA measurements. In line with previous reports we observe a strong photocurrent from α-Fe2O3−x assigned to photoelectrochemical water oxidation, Fig. 1.41,44 It is known that IPCE values for these α-Fe2O3−x films exceed 60% at 1.5 VRHE, leading to photocurrents of ca. 4 mA cm−2 under 100 mW cm−2, however a key concern remains the high onset potential,19 observed here at ca. 1.15 VRHE, Fig. 1. In contrast to α-Fe2O3−x, the air annealed (550 °C) α-Fe2O3 sample shows no significant photocurrent (0.6–1.5 VRHE). The lack of activity of this control α-Fe2O3 sample, chosen due to its similar morphology, preparation route and light harvesting properties, indicates that the presence of VO is critical for enabling photoelectrochemical water splitting in these otherwise un-doped films.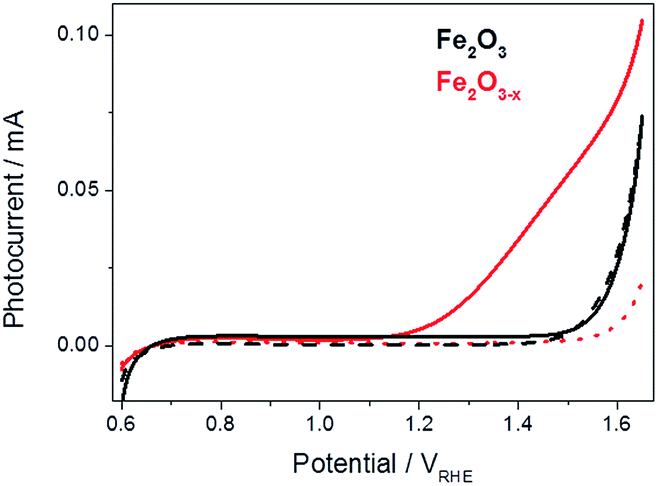 | ||
| Fig. 1 Linear sweep voltammograms for α-Fe2O3 (black) and α-Fe2O3−x (red), 5 mV s−1, under low power white light illumination (solid line, ca. 0.1 sun) and in the dark (dashed line). | ||
In order to rationalize the observed photoelectrochemical response of the hematite samples we have measured TA spectra following UV excitation (355 nm, 6 ns, 100 μJ cm−2) at a range of applied biases, Fig. 2. The excitation energy employed leads to photo-generated carrier densities several orders of magnitude lower than the calculated oxygen vacancy density (ESI, S7†). A broad positive transient absorption is measured at wavelengths greater than 600 nm in all of the spectra. In line with numerous past TA studies on Si-doped and Nb-doped α-Fe2O3 photoelectrodes,40,43,45 the TA feature at λ > 600 nm is assigned to photoholes in α-Fe2O3 (see Fig. S4† for a schematic explanation) and this assignment is further affirmed in TA experiments employing a hole scavenger (H2O2, vide infra) which show a decreased signal at λ > 600 nm. The bias dependent TA spectra also contain a bleach (decrease in optical density) centred at ca. 580 nm which is seen to increase in magnitude with applied bias. It has been proposed that the feature at this wavelength is due to localized states close to the band edge, possibly related to the presence of oxygen vacancies.27,40,46 Under positive bias the trap state occupancy is lowered, enabling the promotion of a valence band electron to the vacant trap state upon absorption of a visible photon (580 nm). Using both fs and μs TA measurements, Pendlebury et al. have shown that following UV excitation of α-Fe2O3 held at potentials significantly positive of the flat band potential, rapid photoelectron trapping can occur leading to a bleaching of this 580 nm feature, (see Fig. S4†).39,40 We also assign the bleach at ∼580 nm to photoelectron trapping at localized states which is primarily occurring on the sub-microsecond timescale.
