DOI:
10.1039/C4RA16784H
(Paper)
RSC Adv., 2015,
5, 20951-20960
Molecularly imprinted solid phase extraction coupled to high performance liquid chromatography for determination of aflatoxin M1 and B1 in foods and feeds
Received
21st December 2014
, Accepted 16th February 2015
First published on 16th February 2015
Abstract
A molecularly imprinted polymer was synthesized by a miniemulsion polymerization method using aflatoxin B1 as the molecular template, methacrylic acid (MAA) as the functional monomer, ethylene glycol dimethacrylate as the cross-linker, Span80 and hexadecyl trimethyl ammonium bromide as the surfactants and n-hexadecane as the hydrophobic reagent in the presence of water. This imprinted polymer was characterized by FT-IR, 1H NMR, scanning electron microscopy, laser light scattering and adsorption experiments, in which the results showed good recognition and selectivity to aflatoxin B1 and M1. Using the prepared polymer as a solid phase extraction sorbent, a highly selective sample pre-treatment method combined with high performance liquid chromatography and fluorescence detection was developed for determination of aflatoxin B1 and M1 in foods and feeds. The limit of detection and limit of quantification of this method for aflatoxins M1 and B1 were 0.05 μg kg−1 and 0.16 μg kg−1, respectively. The average recovery values from barley, peanut oil, feed and beer spiked samples ranged from 83% to 96%. The precision ranged from 2.2% to 5.6% for these samples. The proposed method was found to be more effective and economical as a pre-treatment technique than regulation 2006/40/EC.
1. Introduction
Aflatoxins (AFs) belong to a group of closely related hepato-carcinogenic bisdihydrofurano metabolites produced by certain species of Aspergillus, specifically Aspergillus flavus and Aspergillus parasiticus.1,2 Aspergillus flavus produces only aflatoxin B, whereas Aspergillus parasiticus produces both B and G.3 Among them, aflatoxin B1 (AFB1), the most common and most toxic, has been found to cause human hepatocellular carcinoma and has been classified as a group 1 human carcinogen by the International Agency for Research on Cancer (IARC) in 1993.4 Due to their frequent occurrence and potential threat to human health and animals, the European Commission Regulation 2010/165/EC was therefore established and allows for a maximum residue limit of 8 μg kg−1 for AFB1 in foodstuffs.5 Barley, rice, peanut oils, beer and feeds have been found to be contaminated with AFB1.6,7 To continuously monitor AFB1 levels in these foodstuffs, a sensitive, economical and accurate method is necessary.
Several analytical methods for the determination of AFB1 in foods and feeds include thin layer chromatography,8 enzyme-linked immunosorbent assays,9 biosensor,10,11 capillary electrophoresis12 and high performance liquid chromatography (HPLC).13–15 Among them, an immunoaffinity clean-up step and HPLC with pre-column derivation, and fluorescence detection is often used for routine screening. Immunoaffinity sorbents, based on molecular recognition by antibodies, exhibit high selectivity to target molecules, however, they also display instability, are difficult to prepare, and have a relatively high cost. Therefore, the development of a selective, stable, and economical sorbent material is crucial.
Molecularly imprinted polymers (MIPs) are tailor-made polymers with a predetermined selectivity toward a given analyte or a group of structurally related species.16,17 MIPs are prepared by the polymerization of suitable functional monomers and cross-linking agents in the presence of a molecular template. After polymerization, the template is removed from the polymeric matrix leaving cavities complementary in size and shape to the template. MIPs with binding sites hold many advantages over natural antibodies, including storage stability, low cost, ease of preparation and reusability. Because of these advantages, MIPs are widely used in many different applications, such as affinity separation,18,19 catalysis,20,21 solid-phase extraction (SPE),22–24 drug release25 and sensors.26,27 Recent developments have demonstrated that applications of MIPs as SPE sorbents are the most advanced application area of MIPs and are good alternatives to immunoaffinity sorbents.28,29 To the best of our knowledge, AF molecularly imprinted polymers have not been prepared or employed as SPE sorbents for elimination of AFs from grains, foods and feed samples.
The MIPs used as SPE sorbents are usually prepared by bulk polymerization.30,31 However, it is known that the obtained imprinted polymers are blocks that need to be crushed, ground and sieved to get appropriate polymeric particles. The whole process is tedious and time-consuming, and the shapes and sizes of the obtained particles are usually irregular, resulting in low adsorption capacity and decreased of selectivity.
To address these concerns, a simple molecularly imprinted nanosphere for AFB1 was prepared by a miniemulsion polymerization method and applied in SPE coupled with pre-column derivation and HPLC-FLD. This method was developed and optimized for the determination of AFM1 and AFB1 in barley, beer, peanut oil, and feed samples. The imprinting performance of the AFB1 imprinted nanospheres was evaluated for adsorption capacity and selectivity. The factors affecting the extraction of AFB1 were optimized, and the validation and applicability of this method was evaluated. Compared with an immunoaffinity column, the proposed MISPE column provides a rapid, sensitive, and reliable method for analysis of AFM1 and AFB1 in grains, foods, and feed samples.
