
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural inorganic nanoparticles – formation, fate, and toxicity in the environment†
Virender K.
Sharma
*a,
Jan
Filip
b,
Radek
Zboril
b and
Rajender S.
Varma
*bc
aDepartment of Environmental and Occupational Health, School of Public Health, Texas A&M University, College Station, Texas 77843, USA. E-mail: vsharma@sph.tamhsc.edu
bRegional Centre of Advanced Technologies and Materials, Departments of Experimental Physics and Physical Chemistry, Faculty of Science, Palacký University in Olomouc, 771 46 Olomouc, Czech Republic
cSustainable Technology Division, National Risk Management Research Laboratory, US Environmental Protection Agency, Cincinnati, Ohio 45268, USA. E-mail: Varma.Rajender@epa.gov
First published on 5th October 2015
Abstract
The synthesis, stability, and toxicity of engineered metal nanoparticles (ENPs) have been extensively studied during the past two decades. In contrast, research on the formation, fate, and ecological effects of naturally-occurring nanoparticles (NNPs) has become a focus of attention only recently. The natural existence of metal nanoparticles and their oxides/sulfides in waters, wastewaters, ore deposits, mining regions, and hydrothermal vents, as exemplified by the formation of nanoparticles containing silver and gold (AgNPs and AuNPs), Fe, Mn, pyrite (FeS2), Ag2S, CuS, CdS, and ZnS, is dictated largely by environmental conditions (temperature, pH, oxic/anoxic, light, and concentration and characteristics of natural organic matter (NOM)). Examples include the formation of nanoparticles containing pyrite, Cu and Zn-containing pyrite, and iron in hydrothermal vent black smoker emissions. Metal sulfide nanoparticles can be formed directly from their precursor ions or indirectly by sulfide ion-assisted transformation of the corresponding metal oxides under anaerobic conditions. This tutorial focuses on the formation mechanisms, fate, and toxicity of natural metal nanoparticles. Natural waters containing Ag(I) and Au(III) ions in the presence of NOM generate AgNPs and AuNPs under thermal, non-thermal, and photochemical conditions. These processes are significantly accelerated by existing redox species of iron (Fe(II)/Fe(III)). NOM, metal–NOM complexes, and reactive oxygen species (ROS) such as O2˙−, ˙OH, and H2O2 are largely responsible for the natural occurrence of nanoparticles. AgNPs and AuNPs emanating from Ag(I)/Au(III)–NOM reactions are stable for several months, thus indicating their potential to be transported over long distances from their point of origin. However, endogenous cations present in natural waters can destabilize the nanoparticles, with divalent cations (e.g., Ca2+, Mg2+) being more influential than their monovalent equivalents (e.g., Na+, K+). The toxicity of NNPs may differ from that of ENPs because of differences in the coatings on the nanoparticle surfaces. An example of this phenomenon is presented and is briefly discussed.
 Virender K. Sharma | Virender K. Sharma received his PhD from Rosenstiel School of Marine and Atmospheric Science, University of Miami, Florida. His postdoctoral work was at SUNY, Buffalo and Brookhaven National Laboratory, New York. He is currently a professor at the Department of Environmental and Occupational Health, School of Public Health at Texas A&M University, College Station, Texas. His research focuses on chemistry and environmental applications of ferrates and ferrites and understanding fate, transport, and toxicity of nanoparticles in the aquatic environment. He has published more than 210 peer-reviewed journal publications and authored and edited five books. He has also edited special issues of international journals on environmental aquatic chemistry. He is actively engaged in national and international collaborations. |
 Jan Filip | Jan Filip received his Master's and PhD degrees in Mineralogy at Masaryk University in Brno, Czech Republic (2002 and 2008). His dissertation focused on the crystal chemistry of borosilicate minerals and OH defects in nominally-anhydrous minerals. In 2005 he joined the research group of Prof. Radek Zbořil and currently leads the group “Environmental nanotechnologies” of the Regional Centre of Advanced Technologies and Materials, Palacký University in Olomouc, Czech Republic. He underwent several foreign stays and is actively engaged in national and international collaborations. His research interests cover application of zero-valent iron nanoparticles, ferrates, and iron oxide nanoparticles in technologies of water treatment, identification of nanoparticles in the environment, and application of X-ray based techniques in materials science. He has coauthored over 70 papers in refereed journals. |
 Radek Zboril | Radek Zboril received his PhD degree at the Palacky University, Olomouc, Czech Republic. After his PhD studies, he underwent several foreign stays at universities in Tokyo, Delaware, and Johannesburg. Currently, he is a professor at the Department of Physical Chemistry and a general director of the Regional Centre of Advanced Technologies and Materials at the Palacký University, Olomouc, Czech Republic. His research interests focus on nanomaterials including iron and iron oxide-based nanoparticles, silver nanoparticles, carbon nanostructures, and magnetic nanoparticles and their applications in biomedicine, environmental technologies, sensing and catalysis. He has published more than 250 articles in journals including Chemical Society Reviews, Chemical Communications, Green Chemistry, Accounts of Chemical Research, and Chemical Reviews. |
 Rajender S. Varma | Rajender S. Varma was born in India (PhD, Delhi University, 1976). After postdoctoral research at Robert Robinson Laboratories, Liverpool, U.K., he was a faculty member at Baylor College of Medicine and Sam Houston State University prior to joining the Sustainable Technology Division at the US Environmental Protection Agency in 1999. He has over 40 years of research experience in the management of multidisciplinary technical programs ranging from natural products chemistry to the development of more environmentally friendly synthetic methods using microwaves, ultrasound, etc. Lately, he has focused on greener approaches to the assembly of nanomaterials and the sustainable applications of magnetically retrievable nanocatalysts in benign media. He is a member of the editorial advisory board of several international journals, has published over 415 scientific papers, and has been awarded 14 US Patents. |
Key learning points1. Naturally occurring nanoparticles (NNPs) are often present in all spheres of the Earth (i.e., in the atmosphere, hydrosphere, lithosphere and even in the biosphere), irrespective of human activities.2. Natural organic matter (NOM) could largely contribute to the formation of metal nanoparticles, typically exemplified by silver and gold nanoparticles (AgNPs and AuNPs) in the environment. 3. Mechanistically, the formation of metal nanoparticles entails the reaction of reactive oxygen species and NOM complexes with dissolved metal ions; the reaction being enhanced by elevated temperature and/or exposure to light. 4. Water properties (pH, redox conditions, the presence of ions/ionic strength, and concentrations of various types of NOM) determine the growth and stability of NPs in the aquatic environment. 5. Organic matter-coated natural metal nanoparticles display less toxicity than ENPs that are surface-coated by polymers and/or surfactants. |
1 Introduction
Nanoparticles (NPs) represent a specific type of matter (from about 1 to 100 nm in size). They are intermediate in size between bulk materials and atomic/molecular structures, and possess unique physical and chemical properties. These distinctive properties, related to a high surface area to volume ratio and/or quantum effects, have spawned notable interest from engineers, biologists, chemists, and physicists.1 In the past decade, there has been an exponential growth in the synthesis of NPs, commonly termed as engineered nanoparticles (ENPs), due to their extensive use in emerging technologies and in consumer products such as electronic devices and other products used for personal care, biomedicine, agriculture, water/soil treatment, and renewable energy.2–4 An array of ENPs have been manufactured which include mainly metals, non-metals, metal oxides, lipids, and polymers (Fig. 1) as well as various nanocomposites.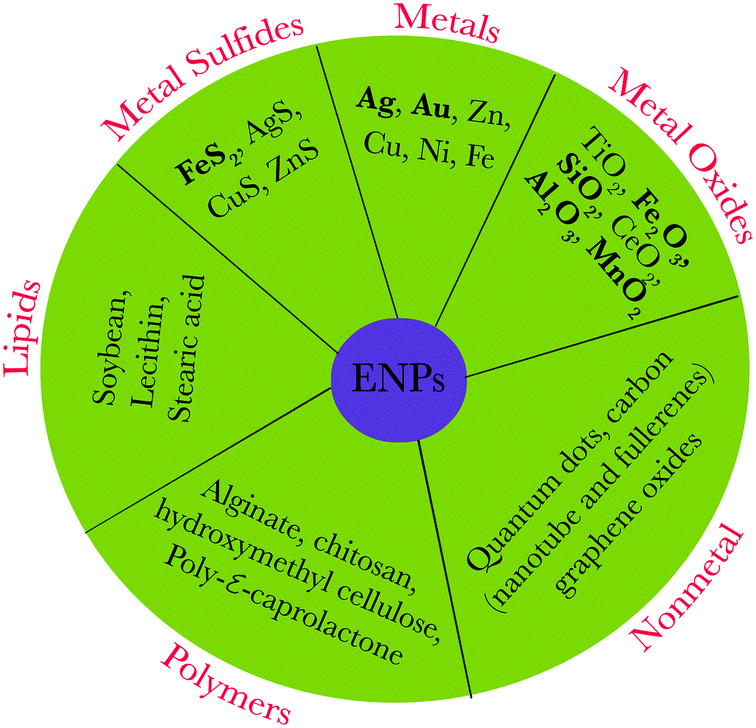 | ||
| Fig. 1 Representative types of engineered/natural nanoparticles; bold letters represent typical naturally occurring nanoparticles. | ||
Besides ENPs, nanoparticles can be formed naturally via processes occurring in all “spheres” of the Earth, thus covering the atmosphere, hydrosphere, lithosphere and even biosphere (Slides 2–8, ESI†). NNPs are being formed by chemical, photochemical, mechanical, thermal, and biological processes separately or in combination,5 including extraterrestrial processes (i.e., production of cosmic dust) as shown in Fig. 2. In addition, NPs are also formed spontaneously as a result of human activities (e.g., during mining, production of wastewaters and wastes in general, and other industrial processes). A recent estimate suggests the formation of NNPs, only from biogeochemical processes alone, occurs in the range of several thousand teragrams per year (1 Tg = 1 million metric tons).5 Comparatively, the mass of ENPs produced per year is orders of magnitude lower, in the range of several hundreds to thousands of Tg per year.5 For example, see the global budget of naturally occurring inorganic nanoparticles (Slide 9, ESI†).