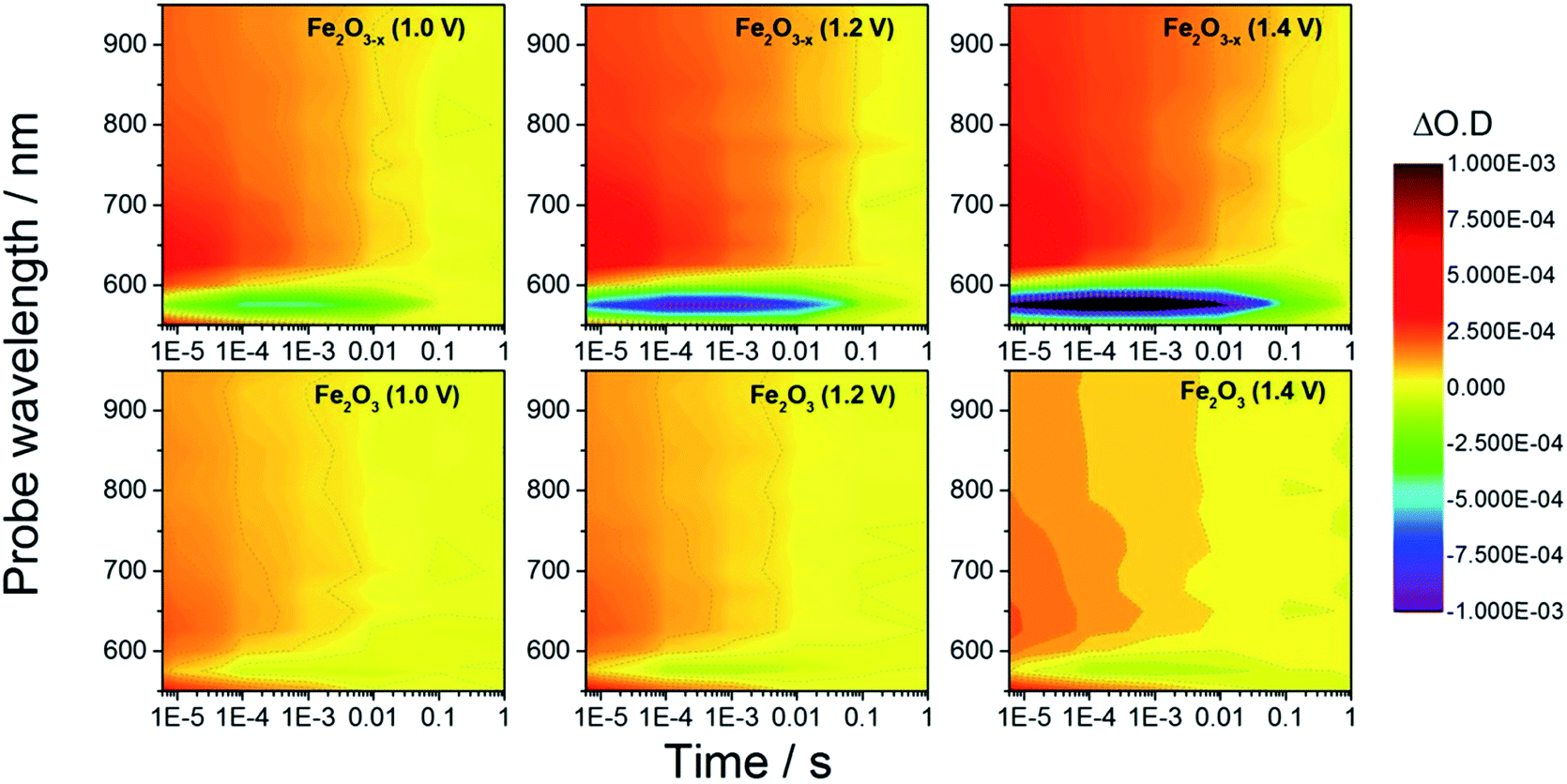 | ||
| Fig. 2 TA spectra of α-Fe2O3 and α-Fe2O3−x at various applied potentials (vs. RHE) in 1 M NaOH with the photoanodes excited from the electrolyte/electrode side (EE) with a 355 nm (6 ns pulse) laser. | ||
Initially we concentrate our study on the yield and kinetics of photoholes in α-Fe2O3 and α-Fe2O3−x. At all potentials investigated we note an increased yield of photoholes in Fe2O3−x at the earliest timescales studied (2 μs), Fig. 2. The increased photohole yield at 2 μs may indicate more efficient initial charge separation in the oxygen deficient hematite, however a detailed study of the photohole kinetics at a single potential (1.4 VRHE) shows only a ∼35% difference in photohole yield at 2 μs (assuming a similar extinction coefficient for both materials), Fig. 3. This suggests that a previously proposed enhancement19,38 in initial charge separation yield in VO rich materials with higher electron densities is not likely to be a significant factor in rationalizing the differences in activity of α-Fe2O3 and α-Fe2O3−x. Instead, of greater significance is the rate of photohole decay in α-Fe2O3 (t50% = 0.27 ms) and α-Fe2O3−x (t50% = 1.20 ms) at 1.4 VRHE.47 Previous studies have indicated that the improved conductivity of α-Fe2O3−x would be expected to aid both initial charge separation and electron transport/extraction which reduce bulk electron–hole recombination and thereby increase the photohole lifetime.21,48
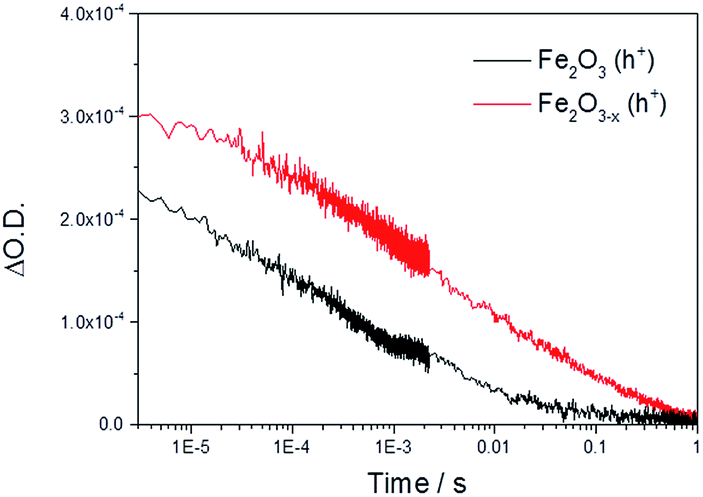 | ||
| Fig. 3 TA decay traces at 700 nm of photoholes on α-Fe2O3 and α-Fe2O3−x at 1.4 VRHE in 1 M NaOH following UV excitation (355 nm). | ||
In order to asses if such a change in photoelectron dynamics is the cause of the improved activity of α-Fe2O3−x we have also measured the transient photocurrent (TPC) between the hematite working electrodes and the counter electrode following UV excitation. At 1.4 VRHE the TPC traces of both α-Fe2O3 and α-Fe2O3−x are relatively similar at early times (<0.5 ms) indicating that, contrary to expectations, initial photoelectron extraction to the external circuit can occur effectively even in the un-doped α-Fe2O3 sample.19 Instead we find that the difference in photohole kinetics and photoelectrochemical activity is due to a slow back electron transfer from the external circuit into the air annealed α-Fe2O3, which we see as a negative current on the timescale of 1–10 ms after photon absorption, Fig. 4. For α-Fe2O3 samples the total charge re-injected from the external circuit approximately matches that initially extracted from the α-Fe2O3 photoelectrode at all potentials studied (0.8–1.4 V), leading to minimal net photocurrent in our transient study, in line with the steady state photocurrent measurements, Fig. 4 and S5.†