2. Experimental
2.1. Materials and reagents
AFB1 and AFM1 (5.0 mg) were purchased from Sigma Chemical (St Louis, MO, USA). Methacrylic acid (MAA) was obtained from Tianjin Kermel Chemical Reagent Co., Ltd, China. Ethylene glycol dimethacrylate (EGDMA) was purchased from Aladdin Chemistry Co., Ltd, China. 2,2′-Azobisiso-butyronitrle (AIBN) was supplied by Tianjin Baishi Chemical Industry Co., Ltd, China. Methanol and acetonitrile (chromatographic grade) were purchased from Tianjin Chemical Reagent Factory (Tianjin, China). Ultrapure water was purified on a Milli-Q system (Millipore Co., USA). All other reagents were analytical-grade and purchased from Guangzhou Chemical Reagent Factory (Guangzhou, China). Northwest barley and Jiangsu barley were obtained from State Farms malt Co., Ltd (Jiangsu province, China). Australian barley was obtained from Cofco Corporation, China. Peanut oil, beer, and feeds were purchased from the local market.
2.2. Instrumentation
HPLC analysis was carried out on an Agilent 1200 LC system (Agilent, Germany) equipped with a fluorescence detector (FLD) and a TC-C18 column (250 mm × 4.6 mm i.d., 5 μm packing). Excitation and emission wavelengths were set at 365 nm and 440 nm, respectively. Pre-column derivatization with trifluoroacetic acid to form a fluorescent intermediate was used to enhance fluorescence intensity. The mobile phase for HPLC experiments was acetonitrile–water (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, v/v), with a flow rate of 1.0 mL min−1. The injection volume was 20 μL, and column temperature was kept at 30 °C. Immunoaffinity columns (AflaTest) were supplied by Vicam (Watertown, MA, USA). Scanning electron microscopy (SEM) was performed on a Philips XL-30 (Japan). FTIR spectra were recorded with a Shimadzu model FTIR-84003 spectrophotometer. Particle size was measured by laser light scattering analyser (Mastersizer, 2000, Malvern, England). 1H NMR spectra measurements were executed on a VARIAN Mercury-Plus 300 NMR spectrometer operating at 250 MHz, using deuterated dimethyl sulfoxide (DMSO-d6) as solvent and tetramethylsilane (TMS) as an internal standard. The temperature was 25 °C.
:25, v/v), with a flow rate of 1.0 mL min−1. The injection volume was 20 μL, and column temperature was kept at 30 °C. Immunoaffinity columns (AflaTest) were supplied by Vicam (Watertown, MA, USA). Scanning electron microscopy (SEM) was performed on a Philips XL-30 (Japan). FTIR spectra were recorded with a Shimadzu model FTIR-84003 spectrophotometer. Particle size was measured by laser light scattering analyser (Mastersizer, 2000, Malvern, England). 1H NMR spectra measurements were executed on a VARIAN Mercury-Plus 300 NMR spectrometer operating at 250 MHz, using deuterated dimethyl sulfoxide (DMSO-d6) as solvent and tetramethylsilane (TMS) as an internal standard. The temperature was 25 °C.
2.3. Emulsion preparation
Fifty mL of a 1% emulsion (mass ratio of 3:7/Span80:CTAB, 0.8 mmol n-hexadecane) was sonicated for 20 min with an ultrasonic sonicator to form a stable emulsion.
2.4. Preparation of molecular imprinted polymers with emulsion methods
1.56 mg of AFB1, 1.72 mg of the functional monomer MAA, 19.82 mg of the cross linking agent EGDMA, and 5 mL of chloroform were added to a flask. The mixture was sonicated for 5 min and vented with N2 for 5 min. The mixture was placed slowly into a reactor containing 50 mL of emulsion, and sonicated for 20 min to form a miniemulsion. Then, 5 mg AIBN was added to the reactor and polymerization was achieved at 75 °C for 17 h under a nitrogen atmosphere with stirring at 400 rpm. Afterwards, the polymer microspheres were filtered and washed with dd-H2O and ethanol. In order to remove the template and residues of nonreactive species, the microspheres were taken into a soxhlet apparatus and refluxed with methanol–acetic acid (9:1, v/v) until no AFB1 was detected by HPLC. Finally, the microspheres were rinsed with water and dried at 60 °C in an oven for 12 h. The non-imprinted polymer microspheres (NIPs) were prepared using the same procedure but without AFB1.
2.5. Binding experiments
10 mg of either MIPs or NIPs was placed into centrifuge tubes and mixed with 2 mL acetonitrile–water (85:15, v/v) solution containing various concentrations (40, 80, 120, 160, 200 and 240 μg mL−1) of AFB1. The mixture was shaken for 1 h at room temperature, centrifuged, and filtered. The free concentration of AFB1 after adsorption was determined by HPLC. Based on the change in concentration of AFB1 in solution before and after binding, the adsorption capacity value Q of nanospheres for AFB1 were calculated by eqn (1)where C0 and Ce are the initial and equilibrium concentration of AFB1, respectively, V is the volume of the solution, and m is the mass of nanospheres.