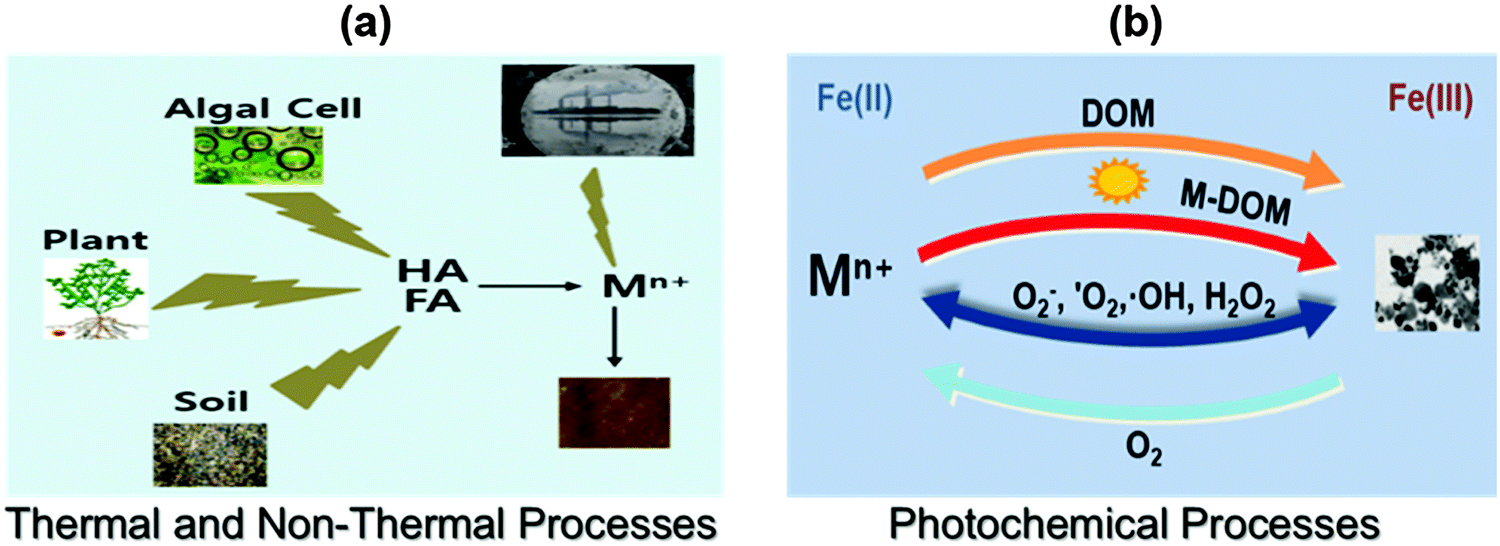 | ||
| Fig. 2 (a) Thermal and non-thermal processes and (b) photochemical processes that generate natural nanoparticles in the environment (for details see Section 2). (HA – humic acid; FA – fulvic acid; DOM – dissolved organic matter; Mn+ – metal ions (e.g., Ag(I) and Au(III)); M–DOM – metal ion complexation with dissolved organic matter). | ||
Reactions of metal salts or dissolved metal ions with vitamins (B1, B2, B12, C), sugars and tea-, plant extracts, coffee- and wine-derived polyphenolic antioxidants6 occur readily wherein these constituents function both as reducing and capping agents (Slide 10, ESI†). These reactions could culminate in simple and fast formation of nanomaterials, thus mimicking natural processes. Learning from nature, such transformations could pave the way for greener assembly of nanoparticles including the sustainable use of agricultural waste residues.6 Light-induced reactive oxygen species (ROS) (e.g., superoxide ion, O2˙−) could reduce metal ions to form nanoparticles7 where the reactions are influenced by the variation of temperature and light in the environment. Iron oxides/sulfides, silver, and gold are some of the representative examples of naturally-occurring nanoparticles in the environment.8,9
Numerous studies have been conducted on the fate and behavior of ENPs released into the environment, especially with the aim of examining their effects on humans and the ecosystems.10 In contrast, the knowledge base on the fate and toxicity to humans and ecosystems of naturally occurring metal nanoparticles is rather sparse. Generally, engineered metal NPs can be synthesized by the reduction of metal ions followed by surface functionalization of nanoparticles; agents such as citric acid, polysaccharides, proteins, surfactants, and polymers have been used to enhance the stability of ENPs.11 It is therefore quite likely that the underlying mechanism and mode of interaction of ENPs with cell surfaces to initiate toxicity is different from the interaction of cell surfaces with natural metal NPs, which are often covered with natural organic matter (NOM) components. Furthermore, the NPs surface-capped by NOM could be affected differently by environmental conditions such as pH, the presence of ions, and light when compared to the typical capping agents deployed for ENPs. It is therefore imperative to comprehend, independently, the formation and fate of naturally occurring metal NPs. The present tutorial focuses on the possibility of the natural existence, among others, of silver and gold nanoparticles (AgNPs and AuNPs) in the aquatic environment as these ions could occur together. Several studies have appeared in the last few years to augment assessments as to how these NPs may impact distinct compartments of the environment.7,12–17
Silver and gold have been known since ancient times as essential components in jewelry and currency coins, and are also present in colorful stained glass displays in cathedrals worldwide. In recent times, nanomaterials have garnered immense attention, and specifically, the closely related nano duo, silver and gold, have found numerous applications. AgNPs are one of the most extensively studied types of nanomaterials due to their unique sensing, catalytic, optical, and antimicrobial properties and as efficient probes for detecting various biomolecules and monitoring biotransformations.18,19 Similarly, AuNPs are exploited in cancer therapy and diagnostics, chemical and biological imaging, catalysis, and sensors.20 The continuing increase in applications of AgNPs and AuNPs has resulted in increasing apprehension regarding their release into the environment and associated potential effects on ecological systems.21–24 However, relatively little attention has been paid to the naturally occurring metal nanoparticles, to assess and model the toxicity of NNPs in the environment.
In the past few years, several studies have appeared to fill the void in understanding the formation of naturally-occurring metal nanoparticles.7,12,13,25 This tutorial contributes to the elucidation of the natural existence of metal nanoparticles, with detailed commentary on their possible formation mechanisms and expected fate, including the associated toxicity under environmentally relevant conditions.
2 Formation of natural nanoparticles (NNPs)
The classification of NNPs and all possible pathways leading to their formation is a complex and massive task as it covers all spheres of the Earth, chemical elements/species, and a vast number of diverse mechanisms, processes and conditions. Therefore, a concise overview is presented with a handful of typical examples of existing groups of NPs and the underlying mechanisms of their formation. The same synthetic principles are valid for both NNPs and ENPs. This synthesis can occur via bottom-up approaches starting from molecular/ionic species, e.g., the formation of ferrihydrite NPs mediated by microbial activity, or the formation of halide and hydrous sulfate nanoparticles from evaporation of sea spray. Synthesis can also occur via top-down approaches starting from larger precursors, e.g., nano-sized mineral fragments generated by wind erosion on deserts, or the formation of carbon NPs from the combustion of biomass.5,15,16On Earth, nano-sized objects are formed and occur within all spheres, thus covering the atmosphere (including the whole troposphere, and some types of NPs can be found at even higher levels), hydrosphere (oceans, lakes and rivers, groundwater, pore water and hydrothermal vents), lithosphere (soils, rocks, lava or magma at certain stages of evolution), and biosphere (mainly in/at microorganisms, but also including higher organisms and even humans).5 From this burgeoning list of possible NP occurrences, the NPs in the atmosphere and hydrosphere, which are reported to occur at concentrations up to 106–107 particles mL−1 have the major effects on biota due to their close contact/interactions with biota.
The main processes leading to the formation of NNPs that are purely inorganic in character may comprise (see Fig. 3):
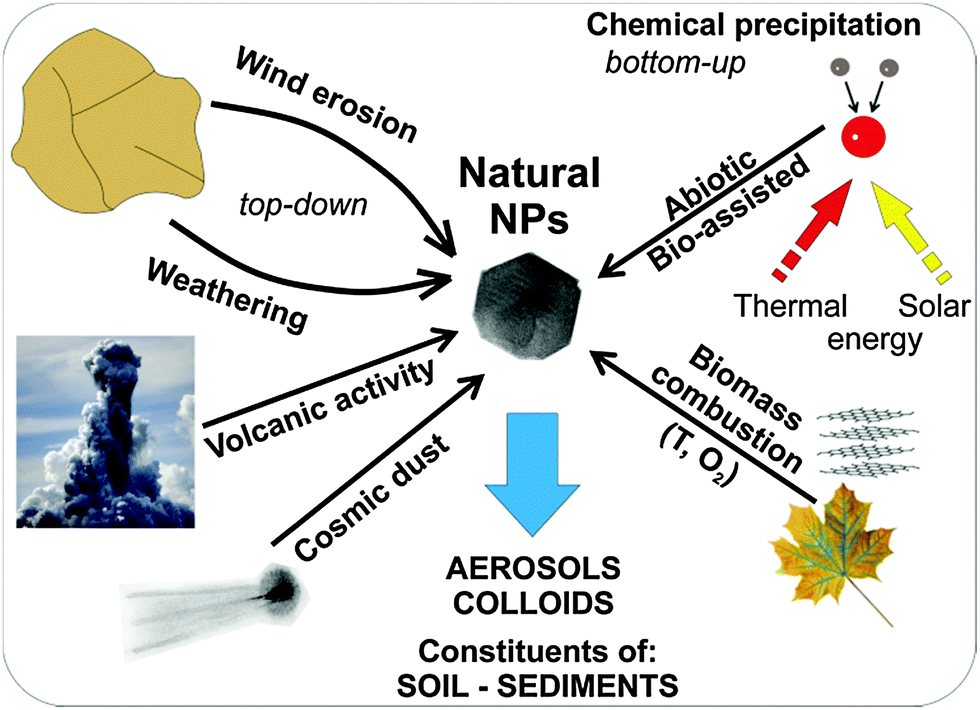 | ||
| Fig. 3 Natural processes leading to the formation of nanoparticles in the environment. (Natural NPs – natural nanoparticles; T – temperature; O2 – oxygen). | ||
– nucleation and growth of various inorganic phases in the atmosphere, hydrosphere (including black smokers and other hydrothermal vents) and even in the lithosphere (melts) as a result of purely inorganic reactions, or with contribution from organic matter.5 The reactions in surface water and hydrothermal vents that contribute to the generation of NNPs in the environment may proceed via non-thermal, thermal, and photochemical processes (Fig. 2). A typical occurrence of Fe(II) in the geochemical environment may thus facilitate the formation of ferrihydrite nanoparticles, which may be stabilized by silicon ions.16 Other NNPs containing Mn, Cr, Cu, Ba, and Pb could also be formed in cold CO2 seeps.
– mechanical processes – exemplified by aeolian erosion by desert wind, deforested lands and un-vegetated farmlands, and particles emanating from events prompted by mechanical grinding of the Earth's crust during earthquakes.
– thermal processes – typified by the most widespread process of combustion of biomass, which is common mainly in equatorial parts of the Earth.
