Direct synthesis of arylboronic pinacol esters from arylamines†
Di
Qiu
,
Yan
Zhang
and
Jianbo
Wang
*
Beijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn
First published on 7th March 2014
Abstract
A metal-free synthetic method for arylboronic pinacol esters from easily available aromatic amines as starting materials has been described. This novel transformation affords borylation products in good yields under mild reaction conditions. This strategy can be easily carried out in gram-scale, demonstrating the practical usefulness of the method. Moreover, the Sandmeyer-type transformation can be followed by the Suzuki–Miyaura cross-coupling reaction without purification of the arylboronate products.
Introduction
Arylboronic compounds have shown wide applications in organic synthesis, pharmaceutical research and other fields.1 In particular, functionalized arylboronic acids and arylboronates are valuable in constructing complex structures of target molecules by using the Suzuki–Miyaura cross-coupling reaction.2 Therefore, the synthesis of various functionalized arylboronic compounds has attracted a great deal of attention in recent years. The traditional method for preparing arylboronates involves the reaction between aryl Grignard reagents or aryllithium species with trialkyl boronates (Scheme 1, path a).3 This general approach needs anhydrous conditions and has poor functional group tolerance. Transition-metal-catalyzed borylation from arylhalides with HBpin or B2pin2 has also been well established in recent decades (path b).4 This method has favourable tolerance to a variety of functional groups. Besides, direct borylation via sp2 C–H bond activation has been developed by Smith, Hartwig and other groups (path c).5 However, expensive ligands and catalysts are necessary for these methods. The heavy metal contamination in the final products is also a serious problem. Recently, Lewis acid-promoted electrophilic borylation from electron-rich arenes has been explored (path d),6 but these reactions have not proved to be practically useful. | ||
| Scheme 1 Synthetic routes for arylboronic compounds. | ||
Sandmeyer reactions and the related reactions have been well-established as valuable tools for synthetic organic chemists.7 Based on the Sandmeyer-type reaction, we have recently developed an entirely new approach toward pinacol arylboronate synthesis by direct conversion of the amino group of aniline derivatives to the boronate group (path e).8 The reaction between arylamines and tert-butyl nitrite (tBuONO), an efficient diazotization reagent,9 generates the aryl diazonium salts in situ, which further reacts with B2pin2 to afford pinacol arylboronates under open air. This borylation method has the following advantages: (1) the transformation is under metal-free conditions; (2) arylamines are inexpensive and ubiquitous starting materials; (3) the reaction has good tolerance to various functional groups, including electron-donating and electron-withdrawing groups, and also heterocyclic amines; (4) the reaction proceeds under very mild reaction conditions and is easy to operate. As a result, this Sandmeyer-type borylation is expected to find wide application in organic synthesis.
Results and discussion
In our previous study, a series of arylboronates have been synthesized by this transformation. To demonstrate the practical usefulness of this borylation strategy, this transformation has been carried out in gram-scale for representative arylamines at room temperature. To our delight, the anilines with either electron-rich or electron-deficient groups have provided the corresponding arylboronates in good yields in gram-scale experiments (Scheme 2).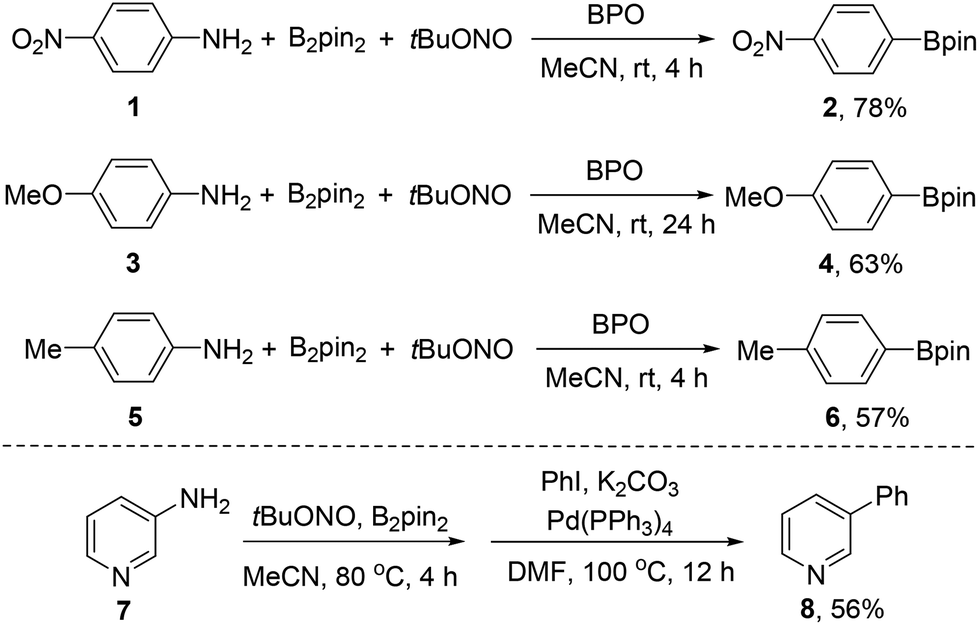 | ||
| Scheme 2 Gram-scale synthesis of arylboronates and the sequential cross-coupling reaction. | ||
Since heterocyclic boronates are not stable on silica gel in some cases, we have further developed a sequential borylation and Pd-catalyzed cross-coupling reaction, in particular for heterocyclic boronates. This sequential reaction can avoid the troublesome purification process and significantly strengthen the practical usefulness of this new borylation method. The sequential borylation and Suzuki–Miyaura cross-coupling reaction has also been scaled-up without the separation of heterocyclic boronate intermediates (Scheme 2).
