DOI:
10.1039/D4SC08443H
(Edge Article)
Chem. Sci., 2025,
16, 3865-3871
Chiral Brønsted acid-catalysed enantioselective allylboration of sterically hindered aldehydes enabled by multiple hydrogen bonding interactions†
Received
13th December 2024
, Accepted 19th January 2025
First published on 28th January 2025
Abstract
Chiral phosphoric acids (CPAs) are an important class of chiral Brønsted acid catalysts that can accomplish highly effective enantioselective allylborations of aldehydes. However, traditional CPA-catalysed allyboration has difficulty utilizing sterically hindered aldehydes, where the corresponding products of enantioenriched secondary alcohols adjacent to a quaternary carbon are important moieties in biologically active natural products. To overcome this issue, we employed a chiral phosphoramide catalyst for allylation and crotylation reactions of the allyl boronic acid pinacol ester with sterically hindered aldehydes to benefit from multiple hydrogen bonding interactions between the chiral phosphoramide and substrates. As a result, not only the sterically hindered aldehydes, but the less sterically hindered ones, could also be subjected to enantioselective allylboration using the chiral phosphoramide catalyst by “interaction strategy”. Indeed, conventional CPAs were ineffective for the reactions presented here, resulting in low conversions and enantioselectivities. Computational studies revealed that the most stable transition state comprises weak attractive interactions between phosphoramide and substrates. These interactions did not exist in traditional allylborations with chiral phosphoric acids. In conclusion, the sum of weak interactions, including S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C and two C–F⋯H–C hydrogen bonding interactions, substantially impacts the enantioselectivity of the allylboration of sterically hindered aldehydes.
O⋯H–C and two C–F⋯H–C hydrogen bonding interactions, substantially impacts the enantioselectivity of the allylboration of sterically hindered aldehydes.
Introduction
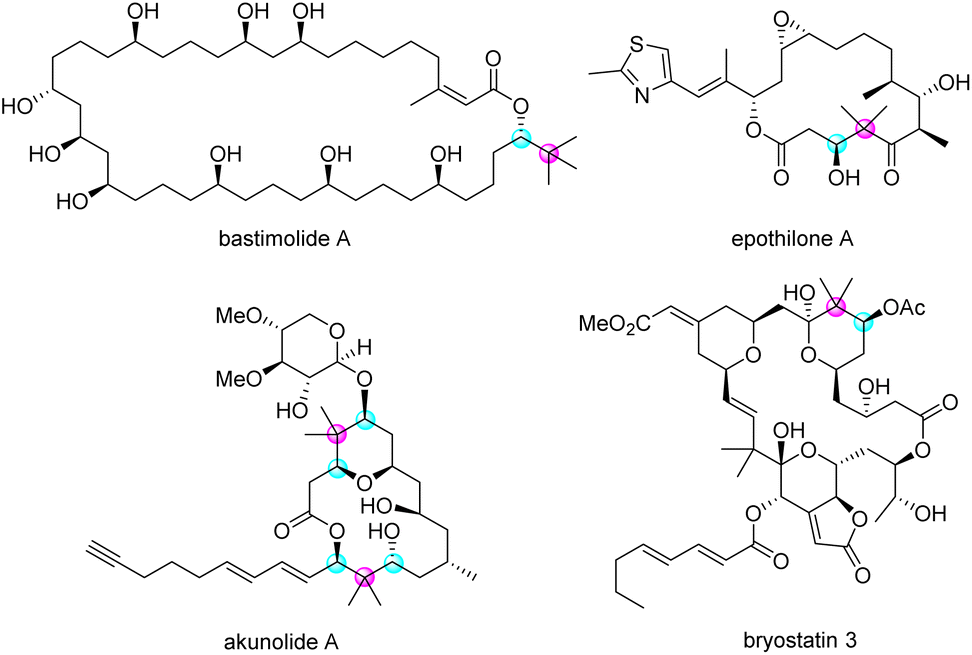
Enantioenriched secondary alcohols carrying sterically hindered groups, such as a quaternary carbon adjacent to a stereogenic centre, are important moieties in biologically active natural products. For instance, many macrolides have this structural feature in their enantioenriched form (Fig. 1).1
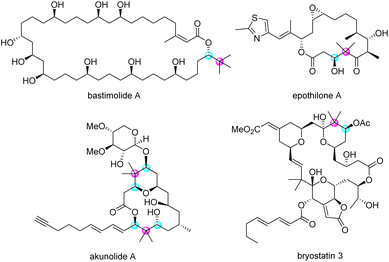 |
| | Fig. 1 Natural product possessing enantioenriched secondary alcohols with neighbouring quaternary carbon. | |