Organisms, particularly microorganisms, extensively generate NPs in the environment.5,15 Biological processes (or biomineralization) in nature produce a number of inorganic nanomaterials such as Fe- and Si-based nanominerals, calcium carbonate, and calcium phosphate.15 Among them, two processes are well understood and are designated as (i) biologically induced mineralization (BIM) and (ii) biologically controlled mineralization (BCM) (Fig. 4). In the BIM process, no function being particularly controlled by microorganisms is involved in nanoparticle formation, except, either an association of a solid substrate attached to bacteria or interaction with bacterial cell wall/membrane (i.e., nanoparticles are formed as a result of metabolic processes). In contrast, nucleation and the growth of the particles are entirely controlled by the organisms during BCM processes and the nanominerals are usually formed in the cells under certain conditions. Therefore, the mineral particles, generated by bacteria based on BCM, are well-defined crystals with narrow particle-size distributions. Nanoparticles produced by these methods have various functions for the organisms, (e.g., the well-known magnetotactic bacteria use magnetite nanoparticles for navigation),26 iron storage and tissue hardening (mainly BCM). Alternatively, NPs are formed indirectly through redox reactions in the microbial environment related to metabolic processes as exemplified in the production of nanocrystalline Mn- and Fe-oxides (a typical BIM process).15 The most representative example is the production by iron-oxidizing bacteria (Leptothrix, Gallionella) of ferrihydrite and bacterially mediated ferric oxyhydroxide that have been identified in sediments, groundwater, and soils. Magnetite NPs act as electron transfer mediators in Geotracer sulurreducens and Thiobacillus denitrificans to promote acetate oxidation coupled to nitrate reduction under anaerobic conditions.5 Interestingly, hematite NPs facilitate respiration of the Shewanella oneidensis MR-1 cell in which reduction of iron occurs.5 The biomineralization processes can also form Cu0 colloids which may be subsequently transformed into copper-rich sulfide particles under sulfate reducing conditions, mediated by bacteria.22 Importantly, large amounts of silica NPs are produced from an assorted group of eukaryotes and prokaryotes.15
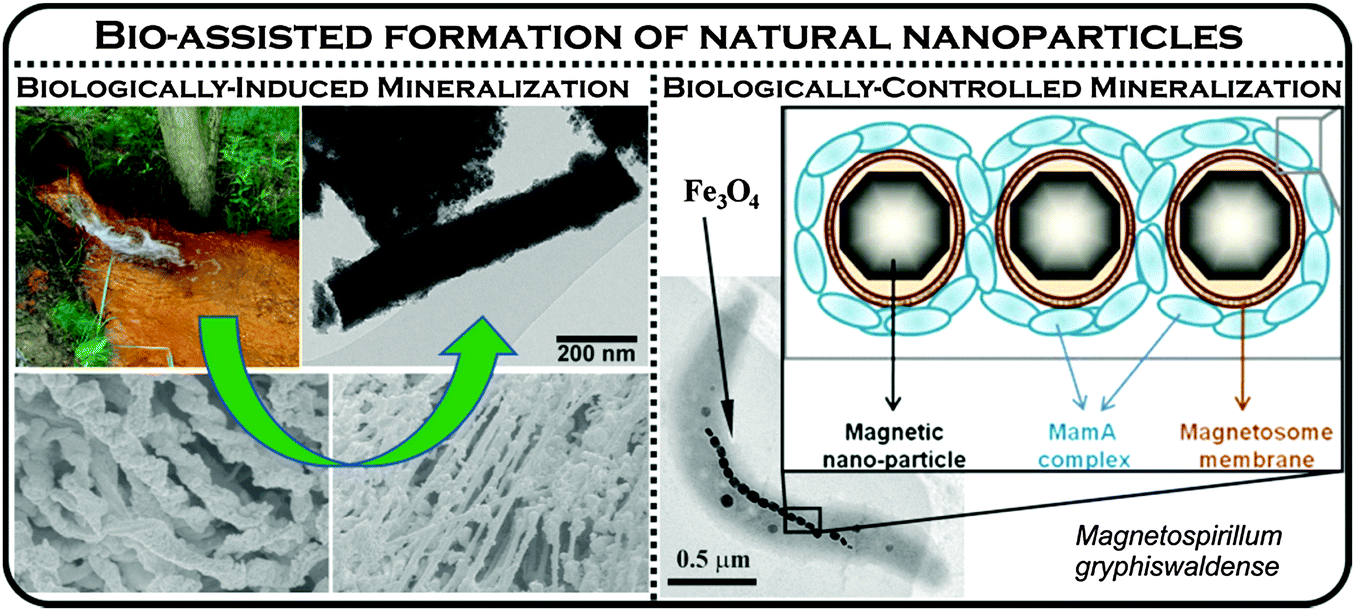 | ||
| Fig. 4 Formation of biogenic iron oxide nanoparticles by two contrasting mineralization processes – biologically-induced and biologically-controlled. The right panel was partly adapted with permission from ref. 27, copyright National Academy of Sciences of the United States of America. | ||
In many cases, the formation of NPs occurs via a combination of various processes. The typical examples are weathering (i.e., mechanical processes combined with dissolution/precipitation), the formation of colloids in rivers, and volcanic activity (fast cooling of fumes and explosions expelling tephra).5 Importantly, NNPs are dominantly formed at phase boundaries (e.g., solid–gas → wind erosion, liquid–gas → evaporation of sea spray, solid–liquid → weathering of rocks/minerals, etc.). From the aforementioned overview, it is evident that NPs can be produced in the form of colloids, aerosols, dust (including cosmic dust), constituents of soils and sediments, hydrothermal/chemical deposits (including evaporites), mineral nuclei, reaction rims, and lamellae. From a chemical compositional viewpoint, natural NPs may represent a very wide spectrum of elements, the most common being:
– metal oxides/hydroxides (e.g., iron oxides/oxyhydroxides, goethite, lepidocrocite, akaganeite, and schwertmannite, green rust, nanocrystalline aluminum hydroxides, manganese oxides and hydroxides),
– metals or alloys (e.g., metal nanoparticles in hydrothermal emissions),
– carbon allotropes and other non-metals,
– silicates (e.g., allophane, shallow spherules and imogolite, fibrous clay minerals including sepolites and palygorskites occurring in the recent sediments and as colloids; asbestos, mineral lamellae and nuclei of minerals in silicate rocks),
– sulfides (for example, the Cu and Zn-containing pyrites FeS2 and ZnS, and nanoframboids in high temperature black smoker hydrothermal vents), and
– sulfates, halides, and carbonates.
Among these chemical groups, metal NPs and mainly noble metal NPs represent an emerging class of NNPs that have many important ecological effects.7,23 Natural AgNPs in water of coastal areas and in silver mine tailings are representative examples of the occurrence of NNPs.12 Natural AuNPs have been observed in both low- and high-temperature locations during ore mining activities.17 Thus, in the next sections, we critically discuss their possible formation, fate, and toxicity in the environment.
2.1 Aerobic environment
Metal salts are known to react with a wide variety of antioxidants, vitamins, sugars, and plant extracts. In addition, the interaction of metal ions with natural organic matter (NOM) and ROS plays a vital role in the formation of metal nanoparticles in the environment.7,14,28–30 NOM is a complex matrix which is made up of constituents such as polysaccharides, proteins, and humic substances (HSs) and comprises an essential element of soil, sediments, river, surface waters, and groundwater at concentrations ranging from sub mg L−1 levels to tens of mg L−1.31 HS have aromatic carbons, conjugated double bonds, and phenolic groups. The exact structure is difficult to describe because of its complexity; the proposed structure is depicted in the ESI† (Slide 11). HSs can be further sub-divided into humic acids (HAs), fulvic acids (FAs), and humin (Slide 12, ESI†). HAs are composed of high molecular weight components, which are normally insoluble at low pH. In contrast, FAs comprise of low molecular weight components, which are soluble over a wide pH range. The humin fraction is insoluble at any pH. The procedures to isolate HAs, FAs, and humin from HS samples are defined by the International Humic Substance Society (http://www.humicsubstances.org/isolation.html). ROS encompass 1O2, O2−˙, H2O2, and ˙OH, which are usually generated via photochemical and Fenton or Fenton-like reactions in natural surface waters.7,29,30The following sections illustrate the possible mechanisms of natural noble-metal NP formation due to the interactions of metal salts or ions with various natural organic components. The interference with NP formation of different environmental parameters such as pH, nature of the NOM, and oxic/anoxic environment is also discussed.
Few studies have been published that focus on the formation of AgNPs through the interaction of Ag+ ions with NOM.12 Natural organic matter, which contains enolic-OH, methoxyls, aldehydes, ketones, phenolic-OH, quinones, and thiols as functional groups, can reduce Ag+ ions to Ag0. Laboratory experiments were conducted to verify the expected natural processes.7,12,13 These experiments were performed by mixing a solution of Ag+ ion with Suwannee river humic acid (SRHA) and fulvic acid (SRFA) in buffered solution at pH 8.0. The color of the solution slowly changed to yellow after a few days and had the characteristics of surface plasmon resonance (SPR) typical of AgNPs (Fig. 5a). The wavelengths of the SPR bands were ∼400 nm and the shapes varied depending on the exact experimental conditions. The heating of the reaction mixtures of Ag+–SRHA (or SRFA) resulted in the formation of AgNPs within a few hours, and the observed spectral bands were narrower than the SPR bands obtained at room temperature (Fig. 5a). The particle growth mechanism explained the thermal effect as was seen on the SPR spectra of AgNPs.12 Similarly, the formation of AuNPs was observed upon heating the mixture of Au(III) in river water to 65 °C (Fig. 5b).30 The characteristic SPR band of AuNPs was found at ∼520 nm with the shape and intensity of the spectral bands being dictated by the organic matter present in different river streams.30 Seaweed, Sargassum muticum, can induce the reduction of Au(III) to AuNPs in seawater.32
