The redox-A3 reaction
Daniel
Seidel
Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA. E-mail: seidel@rutchem.rutgers.edu
First published on 13th March 2014
Abstract
This highlight details the recent emergence of a new type of A3 reaction (three-component condensation of an amine, an aldehyde and an alkyne). In contrast to the classic A3 coupling process, the redox-A3 reaction incorporates an iminium isomerization step and leads to amine α-alkynylation. The overall transformation is redox-neutral by virtue of a combined reductive N-alkylation/oxidative C–H bond functionalization.
The three-component coupling of an amine, an aldehyde and an alkyne, commonly known as the A3 reaction, represents an ideal entry point to synthetically versatile propargylic amines of type 1 (eqn (1)).1 Not surprisingly, therefore, a significant amount of attention has focused on this transformation which has resulted in the development of operationally convenient and high-yielding approaches, including enantioselective variants.1 While cyclic amines readily undergo the A3 reaction to form propargylic amines with the general structure 2, the corresponding ring-substituted regioisomers 3 are inaccessible via this method. A two-component alkynylation approach to compounds 3 is not generally feasible due to the difficulties associated with accessing the requisite aminoaldehydes 4. Here we discuss recent advances that enable the formation of compounds 3via redox-neutral three-component coupling reactions (eqn (2) and (3)).
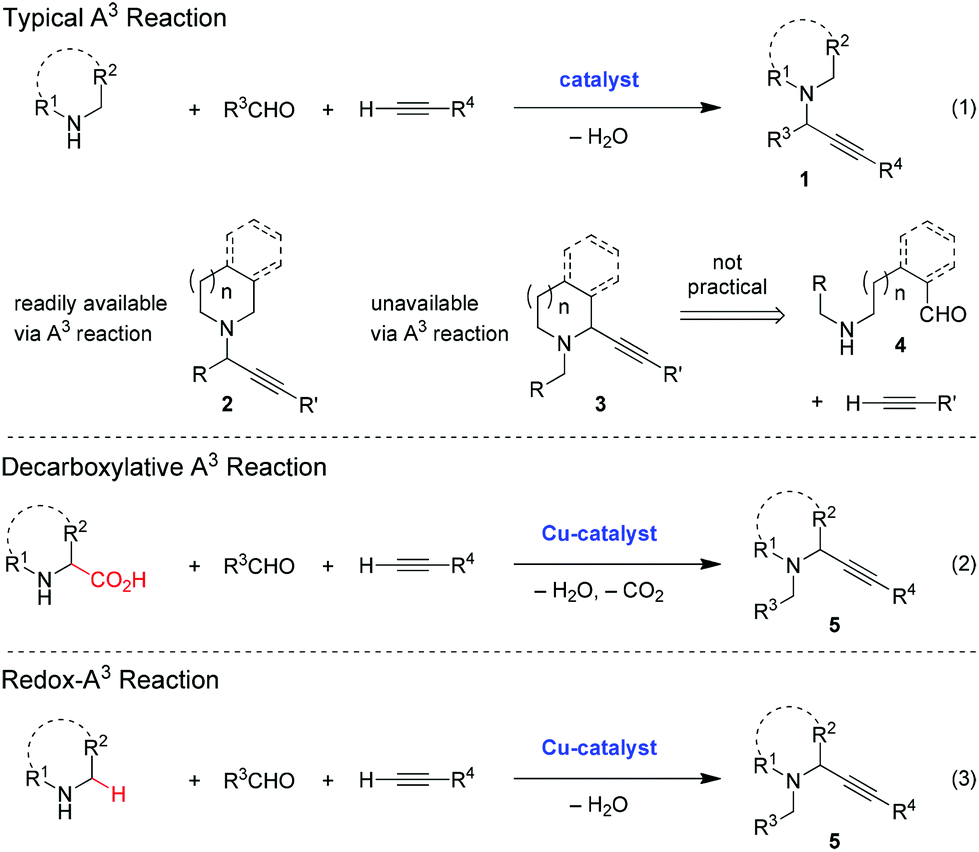
With regard to the mechanism of the two transformations, both the A3 reaction and the redox-A3 reaction likely require the initial formation of iminium ion 6 (Scheme 1). In the classic A3 process, 6 is trapped with a metal acetylide to give product 1. In contrast, the redox-A3 reaction requires the isomerization of iminium ion 6 to its regioisomer 8 which may be achieved via azomethine ylide 7, an intermediate in the decarboxylative A3 reaction.5,6 Through the seminal work of Huisgen and Grigg,14 it is well established that the deprotonation of iminium ions provides a viable entry to azomethine ylides, species that have been used in various [3 + 2] and other pericyclic reactions.15 Once formed, protonation of 7 could result in either the original iminium ion 6 or its desired regioisomer 8. The site of protonation should largely be determined by the charge distribution in 7, in addition