As these natural products have potent biological activities and unique structures, numerous efforts have been made toward their total synthesis, prompting tremendous advancements in synthetic methods over the past decades.2 For example, the Kiyooka group reported an asymmetric aldol reaction of aldehydes with ketene silyl acetal in the presence of stoichiometric valine derivatives and BH3·THF; this reaction produced corresponding aldol adducts possessing an enantioenriched secondary hydroxyl group adjacent to a quaternary carbon in good yields with high enantioselectivities (Scheme 1a).3 Fürstner's group initiated the enzyme-mediated kinetic resolution of a racemic secondary alcohol next to a quaternary carbon centre to furnish the corresponding chiral acetate in the total synthesis of (−)-polycavernoside A (Scheme 1b).4
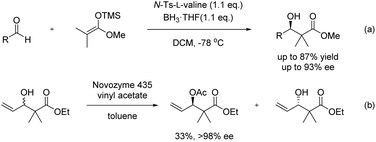 |
| | Scheme 1 Some select examples of the synthesis of enantioenriched secondary alcohols with adjacent quaternary carbon. | |
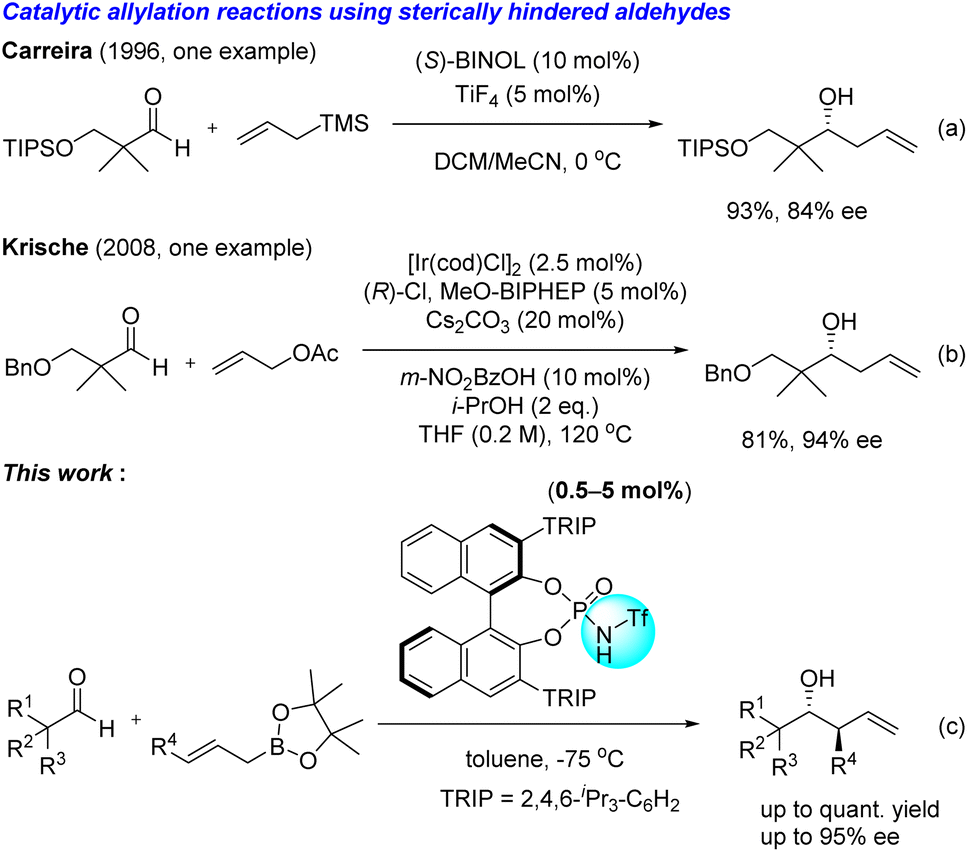
While these conventional methods are reliable, the interest in catalytic and stereoselective carbon–carbon bond-forming reactions has increased in green and sustainable chemistry.5 For instance, Carreira's group described a catalytic and enantioselective allylation reaction of aldehydes with allyltrimethylsilane in the presence of TiF4 and BINOL complex (Scheme 2a).6 This method effectively generates the corresponding allylation product adjacent to a quaternary carbon with good enantioselectivity, despite a hygroscopic and reactive titanium catalyst being required. In 2008, Krische's group published an elegant Ir-catalysed enantioselective allylation reaction to reveal that 3-(benzyloxy)-2,2-dimethylpropanal is an appropriate substrate to obtain the desired product in good yield with excellent enantioselectivity. However, they did not investigate the substrate scope of this methodology (Scheme 2b).7
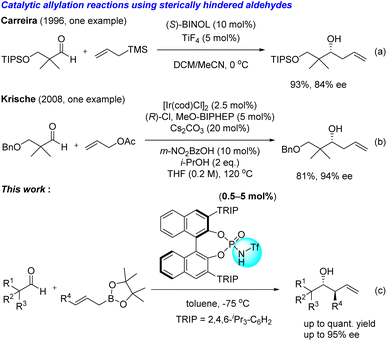 |
| | Scheme 2 Representative examples of catalytic and enantioselective allylation reactions using sterically hindered aldehydes and those presented in the Results and discussion section. | |