As summarized in Scheme 3, this strategy has been applied to synthesize a series of functionalized arylboronates. In most cases, moderate to good yields are obtained for the arylamines with either electron-donating or -withdrawing groups. However, several ortho-substituted substrates afford diminished yields because of the steric hindrance. Notably, the oxidation of diazonium intermediates and the deaminohydrogenation are the competing side reactions in this transformation. A preliminary mechanistic study suggests that this borylation follows a radical mechanism. Thus, in the case when the substrate bearing a vinyl substituent is submitted to this reaction polymerization occurs, resulting in diminished yield of the desired boronate product.
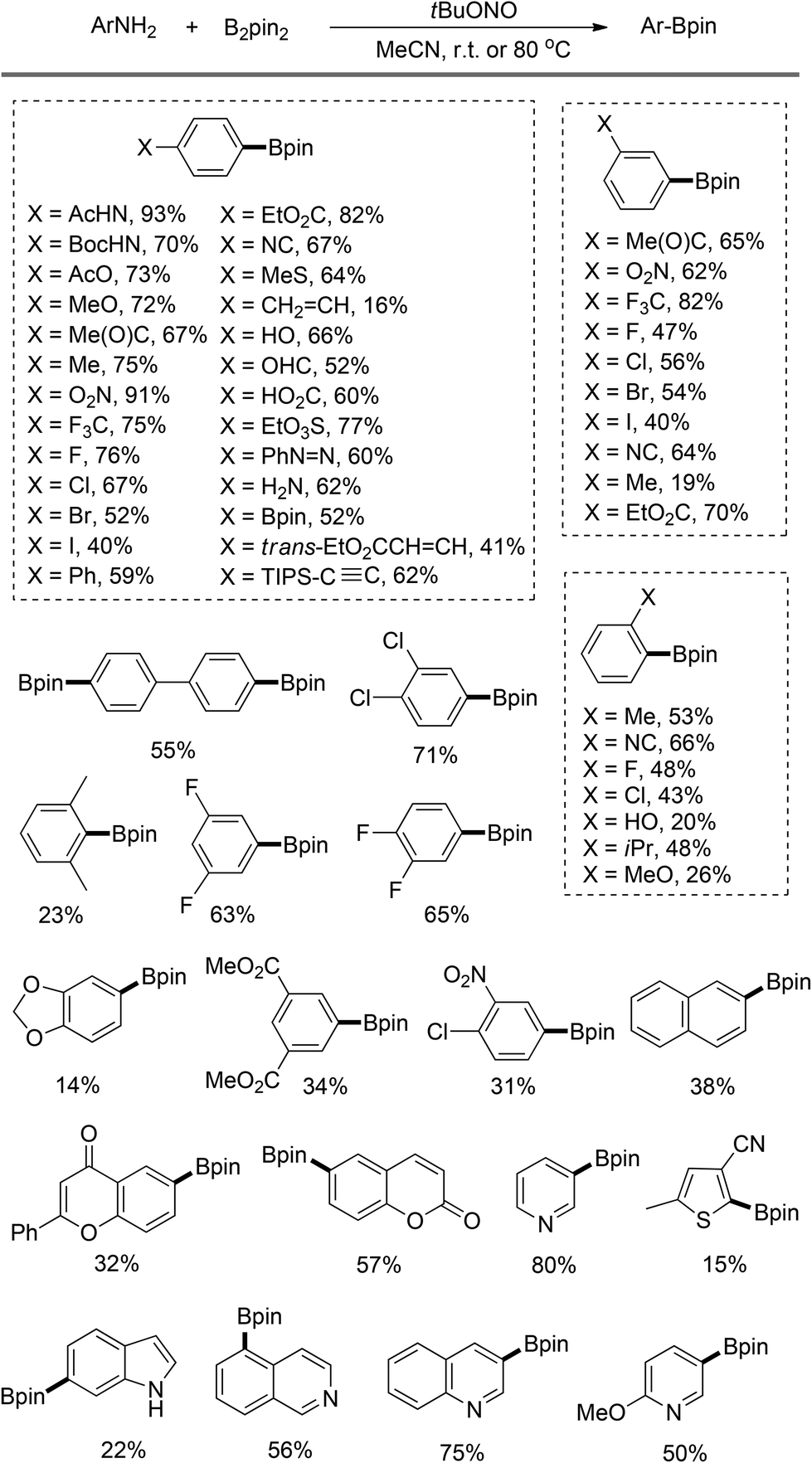 | ||
| Scheme 3 Arylboronates prepared by Sandmeyer-type transformation.8 | ||
Conclusions
We have developed a metal-free transformation for arylboronates from arylamine derivatives under mild reaction conditions. Functionalized arylboronates have been synthesized by this method and the sequential Pd-catalyzed coupling reaction was carried out in gram-scale, affording the desired products in moderate to good yields. The detailed experimental procedure for representative examples is reported in this account.Experimental section
4,4,5,5-Tetramethyl-2-(4-nitrophenyl)-1,3,2-dioxaborolane (2)
A 250 mL, round-bottomed flask equipped with a 2.5 cm rod-shaped, Teflon coated magnetic stir bar is charged with 4-nitroaniline 1 (5.520 g, 40.0 mmol, 1 equiv.), diboron pinacol ester (B2pin2, 10.668 g, 42.0 mmol, 1.05 equiv.), and benzoyl peroxide (BPO, 194 mg, 0.8 mmol, 0.02 equiv.) under ambient atmosphere. The mixture is then dissolved in MeCN (60 mL) in the same flask, and the resulting solution is kept stirred (700 rpm) at room temperature while tert-butyl nitrite (tBuONO, 6.180 g, 60.0 mmol, 1.5 equiv.) diluted by MeCN (40 mL) is added with a glass dropper. A silicone oil seal is used, and the reaction solution is allowed to stir at room temperature maintained by a water bath for 4 hours. The resulting solution is concentrated on a rotary evaporator to remove MeCN (40 °C, 5 kPa). The crude residue is filtered over a flash silica gel plug (60 g) eluting with petroleum ether–ethyl acetate, 20/1 (1000 mL) until TLC analysis shows that no arylboronate 2 remains. The filtrate is then concentrated on a rotary evaporator (25 °C, 5 kPa), and the residue is purified by silica gel column chromatography (eluted with petroleum ether until the product emerges as indicated by TLC, and then eluted with petroleum ether–ethyl acetate, 100/1). The combined elutes are concentrated on a rotary evaporator (25 °C, 5 kPa) and dried over an oil vacuum pump for 10 minutes at room temperature to get the product 