2.6. Selectivity experiment
In order to evaluate the selective recognition ability, AFM1 and griseofulvin (GRI) were selected as competitive agents to estimate selectivity of the imprinted nanospheres for AFB1. 10 mg of MIPs or NIPs were added, respectively, to centrifuge tubes containing 2 mL of 200 μg mL−1 AFB1, 200 μg mL−1 AFM1 and 200 μg mL−1 griseofulvin mixture solutions. The mixture was shaken for 1 h at room temperature, centrifuged and filtered. Free AFB1 and AFM1 were determined by HPLC.
2.7. MISPE procedure
The cartridges were prepared by packing 200 mg of wet polymer into empty SPE-cartridges (Supelco, USA). The cartridges were preconditioned with 5 mL of methanol, 5 mL of methanol–acetic acid (9:1, v/v) and 5 mL of water successively. 2.0 mL of AFB1 (6 ppb, in acetonitrile) and AFM1 (10 ppb, in acetonitrile) mixture solution were loaded onto the cartridges at a flow rate of 2 mL min−1. After loading, the cartridges were washed with 2 mL of water and centrifuged at 4000 rpm for 5 min to avoid incomplete fractions. Finally, the extracts were eluted with 3 × 2 mL of methanol–acetic acid (9:1, v/v). The elution was immediately dried at 60 °C under a nitrogen stream. The residue was dissolved in 200 μL of hexane and 100 μL of trifluoroacetic acid derivative for 30 min. After evaporation of the solution to dryness under a stream of nitrogen at ambient temperature, the residue was dissolved in 200 μL mobile phase solution for subsequent HPLC analysis.
2.8. Official method based on immunoaffinity columns procedure
Immunoaffinity columns (IAC) procedure was performed according to the AOAC official standard method.32 IAC column was equilibrated with 10 mL of PBS solution at a flow rate of 2–3 drops per second. After 2.0 mL of sample extract passed through it, the IAC column was washed with 15 mL dd-H2O, and then the AFs were eluted with 2.5 mL of acetonitrile. The elution was immediately dried at 60 °C under a nitrogen stream. The residue was dissolved in 200 μL of hexane and 100 μL of trifluoroacetic acid derivative for 30 min. After evaporation of the solution to dryness under a stream of nitrogen at ambient temperature, the residue was dissolved in 200 μL mobile phase solution for subsequent HPLC analysis.
2.9. Sample extraction
The powder-type samples were extracted as follows: a 20 g sample was weighed into a 250 mL Erlenmeyer flask and extracted with 80 mL acetone–water (70 + 30) by shaking for 30 min at ambient temperature.33 After extraction, the sample was filtered with folded filter paper. For peanut oil and beer samples, 10 g samples were extracted with 20 mL of hexane (vortexed for 2 min) and 40 mL of acetone–water (70 + 30) and combined for 30 min with shaking. After extraction and separation, a portion of the acetone–water extract was loaded on the MISPE cartridges or immunoaffinity columns. Test portions of blank samples were spiked at levels of 1.2, 2.4 and 4.8 μg kg−1.
3. Results and discussion
3.1. Extraction mechanism of MIP
Template molecule AFB1 with functional monomer MAA based molecular imprinting was synthesized by radical polymerisation. The structural features of the AFB1 indicate that hydrogen bonds and dipole–dipole interactions are expected to be formed between AFB1 and MAA, whereby a carboxylic group of MAA works as a hydrogen bond acceptor interacting with oxygen atom of the AFB1 body, respectively. The aromatic domains can provide structural elements that stabilize intermolecular complexes via π–π interactions and hydrophobic association34 (Fig. 1). After removal of the template, the cavities capable of selectively recognizing and re-binding the AFB1 and its analogues were formed in the polymer (Fig. 1). The MIP as sorbent was packed into a SPE column while AFB1 and its analogues in samples was extracted selectively on the MIP loaded SPE cartridges.
 |
| | Fig. 1 Schematic of the extraction mechanism. | |
3.2. Characterization of MIP and NIP
The chemical structures of the obtained MIP and NIP were confirmed by 1H NMR and FT-IR spectroscopy. 1H NMR spectra are shown in Fig. 2. The signals at 0.8–1.2 and 1.92–2.0 ppm are ascribed to –C–CH3 and –C–CH2– of the MAA unit. The signals at 3.36 ppm and 2.51 ppm are due to O–CH2–CH2–O and C–CH2–C proton of polymer segment respectively. The –COOH active proton peak at 10–12 ppm was not detected in MIP and NIP. This is because the binding of H+ with AFB1 or H2O were destroyed and can be removed during the elution process. Compared with the 1H NMR spectra of NIP, the signals at 5.34 ppm due to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH was clearly seen in MIP, indicated that AFB1 was successfully imprinted in the polymers. The molecular weight of the MIP and NIP was calculated from the NMR spectrum by using the ratio between the peak areas at 2.51 and 1.2. The number-averaged molecular weight of the MAA–EGDMA–EGDMA (NIP) and MAA–EGDMA–EGDMA–AFB1 (MIP) copolymer of 9% and 21% MAA weight content was determined to be 2272.6 and 2463.6, respectively.
CH was clearly seen in MIP, indicated that AFB1 was successfully imprinted in the polymers. The molecular weight of the MIP and NIP was calculated from the NMR spectrum by using the ratio between the peak areas at 2.51 and 1.2. The number-averaged molecular weight of the MAA–EGDMA–EGDMA (NIP) and MAA–EGDMA–EGDMA–AFB1 (MIP) copolymer of 9% and 21% MAA weight content was determined to be 2272.6 and 2463.6, respectively.
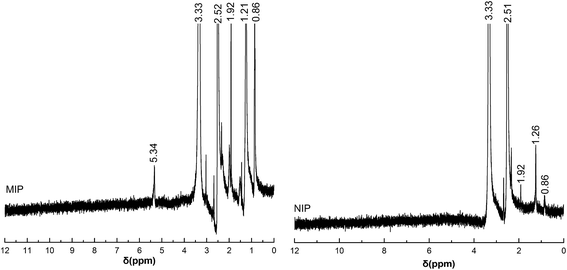 |
| | Fig. 2 1H NMR spectra of MIP and NIP in DMSO-d6. | |
FTIR spectra of MIP and NIP are shown in Fig. 3. The wide and strong absorption bands at about 3518 cm−1 and 953 cm−1 were due to the stretching vibrations of –OH groups from MAA. Vibration bands of CH3 were observed at about 2984, 2947, 1453 and 1387 cm−1, respectively. The strong absorption bands at 1726 cm−1 indicated the existence of carbonyl functional groups (–COO) from EGDMA and MAA. MIP have very weak CC vibrations at 1625 cm−1, suggesting that the CC double bond is broken after polymerization. The strong absorption bands at 1257 cm−1 and 1162 cm−1 indicated the existence of C–O stretching vibrations in the polymers. Compared with the infrared data of NIP, the absorption peaks of MIP at 3518, 1162 and 953 cm−1 corresponding to NIP, showed a slight drift. This indicated that the hydrogen bonds of alpha methyl acrylic acid with molecular template in the MIP were destroyed (Fig. 1), thus causing the electron cloud density of methyl acrylic acid to increase. This, in turn, increased the force constant of the bond, leading to a vibrational frequency shift toward the high frequency end of the spectrum. MIP was thus successfully synthesized based on this hydrogen bonding interaction.
 |
| | Fig. 3 The FTIR spectra of MIP and NIP. | |
The SEM images shown in Fig. 4 revealed that MIP and NIP were regular, spherical with a rough surface, and displayed good dispersion, which is conducive for rapid binding of template molecules. The particle size distribution and average particle size of the microspheres was measured by laser light scattering shown in Fig. 5. The particle size distribution of the MIP and NIP was found to be between 44 and 189 μm, and between 24 and 162 μm, respectively. The average particle size of the MIP and NIP was 108.5 and 98.6 μm, respectively. The obtained MIP and NIP particles with a rather broad size distribution may be attributed to the uneven stirring speed and reaction temperature fluctuations during polymerization, which results in emulsion droplets with size differences. However, the morphology of MIP and NIP and the average particle size showed no significant differences, indicating its adsorption selectivity differences are mainly caused by imprinting effects.
 |
| | Fig. 4 Scanning electron microscopy of MIP and NIP. | |
 |
| | Fig. 5 The particle size distribution and mean particle size of MIPs and NIPs. | |
3.3. Adsorption isotherm
It is important to investigate the adsorption capacity of the imprinted nanospheres. Binding experiments were performed as described in Section 2.5. The adsorption isotherms of MIP and NIP to AFB1 are plotted in Fig. 6, and shows that the binding capacity of MIP increased with increasing concentration of AFB1 until it reached an equilibrium state, which is greater than that of NIP. The maximum, Qmax, adsorption of MIP and NIP for AFB1, was estimated to be 8.2 mg g−1 and 4.9 mg g−1, respectively. The static adsorption capacity of MIP was about two times that of NIP. The results indicated that MIP has a good imprinting effect to AFB1 and is a potential sorbent to enrich tracing of AFB1 in complicated samples.
 |
| | Fig. 6 Binding isotherm of MIPs and NIPs for AFB1. | |
3.4. Adsorption selectivity
AFM1 and GRI were chosen as species for the competitive recognition research. The adsorption amounts, Qe, were examined as described in Section 2.6. The distribution coefficient (Kd), selectivity coefficient (k) and the relative selectivity coefficient (α) of the sorbent were calculated by the following formulas| |
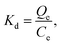 | (2) |
| |
 | (3) |
| |
 | (4) |
where Ce (mg L−1) is the equilibrium concentration of AFB1 or AFM1. The results are shown in Table 1. Kd and k values of MIP are clearly larger than that of NIP. The relative selectivity coefficients of AFB1 and AFM1 were 1.6 and 1.4, respectively, and possibly due to MIP offering more specific recognition sites for the template AFB1 and its structural analogues AFM1. These results indicated that MIP has a strong binding ability and high selectivity for the template molecule AFB1 and its structural analogues AFM1.