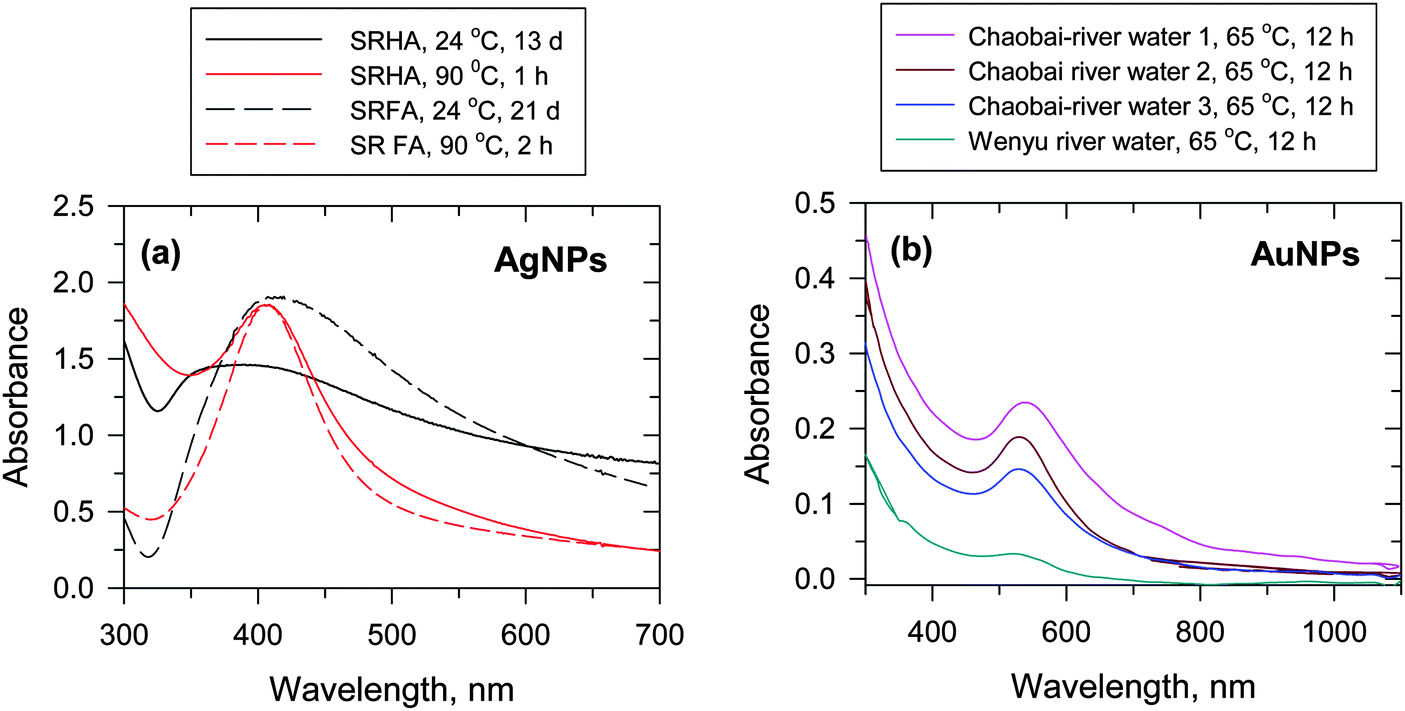 | ||
| Fig. 5 (a) UV-vis absorption spectra of Ag nanoparticles (AgNPs) in Suwannee river humic acid (SRHA) and Suwannee river fulvic acid (SRFA) at 24 °C and 90 °C at pH 8.0 ([Ag+] = 1 × 10−4 mol L−1, [SRFA] = [SRHA] = 100 mg L−1). (Adapted with permission from ref. 12, copyright American Chemical Society). (b) UV-vis absorption spectra of Au nanoparticles (AuNPs) in river water at 65 °C at pH 8.0 ([Au+] = (62.5–1000) × 10−6 mol L−1, [HA] = 5–100 mg DOC L−1, where DOC – dissolved organic carbon). (Adapted with permission from ref. 30, copyright American Chemical Society). | ||
The influence of Fe(II,III) ions on the formation of AgNPs has been explored; the formation of AgNPs was enhanced when mixtures of Ag(I)–fulvic acid were heated at 90 °C.7 In contrast, mixed solutions of Ag(I) and Fe(II) without humic acid did not result in a characteristic SPR peak for AgNPs, thus delineating the role of organic matter in enhancing AgNP formation in Fe(II)/Fe(III)–Ag(I)/NOM reaction mixtures. An additional finding of this study was the change in particle size distribution, which was influenced by the presence of iron species; smaller particle sizes were formed when iron species were present in the reaction mixture, presumably due to the faster growth of the AgNPs. A detailed characterization of the AgNPs was performed using various advanced surface techniques to authenticate the formation of nanoparticles.12 Transmission electron microscope (TEM) images showed that AgNPs were spherical with a relatively broad size distribution ranging from <5 nm to >50 nm; the corresponding selected area electron diffraction (SAED) pattern and high-resolution TEM images confirmed the crystalline character of AgNPs with typical indices of a [111] plane in the cubic lattice of silver.12
The formation of AgNPs and AuNPs under UV/visible-light conditions has been studied.13,28–30 A mixture of Ag(I) or Au(III) in the presence of NOM, upon exposure to either UV or sunlight irradiation, displayed characteristic SPR bands for AgNPs and AuNPs at ∼400 nm and ∼520 nm, respectively.13,28 The formation of nanoparticles in river waters under both simulated and natural sunlight conditions has been shown (Fig. 6).28,30 An absorption peak at ∼410 nm of AgNPs appeared after 20 min of irradiation of the river water sample, which had 2.42 mg carbon (C) L−1 (Fig. 6a).28 In this study, Aldrich humic acid (AHA) and SRHA samples contained about 4 mg C L−1, but displayed less absorption intensity than the river water samples, suggesting the role of other species besides organic carbon in the photoreduction of Ag(I) to Ag0. The presence of diverse inorganic and organic constituents in river water and their role in the formation of AuNPs via the photoreduction of Au(III) are shown in Fig. 6b.30 River waters from different sources had varied intensity of SPR of AuNPs at fixed time intervals of 15 min under sunlight irradiation. More recently, visible light irradiation of a solution containing the bacteria Pseudomonas aeruginosa and extracellular polymeric substances, with and without the addition of NaCl, led to the generation of AgNPs.33
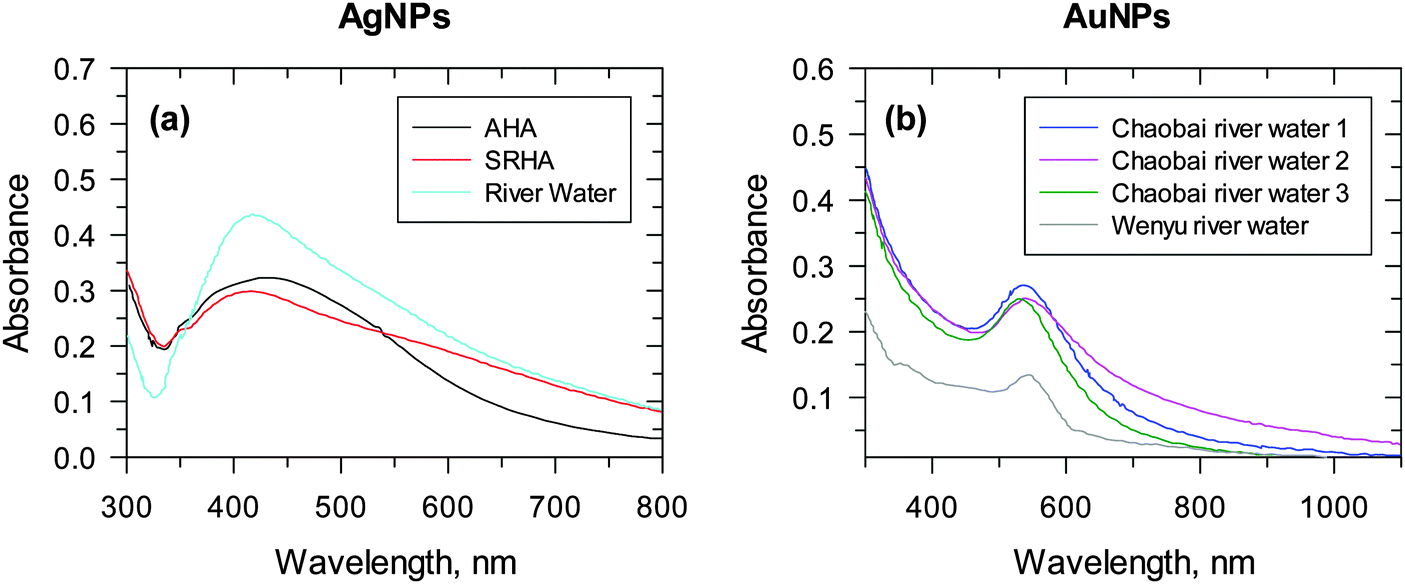 | ||
| Fig. 6 UV-vis spectra of nanoparticles formed under river water conditions. (a) Ag nanoparticles, 4 mg L−1 Suwannee river humic acid (SRHA) (red line), or 4 mg L−1 C/L Aldrich humic acid (AHA) (black line) upon exposure to stimulated sunlight in the CPS+ reactor for 20 min. ([Ag+] = 1 × 10−4 M; pH 8.0). (Adapted with permission from ref. 28, copyright American Chemical Society). (b) Au nanoparticles under natural sunlight. (Cumulative parabolic aluminized reflector (PAR) light = 12.48 E m−2). (Adapted with permission from ref. 30, copyright American Chemical Society). | ||
The comprehensive characterization of AuNPs formed under sunlight conditions has been accomplished (Fig. 7).30 TEM and an energy dispersive spectrum (EDS) confirmed the formation of AuNPs through the reduction of Au(I) in river water (Fig. 7a and b). A high-resolution TEM (HRTEM) image revealed Au lattice planes, whereas the EDS confirmed the presence of gold. Other elements appearing in the EDS represent the common constituents of river water, except Cu, which has its origin from the copper-mesh TEM grid. The SAED pattern (Fig. 7c) displays diffraction rings typical for gold. Similarly, four distinct peaks in the X-ray diffraction (XRD) pattern correspond to four planes of the face-centered cubic Au indexed as [111], [200], [220], and [311] (Fig. 7d). An atomic force microscopy (AFM) investigation confirmed the spherical shape of the particles with less than 10 nm diameter, whereas the larger particles were less spherical.30
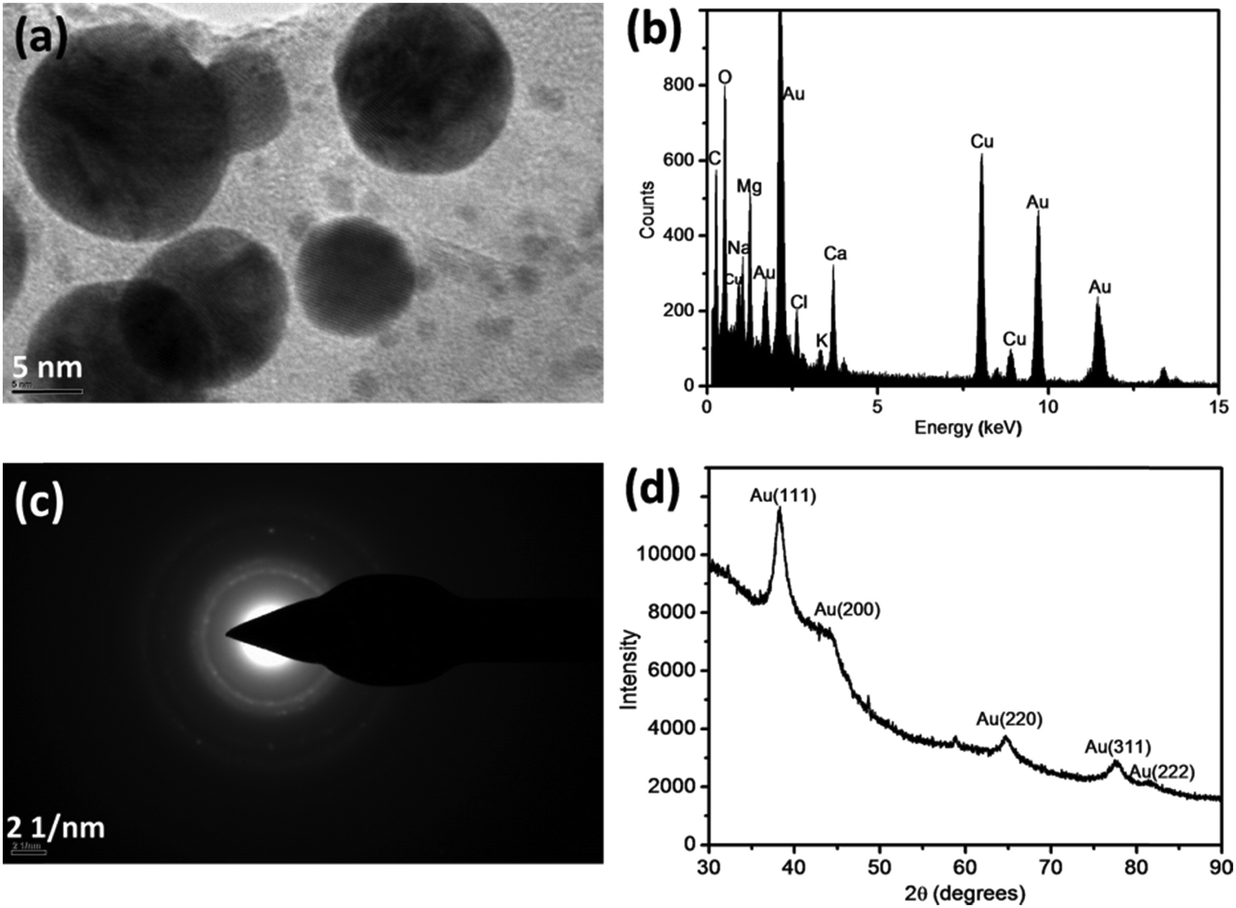 | ||
| Fig. 7 TEM image (a) of Au nanoparticles produced in the Chaobai river water under sunlight irradiation with the corresponding EDS (b) and SAED pattern (c) of the Au nanoparticles. The results of XRD of the as-prepared Au nanoparticles in the Chaobai river water 1 under sunlight irradiation are also shown (d). The concentration of spiked AuCl4− is 200 μmol L−1 and cumulative parabolic aluminized reflector is 7.06 × m−2 (corresponding to 3 h sunlight irradiation). (EDS – energy-dispersive X-ray spectroscopy; SAED – selected-area electron diffraction, XRD – X-ray diffraction). (Adapted with permission from ref. 30, copyright American Chemical Society). | ||
The underlying mechanisms for the formation of AgNPs under different natural conditions are presented in Fig. 8. The growth mechanisms pertaining to AgNP formation are depicted in Fig. 8a while additional reactions that may transpire in the presence of Fe(II)/Fe(III) ions, thus augmenting the formation of AgNPs, are given in Fig. 8b. The possible mechanisms that generate AgNPs via photochemical reactions are shown in Fig. 8c. Analogous reactions may also occur to generate natural AuNPs. The influence of pH and redox potentials of implied reactions has been proposed to describe the reduction of parent ions to their metallic nanoparticles.13,30
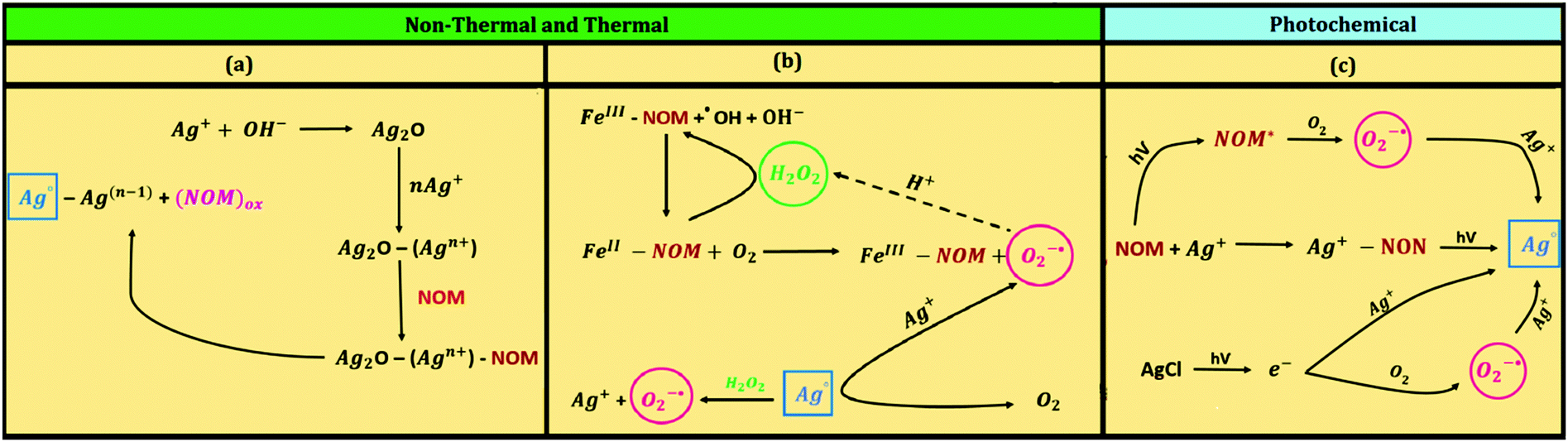 | ||
| Fig. 8 Proposed reaction mechanisms for the generation of Ag nanoparticles in the natural environment (NOM – natural organic matter). | ||
The redox potentials of the involved half-reactions are one of the tools to estimate the thermodynamic feasibility of the formation of AgNPs. This may mechanistically explain the thermally-induced formation of AgNPs from the reduction of Ag+ ions in the presence of NOM.7 The redox potential of Ag+ to an isolated silver atom (Ag0) is highly negative (Ag+ + e− → Ag0; E0 = −1.8 V vs. NHE).34 The redox potential for NOM (e.g., FA) is ∼0.5 V suggesting that the reduction of Ag+ by FAs to Ag0 may not be thermodynamically feasible (Ag+ + FA(red) → Ag0 + FA(ox); E0 = −2.3 V vs. NHE). However, if Ag+ is already deposited onto a solid surface, the potential for the reduction of Ag+ would be different. For example, the redox potential for the reduction of Ag+ onto an Ag electrode has a potential of +0.8 V, which indicates the feasibility of the formation of AgNPs from the reduction of Ag+ ions by FAs (Ag(solid surface)+ + FA(red) → Ag0 + FA(ox); E0 = 0.3 V vs. NHE). This is consistent with the experimentally observed formation of AgNPs in different studies,13,30 and could be easily extrapolated to environmental/natural conditions.