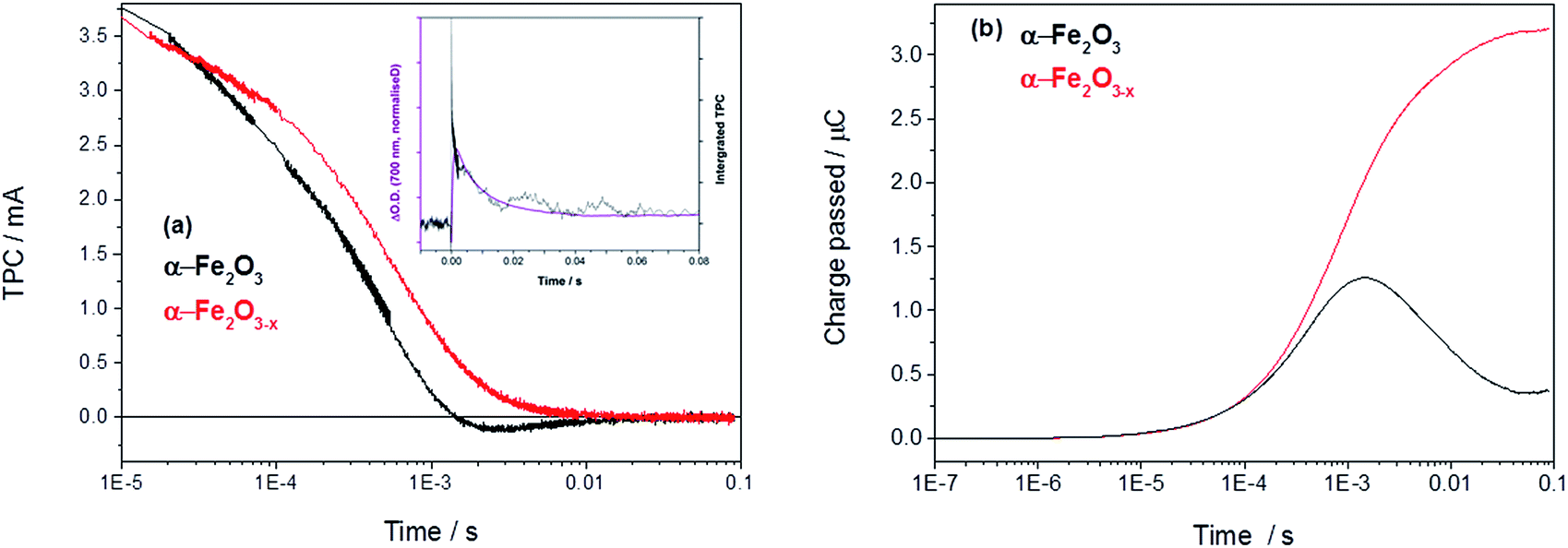 | ||
| Fig. 4 (a) TPC decays of α-Fe2O3 and α-Fe2O3−x held at 1.4 VRHE with an inset showing the overlay of the decay of the TA with the integrated TPC in Fe2O3 at 1.4 VRHE (Fig. S6†) and (b) the converted total charge passed following UV excitation (355 nm, 6 ns). | ||
An overlay of the kinetics of charge reinjection (TPC) and hole decay (TA) on α-Fe2O3 (τ = 10 ms at 1.4 VRHE, Fig. 4 inset and Fig. S6†) shows an excellent agreement indicating that bulk electrons are recombining with the accumulated photoholes measured in the TA experiment, leading to reinjection of electrons from the external circuit. Experiments in the presence of H2O2 in the following section allow us to assign this recombination to be occurring with surface trapped holes. This is in agreement with a very recent study on Si–Fe2O3 where TPC measurements showed the presence of a recombination process between surface trapped holes and bulk α-Fe2O3 electrons on the milliseconds timescale, that led to a flow of electrons back into the photoelectrode from the external circuit.41 In contrast, with α-Fe2O3−x electrodes we do not observe any slow re-injection of electrons into the film, Fig. S5.†
The lack of back electron transfer into α-Fe2O3−x electrodes can be rationalized by Mott–Schottky measurements of the two films (Fig. S7†) that show a higher donor density (Nd) in the VO rich photoelectrode (α-Fe2O3−xNd = 1.2 × 1020 cm−3, α-Fe2O3Nd = 6.7 × 1019 cm−3) with flat-band potentials of 0.4 VRHE (α-Fe2O3−x) and 0.34 VRHE (α-Fe2O3).19 Whilst absolute values of donor densities for a nanostructured film obtained through a Mott–Schottky analysis should be treated with caution, the relative change between these two samples with similar morphologies (Fig. S3†) is significant. This two-fold increase in Nd in the α-Fe2O3−x electrode significantly decreases the width of the space charge layer (Wsc), Fig. S7† for illustrative calculations. Our TAS studies confirm that greater localised band bending in the surface region of α-Fe2O3−x is a significant factor in rationalising the enhanced activity of α-Fe2O3−x, as hypothesised in several previous studies.19,21,30,31,38 However it is not found to be due to more efficient initial charge separation or injection kinetics but instead the blocking of the back flow of electrons from the bulk towards the SCLJ preventing recombination with surface accumulated photoholes.