Table 1 Adsorption selectivity of MIP and NIP
| Adsorbate |
MIP |
NIP |
α |
| Qe (mg g−1) |
Kd (mL g−1) |
k |
Qe (mg g−1) |
Kd (mL g−1) |
k |
| AFB1 |
8.2 |
51.6 |
1.98 |
4.9 |
27.9 |
1.26 |
1.6 |
| AFM1 |
7.4 |
45.4 |
1.75 |
4.8 |
27.3 |
1.23 |
1.4 |
| GRI |
4.6 |
26.0 |
|
4.0 |
22.2 |
|
|
3.5. Desorption and reusability
To investigate the reusability of the MIP sorbents, the adsorption–desorption cycle was checked 10 times using the same MIP sorbents with methanol–acetic acid (9:1, v/v) as the eluting solvent. It was found that the adsorption capacity of MIP for AFB1 and AFM1 remained essentially the same as cycle number increased from 1 to 6. After cycle number 6, however, the adsorption capacity of MIP for AFB1 and AFM1 slowly decreased. After ten cycles, the adsorption capacity of MIP for AFB1 and AFM1 decreased by 11.6% and 14.8%, respectively, indicating that MIP had good reusability and stability for AFB1 and AFM1 adsorption.
3.6. Optimization of the MISPE procedure
The development of a separation and enrichment process was crucial for detecting relatively low concentrations of analytes contained in complex samples. MISPE was the most effective method for the separation and enrichment of the target analytes and the process was optimized with respect to loading sample pH, flow rate, washing solvent and type and volume of the eluting solvent.
The effect of the loading sample pH was investigated within a pH range from 3 to 9. The results show that the adsorption capacity of MIP for AFB1 and AFM1 increased with increasing pH from 3 to 6 and then gradually decreased for pH values from 7 to 9. This might be attributed to the binding of H+ with AFB1 or AFM1 at a low pH, which hindered the combination of AFB1 or AFM1 with MIP. Above pH 7, small amounts of AFB1 or AFM1 bound due to the degradation of AFB1 or AFM1. Thus, a pH of 6.5 was chosen as the optimum loading sample pH.
Different washing solvents, consisting of 5% methanol–water, 10% methanol–water and 20% methanol–water, were selected for this study. As shown in Fig. 7, the best results were obtained with a 10% methanol–water solution. This is probably due to the fact that higher concentrations of methanol (greater than 10%) can wash out different polar interfering compounds, although it may wash out analytes to a certain extent as well. Additionally, washing volume was investigated from a range of 1 mL to 8 mL. The results indicated that washing with a 2 × 2 mL of 10% methanol–water solvent had no obvious effect on the retention of AFB1 and AFM1 on MIP cartridges. Beyond 4 mL, the recovery of AFB1 and AFM1 on the MIP cartridge gradually decreased. Thus, a 2 × 2 mL of 10% methanol–water was selected for washing interfering compounds.
 |
| | Fig. 7 The recovery of washing solution for MISPE experiment. | |
The influence of different eluent solvents was also investigated in order to examine the desorbing properties of AFB1 and AFM1 from the sorbent (Table 2). It was determined that an increase in methanol and acetic acid in the elution solution, increased the recovery of AFB1 and AFM1 in the MIP cartridge. The best results were obtained for 4 mL of methanol–acetic acid (9:1, v/v). This may be because acetic acid competed with AFB1 and AFM1 for MIP in the binding sites and methanol was able to decrease the non-specific interactions between MIP and the two target analytes. The effect of different volumes of eluent (1.0, 2.0, 4.0, 6.0 and 8.0 mL) was also studied. The results are shown in Fig. 8. The recovery of AFB1 and AFM1 increased to 96.4% and 91.9%, respectively, by increasing the volume of eluent up to 6.0 mL and then remained constant. Thus, 6.0 mL of volume was selected for the elution step.
Table 2 Effect of type of eluent on AFB1 and AFM1 recovery
| Eluent |
Volume/mL |
Recovery/% |
| AFB1 |
AFM1 |
| Methanol–water (1:3, v/v) |
4 |
6.0% |
11.5% |
| Methanol–water (1:1, v/v) |
4 |
9.9% |
24.8% |
| Methanol–water (9:1, v/v) |
4 |
23.3% |
38.7% |
| Methanol–acetic acid (95:5, v/v) |
4 |
78.8% |
72.6% |
| Methanol–acetic acid (9:1, v/v) |
4 |
94.1% |
86.9% |
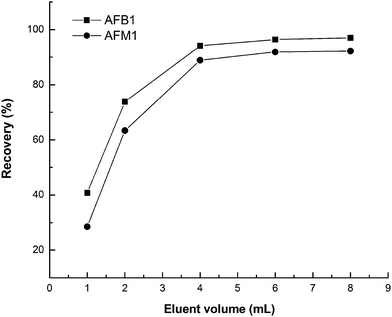 |
| | Fig. 8 Effect of the volume of eluent. | |
The effect of loading sample flow rate was investigated in the range of 0.5 mL min−1 to 4 mL min−1. The results showed that for flow rates higher than 2.0 mL min−1, the recoveries continuously decreased due to the decreased contact time of analytes with the MIP adsorbent. Therefore, 2.0 mL min−1 was chosen as the optimal sample flow rate. The flow rate of eluent solvent was also studied from the range of 0.5 mL min−1 to 4 mL min−1. Maximum recovery was obtained in the range of 0.5 mL min−1 to 1.0 mL min−1. For flow rates higher than 1.0 mL min−1, the recoveries decreased, as the analyte was not completely eluted from the packed column bed. Thus, a 1 mL min−1 flow rate was selected as the optimum eluting flow rate.
3.7. Comparison of retention behaviour of the immunoaffinity column and the MISPE column
Chromatograms of the spiked Australia barley extraction solution from both the immunoaffinity and MISPE columns are shown in Fig. 9. The spike level was 2.4 ppb for aflatoxins M1 and B1. Fig. 9A shows the extraction solution obtained from the immunoaffinity column and Fig. 9B shows the results from the MISPE column. The retention times at 3.99 min and 7.64 min were identified as AFM1 and AFB1, respectively. It was found that AFM1 and AFB1 were retained well for both the immunoaffinity and the MISPE columns. Moreover, the matrix compounds were removed more so for the MISPE column than for the immunoaffinity column. The recoveries of AFM1 and AFB1 were 92.8% and 93.5% on the immunoaffinity column, respectively, and 92.1% and 93.3% for the MISPE column. These results demonstrated that the MISPE column can be used as a substitute for the immunoaffinity column for effective removal of complicated matrix molecules.
 |
| | Fig. 9 Chromatograms of the spiked Australia barley sample extraction solution from the immunoaffinity (A) and MISPE column (B). | |
3.8. Method validation
A method based on MISPE coupled to HPLC was established. Using the determined optimum conditions, an external aqueous calibration and standard addition calibration of a blank sample extract were constructed containing AFB1 and AFM1 between 0.2 ppb to 20 ppb (μg L−1) in order to study possible matrix effects. The calibration equation in aqueous solution was y = 17564cAFM1 − 540 (R2 = 0.9996) and y = 17641cAFB1 − 807 (R2 = 0.9998) for AFM1 and AFB1, respectively. The calibration equation in extract was y = 17189cAFM1 − 282 (R2 = 0.9994) and y = 17296cAFB1 − 756 (R2 = 0.9995) for AFM1 and AFB1, respectively. There were no significant differences in the calibration curves for both AFB1 and AFM1 obtained in aqueous and in the matrix solutions. The results showed that the matrix could be efficiently removed during the MISPE pre-concentration stage. Therefore, the external aqueous calibration can be used to determine AFB1 and AFM1 in practical samples after the MISPE pre-concentration procedure.
Limit of detection (LOD) and limit of quantification (LOQ) were calculated according to LOD = (3SD)/m and LOQ = (10SD)/m, where SD is the standard deviation of 10-replicate measurements on a procedural blank (Milli-Q water treated as a sample), and m is the slope of the external aqueous calibration curve. The LOD of AFM1 and AFB1 was 0.05 μg kg−1. Similarly, the LOQ was found to be 0.16 μg kg−1 for AFM1 and AFB1. These LOD and LOQ values are similar to reports from Chun35 using HPLC with a fluorescence detector (0.08 μg kg−1 and 0.15 μg kg−1 for AFB1) and are slightly less sensitive than what Manetta and Pena reported in previous literature reports12,36 (5 ng kg−1 for AFM1, 0.02 μg kg−1 and 0.07 μg kg−1 for AFB1). However, this developed method using a preliminary MISPE column replacing an immunoaffinity column as a purification step reduces cost. Moreover, the detection limit is far below the current legal national standard limit in China. So the proposed method is feasible for determination of AFM1 and AFB1 in foods and feeds.
Accuracy of the overall procedure was evaluated by the determination of the recoveries of four blank samples spiked with aflatoxins M1 and B1 at concentrations of 1.2, 2.4 and 4.8 ppb. Precision was assessed by analyzing a spiked sample six times and was expressed as the relative standard deviation (RSD). The results are presented in Table 3. The obtained recovery values and RSD from the four spiked samples ranged from 83% to 96% and from 2.2% to 5.6%, respectively, indicating that the developed method is accurate enough for determining AFM1 and AFB1 in food samples.
Table 3 The recovery values and RSD of spiked sample at three different concentration (n = 6)
| Samples |
Analytes |
Spiked (μg kg−1) |
Founda (μg kg−1) |
Recovery (%) |
RSD (%) |
| Found was expressed with the mean ± standard deviation (SD). |
| Barley |
AFM1 |
1.2 |
1.0 ± 0.1 |
83 |
5.6 |
| 2.4 |
2.2 ± 0.1 |
92 |
4.5 |
| 4.8 |
4.5 ± 0.2 |
94 |
3.5 |
| AFB1 |
1.2 |
1.1 ± 0.0 |
92 |
4.5 |
| 2.4 |
2.2 ± 0.1 |
92 |
3.6 |
| 4.8 |
4.5 ± 0.2 |
94 |
3.3 |
| Peanut oil |
AFM1 |
1.2 |
1.0 ± 0.0 |
83 |
4.7 |
| 2.4 |
2.2 ± 0.1 |
92 |
4.5 |
| 4.8 |
4.4 ± 0.2 |
92 |
3.8 |
| AFB1 |
1.2 |
1.1 ± 0.0 |
92 |
5.5 |
| 2.4 |
2.2 ± 0.1 |
92 |
4.1 |
| 4.8 |
4.5 ± 0.2 |
94 |
3.8 |
| Feed |
AFM1 |
1.2 |
1.1 ± 0.0 |
92 |
3.6 |
| 2.4 |
2.2 ± 0.1 |
92 |
3.6 |
| 4.8 |
4.5 ± 0.1 |
94 |
2.2 |
| AFB1 |
1.2 |
1.1 ± 0.0 |
92 |
4.5 |
| 2.4 |
2.3 ± 0.1 |
96 |
4.4 |
| 4.8 |
4.5 ± 0.2 |
94 |
3.5 |
| Beer |
AFM1 |
1.2 |
1.1 ± 0.0 |
92 |
4.6 |
| 2.4 |
2.2 ± 0.1 |
92 |
4.1 |
| 4.8 |
4.4 ± 0.1 |
92 |
2.9 |
| AFB1 |
1.2 |
1.1 ± 0.0 |
92 |
4.5 |
| 2.4 |
2.2 ± 0.1 |
92 |
3.6 |
| 4.8 |
4.5 ± 0.1 |
94 |
3.4 |
3.9. Application
To demonstrate the application of our method, barley, peanut oil, beer and feed samples (Section 2.8) were tested. The results are listed in Table 3. The AFM1 contamination was not detected in all samples tested. The AFB1 contamination was not detected in barley, peanut oil, or beer samples, but was detected in feed. The typical chromatograms of naturally contaminated corn feed samples are shown in Fig. 10. Retention time at 7.67 min was identified as AFB1. Four feed samples out of six (66.7% incidence) were contaminated with AFB1 and 1 feed sample (16.7% incidence) was over 10 μg kg−1, which is the feed hygiene standard (GB13078-2001) tolerance limit for AFB1 in China. High detection rates and high levels of AFB1 in feed samples indicated the need for routine monitoring to maintain AFB1 to the lowest possible levels (Table 4).
 |
| | Fig. 10 Chromatograms of feed (A) and feed spiked with AFB1 at 1.2 ppb (B). | |
Table 4 Levels of aflatoxin M1 and B1 in real samplesa
| Sample no. |
Sample type |
AFM1 |
AFB1 |
| nd: not detected. |
| 1 |
Australia barley |
nd |
nd |
| 2 |
Jiangsu barley |
nd |
nd |
| 3 |
Northwest barley |
nd |
nd |
| 4 |
Luhua peanut oil |
nd |
nd |
| 5 |
Yingmai peanut oil |
nd |
nd |
| 6 |
Jinglongyu peanut oil |
nd |
nd |
| 7 |
Longda peanut oil |
nd |
nd |
| 8 |
Blue ribbon beer |
nd |
nd |
| 9 |
Qingdao beer |
nd |
nd |
| 10 |
Yanjing beer |
nd |
nd |
| 11 |
Budweiser beer |
nd |
nd |
| 12 |
Shell beans feed |
nd |
5.3 ± 0.2 |
| 13 |
Shell beans feed |
nd |
nd |
| 14 |
Corn feed |
nd |
7.3 ± 0.3 |
| 15 |
Corn feed |
nd |
nd |
| 16 |
Formula feed |
nd |
8.2 ± 0.3 |
| 17 |
Formula feed |
nd |
11.7 ± 0.5 |
4. Conclusion
In this study, AFB1-imprinted MIPs were successfully synthesized by a miniemulsion polymerization method and used as sorbents for MISPE of food and feed samples. The sorbent exhibited strong binding ability, high selectivity, good reusability and stability. Under optimized MISPE conditions, the cartridge showed high extraction efficiency and removed matrix interferences from real samples efficiently, suggesting that it can be employed as a substitute to immunoaffinity columns for sample purification steps in the detection of aflatoxin. The developed MISPE coupled with HPLC-FLD showed low detection limits, high precision, a high degree of accuracy, and can serve as a monitoring system for aflatoxin (M1 and B1) contamination in a variety of food and feed samples in a cost-saving manner (immunoaffinity column not required).
Acknowledgements
This work is financially supported by the Science and Technology Innovation Project of Guangdong provincial education department (no. 2012kjcx0104 and no. 2013kjcx0194), the Science and Technology Planning Project of Zhaoqing City, Guangdong province (no. 2013C019) and the Natural Science Foundation of Guangdong Province (no. S2012040007710).
References
- T. Yoshinari, S. Sakuda, M. Watanab, Y. Kamata, T. Ohnishi and Y. Sugita-Konishi, J. Agric. Food Chem., 2013, 61, 7925 CrossRef CAS PubMed.
- R. Zivoli, L. Gambacorta, G. Perrone and M. Solfrizzo, J. Agric. Food Chem., 2014, 62, 5707 CrossRef CAS PubMed.
- E. E. Creppy, Toxicol. Lett., 2002, 127, 19 CrossRef CAS.
- International Agency for Research on Cancer (IARC), IARC Monographs on the Evaluation of Carcinogenic Risk to Humans, 1993, vol. 56, p. 245 Search PubMed.
- Commission Regulation (EU) no. 165/2010 of 26 February 2010 amending Regulation (EC) no. 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards aflatoxins.
- X. Lai, R. Liu, C. Ruan, H. Zhang and C. Liu, Food Control, 2015, 50, 401 CrossRef CAS PubMed.
- K. Benešová, S. Běláková, R. Mikulíková and Z. Svoboda, Food Control, 2012, 25, 626 CrossRef PubMed.
- S. Vijayanandraj, R. Brinda, K. Kannan, R. Adhithya, S. Vinothini, K. Senthil, R. R. Chinta, V. Paranidharan and R. Velazhahan, Microbiol. Res., 2014, 169, 294 CrossRef CAS PubMed.
- Y. K. Wang, Y. X. Yan, S. Q. Li, H. A. Wang, W. H. Ji and J. H. Sun, J. Agric. Food Chem., 2013, 61, 10948 CrossRef CAS PubMed.
- M. Campas, D. Garibo and B. Prieto-Simon, Analyst, 2012, 137, 1055 RSC.
- M. Zangheri, F. D. Nardo, L. Anfossi, C. Giovannoli, C. Baggiani, A. Roda and M. Mirasoli, Analyst, 2015, 140, 358 RSC.
- R. Peña, M. C. Alcaraz, L. Arce, A. Ríos and M. Valcárcel, J. Chromatogr. A, 2002, 967, 303 CrossRef.
- H. Wang, L. Zhao, H. M. Yang, Q. L. Guo, H. L. Shi, H. Y. Pan, L. P. Zhao and C. Qian, Anal. Methods, 2014, 6, 1545 RSC.
- M. Hashemi and Z. Taherimaslak, Anal. Methods, 2014, 6, 7663 RSC.
- W. S. Khayoon and B. Saad, Food Chem., 2010, 118, 882 CrossRef CAS PubMed.
- X. Gao, W. Y. Cao, M. M. Chen, H. Y. Xiong, X. H. Zhang and S. F. Wang, Electroanalysis, 2014, 26, 2739 CrossRef CAS.
- X. Ma, W. Ji, L. Chen, X. Wang, J. Liu and X. Wang, J. Sep. Sci., 2015, 38, 100 CrossRef CAS PubMed.
- K. Nemoto, T. Kubo, M. Nomachi, T. Sano, T. Matsumoto, K. Hosoya, T. Hattori and K. Kaya, J. Am. Chem. Soc., 2007, 129, 13626 CrossRef CAS PubMed.
- R. E. Fairhurst, C. Chassaing, R. F. Venn and A. G. Mayes, Biosens. Bioelectron., 2004, 20, 1098 CrossRef CAS PubMed.
- J. P. Ferreira, R. Viveiros, A. Lourenço, S. M. Soares, A. Rosatella and C. M. Casimiro TAfonso, RSC Adv., 2014, 4, 54948 RSC.
- W. Q. Sun, R. Tan, W. G. Zheng and D. H. Yin, Chin. J. Catal., 2013, 34, 1589 CrossRef CAS.
- H. A. Panahi, A. Mehramizi, S. Ghassemi and E. Moniri, J. Sep. Sci., 2014, 37, 691 CrossRef CAS PubMed.
- S. Manzoor, R. Buffon and A. V. Rossi, Talanta, 2015, 134, 1 CrossRef CAS PubMed.
- Y. Yang, J. Yu, J. Yin, B. Shao and J. Zhang, J. Agric. Food Chem., 2014, 62, 11130 CrossRef CAS PubMed.
- C. Y. Wang, A. Javadi, M. Ghaffari and S. Q. Gong, Biomaterials, 2010, 31, 4944 CrossRef CAS PubMed.
- Y. Tong, H. Li, H. Guan, J. Zhao, S. Majeed, S. Anjum, F. Liang and G. Xu, Biosens. Bioelectron., 2013, 47, 553 CrossRef CAS PubMed.
- R. L. Lei, C. H. Guo, H. Y. Xiong, C. Dong, X. H. Zhang and S. F. Wang, Electroanalysis, 2014, 26, 1004 CrossRef CAS.
- M. Wyszomirski and W. Prus, Mol. Simul., 2012, 37, 1 Search PubMed.
- W. H. Ali, D. Derrien, F. Alix, C. Perollier, O. Lepine, S. Bayoudh, F. Chapuis-Hugon and V. Pichon, J. Chromatogr. A, 2010, 1212, 6668 CrossRef PubMed.
- N. M. Maier, G. Buttinger, S. Welhartizki, E. Gavioli and W. Lindner, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 103 CrossRef CAS PubMed.
- N. Yusof, S. Rahman, M. Hussein and N. Ibrahim, Polymers, 2013, 5, 1215 CrossRef CAS PubMed.
- AOAC Official methods of analysis, Natural Toxins, AOAC International, Gaithersburg, USA, 18th edn, 2005, p. 32, official method 999.07 Search PubMed.
- T. Bertuzzi, S. Rastelli, A. Mulazzi and A. Pietri, Food Anal. Methods, 2012, 5, 512 CrossRef.
- J. Jodlbauer, N. M. Maier and W. Lindner, J. Chromatogr. A, 2002, 945, 45 CrossRef CAS.
- H. S. Chun, H. J. Kim, H. E. Ok, J. B. Hwang and D. H. Chung, Food Chem., 2007, 102, 385 CrossRef CAS PubMed.
- A. C. Manetta, L. D. Giuseppe, M. Giammarco, I. Fusaro, A. Simonella, A. Gramenzi and A. Formigoni, J. Chromatogr. A, 2005, 1083, 219 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 