The solid surface could be in the form of Ag2O, which precipitates rapidly (i.e., 2Ag+ + 2OH− → Ag2O + H2O) (Fig. 8a). Then, the deposition of Ag+ onto Ag2O and subsequent reduction by NOM components would generate Ag0. The proposed mechanistic hypothesis was supported by the analysis of the autocatalytic process for the growth of AgNPs.13 Furthermore, the increase in the growth of AgNPs with increasing pH (i.e., increase in OH− ion concentration from pH 6.1 to 9.0), is accompanied by an enhanced formation of Ag2O.13 This allows more deposition of Ag+ onto solid surfaces and reduction by functional groups of the NOM (see Fig. 8a). In this reaction scheme, NOM is also likely adsorbed onto colloidal particles of Ag2O where it has better contact with Ag+ to reduce it to metallic silver. Such involvement of various moieties of NOM has been probed by Fourier transform infrared spectroscopy (FTIR) measurements on the FA residues on the AgNPs.13 FTIR spectra, with and without the presence of AgNPs, suggested that there was an oxidative damage to FA when AgNPs were formed.13 The formation of AuNPs, in an analogous manner, was also shown to be pH dependent.30 The involvement of redox potentials of Au(III) species and humic acid were invoked to describe the trend seen for the formation of AuNPs with pH.30
The role of various functional groups contained in natural organic matter could be discerned with variation of the growth of AgNPs and with the changing nature of organic matter under both thermal and photoirradiation conditions (Fig. 9). For example, the use of different fulvic acids (Suwannee river fulvic acid – SRFA, Pahokee Peat fulvic acid – PPFA, Nordic lake fulvic acid – NLFA, and Suwannee river humic acid – SRHA) showed the following order for the rate of formation of AgNPs: NLFA > SRHA > PPFA > SRFA. The difference in nitrogen and sulfur content, as well as the difference in the radical character of these various types of organic matter resulted in varying growth of the formation of AgNPs (Fig. 9).13 The important influence of the NOM characteristics was further confirmed when different results were obtained for AgNPs using HA originating from sedimentary and soil sources.12 Humic acid from sediments could reduce Ag+ to AgNPs, but the formation of AgNPs from soil HAs was not possible. Even an increase in temperature to 90 °C failed to produce AgNPs with natural soil HAs.
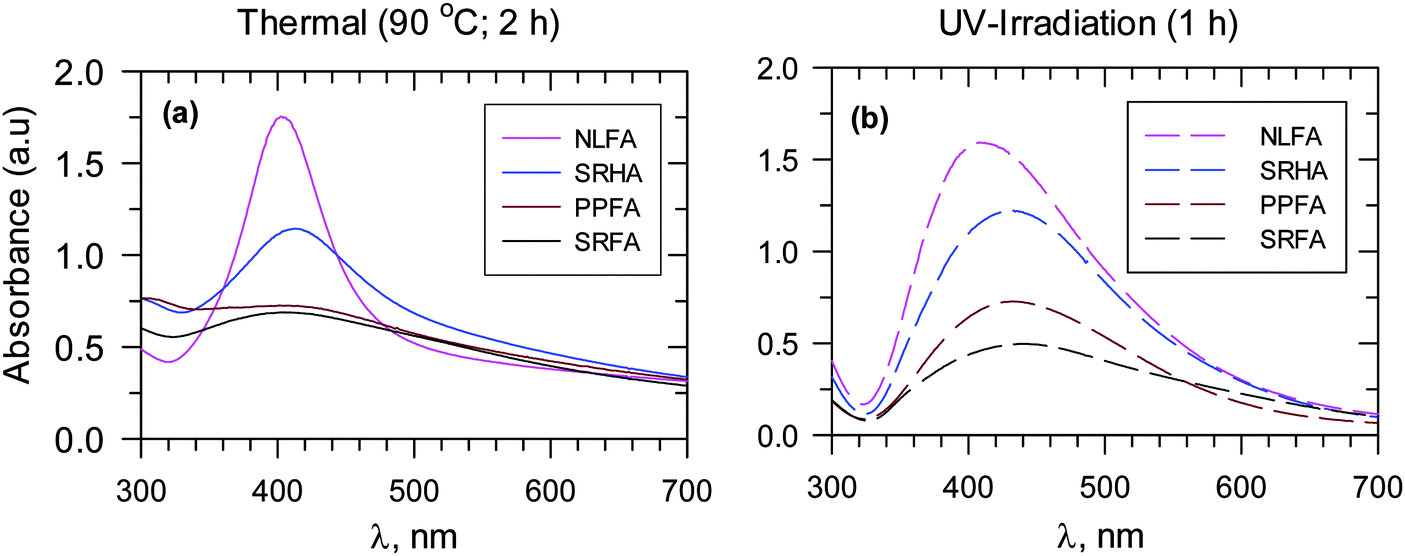 | ||
| Fig. 9 UV-vis absorption spectra of Ag nanoparticles for different fulvic acids and Suwannee river humic acid at pH 8.0. Adapted with permission from ref. 12 and 13, copyright American Chemical Society. (a) Heating at 90 °C for 2 h and (b) UV irradiation for 1 h for in moderately hard reconstituted water (MRHW) synthetic freshwater. (SRFA – Suwannee river fulvic acid I, SRPP – Suwannee river Pahokee Peat II, SRNL – Suwannee river Nordic Lake; SRHA – Suwannee river II humic acid). ([Ag+] = 1 × 10−3 mol L−1, 45 mg L−1 fulvic and humic acids). | ||
The enhanced growth of AgNPs due to the presence of Fe(II) and Fe(III) species in the Ag+–NOM system is rather complex. There are several reaction pathways such as complexation and dissociation of Fe(II)/Fe(III) with organic matter and the generation of reactive species (O2˙− and H2O2) and their subsequent reactions with silver and iron species (Fig. 8b).7 Moreover, the formation of Fe(II)–NOM/Fe(III)–NOM in the system creates positive redox potential (Ag+ + FeII–NOM → AgNPs + FeIII–NOM; E0 = 0.5–1.0 V) thus enhancing the likelihood of the formation of AgNPs.