to potential other factors such as sterics. Importantly, evidence has recently been obtained for the intermediacy of azomethine ylides in iminium isomerizations that lead to non-pericyclic amine α-functionalization.16,17 The realization of a three-component redox-A3 reaction requires for the iminium isomerization pathway to effectively compete with the direct addition of metal acetylide to 6. In favorable cases, 5 may be obtained from 1via stepwise isomerization. This alternate route appears to be viable based on the reversibility of certain iminium alkynylations, as recently demonstrated by Nakamura and co-workers.18
 | ||
| Scheme 1 The A3vs. the redox-A3 reaction, mechanistic considerations. | ||
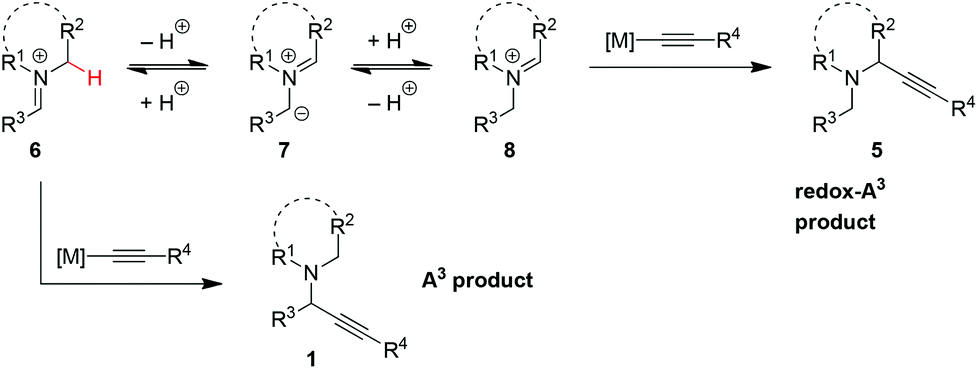
The first redox-A3 reaction was reported by the Seidel group in early 2013.9 Their strategy was based on the notion that the iminium ion isomerization pathway (vide supra) may be accelerated by employing electron-deficient aldehydes. In addition, it was reasoned that sterically demanding aldehydes should slow down the rate of the classic A3 reaction. 2,6-Dichlorobenzaldehyde, an aldehyde that fits these criteria, was evaluated in reactions with pyrrolidine and phenylacetylene. Out of a selection of various commercially available copper(I) and copper(II) compounds, Cu(II) 2-ethylhexanoate (Cu(2-EH)2) was identified as an excellent catalyst. Under the optimized conditions, the redox-A3 product 9a was obtained nearly exclusively over the A3 product 10a (Scheme 2). The nature of the aldehyde was found to have a dramatic effect on the outcome of the reaction. For instance, electronically similar 2,4-dichlorobenzaldehyde showed only a moderate preference for redox-A3 product 9b. In general, for aromatic aldehydes, steric factors appear to outweigh electronics, as illustrated by the observation that mesitaldehyde gave rise to excellent product ratios. However, with aliphatic aldehydes such as cyclohexane-carbaldehyde, the regular A3 products were obtained almost exclusively. Control experiments showed that limited isomerization of 9a to 10a occurs under the reaction conditions, establishing that the product ratios are likely the result of the intrinsic reactivities of the intermediates. Cu(2-EH)2 is thought to play a dual role in the overall process. In addition to forming the copper acetylide, it serves as a source of 2-ethylhexanoic acid which is likely involved in iminium isomerization.