Chiral Brønsted acid catalysts, including chiral phosphoric acids (CPAs),8 are valuable tools for enantioselective reactions. These organocatalytic reactions attract a lot of attention in the context of practical and environmentally benign conditions. Recently, we focused on several interactions of chiral Brønsted acid catalysts to develop the enantioselective intramolecular SN2′ reaction and the intermolecular enantioselective Diels–Alder reaction; both of these are challenging reactions to achieve via conventional methods.9 Herein, we report chiral phosphoramide-catalysed stereoselective allylation and crotylation reactions of allyl boronic acid pinacol ester that contains sterically hindered and less hindered aldehydes, utilizing multiple hydrogen bonding interactions between chiral phosphoramide and substrates (Scheme 2c).10 We found that the sum of weak interactions significantly impacts enantioselectivity in the chiral phosphoramide-catalysed allylation reaction.
Results and discussion
Initial attempts
We investigated the enantioselective allylation reaction using a SPINOL-derived catalyst, (S)-3a (TRIP = 2,4,6-iPr3-C6H2), which generally features the best results in the conventional enantioselective allylboration reaction, as reported by Hu's group (Scheme 3).11 However, the reaction of 1a with 2 in the presence of (S)-3a at −75 °C afforded a product with low enantioselectivity (40% ee), even though the chemical yield was good. The reaction catalysed by BINOL-derived CPA (R)-3b, which also used Antilla's allylboration12 method, also gave an unsatisfactory result, albeit with a slight improvement in enantioselectivity (67% ee). These results show that conventional CPAs are ineffective for sterically hindered aldehydes. Importantly, when the acidic catalyst, (R)-3c, rather than (S)-3a or (R)-3b, was used, the reaction rate was sluggish, and both chemical yield and selectivity dropped. This result suggests that high catalyst acidity is unrelated to the activation of the allylation reaction. Although various enantioselective allylation reactions using CPA13 have been developed due to their importance and utility in organic synthetic chemistry, enantioselective allylation reactions of bulky aldehydes with allylboronic agents have yet to be reported. Therefore, many challenges remain in the use of sterically congested substrates in catalytic and enantioselective allylborations.
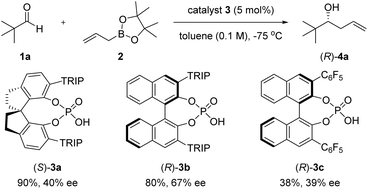 |
| | Scheme 3 The initial attempt of chiral Brønsted acid-catalyzed allylboration of pivalaldehyde (1a) with 2. | |
The Goodman and Houk groups independently performed intensive theoretical studies of the reaction mechanism in the case of benzaldehyde.14 The most stable transition state (TS) in the BINOL-derived (R)-CPA-catalyzed allylation reaction of benzaldehyde with an allylboronic agent is shown in Fig. 2.