A few mechanistic studies have been performed on the formation of AgNPs and AuNPs under UV and visible light irradiation conditions,29 as exemplified by the formation of AgNPs via several reactions under conditions that are relevant in the natural environment (Fig. 8c). The presence of Cl− may enhance the formation of AgNPs from solid AgCl(s) in the presence of visible light, which is important considering the naturally occurring levels of Cl− ions in water. Upon absorption of a photon, AgCl(s) produces an electron, which can reduce Ag+ to Ag0. Also, this electron can react with inherent O2 to produce O2˙−, which subsequently reacts with Ag+ to form Ag0. Significantly, earlier research on the formation of AgNPs from Ag(I)–NOM mixtures under UV-light implored the role of superoxide.29 However, later findings on sunlight-driven formation of AgNPs from the mixture of Ag(I)–NOM ruled out the possibility that superoxide was reducing Ag(I) ions;28 no major role of hydrated electron or triplet NOM was discerned. Instead, it was concluded that sunlight-driven photoreduction of Ag(I) ions to AgNPs occurred through Ag(I)–NOM binding.28
In the case of the formation of AuNPs, a similar mechanism, depicted in Fig. 8, may occur under certain thermal and photochemical conditions.29,30 AuNPs are easier to form compared to AgNPs based on the higher positive redox potentials of the Au(III) ion compared to the Ag(I) ion (E0(Au(III)/Au(s)) = 1.5 V and E0(Ag(I)/Ag(s)) = 0.8 V). This is supported by the near complete conversion of Au(III) to AuNPs by NOM that has been observed by X-ray photoelectron spectroscopy (XPS) following heating or irradiation of an Au(III)–NOM mixture.30 Comparatively, only a fraction of Ag(I) was converted to AgNPs following heating of a mixture of Ag(I)–NOM in solution.30 Because of the high thermodynamic feasibility of the formation of AuNPs, several moieties of NOM (phenol, alcohol, and aldehyde) could reduce Au(III) to elemental gold, whereas a phenolic moiety was involved in reducing Ag(I) to Ag(s).30
2.2 Anaerobic environment
Inorganic sulfide (H2S and HS−) is an important part of the global biogeochemical sulfur cycle under anaerobic conditions, which includes hydrothermal vents, mining water, sediments, terrestrial soils, and sewage treatment plants. The potential formation of noble metal NPs and their sulfides through direct formation or by transformation processes are depicted in Fig. 10.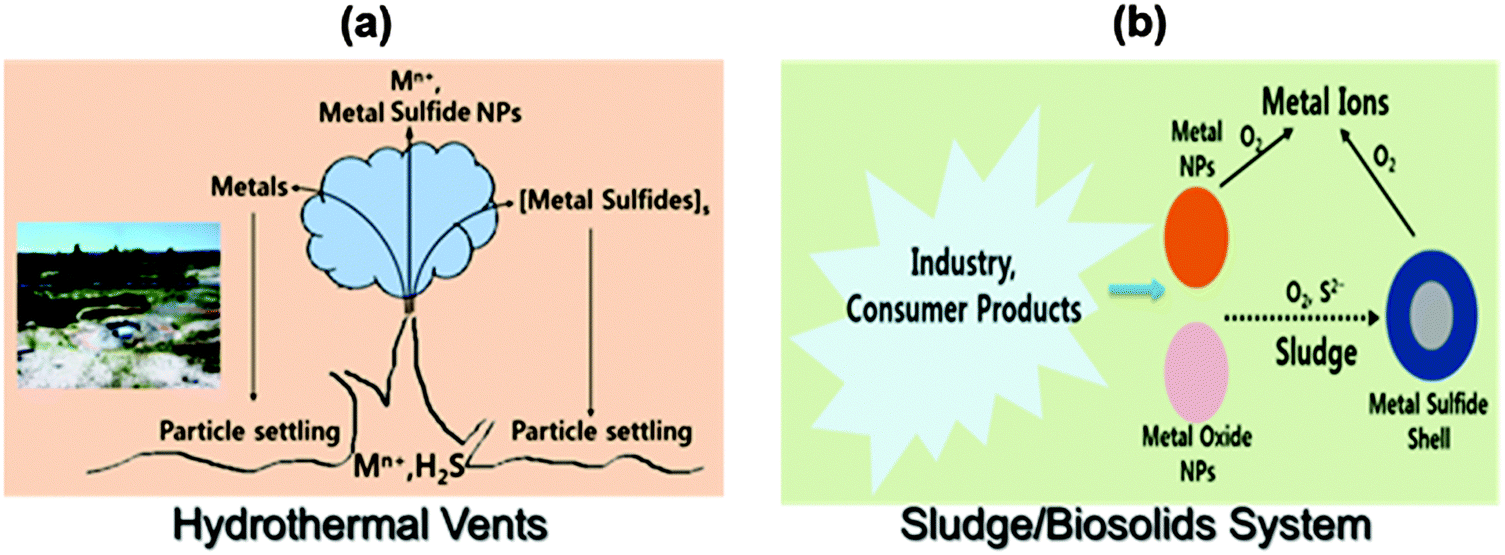 | ||
| Fig. 10 The possible formation of noble metal nanoparticles in the environment: (a) deep ocean and (b) wastewater treatment plants. | ||
Metals and sulfur in the ocean can be emitted from high temperature hydrothermal vents, and these may react with each other to serve as source of metal-bearing sulfide NPs (Fig. 10a).14 The metals and their sulfide NPs may stay suspended. Another example is the sulfide transformation of AgNPs (or AuNPs) into Ag2S (Au2S) (Fig. 10b).35 When AgNPs are oxidized, Ag(0) is transformed to Ag+, which can undergo sulfidation to form very insoluble Ag2S (Ksp = 5.92 × 10−51) that eventually is converted to NPs or core–shell Ag@Ag2S particles.35 The formation of Ag2S from AgNPs depends on the size and morphology of particles, concentration of sulfide, and coating and structure of capping agents on AgNPs. The sulfidation of AgNPs has been observed in sewage sludge and wetland sediments.36 The α-phase of Ag2S (α-Ag2S), formed in sewage sludge, has been found to be similar to the mineral acanthite in nature. In a sewer system, AgCl-NPs can be transformed to Ag2S-NPs. An array of metal sulfide NPs (e.g., Ag2S, CuS, CdS, and ZnS) have been found in sulfidic environments.24,37–39 The natural formation of noble metal sulfides influences the speciation, mobility, and bioavailability of many important metals. For example, sulfide in metal sulfides resists oxidation due to its strong complexation by a metal,40 and thus limits the bioavailability of the metal.
3 Fate of natural metal nanoparticles
In contrast to natural AgNPs, the engineered AgNPs are typically stabilized by saccharides, surfactants and polymers,41 which could lead to altered transformation and environmental fate.42 Work is currently underway to evaluate the stability of AgNPs, formed via the reduction of Ag+ ions by NOM under environmental conditions.7,12,13,25 The results of the stability of particles, monitored for several months, are presented in Fig. 11.7,12,13 AgNPs formed from HAs, obtained from sediments and river water, showed a decrease in the SPR peak up to 25% in 70 days, and the stability of the AgNPs depended on the source of HAs.12 A 7% decrease in stability was observed for sediment HAs (Fig. 11a).12 In the case of river HAs, the decrease in stability was in the range of 6 to 25% during 70 days. More importantly, there was blue-shift in the SPR band from 423 to 410 nm (Fig. 11a). AuNPs, formed via the reduction of Au(III) by NOM, have been observed to have similar stability,30 thus confirming that the stability of naturally formed AuNPs is increased by NOM.7,30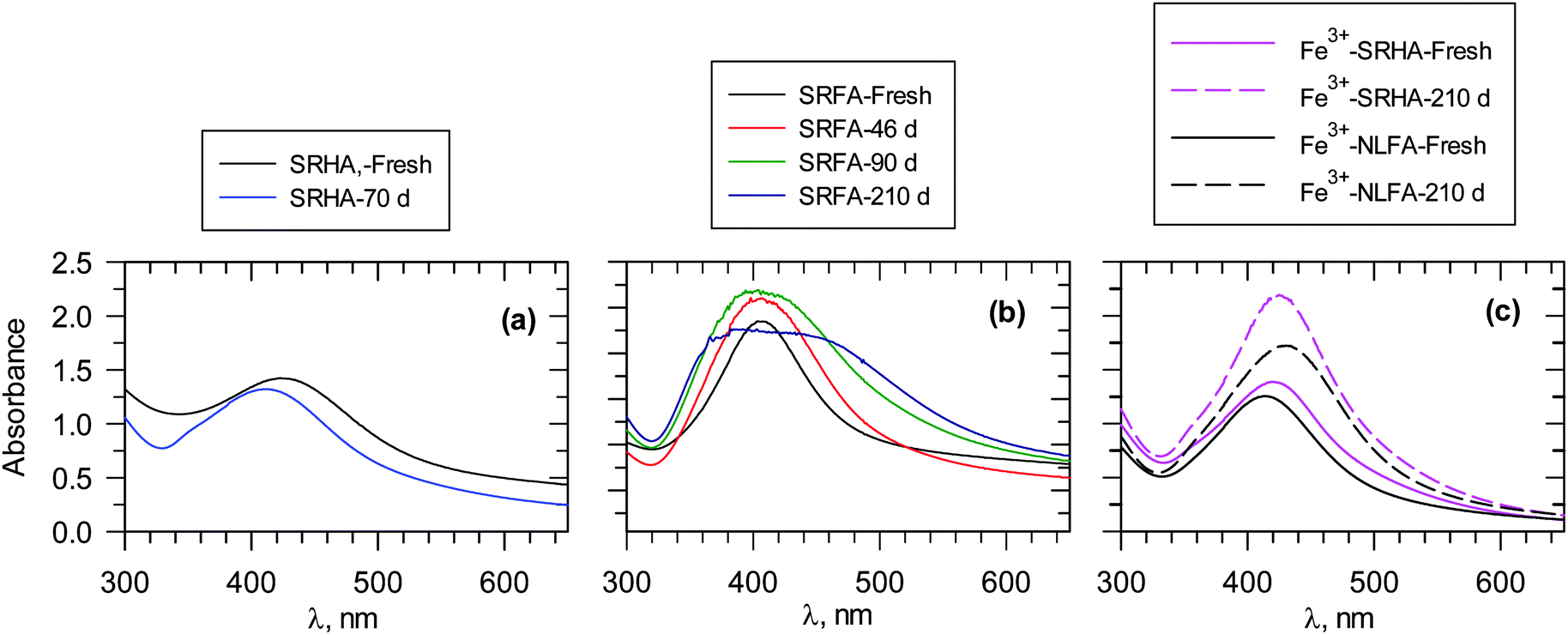 | ||
| Fig. 11 UV-vis measurements of ageing of Ag nanoparticles formed at 90 °C. Days represent time after the formation of particles. (a) 100 mg L−1 SRHA, pH 8.0, (b) 100 mg L−1 SRFA; pH 8.0, and (c) Fe3+–FA reaction mixtures, [SRHA] = [NLFA] = 40 mg L−1 SRFA. [Fe3+] = 13 μM; pH 6.0 ([Ag+] = 1 × 10−3 mol L−1). (SRHA – Suwannee river humic acid; SRFA – Suwannee river fulvic acid; NLFA – Nordic lake fulvic acid). (Adapted with permission from ref. 7, 13 and 25, copyright American Chemical Society). | ||
AgNPs, formed by FAs, follow a similar trend in stability (Fig. 11b). However, broadening of the SPR band with time has been observed.13 Dynamic light scattering (DLS) measurements and TEM images supported broad particle size distribution and increased polydispersity with time.13 Furthermore, the zeta potential of AgNPs varied only from −40 mV to −33 mV during seven months. This indicates the possibility of the persistence of repulsive forces between negatively charged high molecular weight organic matter-coated AgNPs, which would prevent the aggregation of nanoparticles.13 The surface composition and the binding mode of NOM influence the stability of AgNPs and AuNPs. This phenomenon can be noticed whether particles are formed at room temperature (RT) or at elevated temperature (65 °C or 90 °C).12–13,30 The stability of particles produced by light-induced particle formation is different from the stability of particles formed under RT and thermal conditions.13 Additionally, the size and morphology of the particles influence their stability. The surface charge of the AgNP and AuNP particles plays a role in their stability,7,43 as exemplified by a recent study on the fate of polymer-coated Au nanorods in saline estuaries.43 Furthermore, NNPs may possibly dissolve or ionize in the environment. Examples include AgNPs that easily dissolve to yield ionic forms under physiological conditions. The cycles of various inorganic NPs can vary significantly, depending on the surface and near-surface structure of particular NPs.44 The NP cycle in various phases thus play a crucial role in many geochemical processes.
The presence of iron species such as Fe3+ ions in the reaction mixture has been found not to significantly alter the stability of AgNPs (Fig. 11c); an increase in SPR of AgNPs was observed during a seven-month period.7 The zeta potential values were −18 MV and −23 mV with and without Fe3+, respectively, in an Ag+–FA reaction mixture, suggesting that no change in the organic matter surface coating on AgNPs occurred due to Fe3+ ions.7 It appears that the coating of NOM on either AgNPs or AuNPs did not allow the dissolution of NPs to cause their agglomeration.
A large number of studies have been conducted to investigate the effect of ionic strength and background electrolytes on the stability of engineered NPs.18,45 However, the effect of ions on the fate of NOM-formed AgNPs in natural waters has been studied only recently.25 The results are presented in Fig. 12 for Suwannee River natural organic matter (SR-NOM) and SRHA-formed AgNPs.25 The particles were stable for several weeks in low concentrations of moderately hard reconstituted water, which had low concentrations of CaCl2 (0.174 mM) and MgCl2 (0.249 mM).25 However, an increase in the concentrations of salts, including NaCl and KCl, resulted in a significant decrease in the SPR peaks (Fig. 11). This was contingent on the salts used and their ionic strength that varied from 50% to almost complete disappearance of the SRP peak absorbance (Fig. 12).25 AgNPs formed with SR-NOM in CaCl2 solution were relatively less stable than SR-formed AgNPs (Fig. 12aversusFig. 12b). Importantly, the decreases in the SPR peak intensities were more pronounced in chloride salts of divalent cations than those of monovalent ones. Moreover, among the divalent cations, Ca2+ ions caused more instability of NPs than did Mg2+, suggesting a role of ionic radii of cations present in the electrolyte. The equilibrium constant of Ca2+ with humic-type materials is higher than that of Mg2+, indicating that such interactions could also influence the stability of NPs. The measurements of zeta potential and hydrodynamic diameter (HDD) of AgNPs were made to understand the influence of ionic strength on the stability of AgNPs formed with SR-NOM.25 The values of the zeta potentials of AgNPs became less negative with increasing ionic strength in CaCl2 solution (∼−32 mV at 1 mM versus ∼−15 mV at 10 mM).25 The HDD of AgNPs changed from nm to μm in the same ionic strength range, suggesting that agglomeration of NPs occurs with increasing ionic strength of the electrolyte solution. Aggregation of AuNPs formed by thermal and sunlight induced reduction of Au(III) in the presence of Mg2+ and Ca2+ ions has been observed.30 These results further suggest that organic-matter stabilized NPs may become less stable as their transport proceeds from freshwater through estuarine water to seawater.
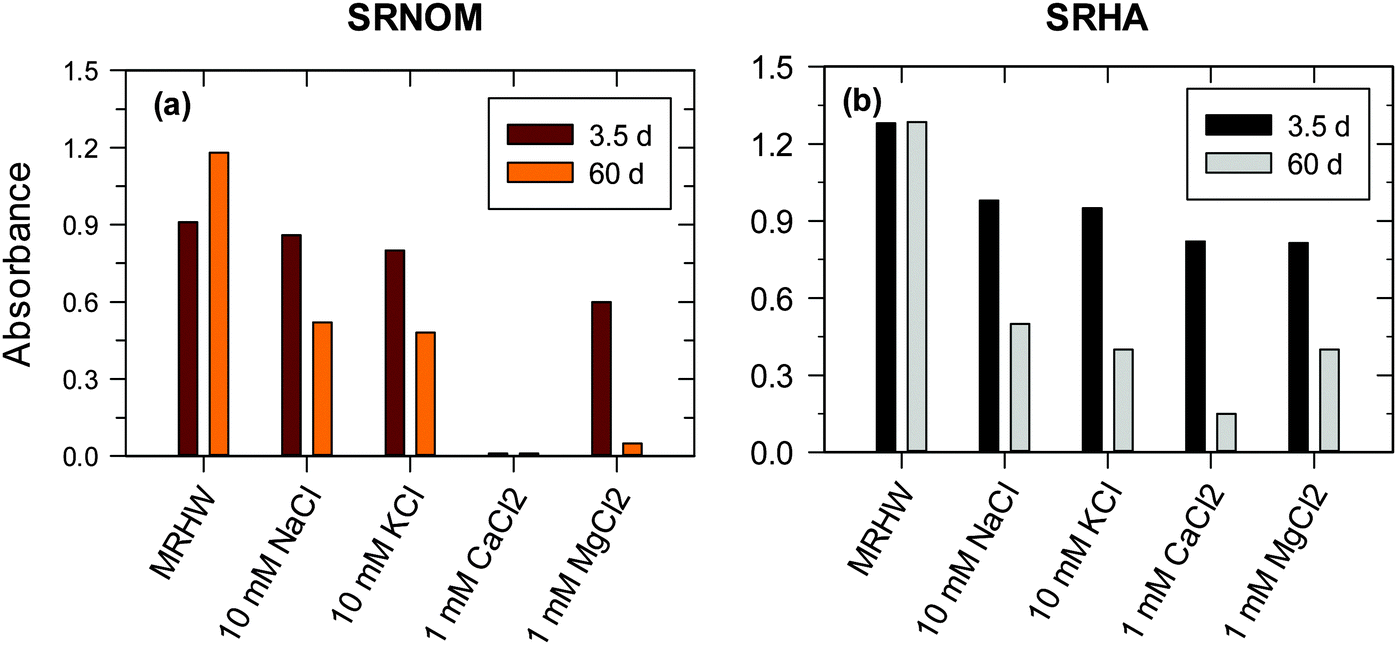 | ||
| Fig. 12 Plot demonstrating the stability of SRNOM- and SRHA-formed Ag nanoparticles in 1 h at 410 nm in chloride solutions by monitoring the absorbance of SPR peaks. ([Ag+] = 1 × 10−3 M, [SRNOM] = [SRHA] = 15 mg L−1 in moderately hard reconstituted water (MRHW) at pH 8.0 and 90 °C). (SRNOM – Suwannee river natural organic matter; SRHA – Suwannee river humic acid). (Adapted with permission from ref. 25, copyright Elsevier Inc.) | ||
In terms of the fate of noble metal nanoparticles, several environmental factors determine the aggregation and dissolution of NPs, which include ionic strength, ionic components, pH, redox conditions, concentration and nature of NOM, and the type of particular nanoparticles. The cumulative effects of these physicochemical factors will determine the potential of NPs to be transformed and transported in the complex environment.
4 Toxicity of natural metal nanoparticles
The concentration of natural Ag and Au nanoparticles is too low to allow the direct investigation of their toxicity. Consequently, several researchers have conducted studies on genotoxicity, cytotoxicity, and ecotoxicity of engineered AgNPs and AuNPs against a number of food chain members such as bacteria, plants, and aquatic and terrestrial organisms,10,18 including the toxicity of engineered AgNPs to marine organisms and algae.23,46,47 The toxic effects of non-stabilized and stabilized (surfactant- and polymer-coated) AgNPs to isolated strains of bacteria and yeasts have been studied.18 The toxic mechanism included the generation of ROS by AgNPs and direct and indirect damage to DNA by AgNPs and/or released Ag+ ions.19,23,48–50 The acute and chronic toxic effects of engineered AgNPs have also been examined using Drosophila melanogaster.51 Genomic and proteomic approaches have been applied to comprehend ecotoxicological impacts of ENPs. Surface chemistry, charge, and the organic coating of NPs play a pivotal role in their toxicity.19,23A very few studies have been conducted that discuss the toxic effects of NNPs. A recent study examined the toxicity of natural AgNPs by determining the minimum inhibitory concentrations (MIC) against Gram-positive (GP) and Gram-negative (GN) bacteria to particles that were formed from the reduction of Ag+ by organic matter (e.g., SRHA).7 Because of distinct differences in these two kinds of bacteria, the toxic effects of AgNPs against GP and GN bacterial species varies.
A comparison of MIC values of ENPs, poly vinylpyrrolidone coated silver nanoparticles (AgNPs–PVP) and sodium dodecyl sulfate coated silver nanoparticles (AgNPs–SDS) with natural NPs (AgNPs–SRHA) against bacterial species is shown in Fig. 13.7,41 MIC values against GN bacteria were lower than those against GP bacteria; again suggesting that the toxicity of AgNPs depends on the species-based difference. More importantly, natural noble metal NPs had higher MIC values than ENPs, indicating that the NNPs were less toxic than the man-made nanoparticles. The difference in toxicity between ENPs and natural AgNPs was much more dramatic for GP than that for GN species. It appears that the organic coating generated in the natural environment produces complex structures, which may be responsible for this decreasing toxicity. Another possibility is that the natural organic matters tend to be inherently less toxic than polymers or surfactants used for ENP production or post-production treatment. The surface coating materials for NNPs and ENPs may thus be responsible for differences in toxicity of these nanoparticles. A detailed mechanistic investigation will be required to comprehend the observed differences in the antibacterial study (Fig. 13).7,41
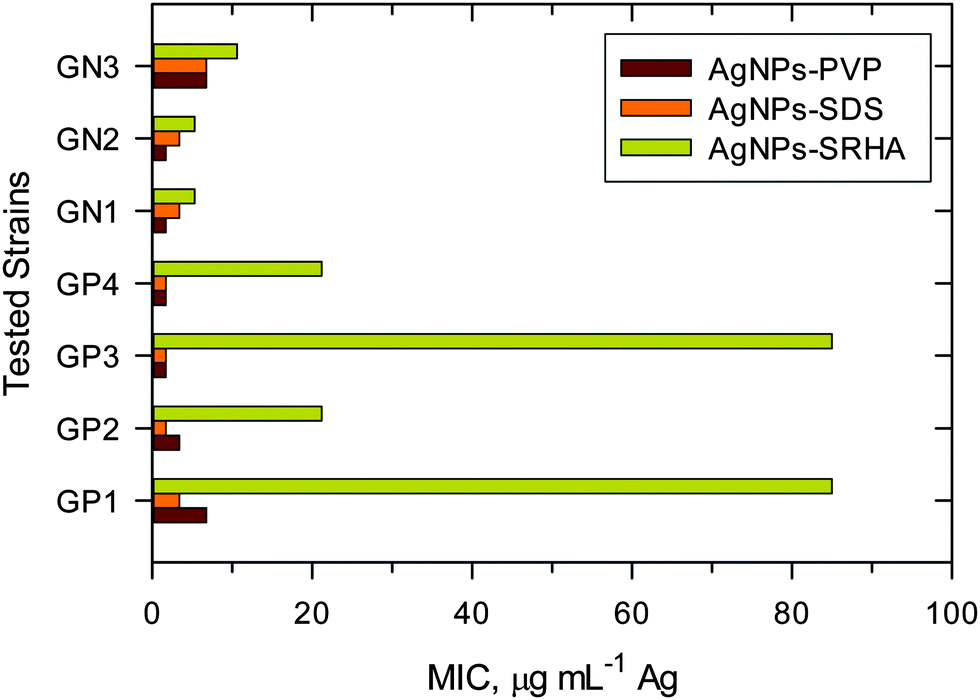 | ||
| Fig. 13 Minimum inhibitory concentrations of engineered nanoparticles (AgNPs–PVP and AgNPS–SDS) and natural Ag nanoparticles (AgNPs–SRHA) against Gram-positive (GP) and Gram-negative (GN) bacteria. (PVP – poly vinylpyrrolidone; SDS – sodium dodecyl sulfate; SRHA – Suwannee River humic acid; GP1 – Enterococcus faecalis CCM 4224; GP2 – Staphylococcus aureus CCM 3953; GP3 – Staphylococcus aureus (MRSA); GP4 – Staphylococcus epidermidis 1; GN1 – Pseudomonas aeruginosa CCM; GN2 – Pseudomonas aeruginosa; GN3 – Klebsiella pneumoniae (ESBL)). (Data were taken with permission from ref. 7 and 52, copyright American Chemical Society). | ||
In the anoxic environment, the rate of dissolution of metallic NPs influences their toxicity. For example, the formation of an insoluble metallic sulfide layer of either Ag or Au on the surface of a metallic metal core can decrease the rate of dissolution of nanoparticles even at a low molar ratio of metal to sulfide. Consequently, there is less possibility of metal ion release from the corresponding nanoparticles in an anoxic environment. This would result in a decrease in toxicity as has been demonstrated for the Ag2S-coated AgNPs against E. coli or nitrifying bacteria.53 The kinetics and mechanism of metal nanoparticle dissolution and also the character of the structure around the metallic nanoparticles under oxic/anoxic conditions control the toxicity of the nanoparticles.
5 Conclusion and outlook
Nanoparticles, produced by industries for numerous applications, can also be formed naturally in aerosols, waters, soil, deep-sea hydrothermal vents, natural ore, and microbial systems. Despite many reports on the natural occurrence of inorganic nanoparticles, the unveiling of their formation mechanisms is still a big challenge. Detailed mechanistic studies on chemical, photochemical, mechanical, thermal, and biological processes delivering natural nanoparticles are lacking. In the environment, more than one reaction pathway may contribute to the formation of natural nanoparticles. For performing measurements on natural nanoparticles, the role of phase boundaries, reaction conditions and microcrystalline substrates should be elucidated.In recent years, some progress has been realized in narrative of formation mechanisms pertaining to noble metal nanoparticles in various natural systems. The interactions of precursor noble metal ions (e.g., Ag(I)/Au(III)) with natural organic matter under thermal and light irradiation conditions of oxygenated waters are largely responsible for the occurrence of noble metal nanoparticles (e.g., AgNPs and AuNPs). The growth of noble metal NPs varies with the source (sedimentary, aquatic, and solid) and type of natural organic matter (fulvic acid versus humic acid). Suggestions have been advanced that alcoholic, aldehyde, and phenolic moieties of humic acid may induce photoreduction and generate noble metal nanoparticles. However, due to the contradictory views on the formation of noble metal nanoparticles under solar light, further mechanistic details must be scrutinized for the complete understanding of reduction processes in the presence of natural organic matter under sunlight irradiation. The roles of reactive oxygen species and transient natural organic matter need to be further investigated in order to advance the understanding of noble metal formation from particular complexes under natural conditions.
Besides the characteristics of dissolved organic matter, the valence state of redox species present in solution mixtures plays an important role in the formation of noble metal NPs. Further investigation is needed regarding how iron species (Fe(II)/Fe(III)) enhance the formation of noble metal NPs under thermal conditions, and in what manner solar light irradiation is involved in the formation mechanisms. Future research should include other redox metal species (e.g., Cu(I)/Cu(II) and Mn(II)/Mn(IV)) to comprehend the formation of other non-noble metal nanoparticles in natural waters under both thermal and sunlight conditions. Also, in situ demonstration of particle formation remains a challenge. Distinguishing natural nanoparticles from engineered nanoparticles is of utmost importance to fully comprehend the distribution and the movement of natural nanoparticles; underlying difficulties associated with identifying sources of nanoparticles (i.e., engineered versus natural) need to be circumvented.
Published results have shown that noble metal NPs such as AgNPs and AuNPs, formed via the reduction of their corresponding ionic salt forms by humic and fulvic acids under thermal and photoirradiation conditions, are stable for a long period of time in the aquatic environment. This high stability of naturally formed noble metal NPs indicates that the migration of such nanoparticles over long distances from their point of origin is highly probable. However, pH, ionic strength, and chemistry of the aqueous and solid phases influence the aggregation, dissolution, and transformation of natural nanoparticles and hence their mobility.
Finally, natural organic matter functional groups that encase naturally formed noble metal NPs would strongly affect their toxicity. Very little is known about how these NPs interact with aquatic organisms. Significantly, these NPs may also exhibit varying toxicity, depending on the functionality of the organic matter and generation of reactive oxygen species. Future studies are thus envisaged to evaluate the underlying mechanisms of toxicity of naturally formed NPs.
Disclaimer
The U.S. Environmental Protection Agency, through its Office of Research and Development, funded and managed, or partially funded and collaborated in, the research described herein. It has been subjected to the Agency's administrative review and has been approved for external publication. Any opinions expressed in this paper are those of the author(s) and do not necessarily reflect the views of the Agency; therefore, no official endorsement should be inferred. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.Acknowledgements
V.K. Sharma, J. Filip, and R. Zboril acknowledge the support by the Ministry of Education, Youth and Sports of the Czech Republic (project LO1305) and Technology Agency of the Czech Republic “Competence Centers” (project No. TE01020218). We thank Dr. Leslie Cizmas and Dr. John Leazer for their comments to improve the article.References
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- W. J. Stark, P. R. Stoesset, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- S. J. Soenen, W. J. Parak, J. Rejman and B. Manshian, Chem. Rev., 2015, 115, 2109–2135 CrossRef CAS PubMed.
- D. M. Mitrano, S. Motellier, S. Clavaguera and B. Nowack, Environ. Int., 2015, 77, 132–147 CrossRef CAS PubMed.
- J. M. F. Hochella, M. G. Spencer and K. L. Jones, Environ. Sci.: Nano, 2015, 2, 114–119 RSC.
- R. S. Varma, Curr. Opin. Chem. Eng., 2012, 1, 123–128 CrossRef CAS PubMed.
- N. F. Adegboyega, V. K. Sharma, K. M. Siskova, R. Vecerova, M. Kolar, R. Zboril and J. L. Gardea-Torresdey, Environ. Sci. Technol., 2014, 48, 3228–3235 CrossRef CAS PubMed.
- A. Gartman, A. J. Findlay and G. W. Luther, Chem. Geol., 2014, 366, 32–41 CrossRef CAS PubMed.
- A. Philippe and G. E. Schaumann, Environ. Sci. Technol., 2014, 48, 8946–8962 CrossRef CAS PubMed.
- P. A. Holden, R. M. Nisbet, H. S. Lenihan, R. J. Miller, G. N. Cherr, J. P. Schimel and J. Gardea-Torresdey, Acc. Chem. Res., 2013, 46, 813–822 CrossRef CAS PubMed.
- J. Virkutyte and R. S. Varma, Chem. Sci., 2011, 2, 837–846 RSC.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Environ. Sci. Technol., 2011, 45, 3895–3901 CrossRef CAS PubMed.
- N. F. Adegboyega, V. K. Sharma, K. Siskova, R. Zbořil, M. Sohn and S. Banerjee, Environ. Sci. Technol., 2013, 47, 757–764 CrossRef CAS PubMed.
- M. Yücel, A. Gartman, C. S. Chan and G. W. Luther III, Nat. Geosci., 2011, 4, 367–371 CrossRef PubMed.
- J. Wu, J. Yao and Y. Cai, Nat. Nanostruct., 2012, 225–247 CAS.
- H. Konishi, H. Xu and H. Guo, Nat. Nanostruct., 2012, 75–113 CAS.
- R. M. Hough, R. R. P. Noble and M. Reich, Ore Geol. Rev., 2011, 42, 55–61 CrossRef PubMed.
- V. K. Sharma, K. Siskova, R. Zboril and J. Gardea-Torresdey, Adv. Colloid Interface Sci., 2014, 204, 15–34 CrossRef CAS PubMed.
- G. E. Batley, J. K. Kirby and M. J. McLaughlin, Acc. Chem. Res., 2013, 46, I854–I862 CrossRef PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- A. D. Dwivedi, S. P. Dubey, M. Sillanpää, Y.-N. Kwon, C. Lee and R. S. Varma, Coord. Chem. Rev., 2015, 287, 64–78 CrossRef CAS PubMed.
- S. Wagner, A. Gondikas, E. Neubauer, T. Hofmann and F. Von Der Kammer, Angew. Chem., Int. Ed., 2014, 53, 12398–12419 CAS.
- A. Quigg, W.-C. Chin, C.-S. Chen, S. Zhang, Y. Jiang, A.-J. Miao, K. A. Schwehr, C. Xu and P. H. Santschi, ACS Sustainable Chem. Eng., 2013, 1, 686–702 CAS.
- C. Levard, E. M. JHotze, G. V. Lowry and J. G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS PubMed.
- N. Akaighe, S. W. Depner, S. Banerjee, V. K. Sharma and M. Sohn, Sci. Total Environ., 2012, 441, 277–289 CrossRef CAS PubMed.
- D. Schüler and R. B. Frankel, Appl. Microbiol. Biotechnol., 1999, 52, 464–473 CrossRef.
- N. Zeytuni, E. Ozyamak, K. Ben-Harush, G. Davidov, M. Levin, Y. Gat, T. Moyal, A. Brik, A. Komeili and R. Zarivach, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E480–E487 CrossRef CAS PubMed.
- W. C. Hou, B. Stuart, R. Howes and R. G. Zepp, Environ. Sci. Technol., 2013, 47, 7713–7721 CrossRef CAS PubMed.
- Y. Yin, J. Liu and G. Jiang, ACS Nano, 2012, 6, 7910–7919 CrossRef CAS PubMed.
- Y. Yin, S. Yu, J. Liu and G. Jiang, Environ. Sci. Technol., 2014, 48, 2671–2679 CrossRef CAS PubMed.
- G. R. Aiken, H. Hsu-Kim and J. N. Ryan, Environ. Sci. Technol., 2011, 45, 3196–3201 CrossRef CAS PubMed.
- P. Lodeiro and M. Sillanpää, Mar. Chem., 2013, 152, 11–19 CrossRef CAS PubMed.
- A. R. Badireddy, B. J. Farner, S. M. Marinakos, S. Chellam and M. R. Wiesner, Environ. Eng. Sci., 2014, 31, 338–349 CrossRef CAS.
- S. T. Gentry, S. J. Fredericks and R. Krchnavek, Langmuir, 2009, 25, 2613–2621 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866–3877 CrossRef CAS PubMed.
- G. V. Lowry, B. P. Espinasse, A. R. Badireddy, C. J. Richardson, B. C. Reinsch, L. D. Bryant, A. J. Bone, A. Deonarine, S. Chae, M. Therezien, B. P. Colman, H. Hsu-Kim, E. S. Bernhardt, C. W. Matson and M. R. Wiesner, Environ. Sci. Technol., 2012, 46, 7027–7036 CrossRef CAS PubMed.
- K. M. Mullaugh and G. W. Luther III, J. Nanopart. Res., 2011, 13, 393–404 CrossRef CAS.
- M. Schaffie and M. R. Hosseini, J. Environ. Chem. Eng., 2014, 2, 386–391 CrossRef CAS PubMed.
- R. Ma, C. Levard, J. D. Judy, J. M. Unrine, M. Durenkamp, B. Martin, B. Jefferson and G. V. Lowry, Environ. Sci. Technol., 2014, 48, 104–112 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, B. Sinnet, E. Morgenroth and R. Kaegi, Environ. Sci. Technol., 2014, 48, 4885–4892 CrossRef CAS PubMed.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- K. L. Garner and A. A. Keller, J. Nanopart. Res., 2014, 16, 2503 CrossRef.
- J. M. Burns, P. L. Pennington, P. N. Sisco, R. Frey, S. Kashiwada, M. H. Fulton, G. I. Scott, A. W. Decho, C. J. Murphy, T. J. Shaw and J. L. Ferry, Environ. Sci. Technol., 2013, 47, 12844–12851 CrossRef CAS PubMed.
- M. F. Hochella Jr., S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef PubMed.
- A. M. El Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS PubMed.
- L. R. Pokhrel, B. Dubey and P. Scheuerman, Environ. Sci. Technol., 2013, 47, 12877–12885 CrossRef CAS PubMed.
- H. Wang, K. T. Ho, K. G. Scheckel, F. Wu, M. G. Cantwell, D. R. Katz, D. B. Horowitz, W. S. Boothman and R. M. Burgess, Environ. Sci. Technol., 2014, 48, 13711–13717 CrossRef CAS PubMed.
- P. Dallas, V. K. Sharma and R. Zboril, Adv. Colloid Interface Sci., 2011, 166, 119–135 CAS.
- H. Wang, F. Wu, W. Meng, J. C. White, P. A. Holden and B. Xing, Environ. Sci. Technol., 2013, 47, 13212–13214 CrossRef CAS PubMed.
- N. Von Moos and V. I. Slaveykova, Nanotoxicology, 2014, 8, 605–630 CrossRef CAS PubMed.
- A. Panacek, R. Prucek, D. Safarova, M. Dittrich, J. Richtrova, K. Benickova, R. Zboril and L. Kvitek, Environ. Sci. Technol., 2011, 45, 4974–4979 CrossRef CAS PubMed.
- L. Kvítek, A. Panáček, J. Soukupová, M. Kolář, R. Večeřová, R. Prucek, M. Holecová and R. Zbořil, J. Phys. Chem. C, 2008, 112, 5825–5834 Search PubMed.
- B. C. Reinsch, C. Levard, Z. Li, R. Ma, A. Wise, K. B. Gregory, J. G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 6992–7000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cs00236b |
| This journal is © The Royal Society of Chemistry 2015 |