 | ||
| Scheme 2 A3vs. redox-A3 reaction with pyrrolidine: dependence of product ratios on the aldehyde. | ||
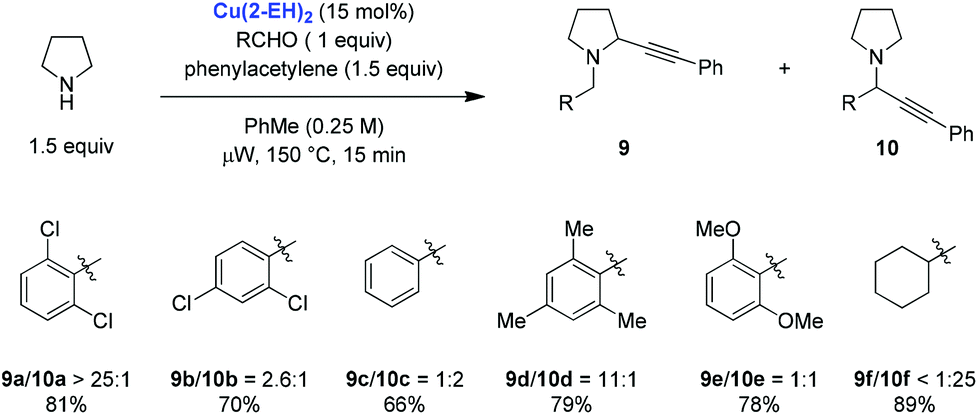
The scope of the Cu(2-EH)2 catalyzed redox-A3 reaction was evaluated with regard to pyrrolidine and various alkynes (Scheme 3). Good to excellent product ratios and moderate to excellent yields of redox-A3 products were obtained. Homologues of pyrrolidine were also evaluated and in the case of piperidine and azepane provided synthetically useful product ratios in favor of the redox-A3 products. Morpholine on the other hand provided mostly regular A3 product in a reaction with phenylacetylene. Interestingly, replacement of phenylacetylene for ortho-tolylacetylene resulted in a slight preference for the redox-A3 product, illustrating that for a bulkier metal acetylide the rate of the standard A3 reaction might be slowed down sufficiently to allow for more effective iminium isomerization.
 | ||
| Scheme 3 Scope of the Cu(2-EH)2 catalyzed redox-A3 reaction by Seidel et al. | ||
Two independent studies from the Yu10 and Ma11 groups, published within a short time frame in late 2013/early 2014, explored the possibility of performing redox-A3 reactions with tetrahydroisoquinoline (THIQ) as the amine component. Both groups employed the same substrate combination in their initial evaluation of this process (Scheme 4). The two studies showed that reactions of THIQ with benzaldehyde and dec-1-yne, conducted between room temperature and 30 °C in the presence of catalytic amounts of CuBr, resulted in near exclusive formation of the standard A3 product 14. Remarkably, Yu et al. found that the simple replacement of CuBr for CuI in a reaction that was conducted under otherwise identical conditions led to the exclusive formation of the redox-A3 reaction product 13. The same effect was observed in the Ma study when CuBr was used in combination with triphenylphosphine (PPh3) as a ligand. These authors also noted a dependence of product ratios on catalyst loading, with lower loadings coupled with higher reaction temperatures giving rise to increased product ratios in favor of the redox-A3 product 13. This was rationalized on the basis that lower catalyst loadings should slow down the rate of addition of the copper acetylide to either iminium ion, allowing more time for iminium isomerization. Both studies nicely illustrate the dramatic effect that a counteranion and/or a ligand can exert on the course of an A3/redox-A3 reaction.