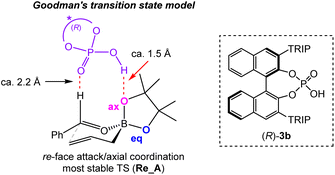 |
| | Fig. 2 Goodman's transition state model. | |
According to this TS model, the CPA catalyst, (R)-3b, does not activate the carbonyl group of the benzaldehyde by protonation. However, it stabilizes the six-membered TS through two-point hydrogen bonding interactions with the axial oxygen of the boronic ester (P–O–H⋯O–B) and the formyl hydrogen of the benzaldehyde (PO⋯H–C). Notably, DFT calculations have shown that the acidic proton of the CPA catalyst and the axial oxygen atom (P–O–H⋯O–B) are approximately 1.5 Å apart, indicating that it is not protonation of the oxygen atom, but the hydrogen bonding instead causing stabilization. From these calculation studies, the unfavorable result observed for the CPA (R)-3c is justifiable because high catalyst acidity is not necessarily a crucial factor stabilizing TS (Re_A) during allylboration reactions. Hence, we focused on chiral phosphoramides, rather than CPAs, to stabilize TS in the major reaction pathway using an “interaction strategy”.9b
Development of a new catalytic system through multiple interactions
Through our previous work, we found that the phosphoramide catalyst with a C6F5 group at the sulfonamide moiety yielded favourable results.9a Thus, we first investigated the allylation reactions of 1a with 2 using (R)-3d having a C6F5SO2NH moiety (Table 1, entry 1). To our delight, the reaction proceeded smoothly to furnish the desired product in 98% yield with good enantioselectivity (84% ee). When we investigated other substituents on the N moiety of the catalyst, we found that the catalyst, (R)-3e, containing a SO2CF3 (Tf) group exhibited better reactivity than (R)-3d, and thus, it helped obtain the desired product in quantitative yield with higher enantioselectivity (95% ee) than that (84% ee) observed using (R)-3d (entry 2). The absolute configuration of compound 4a obtained in this reaction was consistent with those obtained with CPA (R)-3b having the same chiral backbone as phosphoramide (R)-3e. No reaction occurred when the catalyst (R)-3f containing an SO2Me group was employed (entry 3). The substituents on the sulfonamide moiety of the catalyst significantly influenced both reactivity and enantioselectivity. The reaction did not proceed when a more acidic thiophosphoramide catalyst, (R)-3g, that was previously reported by Yamamoto and co-workers,15 was used (entry 4). These results suggest that high catalyst acidity is not essential in the allylation reaction of aldehydes with allylboronic agents; this can also be recognized from the TS model of Goodman (Fig. 2). SPINOL-derived phosphoramide catalyst, (S)-3h, was ineffective in the present reaction, probably because of a narrow reaction space (entry 5).16 After comprehensively investigating reaction conditions, including the solvent, temperature, and concentration, the experimental condition that uses (R)-3e (entry 2) was found to be the best (see ESI†). Finally, catalyst loading could be reduced to 0.5 mol% in a large-scale experiment. The product was quantitatively formed on a 2 g scale by extending the reaction time without losing enantioselectivity (entry 6).
Table 1 Optimization of the reaction conditions using 1a and 2 catalysed by chiral phosphoramide 3a
| Entry |
Catalyst |
Time (h) |
Yield of (R)-4ab/% |
ee/% |
|
Unless otherwise specified, all reactions were carried out using 0.20 mmol of 1a, 0.24 mmol (1.2 eq.) of 2, and 0.010 mmol (5 mol%) of catalyst.
Isolated yield.
Catalyst loading was 0.5 mol%, and 2 g of pivalaldehyde was employed. TRIP = 2,4,6-iPr3-C6H2, nd = not determined.
|
| 1 |
(R)-3d |
72 |
98 |
84 |
| 2 |
(R)-3e |
24 |
Quant. |
95 |
| 3 |
(R)-3f |
72 |
No reaction |
nd |
| 4 |
(R)-3g |
72 |
No reaction |
nd |
| 5 |
(S)-3h |
72 |
No reaction |
nd |
| 6c |
(R)-3e |
48 |
Quant. |
95 |

|
Transition state analysis
After investigating the optimum conditions of phosphoramide-catalysed allylboration reaction with pivalaldehyde, we performed DFT computational studies to gain mechanistic insights into the present reaction system. As the present reaction of pivalaldehyde (1a) afforded (R)-product 4a in the presence of the catalyst, (R)-3e, the TS in the re-face attack had a lower energy barrier than that in the si-face attack.
We calculated four TSs for the re-face and the si-face attacks, considering the axial and equatorial coordination models, respectively (Fig. 3).14 DFT calculation was performed using Gaussian 16 in the M06-2X/6-31G(d) level of theory.17 As a result, TS Re_A, namely, re-face attack and coordination to the axial oxygen of boronic ester, produced the lowest energy. Resembling CPA-catalysed allylborations, two-point interactions were produced through the hydrogen bonding of the phosphoryl oxygen of the catalyst with the proton of the formyl group (2.43 Å); hydrogen bonding was observed between the axial boronate oxygen and the hydrogen atom (N–H) of the catalyst (1.57 Å) in Re_A. We also found that the TS of the major reaction pathway (major-TS) was preferred over the TS of the minor reaction pathway (minor-TS), namely Si_E, by 4.2 kcal mol−1. Single-point energies were conducted with the MN15/6-31G(d)+SMD (toluene) to result in a 3.0 kcal mol−1 energy difference.18 These calculation results are in relatively good agreement with the experimentally observed enantiomeric excess of (R)-3e (95% ee, theoretically ca. 2.2 kcal mol−1).19 In the major-TS (Re_A), the distance for N–H⋯O–B hydrogen bonding was 1.57 Å, which remained almost unchanged from the calculation result in the CPA-catalysed allylboration.14 This suggests that the high acidity of the phosphoramide catalyst does not enhance the N–H⋯O–B hydrogen bonding in this system because the hydrogen bond is not shortened. Interestingly, not only the hydrogen bonding between the sulfonyl moiety and the olefin C–H (SO⋯H–C, 2.47 Å) but also the two weak interactions between a fluorine atom of the CF3 group and the hydrogen atoms of the pinacol Me group (C–F⋯H–C, 2.25 and 2.57 Å) were found in addition to the major two-point hydrogen bonding interactions in Re_A. Conversely, in the minor-TS (Si_E), the PO⋯H–C (formyl) interaction and C–F⋯H–C interactions are not present. Similarly, the C–F⋯H–C interactions were not observed in the minor-TSs (Re_E and Si_A). These observations indicate that the minor-TSs were less stable than Re_A. This was not due to the steric congestion but due to the lack of multiple forces of attraction.20 The sum of the multiple hydrogen bonding interactions, including the relatively weak SO⋯H–C and C–F⋯H–C hydrogen bonding interactions, which are not present in conventional CPA-catalysed allylation reactions, is considered to effectively stabilize the major-TS to give the desired product with excellent enantioselectivity. As the catalyst does not occupy the space around the tBu group in Re_A, we envisioned that various substituents could be introduced to aldehydes. Based on the DFT calculation results, we proceeded to investigate the substrate scope.
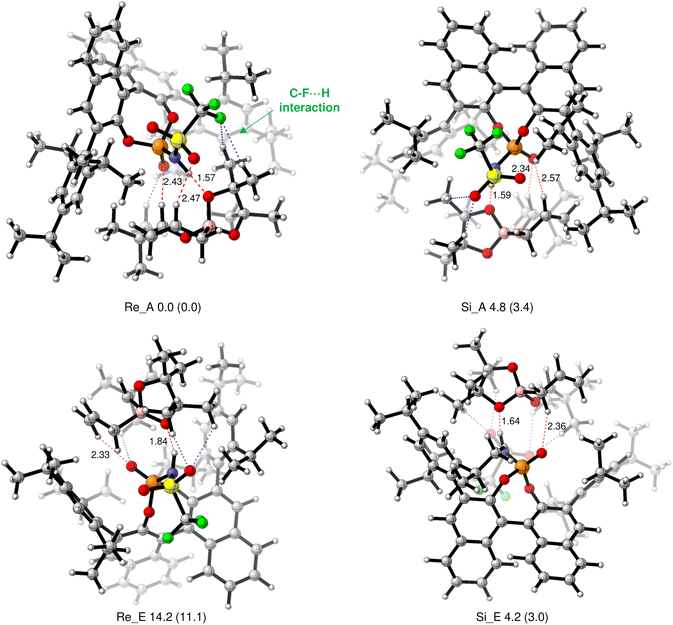 |
| | Fig. 3 Optimized transition states of Re_A, Re_E, Si_A, and Se_E at the M06-2X/6-31G(d) level of theory. Bond lengths are given in Å. Values enclosed in parentheses are energies relative to “Re_A” calculated by MN15/6-31g(d)+SDM (toluene). Energy differences are given in kcal mol−1. See the ESI† on page S38 in detail. | |
Substrate scope
After rationally elucidating the high enantioselectivity of the present allylation reaction under optimal reaction conditions, we explored the substrate scope of the present reaction toward aldehydes with a quaternary centre at the α-position (Scheme 4). Various functional groups at the β-position of the formyl group were found to be suitable for the allylation reaction. They could effectively furnish the corresponding homoallyl alcohols in excellent yields with good enantioselectivities (4b–4d). In contrast, the SPINOL-derived catalyst, (S)-3a, was ineffective for these substrates, resulting in low conversions and enantioselectivities.21a Aldehydes with sensitive functional groups, such as a Ts group, an α,β-unsaturated ester, and an enone, were also tolerated and they produced the corresponding products with a quantitative yield of up to 95% ee (4e–4g). Next, the scope of less hindered aldehydes, such as cyclohexane carboxaldehyde and benzaldehyde, was investigated. The corresponding products were obtained in good yields with high