A simple model of the kinetic competition between the bias dependent back electron–hole recombination pathway and the rate of water oxidation has been shown elsewhere to account for observed photoelectrochemical activity of Si–Fe2O3 electrodes without the need for the inclusion of inter-band trap states.41 Specifically the photocurrent onset potential of a Si-doped α-Fe2O3 photoelectrode, with a similar Nd (∼1020 cm3) to the α-Fe2O3−x samples examined here (1.2 × 1020 cm−3), correlated with the potential at which back electron–hole recombination became slow enough to enable surface hole accumulation and water oxidation to occur (ca. 1.0 VRHE). Here we find that back electron–hole recombination in α-Fe2O3−x is blocked at potentials as low as 0.8 VRHE (Fig. S5†) and no correlation is noted with the photocurrent onset potential, ca. 1.15 VRHE (Fig. 1). This gives rise to an intriguing question, given that our TPC and TA measurements show that holes can be accumulated at potentials as low as 0.8 VRHE and that the slow back electron–hole recombination pathway has been prevented, why is the photocurrent onset potential for water oxidation so positive (1.15 VRHE) with α-Fe2O3−x photoelectrodes?
Site of photohole accumulation in α-Fe2O3−x
It is therefore important to identify if the photoholes measured in α-Fe2O3−x using TA spectroscopy on the μs–ms timescale are accumulating at or close to the SCLJ, as hole trapping elsewhere in this defect rich material may account for the high photocurrent onset potential. In order to distinguish these two cases we explore the response of the hole dynamics to the presence of a hole scavenger. Hydrogen peroxide is a commonly used hole scavenger due to its near unity efficiency for the removal of holes in α-Fe2O3 that are at or close to the SCLJ.49 In the presence of 0.5 M H2O2 we observe a photocurrent at potentials as low as 0.7 VRHE on both α-Fe2O3 and α-Fe2O3−x confirming that (i) initial charge separation is effective in both materials at potentials well below the water oxidation photocurrent onset potential and (ii) that in both samples the photoholes are able to reach the surface to participate in oxidation reactions, Fig. S8.† TA experiments in the presence of H2O2 show a decrease in the hole yield at the earliest time-scales studied (2 μs) in Fe2O3−x when compared to experiments in the absence of a hole scavenger. This indicates that holes are present at the SCLJ and are transferring into solution at time scales earlier than studied here, providing a lower limit for the lifetime of transport and accumulation of holes at the SCLJ (∼2 μs), Fig. 5.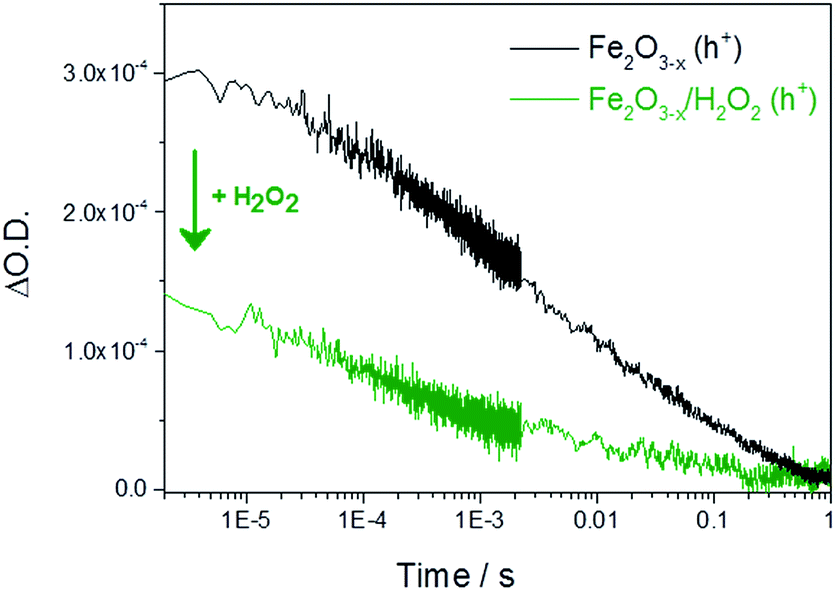 | ||
| Fig. 5 TA decay traces recorded at 700 nm of photoholes on α-Fe2O3−x at 1.4 VRHE in 1 M NaOH in the absence and presence of a hole scavenger (0.5 M H2O2). | ||
Role of trap state mediated recombination
We now turn to the potential role of electron trap states in rationalizing the behaviour of α-Fe2O3−x as it has been proposed that oxygen vacancies may act as a “mixed blessing”38 with the enhanced electrical properties being balanced with a potential increase in trap-mediated electron–hole recombination at the defect sites introduced.30 Following absorption of a UV photon we initially observe a bleaching at 580 nm for α-Fe2O3−x which is assigned to photoelectron trapping at this inter-band state, Fig. 6(b).39 The slow recovery on the millisecond timescale of the trap state feature is due to the subsequent de-trapping of the photoelectrons, as shown by the dynamics in Fig. 6(b). In line with this assignment, the change in occupancy of the 580 nm trap state at 1.4 VRHE is well fitted to the integrated rate equation for an intermediate species in a consecutive reaction scheme (A → B → C) (Fig. 6(b)), eqn (1).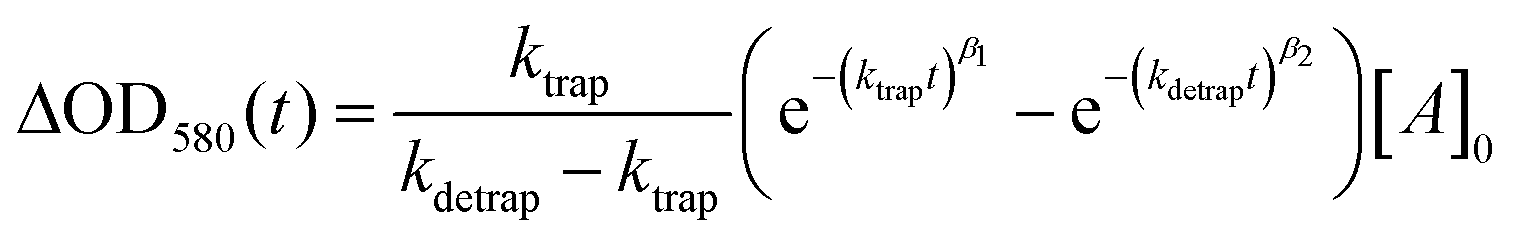 | (1) |
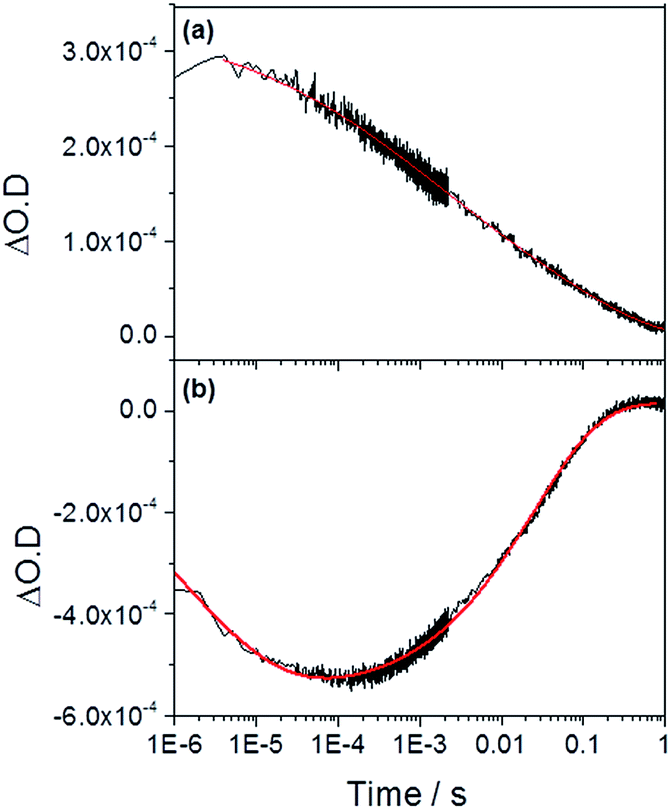 | ||
| Fig. 6 TA traces recorded at (a) 700 nm (photoholes) and (b) 580 nm (electron trap state) in α-Fe2O3−x at 1.4 VRHE following UV excitation (355 nm) in 1 M NaOH. The red lines correspond to the fitted functions identified in the main text. Full parameters can be found in the ESI.† | ||
In previous studies the de-trapping rate of photoelectrons in extrinsically doped hematite correlated to the TPC decay rate as well as a distinct fast decay component in the hole population, leading to an assignment of both electron extraction and electron–hole recombination following de-trapping in the bulk of the electrode.40 In contrast, on α-Fe2O3−x a sample with a high concentration of surface defect sites21,50 it is proposed that a significant level of electron trapping occurs at the surface, closer to the site of photohole accumulation leading to higher electron–hole recombination losses upon detrapping. We are able to assign electron–hole recombination as the fate of the de-trapped electrons due to the lack of correlation between the TPC decay rate and the rate of recovery of the 580 nm signal (Fig. 4, 6 and 7(b)), which indicates that the electrons are unable to reach the external circuit. In the absence of photoelectron extraction it is expected that electron–hole recombination will occur. We can also conclude that a significant level of recombination with trap state electrons occurs close to the SCLJ as in the previous section TA experiments demonstrated that the a large portion of the photohole population reached the surface of α-Fe2O3−x within the time resolution of our experiment (2 μs), i.e. prior to photoelectron detrapping.
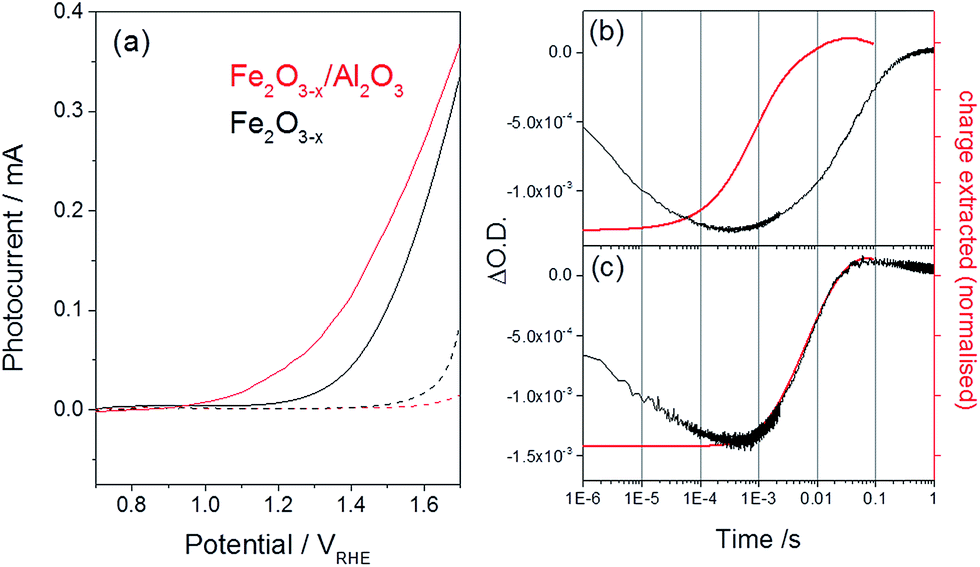 | ||
| Fig. 7 (a) Photocurrent of α-Fe2O3−x before (black) and after deposition of an ALD Al2O3 layer (1 nm, red trace) recorded using a low power 75 W Xe lamp, dark traces are shown as dashed lines. (b) Overlay of the TA trace recorded at 580 nm, assigned to an interband trap state, and the normalized charge extracted for (b) α-Fe2O3−x and (c) α-Fe2O3−x/Al2O3 following UV excitation (355 nm) at 1.4 VRHE. All traces are recorded in 1 M NaOH.51 | ||
We further confirm the presence of a significant level of trap-mediated recombination in the oxygen deficient hematite through a detailed kinetic analysis of the photoholes. In the absence of a recombination process between surface trapped holes and bulk α-Fe2O3 electrons the photohole TA kinetics of α-Fe2O3−x at 1.4 VRHE would consist of three primary kinetic processes on the μs–s timescale corresponding to fast (μs) electron–hole recombination in the bulk, surface electron–hole recombination and hole transfer into solution. An excellent fit of the TA signal of α-Fe2O3−x at 700 nm can indeed be achieved using a triphasic stretched exponential function, with the rate of bulk electron–hole recombination corresponding to the decay of the integrated TPC signal at the same potential (kbulk ∼ 2 × 103 s−1), the surface recombination rate constant matching the rate constant for photoelectron de-trapping (kdetrap ∼ 40 s−1), confirming the occurrence of trap-mediated recombination at the SCLJ, and a hole transfer rate constant into solution of kWO ∼ 3.5 s−1 (full fitting parameters are in the ESI†), Fig. 6(a). The assignment of the slowest kinetic phase of the hole decay is in line with previously measured rate constants for water oxidation on α-Fe2O340 which have ranged from ∼0.2–6 s−142 and is further supported by a plot of the yield of very long-lived photoholes (at 200 ms) versus the applied bias which strongly correlates with the measured photocurrent response of α-Fe2O3−x, (Fig. S9†). Our analysis is also further supported by fitting of the hole trace in Fig. 5, which is well fitted with a biphasic stretched exponential function, with kbulk and kdetrap present, but kWO absent, confirming the nature of this slowest kinetic component, (ESI Fig. S12†). We are able to now construct a full kinetic scheme for the key steps for water oxidation on α-Fe2O3−x, (Scheme 1). Interestingly the amplitude of the fitting components, i.e. the separate populations of holes that are decaying by each pathway, is approximately the same for trap-mediated recombination and hole transfer into solution even at 1.4 VRHE demonstrating that a high level of trap mediated recombination is a critical factor limiting the efficiency of oxygen deficient hematite.
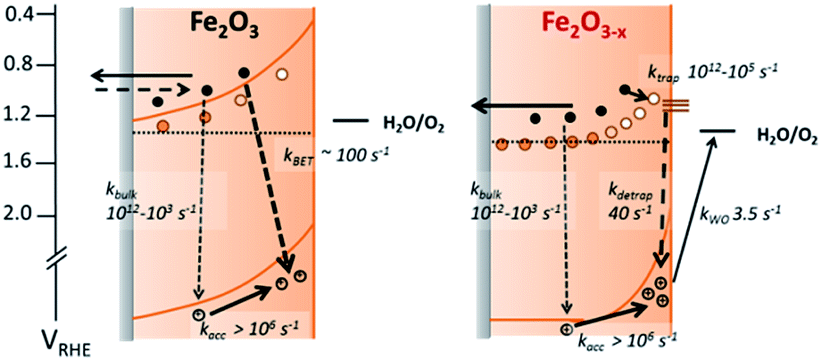 | ||
| Scheme 1 Simplified energy diagrams and processes involving photogenerated charges following UV laser excitation of α-Fe2O3 and α-Fe2O3−x under a positive bias. Rate constants are those determined at 1.4 VRHE as described in the main text. In contrast to α-Fe2O3 which has been air annealed, no slow recombination between surface accumulated holes and bulk electrons (kBET) is observed for α-Fe2O3−x. | ||
In light of our TA studies that highlight the role of α-Fe2O3−x surface states in mediating recombination, and show that the presence of oxygen vacancies also leads to a higher donor concentration and a narrower depletion layer suppressing the back electron injection, we are able to propose a route to both improve the activity of oxygen deficient hematite's and to test the mechanistic model proposed in Scheme 1. It is known that the preparation of α-Fe2O3−x, by thermal decomposition of β-FeOOH in an oxygen deficient atmosphere,19 leads to the formation of VO both throughout α-Fe2O3−x and at the surface.24 Removal of the states solely on the Fe2O3−x surface by passivation, whilst maintaining a suitably high concentration of VO both within the bulk and close to the SCLJ may be anticipated to be a route to obtaining both the desired improved Nd whilst lowering the level of trap-mediated recombination. Previously, passivation of surface states of α-Fe2O3 has been achieved through the use of high temperature annealing steps,29 the deposition of catalytic species including CoPi, IrOx, NiFeOx32,37 and the use of inert metal oxides such as Al2O3 and Ga2O3 overlayers grown by atomic layer deposition (ALD).31,33,38
The deposition of thin Al2O3 layers appear particularly promising as the low temperatures (120 °C) required and the reduced oxygen pressures for ALD deposition are expected to limit the loss of bulk oxygen vacancies in α-Fe2O3−x. ALD Al2O3 layers have been shown to decrease the photocurrent onset potentials of Si-doped α-Fe2O3 electrodes by ∼100 mV,9 with a decrease in the surface capacitance of the Al2O3 coated α-Fe2O3 electrodes also reported,52,53 indicating the passivation of surface trap states. Here we use ALD to form a 1 nm Al2O3 layer. This thickness is chosen as previous experiments with Si–Fe2O39 have indicated that it provides reasonable coverage and stability for the duration of an experiment whilst remaining thin enough to allow for hole tunnelling transfer from the Fe2O3 to water. We find that the addition of an ALD layer on α-Fe2O3−x leads to a large cathodic shift in the photocurrent onset potential (∼200 mV, 0.95 VRHE) and an overall increase in the magnitude of the photocurrent, Fig. 7(a) (150 W Xe lamp). Al2O3 was also deposited onto an air annealed α-Fe2O3 sample (Fig. S10†) however photocurrent measurements showed no improvement in activity confirming that sub-surface oxygen vacancies are required for photoelectrochemical activity in otherwise un-doped samples.
To confirm that the improved photoelectrochemical activity of the α-Fe2O3−x/Al2O3 sample is related to the hypothesised decrease in trap mediated surface electron–hole recombination, we have also measured the TA kinetics of the 580 nm trap states at 1.4 VRHE following surface treatment, Fig. 7(b). To the best of our knowledge this report represents the first TA study of the role of an ALD Al2O3 passivation treatment on hematite. A large bleaching of the TA signal is observed at 580 nm even after the Al2O3 treatment showing that a significant concentration of trap states remains within the bulk of the α-Fe2O3−x/Al2O3 sample. Critically we also note a near four-fold increase in the rate of de-trapping with the α-Fe2O3−x/Al2O3 sample (kdetrap ∼ 150 s−1) indicating that photoelectrons in the ALD treated sample are trapped at energetically shallower sites. In contrast to the untreated α-Fe2O3−x sample, where de-trapping leads to significant levels of recombination with surface trapped holes, we now see an excellent agreement between the recovery of the TA trap state band at 580 nm and the rate of charge extraction as measured by TPC for α-Fe2O3−x/Al2O3, Fig. 7(b and c), further supported by the observation of an increase in the yield of very long-lived photoholes in the presence of the Al2O3 overlayer (Fig. S11†). This leads to the conclusion that detrapped electrons are reaching the external circuit in appreciable quantities. We assign the change in kinetics of the transient photocurrent and trap state occupancy to the passivation of solely the surface trap states. In the absence of a significant concentration of surface trap states, photoelectrons are able to be trapped at interband trap states in the α-Fe2O3−x bulk, which are spatially separated from the population of surface accumulated holes, limiting trap-mediated recombination and enabling electron transport to the external circuit, thus lowering the photocurrent onset potential.
Conclusions
The annealing of hematite in oxygen deficient atmospheres is being increasingly explored as an approach to improving the electrical properties of the photoelectrode, however conflicting reports exist regarding the mechanism of enhancement.40,52 In contrast to previously proposed mechanisms which have often indicated improved charge transport as being a significant factor,18 we find that the primary effect of the introduction of oxygen vacancies is to block the slow recombination of bulk electrons with surface accumulated holes, the so called “back electron recombination” pathway at even moderate applied biases (0.8 VRHE).A key target of mechanistic research is to provide design rules for rational material development and here we achieve that goal, by both proposing and verifying a modification to address the very anodic photocurrent onset potential of α-Fe2O3−x. We have investigated the effect of an ALD surface passivation treatment on both the photoelectrochemistry of α-Fe2O3−x leading to validation of our mechanistic model and an improvement in the photocurrent onset potential of 0.2 V. Furthermore we report, to the best of our knowledge, the first TA measurements on ALD Al2O3 coated hematite providing important insights into this widely used surface treatment.
Acknowledgements
MF thanks the University of Liverpool for a GTA award. AJC gratefully acknowledges a fellowship from the EPSRC (EP/K006851/1).Notes and references
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- J. Y. Kim, G. Magesh, D. H. Youn, J.-W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 Search PubMed.
- F. J. Morin, Phys. Rev., 1951, 83, 1005–1010 CrossRef CAS.
- N. J. Cherepy, D. B. Liston, J. A. Lovejoy, H. Deng and J. Z. Zhang, J. Phys. Chem. B, 1998, 5647, 770–776 CrossRef.
- M. Dare-Edwards, J. Goodenough, A. Hamnett and P. Trevelick, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2027–2041 RSC.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CAS.
- I. Cesar, A. Kay, J. A. Gonzalez Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- S. Saremi-Yarahmadi, K. G. U. Wijayantha, A. A. Tahir and B. Vaidhyanathan, J. Phys. Chem. C, 2009, 113, 4768–4778 CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- C. D. Bohn, A. K. Agrawal, E. C. Walter, M. D. Vaudin, A. A. Herzing, P. M. Haney, A. A. Talin and V. A. Szalai, J. Phys. Chem. C, 2012, 116, 15290–15296 CAS.
- N. T. Hahn and C. B. Mullins, Chem. Mater., 2010, 22, 6474–6482 CrossRef CAS.
- G. Wang, Y. Ling, D. A. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503–3509 CrossRef CAS PubMed.
- R. Franking, L. Li, M. A. Lukowski, F. Meng, Y. Tan, R. J. Hamers and S. Jin, Energy Environ. Sci., 2013, 6, 500 CAS.
- Y. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- A. Mao, N.-G. Park, G. Y. Han and J. H. Park, Nanotechnology, 2011, 22, 175703 CrossRef PubMed.
- G. Wang, Y. Ling and Y. Li, Nanoscale, 2012, 4, 6682–6691 RSC.
- Y. Ling, G. Wang, J. Reddy, C. Wang, J. Z. Zhang and Y. Li, Angew. Chem., Int. Ed. Engl., 2012, 51, 4074–4079 CrossRef CAS PubMed.
- A. Pu, J. Deng, M. Li, J. Gao, H. Zhang, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 2491 CAS.
- Y. Ling, G. Wang, H. Wang, Y. Yang and Y. Li, ChemSusChem, 2014, 7, 848–853 CrossRef CAS PubMed.
- T.-Y. Yang, H.-Y. Kang, U. Sim, Y.-J. Lee, J.-H. Lee, B. Koo, K. T. Nam and Y.-C. Joo, Phys. Chem. Chem. Phys., 2013, 15, 2117–2124 RSC.
- J. Engel and H. L. Tuller, Phys. Chem. Chem. Phys., 2014, 16, 11374–11380 RSC.
- M. Li, J. Deng, A. Pu, P. Zhang, H. Zhang, J. Gao, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 6727 CAS.
- J. Lee and S. Han, Phys. Chem. Chem. Phys., 2013, 15, 18906–18914 RSC.
- J. Deng, X. Lv, J. Gao, A. Pu, M. Li, X. Sun and J. Zhong, Energy Environ. Sci., 2013, 6, 1965 CAS.
- K. Kobayashi, G. Okada and J. Kumanotani, J. Mater. Sci. Lett., 1988, 7, 3–4 CrossRef.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626 CAS.
- O. Zandi and T. W. Hamann, J. Phys. Chem. Lett., 2014, 5, 1522–1526 CrossRef CAS PubMed.
- K. Sivula, J. Phys. Chem. Lett., 2013, 4, 1624–1633 CrossRef CAS PubMed.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tétreault, K. Sivula and M. Grätzel, Energy Environ. Sci., 2011, 4, 2512 CAS.
- T. Wang, Z. Luo, C. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7469–7484 RSC.
- F. Le Formal, N. Tétreault, M. Cornuz, T. Moehl, M. Grätzel and K. Sivula, Chem. Sci., 2011, 2, 737 RSC.
- A. Yamakata, J. J. M. Vequizo and M. Kawaguchi, J. Phys. Chem. C, 2014, 119, 1880–1885 Search PubMed.
- K. Furuhashi, Q. Jia, A. Kudo and H. Onishi, J. Phys. Chem. C, 2013, 117, 19101–19106 CAS.
- F. M. Pesci, G. Wang, D. R. Klug, Y. Li and A. J. Cowan, J. Phys. Chem. C Nanomater Interfaces, 2013, 117, 25837–25844 CrossRef CAS PubMed.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 CAS.
- L. Steier, I. Herraiz-cardona, S. Gimenez, F. Fabregat-santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 2681–7688 CrossRef.
- S. R. Pendlebury, X. Wang, F. Le Formal, M. Cornuz, A. Kafizas, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 9854–9857 CrossRef CAS PubMed.
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724 RSC.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- H. K. Dunn, J. M. Feckl, A. Müller, D. Fattakhova-Rohlfing, S. G. Morehead, J. Roos, L. M. P. Peter, C. Scheu and T. Bein, Phys. Chem. Chem. Phys., 2014, 16, 24610–24620 RSC.
- A. J. Cowan, C. J. Barnett, S. R. Pendlebury, M. Barroso, K. Sivula, M. Grätzel, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2011, 133, 10134–10140 CrossRef CAS PubMed.
- S. R. Pendlebury, M. Barroso, A. J. Cowan, K. Sivula, J. Tang, M. Grätzel, D. Klug and J. R. Durrant, Chem. Commun., 2011, 47, 716–718 RSC.
- S. R. Pendlebury, A. J. Cowan, M. Barroso, K. Sivula, J. Ye, M. Grätzel, D. R. Klug, J. Tang and J. R. Durrant, Energy Environ. Sci., 2012, 5, 6304 CAS.
- We note that a similar spectral feature, measured using steady-state spectroelectrochemical measurements has been alternatively assigned to a surface trap species that is an intermediate in the water splitting mechanism, potentially Fe(V)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ref. 49) and this specific question is to be addressed in a further forthcoming manuscript.
O (ref. 49) and this specific question is to be addressed in a further forthcoming manuscript. - t 50% is defined as the time required for the change in optical density to decay to 50% of the magnitude measured at 2 microseconds.
- N. Iordanova, M. Dupuis and K. M. Rosso, J. Chem. Phys., 2005, 122, 144305 CrossRef CAS PubMed.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958 CAS.
- D. L. A. De Faria, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- The small positive signal at very long times in the TA study at 580 nm of α-Fe2O3−x/Al2O3 is assigned to the known spectral overlap with the broad hole absorption feature, which we monitor at 700 nm as has been previously noted elsewhere (ref. 43).
- B. Klahr and T. Hamann, J. Phys. Chem. C, 2014, 118, 10393–10399 CAS.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and sample characterization data can be found in the ESI. See DOI: 10.1039/c5sc00423c |
| This journal is © The Royal Society of Chemistry 2015 |