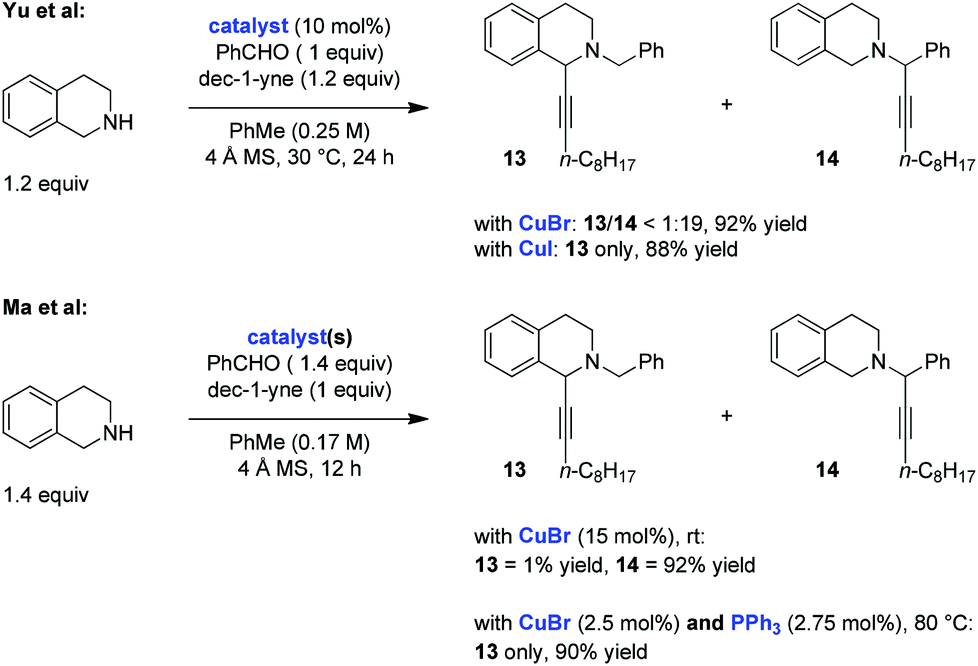 | ||
| Scheme 4 Evaluation of catalysts in the redox-A3 reaction with THIQ. | ||
The Yu group studied the scope of the redox-A3 reaction of THIQ with CuI as the catalyst, using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio of substrates (Scheme 5).10 Excellent results were obtained for a range of different alkynes and aldehydes. In many instances, the redox-A3 product 15 was formed exclusively over the standard A3 product 16. However, a drop in product ratios was observed for aliphatic aldehydes. It was further established that 1,2,3,4-tetrahydro-β-carboline is also amenable to the redox-A3 reaction (not shown). The authors noted that, under their optimized conditions, pyrrolidine provided exclusively the standard A3 products.
:1:1 ratio of substrates (Scheme 5).10 Excellent results were obtained for a range of different alkynes and aldehydes. In many instances, the redox-A3 product 15 was formed exclusively over the standard A3 product 16. However, a drop in product ratios was observed for aliphatic aldehydes. It was further established that 1,2,3,4-tetrahydro-β-carboline is also amenable to the redox-A3 reaction (not shown). The authors noted that, under their optimized conditions, pyrrolidine provided exclusively the standard A3 products.
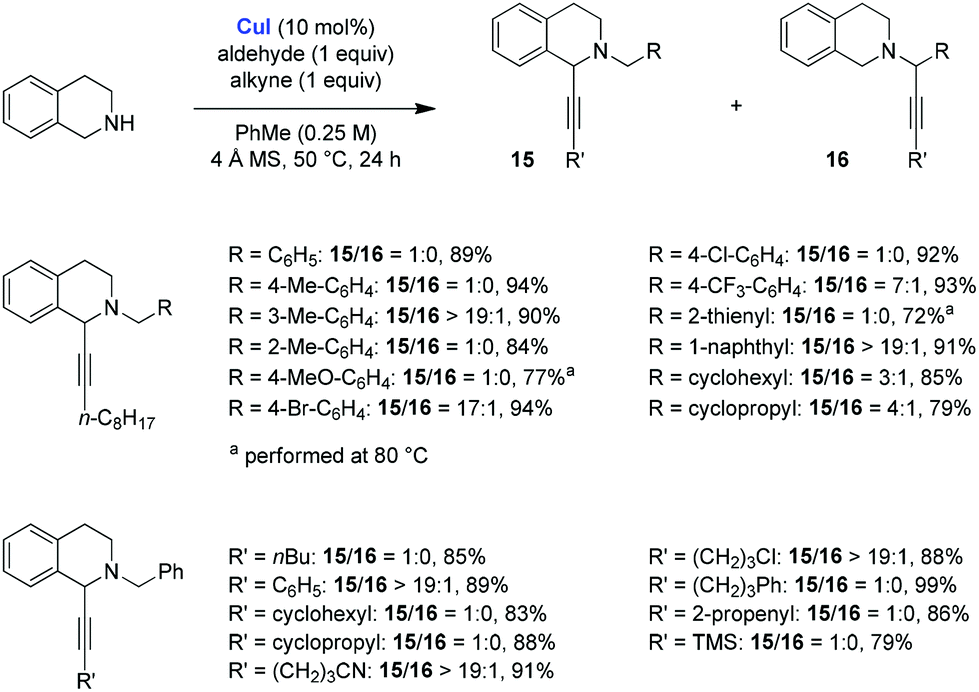 | ||
| Scheme 5 Scope of the CuI catalyzed redox-A3 reaction by Yu et al. | ||
Ma and coworkers, in addition to exploring the scope of the THIQ-based redox-A3 reaction with the CuBr/PPh3 catalyst system, developed a remarkably efficient enantioselective variant of this process (Scheme 6).11 A pinap ligand, originally developed by Carreira et al. and used in the regular A3 reaction,19 was shown to be ideally suited to facilitate asymmetric redox-A3 reactions. A low catalyst loading was sufficient and all products were obtained in excellent yields and enantioselectivities. Interestingly, the addition of benzoic acid as a cocatalyst was shown to result in improved yields. The scope of this reaction was further extended to 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline, which was shown to undergo a highly enantioselective redox-A3 reaction (eqn (4)). Interestingly, Yu et al. reported that under their conditions of CuI catalysis, 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline failed to undergo a reaction with benzaldehyde and dec-1-yne; neither A3 nor redox-A3 products were isolated.