enantioselectivities (4h, 4i). Unfortunately, the allylboration of benzaldehyde was inferior to that of the CPA-catalysed reactions using (S)-3a and (R)-3b (99% ee and 98% ee, respectively), whereas the enantioselectivity of the reaction using cyclohexane carboxaldehyde was similar to that with (S)-3a (91% ee) and superior to that with (R)-3b (73% ee).11,12 Next, we applied less sterically hindered alkyl aldehydes to our system, and found that these afforded 4j and 4k in good yields and ee values (85% ee and 83% ee, respectively). A similar tendency to reduce enantioselectivities in the case of less hindered aldehydes was observed in our reaction. Diastereoselective allylation reactions of enantioenriched aldehydes with substituents at the α- or β-position under optimized conditions produced the desired products in excellent yields with moderate to high diastereoselectivities (4l–4o). In contrast, the conventional CPA-catalysed reactions produced 4n and 4o with low diastereoselectivities.21b Subsequently, we extended this protocol to crotylation reactions using diverse aldehydes, including those having a stereogenic centre. Various enantioenriched substrates worked well and afforded products, 4p–4s, in good yields and moderate to reasonably high diastereomeric ratios (drs). Notably, the anti-selective crotylation products, 4p and 4q, with three continuous stereogenic centres, commonly found in natural products, such as marine macrolides, were obtained with excellent diastereoselectivities. Not only the enantioenriched aldehydes but also simple aldehydes, such as benzaldehyde and 3-phenylpropionaldehyde, were suitable crotylation, furnishing corresponding anti-products in good yields (4t and 4u). In contrast, sterically hindered 3-(benzoyloxy)-2,2-dimethylpropanal did not yield the desired product, and there was no reaction even when the temperature was elevated to 0 °C (4v).
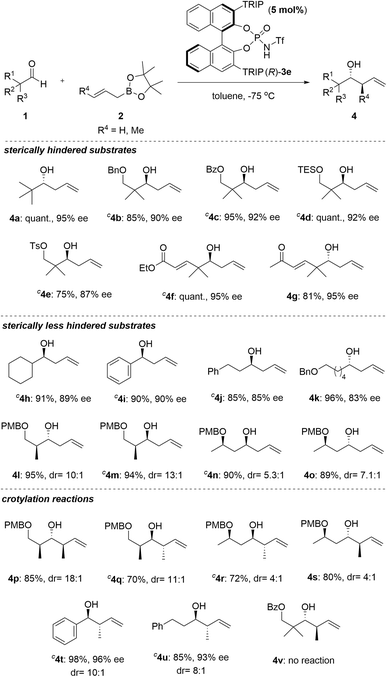 |
| | Scheme 4
a
Unless otherwise specified, all reactions were carried out using 0.20 mmol of 1a, 0.24 mmol (1.2 eq.) of 2, and 0.010 mmol (5 mol%) of catalyst. bIsolated yield. c(S)-3e was used instead of (R)-3e. TRIP = 2,4,6-iPr3-C6H2. Chiral phosphoramide-catalysed allylation and crotylation of aldehydes.a,b | |
Derivatization
A derivatization of products was attempted to show the synthetic utility of the present allylation and crotylation reactions to construct functional chiral building blocks (Fig. 4). The enantioenriched alcohol 4g was treated with p-nitrobenzaldehyde in the presence of catalytic trifluoromethanesulfonic acid (TfOH) to afford sterically congested 1,3-syn-dioxane derivative 5 with 87% yield and excellent diastereoselectivity (eqn (1)).22 Alternatively, the esterification of compound 4q using acryloyl chloride, followed by ring-closing metathesis, produced functionalized α,β-unsaturated lactone 6 in 91% yield in two steps (eqn (2)).
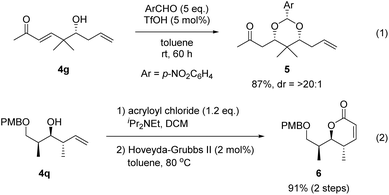 |
| | Fig. 4 Derivatization of products. | |
Conclusions
We have developed an enantio- and diastereoselective allylboration of sterically hindered aldehydes that utilize multiple hydrogen bonding interactions between the chiral phosphoramide catalyst and substrate to furnish synthetically useful secondary alcohols with a quaternary carbon adjacent to a stereogenic centre. Computational studies suggest that the major-TS is stabilized by weak attractive interactions, including SO⋯H–C and two C–F⋯H–C hydrogen bonding interactions. In contrast, minor-TSs lack some of these multiple interactions. Hence, the sum of weak interactions substantially impacts the enantioselectivity in the allylboration of sterically hindered aldehydes. Using phosphoramide 3e as a multiple interaction catalyst, 0.5 mol% catalyst loading is sufficient to complete the allylboration of pivalaldehyde on a gram-scale. The present allylation and crotylation reactions can be applied to both sterically hindered and less hindered aldehydes to obtain the corresponding homoallylic alcohols in excellent yields and high enantioselectivities. Further studies on the efficient synthesis of important natural products using the present methodology are in progress in our laboratory.