2 (7.787 g, 78%) as a pale yellow solid, mp = 109–110 °C, 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 1.37 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 149.8, 135.6, 122.3, 84.6, 24.8; IR (film): 2975, 1788, 1518, 1363, 1349, 1147, 860, 851, 697 cm−1; EI-MS (m/z, relative intensity): 249 (M+, 18), 234 (100), 163 (85), 150 (43), 149 (22), 104 (22), 85 (23), 58 (46), 43 (46), 42 (59).2-(4-Methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4)
A 250 mL, round-bottomed flask equipped with a 2.5 cm rod-shaped, Teflon coated magnetic stir bar is charged with 4-methoxyaniline 3 (4.920 g, 40.0 mmol, 1 equiv.), B2pin2 (10.668 g, 42.0 mmol, 1.05 equiv.), and benzoyl peroxide (BPO 194 mg, 0.8 mmol, 0.02 equiv.) under ambient atmosphere. The mixture is then dissolved in MeCN (100 mL) in the same flask, and the resulting solution is kept stirred (700 rpm) at room temperature when tert-butyl nitrite (tBuONO, 6.180 g, 60.0 mmol, 1.5 equiv.) is directly added. A silicone oil seal is used, and the reaction solution is allowed to stir at room temperature maintained by a water bath for 24 hours. The resulting solution is concentrated on a rotary evaporator to remove MeCN (40 °C, 5 kPa). The residue is filtered over a flash silica gel plug (60 g) eluting with petroleum ether–ethyl acetate, 20/1 (1000 mL) until TLC analysis shows that no arylboronate 4 remains. The filtrate is then concentrated on a rotary evaporator (25 °C, 5 kPa), and the residue is purified by silica gel column chromatography (eluted with petroleum ether). The combined elutes are concentrated on a rotary evaporator (25 °C, 5 kPa) and evaporated over an oil vacuum pump for 10 minutes at room temperature to get the product 4 (5.930 g, 63%) as a pale yellow liquid, 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 3.82 (s, 3H), 1.33 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 162.1, 136.4, 113.2, 83.5, 55.0, 24.8; IR (film): 2978, 1604, 1396, 1360, 1318, 1277, 1247, 1175, 1143, 1091, 1030, 962, 860, 831, 736 cm−1; EI-MS (m/z, relative intensity): 234 (M+, 62), 219 (27), 148 (39), 135 (86), 134 (100), 133 (22), 43 (30), 41 (33).4,4,5,5-Tetramethyl-2-p-tolyl-1,3,2-dioxaborolane (6)
A 500 mL, round-bottomed flask equipped with a 2.5 cm rod-shaped, Teflon coated magnetic stir bar is charged with p-toluidine 5 (5.350 g, 50.0 mmol, 1 equiv.), B2pin2 (13.335 g, 52.5 mmol, 1.05 equiv.), and benzoyl peroxide (BPO 242 mg, 1.0 mmol, 0.02 equiv.) under ambient atmosphere. The mixture is then dissolved in MeCN (120 mL) in the same flask, and the resulting solution is kept stirred (700 rpm) at room temperature when tert-butyl nitrite (tBuONO, 7.730 g, 75.0 mmol, 1.5 equiv.) is directly added. A silicone oil seal is used, and the reaction solution is allowed to stir at room temperature maintained by a water bath for 4 hours. The resulting solution is concentrated on a rotary evaporator to remove MeCN (40 °C, 5 kPa). The residue is filtered over a flash silica gel plug (60 g) eluting with petroleum ether–ethyl acetate, 20/1 (1000 mL) until TLC analysis shows that no arylboronate 6 remains. The filtrate is then concentrated on a rotary evaporator (25 °C, 5 kPa), and the residue is purified by silica gel column chromatography (eluted with petroleum ether until the product emerges, which is verified by TLC and then eluted with petroleum ether–ethyl acetate, 100/1). The combined eluates are concentrated on a rotary evaporator (25 °C, 5 kPa) and evaporated over an oil vacuum pump for 10 minutes at room temperature to get the product 6 (6.162 g, 57%) as a pale yellow liquid, 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.9 Hz, 2H), 7.18 (d, J = 7.9 Hz, 2H), 2.36 (s, 3H), 1.33 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 141.3, 134.7, 128.4, 83.5, 24.8, 21.7; IR (film): 2978, 1613, 1398, 1359, 1144, 1089, 859, 656 cm−1; EI-MS (m/z, relative intensity): 218 (M+, 34), 203 (38), 132 (60), 119 (100), 118 (55), 41 (15).3-Phenylpyridine (8)
B2pin2 (5.334 g, 21 mmol, 1.05 equiv.) and 3-aminopyridine 7 (1.880 g, 20 mmol, 1 equiv.) were weighed in a 250 mL round-bottomed flask. MeCN (60 mL) and tert-butyl nitrite (tBuONO, 3.090 g, 30.0 mmol, 1.5 equiv.) were then added in succession. The resulting reaction solution was stirred for 4 h at 80 °C (N2 evolution completed within 5 min to 15 min). The resulting solution is concentrated on a rotary evaporator to remove MeCN (40 °C, 5 kPa). The system was degassed 3 times and was set under a nitrogen atmosphere. Then Pd(PPh3)4 (1.154 g, 1 mmol, 0.05 equiv.), K2CO3 (5.440 g, 40 mmol, 2 equiv.), DMF (60 mL) and PhI (4.896 g, 24 mmol, 1.2 equiv.) were added and the solution was stirred at 100 °C for 12 h. The solution was then concentrated by rotovap under reduced pressure to leave a crude residue which was purified by silica gel column chromatography (petroleum ether–EtOAc = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, then 5:1). The combined elutes are then concentrated on a rotary evaporator (40 °C, 5 kPa) and then dried over an oil vacuum pump for 10 minutes at room temperature to give 3-phenylpyridine 8 (1.739 g, 56%) as a pale yellow liquid. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.59 (d, J = 4.1 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 7.5 Hz, 2H), 7.47 (t, J = 7.5 Hz, 2H), 7.42–7.33 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 148.4, 148.3, 137.7, 136.5, 134.2, 129.0, 128.0, 127.0, 123.4; IR (film): 3031, 1582, 1473, 1407, 1024, 1006, 754, 697 cm−1. EI-MS (m/z, relative intensity): 155 (M+, 100), 154 (50), 127 (14), 102 (10), 63 (8), 51 (13).
:1, then 5:1). The combined elutes are then concentrated on a rotary evaporator (40 °C, 5 kPa) and then dried over an oil vacuum pump for 10 minutes at room temperature to give 3-phenylpyridine 8 (1.739 g, 56%) as a pale yellow liquid. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.59 (d, J = 4.1 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 7.5 Hz, 2H), 7.47 (t, J = 7.5 Hz, 2H), 7.42–7.33 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 148.4, 148.3, 137.7, 136.5, 134.2, 129.0, 128.0, 127.0, 123.4; IR (film): 3031, 1582, 1473, 1407, 1024, 1006, 754, 697 cm−1. EI-MS (m/z, relative intensity): 155 (M+, 100), 154 (50), 127 (14), 102 (10), 63 (8), 51 (13).
References
- (a) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed; (b) A. Suzuki, Acc. Chem. Res., 1982, 15, 178 CrossRef CAS; (c) N. Miyaura, Bull. Chem. Soc. Jpn., 2008, 81, 1535 CrossRef CAS.
- (a) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2009, 48, 3565 CrossRef CAS PubMed; (b) C. Wang and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 5240 CrossRef CAS PubMed; (c) P. Merino and T. Tejero, Angew. Chem., Int. Ed., 2010, 49, 7164 CrossRef CAS PubMed.
- (a) H. C. Brown, M. Srebnik and T. E. Cole, Organometallics, 1986, 5, 2300 CrossRef CAS; (b) H. C. Brown and T. E. Cole, Organometallics, 1983, 2, 1316 CrossRef CAS.
- (a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS; (b) M. Murata, S. Watanabe and Y. Masuda, J. Org. Chem., 1997, 62, 6458 CrossRef CAS; (c) W. Zhu and D. Ma, Org. Lett., 2006, 8, 261 CrossRef CAS PubMed; (d) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 CrossRef CAS PubMed.
- For reviews, see: (a) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3 CrossRef CAS; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. C. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- (a) M. J. Ingleson, Synlett, 2012, 1411 CrossRef CAS PubMed; (b) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246 CrossRef CAS PubMed.
- (a) C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS; (b) E. B. Merkushev, Synthesis, 1988, 923 CrossRef CAS PubMed; (c) R. Bohlmann, in Comp. Org, Synth., ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 6, p. 203 Search PubMed; (d) G. Balz and G. Schiemann, Ber., 1927, 60B, 1186 CrossRef CAS.
- (a) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846 CrossRef CAS PubMed; (b) D. Qiu, L. Jin, Z. Zheng, H. Meng, F. Mo, X. Wang, Y. Zhang and J. Wang, J. Org. Chem., 2013, 78, 1923 CrossRef CAS PubMed.
- (a) M. P. Doyle, B. Siegfried and J. F. Dellaria, Jr., J. Org. Chem., 1977, 42, 2426 CrossRef CAS; (b) M. P. Doyle, J. F. Dellaria, Jr., B. Siegfried and S. W. Bishop, J. Org. Chem., 1977, 42, 3494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR spectra of compounds 2, 4, 6, and 8. See DOI: 10.1039/c4qo00009a |
| This journal is © the Partner Organisations 2014 |