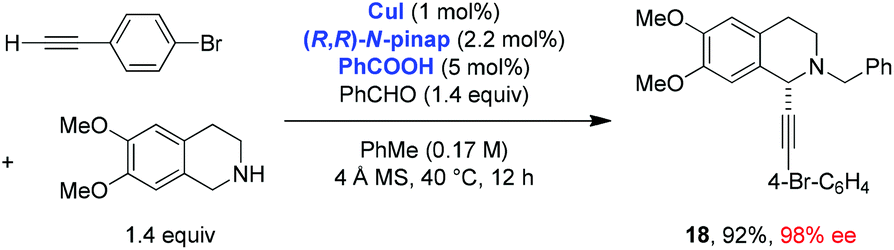 | (4) |
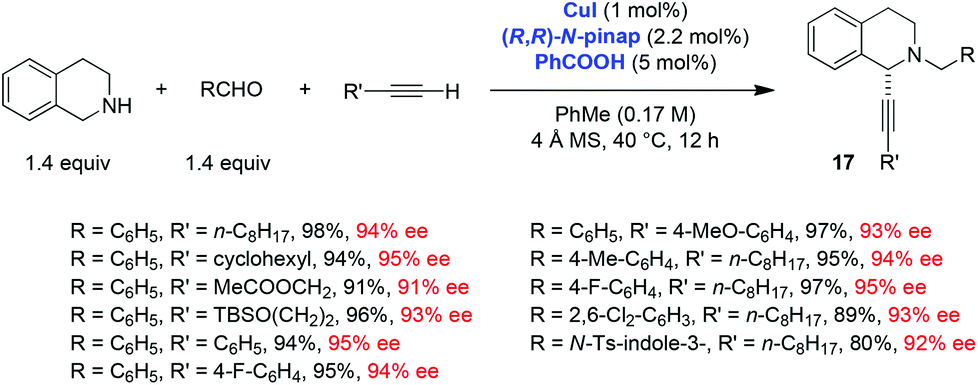 | ||
| Scheme 6 Scope of the enantioselective redox-A3 reaction by Ma et al. | ||
In conclusion, the recently developed redox-A3 reaction provides a useful complement to the traditional A3 coupling process. Ring-substituted propargylic amines not readily available by other means can now be prepared in a single step and in redox-neutral fashion. Some of these reactions proceed under remarkably mild conditions which undoubtedly will encourage their widespread application. The observation that a simple change in counteranion and/or the addition of appropriate ligands can change the course of the reaction from exclusively A3 to exclusively redox-A3 bodes well for the development of related redox-transformations.
Acknowledgements
Financial support from the NIH-NIGMS (grant R01GM101389-01) is gratefully acknowledged.Notes and references
- Selected reviews on the A3 reaction: (a) C. Wei, Z. Li and C.-J. Li, Synlett, 2004, 1472 CAS; (b) L. Zani and C. Bolm, Chem. Commun., 2006, 4263 RSC; (c) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790 RSC.
- Selected recent reviews on amine α-functionalization: (a) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092 CrossRef CAS PubMed; (b) B. Peng and N. Maulide, Chem. – Eur. J., 2013, 19, 13274 CrossRef CAS PubMed; (c) Y. Qin, J. Lv and S. Luo, Tetrahedron Lett., 2014, 55, 551 CrossRef CAS PubMed; (d) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC.
- Selected recent reviews on the CDC reaction: (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- (a) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed; (b) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem. – Eur. J., 2012, 18, 5170 CrossRef CAS PubMed.
- H.-P. Bi, Q. Teng, M. Guan, W.-W. Chen, Y.-M. Liang, X. Yao and C.-J. Li, J. Org. Chem., 2010, 75, 783 CrossRef CAS PubMed.