Data availability
The exploratory investigation results, experimental procedures, computational data, and characterization data are available.
Author contributions
S. U. contributed to the conceptualization, project administration, and design of the work, as well as data curation, formal analysis, investigation (experimental and theoretical studies), writing – original draft. S. O., N. S., and T. H. contributed to the formal analysis and investigation (experimental). M. T. contributed to the conceptualization, writing – review and editing, supervision, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The computation was performed at the Research Center for Computational Science, Okazaki, Japan. This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) and a Grant-in-Aid for Transformative Research Areas (A) “Green Catalysis Science for Renovating Transformation of Carbon-Based Resources” (JP23H04908) from MEXT, Japan (M. T.), a Grant-in-Aid for Scientific Research (C) (23K04730) from JSPS, Japan (S. U.) and a Grant-in-Aid for Young Scientists (24KJ0444) from JSPS, Japan (N. S.).
Notes and references
- Isolations of these natural products, see:
(a) C.-L. Shao, R. G. Linington, M. J. Balunas, A. Centeno, P. Boudreau, C. Zhang, N. Engene, C. Spadafora, T. S. Mutka, D. E. Kyle, L. Gerwick, C.-Y. Wang and W. H. Gerwick, J. Org. Chem., 2015, 80, 7849–7855 CrossRef CAS PubMed;
(b) K. Gerth, N. Bedorf, G. Hofle, H. Irschik and H. J. Reichenbach, J. Antibiot., 1996, 49, 560–563 CrossRef CAS PubMed;
(c) K. Umeda, A. Iwasaki, R. Taguchi, N. Kurisawa, G. Jeelani and T. N. K. Suenaga, J. Nat. Prod., 2023, 86, 2529–2538 CrossRef CAS PubMed;
(d) G. R. Pettit, J. F. Day, J. L. Hartwell and H. B. Wood, Nature, 1970, 227, 962–963 CrossRef CAS PubMed;
(e) G. R. Pettit, C. L. Herald, D. L. Doubek and D. L. Herald, J. Am. Chem. Soc., 1982, 104, 6846–6848 CrossRef CAS.
- Selected examples of total syntheses of natural products possessing quaternary carbon adjacent to enantioenriched secondary alcohol moieties, see:
(a) A. Balog, D. Meng, T. Kamenecka, P. Bertinato, D.-S. Su, E. J. Sorensen and S. J. Danishefsky, Angew. Chem., Int. Ed., 1996, 35, 2801–2803 CrossRef CAS;
(b) K. Ohmori, Y. Ogawa, T. Obitsu, Y. Ishikawa, S. Nishiyama and S. Yamamura, Angew. Chem., Int. Ed., 2000, 39, 2290–2294 CrossRef CAS;
(c) B. M. Trost, Y. Wang, A. K. Buckl, Z. Huang, M. H. Nguyen and O. Kuzmina, Science, 2020, 368, 1007–1011 CrossRef CAS PubMed;
(d) J. B. Cox, A. A. Kellum, Y. Zhang, B. Li and A. B. Smith III, Angew. Chem., Int. Ed., 2022, 61, e202204884 CrossRef CAS PubMed.
- S. Kiyooka, Y. Kaneko, M. Komura, H. Matsuo and M. Nakano, J. Org. Chem., 1991, 56, 2276–2278 CrossRef.
- L. Brewitz, J. Llaveria, A. Yada and A. Fürstner, Chem.–Eur. J., 2013, 19, 4532–4537 CrossRef CAS PubMed.
- For selected reviews, see:
(a) P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3 CrossRef CAS;
(b) J. F. Jenck, F. Agterberg and M. J. Droescher, Green Chem., 2004, 6, 544 RSC;
(c) I. Delidovich and R. Palkovits, Green Chem., 2016, 18, 590 RSC.
- D. R. Gauthier Jr and E. M. Carreira, Angew. Chem., Int. Ed., 1996, 35, 2363–2365 CrossRef.
-
(a) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891–14899 CrossRef CAS PubMed;
(b) I. S. Kim, S. B. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2514–2520 CrossRef CAS PubMed;
(c) When more inexpensive BINAP ligand than MeO-BIPHEP ligand was used for the Krishche allylation, decrease of enantioselectivity was observed (77% ee) in our investigation. See the ESI† in detail.;
(d) Recently, the Krische group reported an enantioselective tert-prenylation using chiral ruthenium catalyst, see: J. Am. Chem. Soc., 2023, 145, 18676−18683..
- For seminal studies of chiral phosphoric acid catalysts, see:
(a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed;
(b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
-
(a) M. Shimizu, J. Kikuchi, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 5747–5757 RSC;
(b) T. Nakanishi and M. Terada, Chem. Sci., 2023, 14, 5712–5721 RSC.
- For seminal study of chiral phosphoramide catalyst, see:
(a) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed. For a recent review, see:
(b) G. Caballero-García and J. M. Goodman, Org. Biomol. Chem., 2021, 19, 9565–9618 RSC.
- C.-H. Xing, Y.-X. Liao, Y. Zhang, D. Sabarova, M. Bassous and Q.-S. Hu, Eur. J. Org Chem., 2012, 1115–1118 CrossRef CAS.
- P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884–11886 CrossRef CAS PubMed.
- Selected recent examples of enantioselective allylboration catalyzed by CPA, see:
(a) E. Rodríguez, M. N. Grayson, A. Asensio, P. Barrio, K. N. Houk and S. Fustero, ACS Catal., 2016, 6, 2506–2514 CrossRef;
(b) S. Gao, M. Duan, Q. Shao, K. N. Houk and M. Chen, J. Am. Chem. Soc., 2020, 142, 18355–18368 CrossRef CAS PubMed;
(c) S. Gao, M. Duan, L. R. Andreola, P. Yu, S. E. Wheeler, K. N. Houk and M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202208908 CrossRef CAS PubMed.
-
(a) M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2012, 134, 2716–2722 CrossRef CAS PubMed;
(b) H. Wang, P. Jain, J. C. Antilla and K. N. Houk, J. Org. Chem., 2013, 78, 1208–1215 CrossRef CAS PubMed.
- M. Sai and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 7091–7094 CrossRef CAS PubMed.
- H. Ye, T. Sato, T. Nakanishi, S. Ito, S. Umemiya and M. Terada, Catal. Sci. Technol., 2024, 14, 6869–6881 RSC.
- Whereas Goodman and Houk used the B3LYP density function, we decided to use M06-2X because of the importance of weak attractive interactions in our system. Indeed, after some calculation studies, we found that B3LYP was unsuitable for the present reaction that includes multiple weak interactions. see the ESI.†.
- H. S. Yu, X. He, S. L. Lia and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- Similar tendency of energy differences was observed by using B3LYP-D3(BJ)/6-311G(d, p) and wB97XD/6-31G(d). See the ESI† in details.
-
(a) Interestingly, TS Si_E was more stable than TS Si_A despite the absence of hydrogen bonding between the phosphoryl oxygen and the hydrogen atom of the formyl group. In TS Si_E, weak attractive interactions of SO···H–C (sp3) in the pinacol moiety of 2 and the tBu group of pivalaldehyde were observed in DFT calculations.;
(b) Goodman's group performed the asymmetric propargylation of cyclohexanecarboxaldehyde with allenylboronic acid pinacol ester in the presence of (R)-3e to give a product with only 39% ee. They did not mention the multiple hydrogen bonding interactions between phosphoramide and substrates. See: M. N. Grayson and J. M. Goodman, J. Am. Chem. Soc., 2013, 135, 6142–6148 CrossRef CAS PubMed.
-
(a) For instance, the allylation reaction using SPINOL-derived catalyst (S)-3a afforded product 4b in 7% yield and 33% ee. See the ESI† for experimental results in detail.;
(b) The reaction using (R)-3a gave 1,3-syn product 4n with dr = 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the reaction using (S)-3a afforded 1,3-anti product 4o with dr = 2:1..
:1, and the reaction using (S)-3a afforded 1,3-anti product 4o with dr = 2:1..
- Selected examples of synthesis of 1,3-syn diol derivatives via hemiacetalization/intramolecular oxy-Michael reactions, see:
(a) P. A. Evans, A. Grisin and M. J. Lawler, J. Am. Chem. Soc., 2012, 134, 2856–2859 CrossRef CAS PubMed;
(b) Y. Hayashi, T. Saitoh, H. Arase, G. Kawauchi, N. Takeda, Y. Shimasaki and I. Sato, Chem.–Eur. J., 2018, 24, 4909–4915 CrossRef CAS PubMed;
(c) K. Murata, H. Takeshita, K. Sakamoto and H. Fuwa, Chem.–Asian. J., 2020, 15, 807–819 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sotaro
Osaka
b,
Naoya
Shinagawa
b,
Takumi
Hirata
b and
Masahiro
Terada
a,
Sotaro
Osaka
b,
Naoya
Shinagawa
b,
Takumi
Hirata
b and
Masahiro
Terada