- C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798 CrossRef CAS PubMed.
- For an oxidative/decarboxylative variant, see: H.-P. Bi, L. Zhao, Y.-M. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2009, 48, 792 CrossRef CAS PubMed.
- Examples of other types of decarboxylative A3 reactions: (a) H. D. Feng, D. S. Ermolat'ev, G. H. Song and E. V. Van der Eycken, J. Org. Chem., 2011, 76, 7608 CrossRef CAS PubMed; (b) H. D. Feng, D. S. Ermolat'ev, G. H. Song and E. V. Van der Eycken, Org. Lett., 2012, 14, 1942 CrossRef CAS PubMed; (c) H. Feng, D. S. Ermolat'ev, G. Song and E. V. Van der Eycken, J. Org. Chem., 2012, 77, 5149 CrossRef CAS PubMed.
- D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765 CrossRef CAS PubMed.
- Q.-H. Zheng, W. Meng, G.-J. Jiang and Z.-X. Yu, Org. Lett., 2013, 15, 5928 CrossRef CAS PubMed.
- W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277 CrossRef CAS PubMed.
- For conceptually different α-alkynylations of tertiary amines, see: T. Sugiishi and H. Nakamura, J. Am. Chem. Soc., 2012, 134, 2504 CrossRef CAS PubMed.
- For Ir-catalyzed α-alkenylations of imines with alkynes, see: S. Sakaguchi, T. Kubo and Y. Ishii, Angew. Chem., Int. Ed., 2001, 40, 2534 CrossRef CAS.
- (a) R. Huisgen, R. Grashey and E. Steingruber, Tetrahedron Lett., 1963, 1441 CrossRef CAS; (b) H. Ardill, R. Grigg, V. Sridharan, S. Surendrakumar, S. Thianpatanagul and S. Kanajun, J. Chem. Soc., Chem. Commun., 1986, 602 RSC; (c) H. Ardill, X. L. R. Fontaine, R. Grigg, D. Henderson, J. Montgomery, V. Sridharan and S. Surendrakumar, Tetrahedron, 1990, 46, 6449 CrossRef CAS.
- Selected reviews on azomethine ylides: (a) A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, Wiley, Chichester, U. K., 2002 CrossRef PubMed; (b) C. Najera and J. M. Sansano, Curr. Org. Chem., 2003, 7, 1105 CrossRef CAS; (c) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765 CrossRef CAS PubMed; (d) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484 CrossRef CAS PubMed; (e) T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2006, 2873 CrossRef CAS PubMed; (f) C. Najera and J. M. Sansano, Top. Heterocycl. Chem., 2008, 12, 117 CAS; (g) M. Nyerges, J. Toth and P. W. Groundwater, Synlett, 2008, 1269 CrossRef CAS PubMed; (h) J. Adrio and J. C. Carretero, Chem. Commun., 2011, 47, 6784 RSC.
- (a) I. Deb, D. Das and D. Seidel, Org. Lett., 2011, 13, 812 CrossRef CAS PubMed; (b) A. Dieckmann, M. T. Richers, A. Y. Platonova, C. Zhang, D. Seidel and K. N. Houk, J. Org. Chem., 2013, 78, 4132 CrossRef CAS PubMed; (c) M. T. Richers, I. Deb, A. Y. Platonova, C. Zhang and D. Seidel, Synthesis, 2013, 1730 CAS.
- Although mechanistically distinct, the overall isomerization mechanism shares certain features with the biologically important transamination process: (a) S. Mathew and H. Yun, ACS Catal., 2012, 2, 993 CrossRef CAS. For leading references on recent synthetic applications, see: (b) X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914 CrossRef CAS PubMed; (c) X. Xiao, M. Liu, C. Rong, F. Xue, S. Li, Y. Xie and Y. Shi, Org. Lett., 2012, 14, 5270 CrossRef CAS PubMed; (d) Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334 CrossRef CAS PubMed; (e) C. Guo, J. Song and L.-Z. Gong, Org. Lett., 2013, 15, 2676 CrossRef CAS PubMed.
- T. Sugiishi, A. Kimura and H. Nakamura, J. Am. Chem. Soc., 2010, 132, 5332 CrossRef CAS PubMed.
- T. F. Knoepfel, P. Aschwanden, T. Ichikawa, T. Watanabe and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 5971 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |