
Pyridyl disulfide-based thiol–disulfide exchange reaction: shaping the design of redox-responsive polymeric materials
Ismail
Altinbasak
a,
Mehmet
Arslan
 *b,
Rana
Sanyal
ac and
Amitav
Sanyal
*ac
*b,
Rana
Sanyal
ac and
Amitav
Sanyal
*ac
aDepartment of Chemistry, Bogazici University, Bebek 34342, Istanbul, Turkey. E-mail: amitav.sanyal@boun.edu.tr
bYalova University, Faculty of Engineering, Department of Polymer Materials Engineering, 77100 Yalova, Turkey. E-mail: mehmet.arslan@yalova.edu.tr
cCenter for Life Sciences and Technologies, Bogazici University, Bebek 34342, Istanbul, Turkey
First published on 25th November 2020
Abstract
Recent years have witnessed an increase in the utilization of stimuli-responsive polymers in various areas of materials and biological sciences. In particular, disulfide linkage containing redox-responsive polymers have attracted interest in various biomedical applications ranging from fabrication of drug delivery vehicles to diagnostic interfaces. Cleavage of the disulfide linkage in the presence of an endogenous reducing agent, namely, glutathione, also found in increased amounts in diseased tissues has led to the incorporation of this particular linkage into several therapeutic platforms. Among the various methods available for introducing the redox-sensitive disulfide unit, the pyridyl disulfide (PDS) moiety has been one of the most widely employed building blocks. The rapid thiol–disulfide exchange reaction of the PDS group with thiol functional groups has been exploited from the reversible conjugation of therapeutic agents to the fabrication of redox-responsive crosslinked materials such as hydrogels and nanogels. This review provides an overview of various synthetic approaches utilized to incorporate this particular thiol-reactive motif into different types of polymeric materials and briefly highlights its utilization to obtain functional materials.
 Ismail Altinbasak | Ismail Altinbasak received his undergraduate degree from the Department of Chemistry at Bogazici University, Istanbul, Turkey. Thereafter, he obtained his M.S. degree in chemistry in 2016 under the supervision of Prof. Amitav Sanyal, working in the area of responsive hydrogels for protein delivery. Currently, Ismail is pursuing his doctoral studies in the same group, and focussing on the design and fabrication of novel polymeric biomaterials ranging from nanofibers to nanogels for various biomedical applications such as combating bacterial infection to targeted drug delivery to cancer cells. |
 Mehmet Arslan | Mehmet Arslan obtained his undergraduate degree from Bilkent University, Department of Chemistry, Ankara, Turkey. He completed his PhD in 2015 from Bogazici University, Istanbul, Turkey. He currently works at Yalova University, Department of Polymer Engineering, Turkey as an academician. His research primarily focuses on the fabrication of reactive polymeric materials for key biomedical applications such as drug delivery and biomolecular immobilization. He holds interest in developing new condensation and multi-component polymerization methods by adapting versatile synthetic organic transformations into polymer chemistry. |
 Rana Sanyal | Rana Sanyal received her undergraduate degree from the Department of Chemical Engineering at Bogazici University, Istanbul, Turkey. After completing her PhD from Boston University (USA), she worked in Amgen Inc., Thousand Oaks (USA) as a Research Scientist and then joined Bogazici University in 2004 as an assistant professor. Currently, she is professor of chemistry and the director of the Center for Life Sciences and Technologies. Her research focuses on the development of targeted drug-delivery systems for cancer and hydrogels for biomedical applications from dendritic scaffolds. She was the recipient of L'Oréal Turkey for Women in Science award and young investigator award administered by the Turkish Academy of Sciences. |
 Amitav Sanyal | Amitav Sanyal obtained his undergraduate degree from the Indian Institute of Technology at Kanpur, India. He received his Ph.D. from Boston University (USA), in the area of asymmetric organic synthesis. During post-doctoral work with Prof. Vincent M. Rotello at the University of Massachusetts at Amherst, USA, he worked in the area of renewable polymeric coatings. Currently, he is a professor in the Department of Chemistry at Bogazici University, Istanbul, Turkey. His research focuses on the design of novel polymeric materials such as coatings, hydrogels and nanofibers for biomedical applications. His awards include the young investigator award from TUBA (The Turkish Academy of Sciences) and from TUBITAK (The Scientific and Technological Research Council of Turkey). |
1. Introduction
Conjugation of bioactive agents ranging from small molecules such as drugs, imaging agents, and receptor-specific targeting groups, to large bio-macromolecules such as proteins, oligonucleotides, and growth factors onto polymeric platforms is actively pursued in many areas of biomedical sciences such as diagnostics, therapeutics, and tissue engineering.1 In all of these applications, the nature of the specific chemical linkages employed to conjugate the biologically relevant motif to the polymeric material plays a critical role in their eventual performance. Though seemingly trivial, the chemical attachment of biomolecules to polymeric materials has always been a challenge. The high efficiency of such conjugations under mild conditions, as well as their execution with high specificity, without the formation of by-products, is important. Enhanced steric hindrance posed by the large structure of polymers, as well as several biological molecules, makes such conjugations demanding. Furthermore, the multitude of several functional groups present on most biomolecules can lead to several undesirable reactions leading to eventual crosslinking or non-specific functionalization of these materials. For several decades, a lot of effort has been dedicated to developing versatile, effective, and easy to implement conjugation strategies. In recent years, click chemistry-based methodologies have expanded the conjugation tool-box and opened new avenues in the design, synthesis, and applications of functional polymeric materials.2–12 While efficient conjugation of (bio) molecules of therapeutic interest to polymeric materials can be a boon, robust conjugation to macromolecules may also compromise their performance. Thus conjugation of the therapeutic agent through a reversible linkage, which would release it from the polymeric component, preferably, at the site of action, would be highly desirable. While many reversible linkages such as hydrazones,13 acetals,14 carbamates,15,16 and Diels–Alder cycloadducts17 have been investigated to date, interest in the utilization of a disulfide-based linker has been ever increasing. The presence of an increased amount of glutathione, a thiol-containing tripeptide, in inter and intra-tumoral environments as an endogenous disulfide cleaving agent has propelled the incorporation of the disulfide group into therapeutically relevant polymer conjugates.18–21 Examples in subsequent sections will highlight that one of the most effective approaches to attain disulfide linkage based conjugation is mediated through the utilization of the pyridyl-disulfide (PDS) moiety.Another actively pursued area with emerging utilization of the disulfide linkage is fabrication of redox-responsive crosslinked polymeric materials.22,23 Crosslinked materials from nanogels, microgels to bulk hydrogels find widespread use in various applications related to diagnostics and therapy. In many of these applications, degradation of the crosslinked structure can be beneficial, e.g. promoting release of therapeutic agents when exposed to thiol-containing compounds, present either endogenously or exogenously. Facile fabrication of such disulfide linkage containing polymeric materials has been often undertaken by employing the thiol–disulfide exchange reaction. In particular, as discussed in sections below, the PDS based motif has been employed as the disulfide component due to its fast reaction with thiol-containing polymers or crosslinkers.
In this review, we aim to provide an overview of the vital role played by the thiol-reactive PDS group in the fabrication of various redox-responsive functional polymeric materials (Scheme 1). The range of polymeric materials where the PDS functional group has been employed spans from soluble polymers to crosslinked ones like nanoparticles, gels and surface coatings. This review highlights the synthetic approaches used for incorporating the PDS group, as well as discusses briefly the potential application of these PDS-containing polymeric materials. In particular, various approaches used over the past two decades are summarized. While earlier approaches involved introduction of the PDS-moiety through post-polymerization modifications, recent years have witnessed increased utilization of PDS-containing initiators, chain transfer agents (CTAs) and monomers.
 | ||
| Scheme 1 Schematic representation of common applications of the PDS functional group based exchange chemistry in fabrication and functionalization of polymeric materials. | ||
2. Thiol-based conjugations: the thiol–pyridyl disulfide exchange reaction
The thiol functional group has been exploited for decades in bioconjugation processes ranging from modification of proteins with dyes to immobilization of ligands and proteins on solid supports for chromatographic purposes. In recent years, interest in thiol-based modifications has intensified due to high efficiency of the thiol-based click reactions. The thiol functional group is known to undergo efficient reactions24 with a variety of functional groups such as electron-rich alkenes,25 electron-deficient alkynes,26,27para-fluoro benzenes,28,29 epoxides,30,31 thioesters32–34 and disulfides.35–38 In particular, the nucleophilic thiol–maleimide and radical thiol–ene reactions have been widely employed in fabrication and functionalization of polymeric materials.39–41 In particular, the nucleophilic Michael addition reaction between cysteine-containing proteins or peptides and highly electron-poor double bond containing maleimide functionalized polymers has been widely studied. While these thiol–ene reactions are quite efficient, other than the retro Michael-type reversal of phenylthio-maleimide adducts,42 these linkages are in general not easily reversible. While not so common in the polymer community until recently, another reaction that the thiol function group undergoes with high efficiency is the thiol–disulfide exchange reaction. Biologists and biochemists have long used the thiol–disulfide exchange reactions for cleavage of disulfide bridges in proteins using dithiothreitol (DTT) as a reducing agent,43 as well as for quantification of thiol groups using Ellman's assay.44 The thiol–disulfide exchange is a SN2-type nucleophilic substitution where a thiol-bearing molecule reacts with either a symmetric or mixed disulfide group to form a new thiol compound and a mixed disulfide (Scheme 2). Additionally, this exchange reaction is quite pragmatic since it can be generally implemented under mild conditions, at ambient temperature and physiological pH. The exchange reaction can take place reversibly in various solvents including water, and can proceed in a broad range of reaction temperatures and pH.45 Usually, thiol–disulfide exchange is promoted at high pH due to the significant formation of thiolate ions which enhances the active nucleophilic attacks to the disulfide bond. Additionally, the rate constant of the thiol-exchange reaction increases with increasing pKa of the thiol group due to the increased nucleophilicity.46 | ||
| Scheme 2 General representations of (a) thiol–disulfide, and (b) thiol–pyridyl disulfide exchange reactions. | ||
The PDS moiety has some characteristic attributes, which makes it a functional group of choice for the thiol-based exchange chemistry. In particular, the exchange reaction of the thiol group with a pyridyl disulfide functional group proceeds with very high efficiency. The reaction is driven to the right due to the formation of the more stable tautomeric pyridothione fragment from the pyridylthiol released upon exchange, which also precludes any thiol–disulfide exchange from the released thiol. Furthermore, the unique chemical structure of the PDS functional group i.e. the presence of a basic pyridine group ortho to the disulfide sulfur atoms efficiently facilitates the activation of disulfide groups in acidic medium through protonation of the pyridine nitrogen. Compared to other disulfide containing molecules, such as 5,5′-dithio-bis-(2-nitrobenzoic acid) (Ellman's reagent) which has applications in quantifying free thiol groups in a 7.8–10.4 pH range, the PDS functional group can react with free thiols under relatively low pH conditions (3.4–8.1) due to the favoured release of the 2-thiopyridone group.47 Of pragmatic importance, the efficiency of the thiol-PDS exchange can be quantitatively followed using UV-Vis spectroscopy by determining the amount of released 2-thiopyridone through its characteristic absorbance e.g. near 343 nm in Krebs–Ringer phosphate buffer (pH 7.2).48 The evolution of such an absorbance provides indirect evidence of the thiol-PDS exchange based functionalization, which is quite important for cases where it is either difficult or impossible to analyse the realization of reaction since the amounts of materials are too low for routine analytical techniques or in cases of functionalization on crosslinked materials and surfaces. The commercial availability of the commonly utilized starting material 2,2′-dithiodipyridine (2,2′-DTDP) has also facilitated the widespread adaptation of the PDS group for the fabrication of redox-responsive polymeric materials.
3. Incorporation of the pyridyl disulfide group into soluble polymers
In general, three different approaches have been employed for the synthesis of PDS group containing polymeric materials. These are categorized in the subsequent sections according to the chemical strategies used to introduce the PDS functionality. The widely employed methods are: (a) polymerizations with PDS-containing initiators and CTAs, (b) polymerization of PDS-containing monomers, and (c) post-polymerization modification with PDS-containing small molecules (Scheme 3). The sections below outline the synthetic details to highlight the facile accessibility of PDS group bearing polymeric materials, along with a brief description of their possible applications. | ||
| Scheme 3 General approaches for incorporating the PDS functional group into polymeric materials. | ||
3.1. Installation of the pyridyl disulfide group through post-polymerization modification
Modification of macromolecular constructs is challenging as a result of the encountered steric hindrance. Over the past decade, several studies demonstrate that employment of ‘click’ chemistry based transformations addresses such issues with post-polymerization functionalization. Efficient functionalization of polymeric materials can be achieved using chemical transformations that proceed with fast kinetics under mild conditions.49 Along similar lines, the high efficiency and fast kinetics of the PDS based thiol–disulfide exchange reaction make it an attractive method for achieving a disulfide based post-polymerization functionalization of polymers appended with the PDS group. The most commonly used method to prepare PDS-functionalized polymers is to modify the polymers with a PDS-containing small molecule. This method requires appropriate reactive moieties as terminal end groups or side chains of polymers. In many of the early examples, the PDS group was used as a common moiety in heterobifunctional polymers because of its selective and orthogonal reactivity with thiol groups in the presence of other reactive groups.50 With the advent of polymerization methods such as controlled living radical polymerization and ring opening polymerization, the incorporation of reactive functionalities into polymers to afford subsequent PDS attachment could be modularly achieved. The reaction conditions such as the type of polymer, temperature, pH, solvent and reaction stoichiometry between the polymer reacting moiety and PDS-containing molecule obviously have utmost importance to attain a high degree of functionalization. One of the widely used approaches for incorporating the PDS functional group onto polymers involves treatment of the thiol group on the polymer with 2,2′-DTDP (Table 1). Although this method proceeds with high efficiency by virtue of the facile nature of this particular thiol–disulfide exchange reaction, due to the facile oxidative coupling of the thiol group, the thiol-based precursors should be generated either in situ or just before the exchange reaction. As an alternative approach, the PDS group is incorporated into non-thiolated polymers through post-polymerization modification with a PDS-containing fragment (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Polymer | Reacting moiety | Strategy | Modification method | Application/significance | Ref. |
| Abbreviations: PLA: poly(lactic acid); PMMA: poly(methyl methacrylate); PHPMA: poly(hydroxypropylacrylamide); POEGA: poly(oligoethylene glycol acrylate); PS: polystyrene; PGA: poly(glutamic acid); PF-DBT-PEG: poly(1,4-dithienyl benzothiadiazole)-poly(ethylene oxide). | |||||
| PEG |

|
1 | Deprotection of the trityl group, followed by exchange with pyridylsulfenyl chloride | Synthesis of heterobifunctional PEG derivatives | 50 |
| PLA | 1 | Deprotection of the trityl group, followed by exchange with 2,2′-DTDP | Synthesis of side chain thiol reactive polyesters | 60 | |
| PEG |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Synthesis of heterobifunctional PEG derivatives | 51 |
| PEG |
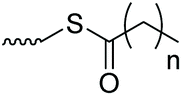
|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Synthesis of heterobifunctional PEG derivatives | 52 |
| PMMA, PHPMA, PNIPAAm, POEGA, PS |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Synthesis of end chain thiol reactive RAFT polymers | 54 |
| PHPMA | 1 | Aminolysis, followed by exchange with 2,2′-DTDP | Preparation of polymeric carbohydrate scaffolds for the attachment of biomolecules | 55 | |
| PNIPAAm |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Synthesis of thiol end chain reactive RAFT polymers | 56 |
| PEOz |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Preparation of diblock copolymers as nonviral gene carriers | 57 |
| PNIPAAm |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Preparation of thermally-responsive protein nanocapsules | 58 |
| PolyPOEGA Stars |

|
1 | Aminolysis, followed by exchange with 2,2′-DTDP | Synthesis of biodegradable star polymers with β-cyclodextrin complexes | 59 |
| PNIPAAm |

|
2 | Amidation by NHS functional PDS molecules | Preparation of thermo-responsive polymer–protein conjugates through disulfide bonding | 61 |
| PGA | 2 | Amidation by amine functional PDS molecule | Preparation of redox-responsive polymer–protein conjugates | 62 | |
| PF-DBT-PEG |

|
2 | Amidation by amine functional PDS molecules | Preparation of conducting polymers with solubility-induced fluorescence conversion for detection of thiols | 63 |
| Hyaluronic acid | 2 | Amidation by amine functional PDS molecules | Preparation of redox-responsive shell-crosslinked nanoparticles for tumor-targeted drug delivery | 64 | |
| PCl | 2 | Amidation by amine functional PDS molecules | Side chain functionalization of PCl with thiol containing peptides and resulting self-assembly | 67 | |

One of the very first examples of trapping of an in situ generated thiol group as a PDS group was reported by Kataoka and coworkers.51 In this study, acid sensitive initiator potassium 4-(diethoxymethyl)benzylalkoxide was used in ring-opening polymerization of ethylene oxide to synthesize a linear polyethylene glycol (PEG) polymer, followed by addition of methanesulfonyl chloride to terminate the polymerization. The methylsulfonyl end group was then converted to a dithiocarbonate group with addition of potassium O-ethyldithiocarbonate. Thereafter, the ethyldithiocarbonate end group was converted to the PDS group in a one pot procedure involving reduction to the thiol group by aminolysis with addition of n-propylamine, followed by treatment with 2,2′-DTDP. Based on 1H NMR analysis, 99% of ω-end conversion to the 2-pyridyldithio group was confirmed. As an alternative, aminolysis of thioesters also generates reactive thiol groups on the polymer chain. Thioester terminated polymers can be readily obtained by subjecting an alkene terminated polymer to the thiol–ene reaction with thioacetic acid. For example, Nagasaki and coworkers synthesized linear PEG from ethylene oxide using allyl alcohol as an initiator, and terminated it with succinic anhydride to obtain a carboxylic acid and alkene terminated hetero-telechelic polymer.52 To generate the free thiol group on the polymer, the double bond was first functionalized with thioacetic acid through the thiol–ene reaction under UV irradiation, and then the thioester group was converted to the PDS group in the presence of 2,2′-DTDP and n-propylamine to give pyridyl disulfide and acid terminated hetero-bifunctional linear PEG. These studies also demonstrated orthogonal reactivity of the PDS groups in the presence of other functional groups. More recently, Wandrey and coworkers reported synthetic pathways for obtaining azide/PDS and amine/PDS terminated linear PEGs through modification of commercially available hydroxyl group terminated linear PEG (Fig. 1a).53 In one approach, a PDS group was introduced to one end of the polymer chain by converting one of the hydroxyl groups to a tosylate leaving group, followed by its replacement with a thioester functional group. At this point, the other hydroxyl group was transformed to a mesylate leaving group. Aminolysis was used to convert the mesylate group to an amine moiety, while reducing the thioester group to a thiol functionality that was reacted with 2,2′-DTDP to install the PDS end group. Using a similar approach a hetero-telechelic PEG containing an azide and PDS group was also obtained.
 | ||
| Fig. 1 Examples of synthetic approaches to obtain PDS-containing polymers using 2,2′-DTDP: (a) from commercial PEG polymers,53 (b) from thioester terminated polymers obtained using RAFT polymerization,56 and (c) from polymers containing a protected-thiol group side chain.60 Adapted from Wandrey et al.53 with permission. Adapted from Davis et al.56 with permission. Copyright © 2009 by Wiley-VCH Verlag GmbH & Co. Adapted from Pappalardo et al.60 with permission. Copyright © 2016 by American Chemical Society. | ||
Over the past two decades, polymers synthesized through reversible addition fragmentation chain transfer (RAFT) polymerization have been widely used with PDS chemistry. As mentioned earlier, trithiocarbonate and thioester groups from RAFT chain transfer agents can be reduced to a free thiol group in the presence of primary or secondary amines, and the freshly generated thiol group can be converted to a PDS functionality in the presence of 2,2′-DTDP through the thiol–disulfide exchange reaction (Fig. 1b). Bulmus, Davis and coworkers have made several vital contributions in the field of post-modification of polymers with pyridyl disulfide.54,55 As one of the early examples, they reported introduction of a PDS end group on a polymer synthesized using RAFT polymerization.56 They synthesized poly(N-isopropylacrylamide) (PNIPAAm) using RAFT polymerization for subsequent conjugation with a biomolecule. 2-Aminoethanol was used to reduce the trithiocarbonate group on the RAFT agent to a thiol, which upon treatment with 2,2′-DTDP furnished a PDS group containing polymer, which was subsequently functionalized with a hexapeptide through the thiol-PDS exchange reaction. This approach of installation of the PDS end group was also utilized for obtaining diblock copolymers consisting of a disulfide group at the block junction. Hsiue and coworkers reported a diblock copolymer system by coupling of poly(2-ethyl-2-oxazoline) (PEOz) and linear polyethylenimine (LPEI) through the thiol exchange reaction.57 Briefly, thioacetate terminated PEOz was reacted with 2,2′-DTDP in the presence of ammonia to yield a PDS group terminated PEOz. This polymer is converted to LPEI in acidic solution at high temperature. Thereafter, thus obtained PDS-terminated LPEI was reacted with a thioacetate terminated PEOz using aminolysis to yield a disulfide junction containing diblock copolymer PEOz-b-LPEI. These diblocks were utilized to condense DNA into stable polyplexes. While the transfection ability of PEOz-b-LPEI was as high as LPEI, it exhibited lower toxicity toward cells, presumably due to lower cationic charge density. A similar approach was utilized by Maynard and coworkers for obtaining temperature-responsive protein cages using PNIPAAm synthesized via RAFT polymerization.58 PDS-terminated PNIPAAm was obtained through post-polymerization aminolysis followed by treatment with 2,2′-DTDP. Conjugation of PDS-terminated PNIPAAm with a thiol-containing protein cage yielded thermally-responsive protein nanocapsules. A similar strategy for installation of the PDS group at the chain end in the three-arm star polymer was used by Davis and coworkers.59 Apart from post-polymerization modification of end groups of polymers, side chains of polymers can also be modified to incorporate the PDS group. For example, Pappalardo and coworkers utilized a trityl (trt) protected thiol-containing lactide-based monomer, which they copolymerized with caprolactone (CL) and L-lactide to obtain degradable copolymers (Fig. 1c).60 Removal of the protecting groups using triethylsilane and trifluoroacetic acid generated free thiol groups, which upon treatment with 2,2′-DTDP installed PDS groups along the side chain.
Another widely used approach to introduce the PDS group involves treatment of polymers containing reactive functional groups e.g. activated esters with the PDS group bearing small molecules containing complementary reactive groups such as amines (Table 1).61–65 One of the earliest examples of such modification was reported by Caldwell and coworkers.66 Activated carbonate group terminated poloxamer coated surfaces were modified with 2-(2-pyridyldithio)ethylamine to yield a PDS containing polymeric surface. It was shown that binding efficiency of β-galactosidase was significantly higher on the PDS group modified surface compared to the non-modified counterpart. In a recent study, they reported a post-polymerization modification of the NHS-activated ester side chain bearing polycaprolactone (PCL) copolymer with 2-(2-pyridyldithio)ethylamine.67 The pendant PDS groups were functionalized with GSH and L-carnosine-SH through the thiol-PDS exchange reaction to facilitate self-assembly of peptide functionalized PCL to form vesicles or micelles depending on the pH.
To collectively demonstrate the preferred post-polymerization modification strategies, Table 1 summarizes a list of studies that utilize either end-chain or side chain functional polymers for installation of the PDS group.
As shown by the abovementioned examples, a variety of conjugation methods are available for introducing the PDS functional group onto polymers as terminal or side chain groups. While such approaches are simple and may be pragmatic for modification of polymers, earlier it was mostly used for modification of commercially available polymers. As with any post-polymerization modification, the extent and efficiency of the process will also depend on the structure of the macromolecule. In order to gain access to functional polymers with precise positioning, as well as control over the amount of PDS groups, direct incorporation through utilization of PDS-containing initiators or monomers has emerged as a widely used approach. Utilization of appropriately designed initiators or monomers, coupled with their implementation in different types of polymerizations, allows access to a large variety of compositionally different polymers. Subsequent sections focus on the synthesis of polymers where this niche thiol-reactive functional group is incorporated directly during polymerization and highlight examples that exploit the PDS mediated thiol–disulfide exchange reaction for obtaining functional materials.
3.2. Utilization of PDS-functionalized initiators and chain transfer agents
Instead of incorporating the reactive functional group of interest through post-polymerization modification steps, one can introduce the reactive group directly into the polymeric structure by employing initiators, CTAs or monomers bearing the reactive group. In recent years, polymers bearing the PDS group at their chain-end have been increasingly synthesized by employing appropriate PDS functional group containing initiators or CTAs using a variety of polymerization techniques such as atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer (RAFT) polymerization and ring opening polymerization (ROP).Over the past several years, Maynard and coworkers have utilized the PDS-thiol exchange chemistry to design several polymer–biomolecule conjugates.68,69 In one of their seminal contributions, a PDS-containing ATRP initiator was prepared by reaction of a PDS-containing alcohol with 2-bromo-2-methylpropionic acid through N,N‘-dicyclohexylcarbodiimide (DCC) coupling to yield a PDS group bearing ATRP initiator (Fig. 2a).69 A 2-hydroxyethyl methacrylate (HEMA) based hydrophilic polymer was synthesized, and utilized for conjugation with bovine serum albumin (BSA) through a thiol-PDS exchange reaction. Few years later, the same group polymerized a glycomonomer using the same PDS-containing initiator for conjugation of biomolecules and patterning on gold surfaces. Obtained glycopolymers were used for conjugation of small interfering RNA (siRNA) through the disulfide exchange reaction.70
 | ||
| Fig. 2 (a) Synthesis of pHEMA with PDS containing the ATRP initiator,69 and (b) synthesis of reversible siRNA–polymer conjugates72 mediated by thiol–pyridyl disulfide exchange reactions. Adapted from Maynard et al.69 with permission. Copyright © 2018 by American Chemical Society. Adapted from Bulmus, Maynard et al.72 with permission. Copyright © 2008 by Royal Society of Chemistry. | ||
Due to concerns with residual amounts of metal salts that pose challenges in their complete removal and thus may possibly interfere with the activity of biomolecules or pose cellular toxicity, metal-free polymerization techniques such as RAFT have gained popularity.71,73 In the following years, several PDS group containing CTAs for utilization in RAFT polymerization were synthesized through esterification or amidation reaction between a RAFT CTA and with an amine or alcohol group bearing PDS moiety containing molecules.74 A great deal of contributions with several innovative examples of PDS containing RAFT agents have been reported by Davis and coworkers. Using a ‘graft from’ approach Davis, Bulmus and coworkers reported a clever approach to obtain a hetero-biofunctional polymer–protein conjugate.75 A bifunctional RAFT agent containing two PDS groups was reacted with BSA in a site specific manner to obtain a BSA-macro RAFT agent. Polymerization of oligo(ethylene glycol) acrylate (OEGA) afforded polymers containing a BSA–polymer conjugate with a PDS group at the polymer chain end, which could be modified using thiol-containing cholesterol or fluorescent dye. In an early study, in 2007, Bulmus, Davis and coworkers reported RAFT polymerization mediated synthesis of a block copolymer of PEG acrylate and butyl acrylate (BA) using a PDS group containing CTA. As a demonstration of end-group modification, the obtained polymer was treated with 11-mercapto-1-undecanol, and the high efficiency (ca. 90%) of the thiol–disulfide exchange reaction was confirmed from the release of pyridine-2-thione, as monitored by evolution of its characteristic UV-Vis absorption.76 Subsequently, the potential application of similar PDS-containing polymers for reversible conjugation of small interfering RNA (siRNA) in an efficient manner was reported by Bulmus, Maynard and coworkers (Fig. 2b).72 A PDS-containing RAFT agent was utilized to obtain poly(PEG acrylate) based hydrophilic polymers. A 5′-thiol modified siRNA fragment was efficiently conjugated (ca. 88.3 ± 6.5%) to the chain end of polymers using the thiol–disulfide exchange reaction. In a subsequent study, it was demonstrated that conjugation to the polymer significantly enhanced in vitro serum and nuclease stability, compared to the free siRNA.77 As another example of biological applications of the PDS-based polymers, Stayton and coworkers reported the synthesis of a proapoptotic peptide polymer conjugate where the bioactive peptide was linked to an endosomolytic polymer through a disulfide bond.78 A PDS-containing CTA was obtained through coupling of an aminoethyl pyridyl disulfide with an N-hydroxysuccinimide-activated RAFT agent. A copolymer composed of a hydrophilic N-(2-hydroxypropyl) methacrylamide based first block, and a second pH-responsive and ampholytic block composed of dimethylaminoethyl methacrylate, propylacrylic acid and butyl methacrylate (BMA) monomers was synthesized using the PDS group containing RAFT agent. The obtained diblock copolymer was functionalized with a therapeutic peptide through the thiol-PDS exchange reaction to yield a peptide–polymer conjugate that demonstrated significantly higher apoptotic activity against HeLa cervical carcinoma cells, when compared to the free peptide (Fig. 3).
 | ||
| Fig. 3 Synthesis of the pH-responsive diblock-copolymer using the PDS-bearing RAFT initiator for peptide conjugation.78 Adapted from Stayton et al.78 with permission. Copyright © 2009 by American Chemical Society. | ||
An early study that demonstrates an interesting application of polymers containing a PDS group at their chain end for protein purification was reported by the Maynard group.79 A strategy for purification of glutathione S-transferase (GST) protein with a glutathione (GSH) conjugated thermoresponsive PNIPAAm polymer was developed. Briefly, a PNIPAAm polymer synthesized using a PDS group bearing RAFT agent was modified with the GSH peptide through the thiol–disulfide exchange reaction. This GSH-containing thermoresponsive peptide–polymer conjugate binds selectively to the GST protein. The polymer–GST conjugates were isolated by precipitation upon heating the polymer above its lower critical solution temperature (LCST) of 37 °C. Thus precipitated protein–polymer conjugates were separated using rapid high speed centrifugation. Recently, the Maynard, Terashima and coworkers have extended the approach to obtain lysozyme conjugates with amphiphilic/fluorous polymers that form protein encapsulated aggregates in aqueous media.80
It is well known that conjugation of polymers to bioactive molecules like proteins enhances their stability.81 Such conjugations also allow recyclability, as recently demonstrated by De, Datta and coworkers.82 The authors prepared a thermoresponsive PNIPAAm conjugate of thermophilic β-glucosidase (B8CYA8) which showed high enzyme activity, thermal stability, long half-life and reusability. A β-glucosidase-macro CTA obtained using the thiol-PDS exchange reaction between a cysteine residue on the enzyme and PDS-containing RAFT CTA was used for growing the PNIPAAm chain using the ‘grafting from’ approach (Fig. 4a). Another interesting contribution involving PDS-containing polymers was reported by Maynard and coworkers who prepared a protein–polymer conjugate using the ‘graft to’ approach.83 While trehalose has been long used as a stabilizing excipient to retain protein activity upon lyophilization, a creative demonstration of using a trehalose-based protein–polymer conjugate was reported. The trehalose-containing monomer based glycopolymers containing the activated disulfide group at their chain end were obtained using RAFT polymerization by employing PDS-containing CTAs (Fig. 4b). Conjugation of obtained polymers to thiol-modified hen egg lysozyme imparted them with significantly increased stability when subjected to lyophilization or heat treatment, compared to the unmodified protein.
 | ||
| Fig. 4 (a) ‘Graft-from’ strategy for the synthesis of the thermoresponsive enzyme-polymer conjugate,82 and (b) ‘graft-to’ strategy for the synthesis of the trehalose based protein–polymer conjugate for protein stabilization.83 Adapted from De, Datta et al.82 with permission. Copyright © 2018 by American Chemical Society. Adapted from Maynard et al.83 with permission. Copyright © 2012 by American Chemical Society. | ||
Another important study that highlights the prominent role of the thiol-PDS exchange based conjugation in accessing protein–polymer conjugates was reported by Hoogenboom, De Geest and coworkers.84 In this extensive study, authors compared the efficiency and suitability of polymers containing different reactive groups at the chain end, including the PDS group, to obtain protein–polymer conjugates using the ‘graft-to’ approach. N-Hydroxysuccinimide (NHS), pentafluorophenyl (PFP), PDS and maleimide (MAL) modified RAFT agents were prepared, and utilized to obtain hydrophilic pHEMA polymers. Obtained results suggested that the PDS group acts as one of the most efficient groups that enables efficient thiol–disulfide exchange based coupling of the investigated polymers with proteins like BSA and ovalbumin (OVA).
The Maynard group has also demonstrated that polymers containing the PDS group at the chain end can be conjugated to vault nanoparticles, which are natural non-immunogenic ribonucleoprotein particles present in most eukaryotes.85 Authors fabricated protein nanoparticles responsive to both temperature and pH by using polymers composed of NIPAAm and acrylic acid (AA) monomers. Thiol-reactive polymers were conjugated with the human major vault protein through the thiol exchange reaction with thiol groups on the surface of the vault protein nanoparticles. In this design the temperature responsiveness of nanoparticles was dependent on pH of the media. While there was no response below 60 °C at pH 7.0, nanoparticles started to respond reversibly to the temperature below 60 °C at pH 6 and started to aggregate.
Hetero-telechelic polymers containing the PDS group at the chain end have been reported by employing appropriately designed initiators or chain transfer agents. Bulmus, Davis and coworkers reported an elegant strategy for direct access of α,ω-hetero-telechelic copolymers bearing an azide and a PDS group at chain ends.86 A trithiocarbonate RAFT CTA containing an azide and a PDS group was used for polymerization of various monomers such as styrene, NIPAAm, HPMA, OEGA and methyl methacrylate (MMA). Efficient hetero-bifunctionalization was demonstrated with the α-azide, ω-PDS containing PNIPAAm polymer through functionalization with an alkyne-containing biotin ligand and thiol-containing tripeptide GSH, among other biomolecules.
In recent years, a set of elegant reports by Ghosh and coworkers have demonstrated that the versatile chemistry of the thiol-PDS exchange reaction can be extended to functionalization of biodegradable polymers. To obtain PDS end group containing polyesters, hydroxyethyl pyridyl disulfide was used to initiate ROP of lactide.87 Obtained polymers could be easily modified with thiol-containing bioactive ligands like carbohydrates, and chromophores like naphthalene-diimide. Apart from using the thiol–disulfide exchange based conjugations, the free thiol groups generated by addition of DTT to the PDS terminated polylactide allow subsequent functionalization using radical and nucleophilic thiol–ene conjugations (Fig. 5).
 | ||
| Fig. 5 Synthesis of thiol-reactive PDS-containing biodegradable polymers.87 Adapted from Ghosh et al.87 with permission. Copyright © 2012 by American Chemical Society. | ||
While most of the reports employ PDS-group containing initiators to install this thiol-reactive group at the chain end of the linear polymers, the use of appropriately designed initiators can yield multiarm polymers with the thiol-reactive group at their core. As one of the examples, Davis and coworkers reported a branched PHPMA polymer containing the PDS group at the mid-chain location.88 A PDS group containing CTA bearing two thiocarbonate moieties was synthesized to obtain polymers using RAFT polymerization. The thiol exchangeable disulfide group was thus located at the midpoint of the obtained polymers. Successful conjugation of the obtained polymer with the thiol-bearing protein BSA was demonstrated.
Another example of installing the PDS group at the core of multiarm polymers was reported by Sanyal and coworkers.89 A tetra-arm dendron based initiator was designed to contain a PDS group at the focal point and radical initiators at its periphery. The PDS-modified core allows for redox-responsive conjugation of thiol containing small molecules or peptides to obtain multiarm star polymers. The hydroxyl groups at the periphery of dendrons were converted to bromine-containing ATRP initiators to grow PEGMA-based multiarm star polymers. Water soluble multi-arm star polymers bearing the PDS at the core were efficiently modified with thiol-containing dyes and bioactive ligands. In particular, a thiol bearing KLAK-sequence containing apoptosis inducing peptides was conjugated and enhanced cytotoxicity of the polymer–peptide conjugate compared to the naked peptide was demonstrated. Instead of incorporation of the PDS group at the core of the multi-arm star polymers, recent work by Kent, Davis and coworkers shows that it is also possible to fabricate star copolymers where the PDS groups are located at the chain termini of the arms emanating from a crosslinked core.90 First, polymers containing PDS groups at chain ends were synthesized using RAFT polymerization. The hetero-telechelic arms were core-crosslinked in the presence of an amine-reactive monomer (to subsequently attach a fluorescent dye) and a bis-acrylamide crosslinker. Enhanced association with particular subsets of white blood cells was observed for the thiol-reactive nanoparticles. The authors postulated that such thiol-reactive PDS-containing nanoparticles may provide a viable strategy for targeting subsets of primary human immune cells.
While a lot of focus has been given toward utilization of the PDS group containing polymers at the chain end for the fabrication of polymer-biomolecule conjugation, this particular thiol–disulfide exchange reaction has been also demonstrated as a powerful methodology to furnish redox-responsive diblock and multiblock copolymers. Thayumanavan and coworkers exploited the high efficiency of this coupling to obtain cleavable diblock copolymers (Fig. 6).91
 | ||
| Fig. 6 Synthesis of a redox-responsive diblock copolymer obtained using thiol–disulfide mediated polymer–polymer coupling.91 Adapted from Thayumanavan et al.91 Copyright © 2007 by American Chemical Society. | ||
Polymers with the PDS group located at one of the chain ends were synthesized using ATRP. The PDS end group in these polymers could be reduced to yield thiol-terminated polymers. Coupling of polymers bearing the PDS and thiol groups proceeded under mild conditions in an efficient manner to provide diblock copolymers with a redox-responsive linkage at the junction point. Thayumanavan and coworker utilized this strategy in a subsequent study to obtain block copolymer assemblies that were sensitive to changes in three different stimuli: temperature, pH and redox potential sensitive.92 The amphiphilic diblock copolymer with a redox-sensitive disulfide junction was obtained through combination of the thiol-end group containing tetrahydropyran-protected HEMA polymer as the acid-sensitive hydrophobic block and the PDS end group containing PNIPAAm polymer as the thermo-sensitive hydrophilic block. Authors note that such multi-responsive systems not only allow fine-tuning of release kinetics but also would provide delivery of therapeutics in a location-specific manner.
In an elegant study, Gibson and coworkers employed the reversible nature of the disulfide linkage to modulate the solubility of hydrophilic polymers. They demonstrated that the LCST of the polymer bearing the PDS group at the chain end changes upon treatment with glutathione. In this study, PEGMA and NIPAM were polymerized using a PDS-bearing RAFT agent.93 Interestingly, it was observed that in reducing environments, the LCST of these polymers changes because of the exchange of the 2-mercaptopyridine end group with a thiol group. Thus a coil-to-globule transition could be attained for the polymer under ‘isothermal conditions’ i.e. without changing the temperature. Using these types of polymers, another interesting approach to redox-responsive polymers was reported by Gibson and coworkers. They polymerized a series of acrylamides, methacrylates and acrylates with a PDS-bearing RAFT agent.94,95
Aminolysis of the sulphur containing functional group of the RAFT CTA fragment at the chain end of these polymers induced a poly(condensation) reaction (Fig. 7). The thiol group containing polymers reacted in situ with each other through thiol-PDS exchange reaction to result in step-growth polymerization. Using this approach, dual responsive polymers consisting of thermoresponsive PNIPAAm based domains connected to each other through redox-responsive disulfide bonds formed during polycondensation were obtained.
 | ||
| Fig. 7 Synthesis of redox-responsive thermoresponsive polymers through in situ polycondensation.95 Adapted from Gibson et al.95 with permission. Copyright © 2012 by Royal Society of Chemistry. | ||
A variety of examples that utilize appropriately designed PDS containing initiators and chain transfer agents in preparation of functional polymeric materials are summarized in Table 2.
| Monomer | Initiator (ATRP and ROP)/chain transfer agent (RAFT) | Polymerization method | Application/significance | Ref. |
|---|---|---|---|---|
| Abbreviations: NHSMA: N-hydroxysuccinimide methacrylate; PEGA: poly(ethylene glycol) acrylate, HEA: 2-hydroxyethyl acrylate; HPMA: N-(2-hydroxypropyl) methacrylamide; DMAEMA: 2-dimethylaminoethyl methacrylate; DEGMA: diethylene glycol methacrylate; OEGMA: oligoethylene glycol methacrylate; DMA: N,N-dimethyl acrylamide; tBuA: tert-butyl acrylate; DEGMEMA: diethylene glycol methyl ether methacrylate. | ||||
| NIPAAm |

|
ATRP | Modification of reduced BSA for polymer grafting | 68 |
| HEMA | Cysteine-reactive polymers for conjugation to BSA | 69 | ||
| Glycomonomer | Reactive polymers for conjugation to siRNA and patterning on gold surface | 70 | ||
| NHSMA, MMA | Synthesis of disulfide bridged block copolymers | 91 | ||
| NIPAAm, HEMA | Synthesis of multi stimuli-responsive block copolymers | 92 | ||
| PEGA, BA |

|
RAFT | Synthesis of end group pyridyl disulfide-functionalized homo- and amphiphilic block copolymers | 76 |
| NIPAAm, PEGA |

|
RAFT | Mechanistic study to elucidate the stability of PDS group under radical polymerization conditions | 74 |
| NIPAAm, HEA | RAFT | Modification of reduced BSA for polymer grafting | 96 | |
| PEGA |
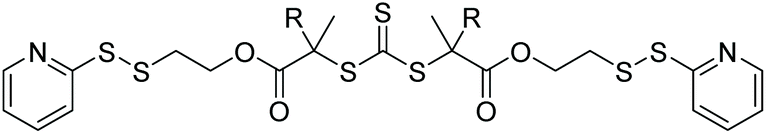
|
RAFT | Synthesis of heterotelechelic protein–polymer–biomolecule conjugates | 75 |
| HPMA, AA, BMA, DMAEMA |
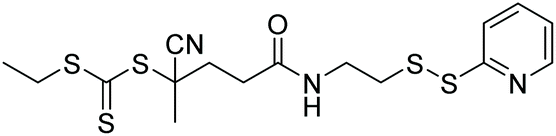
|
RAFT | Preparation of peptide block copolymer conjugates | 78 |
| Glycomonomer |

|
RAFT | Synthesis of protein–trehalose glycopolymer conjugates | 83 |
| PEGA | Synthesis of siRNA–polymer conjugates | 72 | ||
| NIPAAm | Synthesis of glutathione–polymer conjugates for glutathione; S-transferase protein purification | 79 | ||
| PEGA | Synthesis of siRNA–polymer conjugates | 77 | ||
| NIPAAm, DEGMA, OEGMA | Synthesis of chain termini thiol reactive polymers displays glutathione triggered LCST change | 93 | ||
| PEGMA, fluoromonomer | Synthesis of fluorous copolymers for protein bioconjugation | 80 | ||
| OEGMA | Synthesis of thiol-reactive star polymers for biocompatible polymeric nanoparticle fabrication | 90 | ||
| HEA, HPMA | Synthesis of different end group reactive polymers and comparative study to assign their protein conjugation efficiencies | 84 | ||
| NIPAAm, AA | Preparation of pH- and temperature-responsive vault protein nanoparticles | 85 | ||
| NIPAM, DMA, OEGMA, MMA, DEGMA, OEGA, tBuA | Synthesis of biodegradable poly(disulfide)s | 94 | ||
| NIPAM | Synthesis of recyclable thermoresponsive polymer-β-glucosidase conjugate | 82 | ||
| NIPAAm |

|
RAFT | Synthesis of thermoresponsive polymers displaying redox-responsive LCST behaviour | 95 |
| Lactide |

|
ROP | Synthesis of end group functional polylactide | 87 |
| HPMA |

|
RAFT | Synthesis of branched polymer–protein conjugates | 88 |
| DEGMEMA |

|
ATRP | Synthesis of dendritic multi-arm polymers with a thiol-reactive PDS core | 89 |
| HPMA, MMA, sytrene, OEGA, NIPAAm |
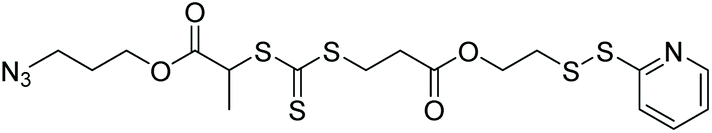
|
RAFT | Synthesis of heterobifunctional polymers | 86 |
The above examples demonstrate that a wide variety of semi-telechelic or hetero-telechelic polymers bearing the PDS group can be obtained through clever design of initiators and CTAs. While most examples above demonstrate the usage of the PDS group bearing polymer for obtaining a polymer–biomolecule conjugate using the ‘graft-to’ approach, the PDS group has also been employed to facilitate the synthesis of polymer–biomolecule conjugates using the ‘graft-from’ approach. For example, Davis, Bulmus and coworkers utilized a PDS group bearing short PEG based water-soluble RAFT agent for undertaking polymerization of NIPAAm from BSA.96 The PDS group containing a hydrophilic RAFT agent was conjugated to the protein using the thiol-PDS exchange reaction with the available free cysteines on BSA. The obtained BSA-RAFT agent was used to grow PNIPAAm, which rendered the protein with ability to undergo thermo-responsive aggregation. Likewise, in another study, Maynard and coworkers demonstrated that the ‘grafting from’ strategy can be successfully used to obtain polymer–siRNA conjugates, where the siRNA conjugation with an ATRP initiator was mediated through exchange between the thiol group on the oligonucleotide and a PDS group bearing the ATRP initiator.97 Overall, the wide range of applications of the PDS end group containing polymers as discussed above highlights the versatile nature of this reactive group.
3.3. Installation through utilization of PDS-functionalized monomers
The thiol-reactive PDS functional group can be directly incorporated into polymers as side chains using PDS-containing monomers. In recent years, polymers containing multiple PDS groups as chain units have attracted attention since they allow conjugation of several therapeutically active molecules which can be subsequently cleaved under reducing conditions, and thus provide a selective pathway for release.98 Also, as mentioned above, due to the orthogonal reactivity of the PDS group to many other reactive monomers, copolymers bearing the PDS group along with other reactive groups can undergo multi-functionalization in a selective manner.99 In this section, we review approaches taken to incorporate the PDS functional group as side chain moieties into different classes of polymers, obtained by using PDS-containing monomeric building blocks.An early example of PDS-containing monomer was reported by Stayton, Hoffman and coworkers.100 A novel pyridyl disulfide acrylate (PDSA) monomer was synthesized through reaction between hydroxypropyl pyridyl disulfide and acryloyl chloride in the presence of pyridine. Later, this monomer was polymerized with methacrylic acid and butyl acrylate using AIBN-initiated free-radical polymerization to yield a redox- and pH-sensitive amphiphilic copolymer. Peptides containing cysteine residues were conjugated to this terpolymer using the thiol–disulfide exchange reaction. Antisense oligonucleotides are ionically complexed with hexalysine conjugated polymer and cellular uptake of the thus-obtained GSH sensitive complex was investigated. Shortly after, Thayumanavan and coworkers reported the PDS group bearing methacrylic monomer (PDSMA), which was polymerized using ATRP to yield homopolymers and copolymers, the latter upon polymerization in the presence of N-hydroxysuccinimide methacrylate.101 The PDSMA monomer was obtained in two steps: 2,2′-DTDP was reacted with 2-mercaptoethanol to yield a PDS group bearing alcohol, which was subsequently treated with methacryloyl chloride in the presence of a base. The orthogonally reactive copolymers could be functionalized in a quantitative manner using thiol and amine containing molecules either in a one-step or sequential reaction. Such orthogonal functionalization provides an attractive method to obtain multi-functional systems given the presence of amine and cysteine groups in various peptides and proteins. The versatility of the PDS group containing polymers was also demonstrated by Bulmus and coworkers, who synthesized polymers using RAFT polymerization of the PDS containing methacrylate monomer.102 They showed that thiol-reactive homopolymers could undergo simultaneous multi-functionalization in a highly efficient manner upon treatment with mixtures of thiol-containing molecules. They also observed that the PDS-containing hydrophobic homopolymers upon treatment with a hydrophilic peptide like GSH yield amphiphilic copolymers that assemble into spherical aggregates in aqueous media, as suggested through dynamic light scattering (DLS) and atomic force microscopy (AFM) studies. The same group exploited the PDS group containing homopolymers obtained using RAFT polymerization as a macroinitiator to obtain diblock copolymers for fabrication of crosslinked micellar drug delivery systems,103 as discussed in the subsequent section. Along with these contributions, synthesis of other multiblock copolymers using the PDSMA monomer has also been reported.104 As expected, amphiphilic multi-block copolymers assemble to provide micellar structures that can act as delivery vehicles for therapeutic agents. Stayton and coworkers designed a diblock copolymer with HPMA and PDSMA containing hydrophilic blocks and pH-responsive PAA, DMAEMA and BMA containing hydrophobic blocks.105 The obtained pH-responsive polymeric micellar carrier with PDS side chains was used to conjugate ovalbumin through the thiol-PDS exchange reaction (Fig. 8). Compared to free ovalbumin, the micelle–ovalbumin conjugate showed higher antigen cross-penetration during in vitro experiments. Moreover, 90% of micelle–ovalbumin conjugates were internalized in cells after 1.5 h, compared to 40% internalization of free albumin.
 | ||
| Fig. 8 Antigen conjugation on PDS-bearing pH-responsive polymers.105 Adapted from Stayton et al.105 with permission. Copyright © 2014 by Elsevier. | ||
The PDS moiety has been also integrated as side chains in biodegradable constructs, as reported by Zhong and coworkers.106 In their study, a PDS group containing a cyclic carbonate monomer and poly(ε-caprolactone) were copolymerized using ROP. Thereafter, the PDS groups were exchanged with PEG-thiol to install hydrophilic side chains. DOX-loaded micelles were fabricated using this amphiphilic copolymer and redox-triggered release was demonstrated. As expected, DOX release was enhanced in the presence of DTT (10 mM), in contrast to release in a non-reductive environment. Another interesting approach to incorporate the PDS group as side chains into poly(β-amino ester)s was reported by Langer and coworkers. Poly(β-amino ester)s can be readily synthesized by the reaction of primary amines and diacrylates. In this study, 2-(pyridyldithio)-ethylamine was used as an amine-bearing monomer and polymerized with a series of diacrylates to yield poly(β-amino ester)s bearing PDS based side chains. Obtained polymers were functionalized with thiol-containing peptide RGDC and mercaptoethylamine through the thiol–disulfide exchange reaction. Thus obtained polymers showed DNA-binding ability, which decreases upon fragmentation when exposed to intercellular glutathione concentration, and thus can be anticipated to facilitate release of DNA inside the cell.107
In an interesting approach, the thiol–disulfide exchange reaction was used in an elegant manner by Ghosh and coworkers to obtain telechelic poly(disulfide) polymers bearing the PDS group at chain termini.108 The polycondensation reaction of aldrithiol and 1,6-hexanedithiol provides access to polymers with disulfide groups along the backbone and PDS group at chain ends. Importantly, the selective thiol-PDS exchange reaction allows modification of the chain ends to install desired groups. For example, hydroxyl groups were obtained at chain ends by treatment with 2-mercaptoethanol, followed by ROP of cyclic lactide to yield triblock copolymers. In another study, the authors demonstrated that the end groups could be capped with a variety of functional groups using PDS-containing capping reagents during the polymerization step.109 In a subsequent study, the authors utilized this strategy to obtain a redox-responsive micellar construct with tunable degradation.110 The triblock copolymers composed of a middle block possessing different hydrophobicity (attained by using 1,6-hexanedithiol or 2,2′-(ethylenedioxy)diethane thiol), and outer blocks based on poly(triethylene glycol monomethyl ether) methacrylate (PTEGMA) were used (Fig. 9). The outer blocks were grown using ATRP, after installation of the required bromine initiator on the telechelic hydroxyl group containing polymer obtained using their previous protocol. Triblock copolymers thus obtained were assembled into micelles in aqueous media and were loaded with a hydrophobic dye, namely, Nile Red. It was demonstrated that the overall hydrophobic character of the inner block has a noteworthy effect on release of the cargo. An increased hydrophobicity of the micelle interior leads to slower disintegration and release of cargo in the presence of GSH, presumably due to the slow diffusion of the reducing agent into the non-polar interior. In a recent study, authors extended the thiol-PDS exchange based condensation polymerization to the synthesis of hyperbranched polydisulfides by employing an A2 + B3 type condensation approach of complementary monomers.111 In another related study, PDS incorporation via termination of polydisulfide synthesis via 2,2′-DTDP was later employed by Lu and coworkers to obtain redox-responsive amphiphilic triblock mPEG-b-PDS-b-mPEG copolymers.112
 | ||
| Fig. 9 Synthesis of poly(disulfide) polymers using the PDS-thiol exchange reaction.110 Adapted from Ghosh et al.110 with permission. Copyright © 2020 by Royal Society of Chemistry. | ||
Another elegant and mechanistically interesting approach to synthesize redox-degradable polyamides was recently reported by Thayumanavan and coworkers by employing cascaded step-growth polymerization methodology based on thiolactone ring opening and disulfide–thiol exchange reactions. A set of backbone disulfide containing polymers with variable side chain groups were obtained by polymerization of an amine containing PDS monomer along with thiolactone monomers carrying diverse functionalities.113
An overview of the preparation and applications of a variety of PDS-containing polymeric materials obtained using PDS functional monomers is shown in Table 3.
| Comonomer | PDS monomer | Polymerization method | Application/significance | Ref. |
|---|---|---|---|---|
| Abbreviations: OEMA: oligoethyl acrylate; AzEMAm: 2-azidoethylmethyacrylamide; TrMA: trehalose methacrylate; VP: N-vinylpyrrolidone; VPI: N-vinylpiperidone; VCL: N-vinylcaprolactam; EAMFB: N-ethylacrylamide-2-(4-formylbenzamide); TBCPAM: N-(tert-butoxycarbonyl)-propylaminoacrylamide; MAP: Michael addition polymerization. | ||||
| — |

|
FRP | Synthesis of pH and redox responsive nanoparticles for drug delivery | 98 |
| AA, BA | FRP | Synthesis of pH and glutathione responsive polymers | 100 | |
| OEMA, PEGA | RAFT | Preparation of graphene/copolymer composites for pH and biodegradation controlled drug delivery | 104 | |
| HPMA, DMAEMA, AzEMAm |

|
FRP | Preparation of peptide-functionalized mRNA polyplexes | 99 |
| HPMA, AA, DMAEMA, BMA | RAFT | Synthesis of pH-responsive polymer micelle carriers | 105 | |
| NHSMA |

|
ATRP | Synthesis of orthogonally functionalizable copolymers | 101 |
| — | RAFT | Synthesis of side chain thiol reactive polymers | 102 | |
| HPMA | RAFT | Preparation of doxorubicin conjugated acid- and redox sensitive crosslinked micelles | 103 | |
| OEGMA | RAFT | Synthesis of surface-functionalizable polymer nanogels | 122 | |
| OEGMA | RAFT | Synthesis of polymer nanogels for concurrent binding and delivery of proteins and lipophilic small molecules | 123 | |
| OEGMA | RAFT | Synthesis of self-crosslinked polymer nanogels for drug delivery | 124 | |
| OEGMA | RAFT | Synthesis of self-crosslinked polymer nanogels | 125 | |
| OEGMA | RAFT | Synthesis of polymer nanogels for intracellular drug delivery | 126 | |
| OEGMA, DEGMA | RAFT | Synthesis of enzyme-polymer hybrid nanogels | 127 | |
| TrMA | FRP | Synthesis of trehalose glycopolymer nanogels for glucagon stabilization | 128 | |
| HPMA | RAFT | Fabrication of gelatin based dynamic hydrogels | 134 | |
| PEGMA, DEGMA | FRP | Fabrication of bulk and patterned hydrogels for reversible protein capture and cell attachment | 137 | |
| VP, VPI, VCL | RAFT | Synthesis of N-vinyl lactam based reactive copolymers | 138 | |
| Diacrylates |

|
MAP | Synthesis of cationic polymers with thiol-reactive side chains for DNA delivery | 107 |
| Thiolactones |

|
ROP/disulfide exchange | Synthesis of polyamides with backbone disulfide units | 113 |
| EAMFB, TBCPAM, DMA |
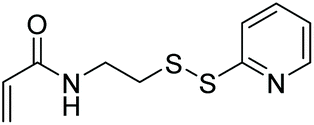
|
RAFT | Synthesis of pH and redox responsive polymer nanoparticles | 114 |
| CBMA |
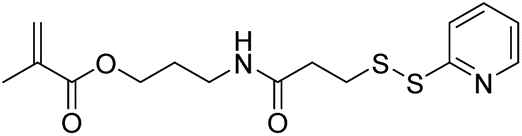
|
RAFT | Fabrication of cell viable degradable hydrogels | 135 |
4. Pyridyl disulfide chemistry in fabrication and functionalization of crosslinked materials: polymer nanoparticles, nanogels and hydrogels
Apart from obtaining redox-responsive attachment of therapeutic agents to soluble polymers, the thiol-PDS exchange reaction has also been exploited for reversible crosslinking or attachment i.e. for fabrication and/or functionalization of crosslinked materials such as micellar nanoparticles, nanogels, hydrogels, and surface coatings. In particular, the incorporation of redox-sensitive crosslinks into bulk or particle based drug delivery platforms is quite important since it enhances the release of the therapeutic agent under enhanced GSH environments specific to cancerous tumors and cells. The breakdown of the crosslinked material enhances release of encapsulated therapeutic agents, as compared to release pathways such as diffusion or erosion of the matrix. In this part of the review, we highlight different types of chemically crosslinked materials that have been obtained using the PDS group mediated thiol–disulfide exchange reactions.4.1. PDS-functional group-based fabrication and functionalization of polymer nanoparticles
One of the earliest examples of a redox-sensitive crosslinked micellar nanoparticle system was reported by Bulmus and coworkers.103 An amphiphilic diblock copolymer was synthesized using RAFT polymerization of HPMA and PDS-containing methacrylate monomers. Treatment of this diblock copolymer with tris(2-carboxyethyl)phosphine (TCEP), in the presence of a maleimide-containing hydrazine based doxorubicin derivative, resulted in the in situ generation of drug conjugated disulfide crosslinked micellar nanoparticles with an average diameter of 60 nm in aqueous media. Redox-responsive degradation of nanoparticles was observed upon treatment with an excess amount of TCEP. The clever design of this drug delivery system benefits from the pH-dependent drug release due to the presence of the hydrazone linkage between the drug and the carrier, as well as from redox-responsive degradation of the carrier to warrant eventual clearance. Another example of incorporation of the PDS group onto crosslinked polymeric nanoparticles was reported by Hubbell and coworkers.114 They reported the synthesis of PDS-containing nanoparticles for reversible conjugation of thiol-containing small molecules, peptides and proteins. Using emulsion polymerization in the presence of a carboxylate-pluronic, followed by treatment with a cysteamine-PDS derivative, they obtained crosslinked polymeric nanoparticles bearing the thiol-reactive PDS groups on their surface. Reversible conjugation of these nanoparticles with a thiol-containing biotin, antigenic peptides and ovalbumin was also demonstrated. An example of polymeric nanoparticles responsive to both acidic pH and reducing environments was reported by Fulton and coworkers.115 A set of copolymers both bearing the PDS group along with either aldehyde or amine functional groups were synthesized using a PDS-containing acrylamide monomer. These copolymers were crosslinked through imine- and disulfide-linkages in a sequential manner to afford dual-crosslinked polymeric nanoparticles. Complete dissociation of nanoparticles was obtained only in the presence of both low acidic pH and reducing environments obtained using TCEP. While examples of such redox-responsive nanoparticles are limited, a surge of recent interest in nanogels has led to many reports where the PDS group has played an important role, as discussed in the following section.It should be noted that the PDS functional group has been also used to modify inorganic surfaces. For example, thiol-modified surfaces of silica nanoparticles can be converted to PDS group containing surfaces which can be further functionalized with thiol-containing polymers and proteins.116,117 Alternatively, using a slightly different approach, thiol-modified surfaces of nanoparticles can be directly functionalized with PDS-bearing polymers through the thiol-PDS exchange reaction. Surface modification using the latter strategy was reported by Oupicky, Brock and coworkers, who reported PNIPAM coated mesoporous silica nanoparticles for temperature-depending uptake and release of small molecules.118 In particular, thiol-functionalized silica nanoparticles were coated with PNIPAM containing a PDS group at the chain end. Results showed that polymer-coated nanoparticles exhibited a low level of leakage at 38 °C, a temperature above the LCST of the polymer.
4.2. PDS-functional group-based fabrication and functionalization of redox-responsive nanogels
In recent years, PDS-containing nanogels have been used for protein/peptide delivery applications since these biologically active agents can be attached in a reversible fashion, and thus can be transported and released at the site of interest.119,120 Naturally occurring or engineered cysteine containing proteins and peptides can be conjugated on PDS-containing nanogels under mild conditions using the thiol-PDS exchange reaction. Besides protein/peptide conjugation, the PDS functional group has also been used to introduce crosslinking to obtain stable nanogels that are degradable under a reducing environment. Two different approaches for crosslinking have been investigated. One of the methods involves in situ generation of thiols by cleaving few of the PDS groups on the polymer, which leads to crosslinking by reaction with the residual PDS units on polymers. The alternative method involves formation of self-assembled nanoaggregates from PDS-containing polymers, followed by crosslinking with dithiol-containing molecules through the thiol exchange reaction.Using the first approach, Thayumanavan and coworkers have made seminal contributions to the fabrication of redox-responsive nanogels using PDS-containing copolymers.121–123 In one of their work, PDSMA and PEGMA were randomly copolymerized using RAFT polymerization.124 These PEGMA-based polymers have relatively low LCST and give nanoaggregates above their LCST. After formation of nanoaggregates, DTT is added to the solution to induce crosslinking to yield nanogels (Fig. 10). In situ encapsulation of hydrophobic molecules such as drugs can be achieved during formation of the nanogels, which can be released when exposed to biological reducing agents such as GSH. Facile fabrication of the drug encapsulated delivery vehicle, where crosslinking can be disrupted under biological conditions found in cancerous tissues and cells, makes this nano-drug delivery system a promising platform for addressing challenges faced in diseases such as cancer.
 | ||
| Fig. 10 (a) Synthetic approach, and (b) crosslinking strategy of PDS bearing polymers to fabricate a redox-responsive nanogel based drug delivery system.124 Adapted from Thayumanavan et al.124 with permission. Copyright © 2010 by American Chemical Society. | ||
In another study, Thayumanavan, Maynard and coworkers used a similar strategy to fabricate dye encapsulated nanogels that were conjugated with the thiol-containing protein BSA.125 Lipophilic small dye molecules were loaded into nanoaggregates and were encapsulated during nanogel formation. Thereafter, unreacted PDS groups on the nanogels were utilized for conjugation of thiol-modified BSA through the thiol-PDS exchange reaction. Recently, in an elegant approach, Thayumanavan and Liu demonstrated that self-assembled nanogels can be obtained through simultaneous crosslinking and functionalization of nano-aggregates.126 A copolymer containing two reactive components was synthesized using PDSMA, PEGMEMA and a thio-lactone based monomer. Reaction with an amine-containing molecule leads to the opening of the thio-lactone ring which results in the formation of a thiol-functional group. This newly formed thiol group attacks the PDS group on the polymer and thus leads to crosslinking of the aggregate. This three-component reaction based fabrication of nanogels does not utilize DTT as in previous cases and leads to desired functionalization in the same step.
Using the alternative approach, which involves crosslinking of PDS-containing copolymers with a di-thiol based crosslinker, Zhao, Liu and coworkers prepared nanoparticles using the LCST behaviour of the PDS-containing PEG based polymer, in the presence of PDS modified porcine pancreatic lipase (PPL).127 Assembled polymers and PDS-PPL were crosslinked using meso-2,3-dimercaptosuccinic acid while they are in the nanoaggregate form. The resulting PPL containing nanogels showed enhanced heat resistance and reusability compared to native PPL, which could have been a promising candidate for biocatalyst applications. As another example of nanogel based biomolecular stabilization, Maynard and coworkers reported a PDS-containing glycopolymers based nanogel for delivery of glucagon (Fig. 11). Glucagon is a peptide hormone which hypoglycemia patients used to regulate the blood sugar level. However, instability and poor solubility of glucagon limit the use of drugs. In particular, PDSMA and trehalose based monomers were polymerized and crosslinked by PEG-dithiol to form a nanogel. The residual PDS groups in the nanogels were used for conjugation of thiolated glucagon. The glucagon–nanogel conjugate showed high stability in solution compared to non-encapsulated glucagon.128 A recent study by Zhao and coworkers reported that thiolated proteins can be employed as crosslinkers to obtain nanogels using PDSMA-containing copolymers.129 A P(NIPAM-co-PDSMA) copolymer was crosslinked using thiolated lipase to obtain protein containing nanogels. It was demonstrated that subsequent treatment of nanogels with reduced BSA, followed by bio-mineralization of CaCO3, increased the stability of encapsulated proteins against trypsin.
 | ||
| Fig. 11 Glucagon conjugation on a nanogel obtained using a thiol–disulfide exchange reaction.128 Adapted from Maynard et al.128 with permission. Copyright © 2011 by Wiley-VCH Verlag GmbH & Co. | ||
4.3. PDS-functional group in fabrication and functionalization of hydrogels
Hydrogels are three dimensional structures of crosslinked hydrophilic polymers that have applications in a wide range of biomedical applications. While their ability to undergo facile functionalization is important, incorporation of degradable junctions augments their performance in many applications. The dynamic nature of the disulfide linkage can impart hydrogels with either or both of these characteristics.130,131 Hence, PDS groups have been used to install crosslinking junctions in polymeric networks. For example, Wang and coworkers reported an injectable hydrogel system which undergoes fast gelation through the thiol-PDS exchange reaction.132 In their study, hyaluronic acid (HA) was first modified with 1,6-diaminohexane to introduce free amine groups on the polymer, which were reacted with succinimidyl 3-(2-pyridyldithio)propionate (SPDP) to generate PDS groups. Hydrogels were fabricated upon mixing of the PDS-containing HA and PEG-dithiol at 37 °C in PBS. A cysteine-containing chemokine and stromal cell-derived factor 1Rα were encapsulated inside the hydrogel and cell viability tests were performed with various cell lines. Results showed that cell viability in the hydrogels was sustained up to 7 days. Likewise, Ossipov, Redl and coworkers utilized a PDS-conjugated HA derivative to obtain a HA-fibrin interpenetrating double network hydrogel through fibrogenesis and disulfide-linkage formation.133The PDS group is often used as a masked thiol group; in particular, for cases where multi-thiol systems are needed. Generation of free thiol groups is achieved quite rapidly under mild reducing conditions. For example, Ayres and coworkers synthesized the PDS bearing polymer via RAFT polymerization of PDSMA and HPMA.134 Reactive thiol groups were generated after cleaving the PDS groups with TCEP. The thiolated polymer was crosslinked with norbornene functionalized gelatin using the radical mediated thiol–norbornene click reaction in the presence of UV irradiation to afford gelatin based hydrogels. As another example, poly(carboxybetaine) based hydrogels were reported by Jiang, Tsai and coworkers using a copolymer obtained from carboxybetaine methacrylate and a PDS-containing monomer, namely, N-(3-(2-(pyridyldithio)propanamido)ethyl methacrylate (PDPMA), using RAFT polymerization.135 A telechelic dithio-terminated poly(carboxybetaine) polymer synthesized using RAFT polymerization was used as the crosslinker. Hydrogels were formed upon mixing the polymer and crosslinker within a minute. Enhancement in cell proliferation was observed for hydrogels containing RGD-based peptide crosslinkers, also integrated within the hydrogels using the thiol–disulfide exchange reaction.
Apart from utilization of hydrogels as bulk materials, these soft materials find applications in the form of surface coatings, in areas ranging from diagnostics to implant surface modifications for better bio-assimilation. The reversibility of the disulfide bond affords a dynamic linkage for fabrication of ‘catch and release’ interfaces. Such systems can be used to isolate certain biomolecules upon modification of PDS-containing hydrogels with specific ligands which can capture those biomolecules from a mixture, and subsequently release them for further analysis. For example, Iwasaki and coworkers immobilized antibody fragments onto polymeric brushes.136 In this study, 2-methacryloyloxyethyl phosphorylcholine (MPC) and glycidyl methacrylate (GMA) were polymerized via ATRP. Epoxy groups on the polymer brushes were modified with PDS groups which were modified with Fab’ fragments through the thiol-PDS exchange reaction, and higher affinity of the thus modified surface against FITC-conjugated mouse IgG was demonstrated. In a recent example, Sanyal and coworkers reported PDS-containing hydrogels for capture and release of proteins.137 A surface coating was fabricated using photopolymerization of the PDSMA monomer to afford a soft interface that could be modified with a thiol-containing protein binding ligand, namely, a thiolated biotin. Upon exposure to a mixture of extravidin and BSA, only extravidin was captured on the surface through specific biotin–avidin interaction. Subsequently, the bound protein was released through treatment with a solution containing DTT and the purity of the isolated protein was analysed using gel electrophoresis. Furthermore, upon using a cell-adhesive peptide decorated hydrogel, cells adhered and proliferated on the surface, and could be released upon treatment with DTT, while preserving their viability (Fig. 12).
 | ||
| Fig. 12 Cell attachment and release using thiol–disulfide exchange reaction on a hydrogel interface.137 Adapted from Sanyal et al.137 Copyright © 2018, American Chemical Society. | ||
In recent years, thin polymeric coatings have been extensively used to obtain functional interfaces that can be used for attachment or detection of biomolecules. While most reports utilize the amine-activated ester or thiol–maleimide conjugations, a recent example by Pich and coworkers reported the PDS group mediated exchange reaction as a viable tool.138 They synthesized various poly(N-vinyl lactam)-based copolymers with PDS groups as side chains using RAFT polymerization. Contact angle measurement results and a fluorescence microscopy study indicated that the reactive films based on the PDS-functionalized copolymers allowed facile, direct, and environmentally friendly modification of surfaces from aqueous solution suggesting potential application in surface decoration of tissue-engineering scaffolds and medical implants. Another interesting application of the disulfide based surface obtained using thiol–disulfide exchange was reported by Akimoto and coworkers who modified cell-culture glass surfaces with disulfide linked polyethyleneimine (PEI).139 In this study, PEI, a transfection reagent, was modified with the PDS functional group through amidation with a PDS-containing activated carboxylic acid, namely, N-succinimidyl-3-(2-pyridyldithio)propionate. Thus obtained PDS-conjugated PEI was coated onto the glass surface which was previously modified with thiol- and amine groups using appropriate trialkoxysilane reagents. In vitro studies showed that attached PEI was released from the surface upon exposure to a cysteine containing buffer solution (1 mM), which was not toxic against 293T cells. Authors note that such modified redox-responsive cell culture surfaces can find applications in reverse transfection.
5. Conclusions
The diverse examples of disulfide bearing redox-responsive polymeric materials fabricated using the thiol-PDS exchange reaction surveyed in this review clearly highlight the powerful role that this transformation has played in shaping the current state of the art of such materials. Utilization of PDS-based initiators and monomers allows one to incorporate this exchangeable unit at precise locations such as chain ends and side chains, which in turn facilitates subsequent programmed installation of the reversible disulfide linkage within polymeric materials. As noted in various examples, such predetermined incorporation of the disulfide unit can either lead to dissociations that may result in the release of a single therapeutic agent like drugs or biomolecules from the polymer or result in complete dissociation of the polymeric material to release the encapsulated materials. While the advances in contemporary polymer synthesis have played a critical role in the fabrication of PDS-containing materials, it is the stable and compatible nature of the PDS functional group toward various polymerization techniques that have enabled its facile incorporation. Perhaps the most important reason that is responsible for its widespread utilization is the high specificity and fast kinetics of the exchange reaction with thiols, under extremely mild conditions. Given the increasing sophistication of emerging therapeutic platforms, one can only expect that the PDS-based exchange reaction will continue to play a crucial role in the design and fabrication of future redox-responsive smart materials.Conflicts of interest
There are no conflicts to declare.References
- E. M. Pelegri-Oday, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef CAS
.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS
- R. M. Arnold, N. E. Huddleston and J. Locklin, J. Mater. Chem., 2012, 22, 19357 RSC
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS
- Y. Zou, L. Zhang, L. Yang, F. Zhu, M. Ding, F. Lin, Z. Wang and Y. Li, J. Controlled Release, 2018, 273, 160–179 CrossRef CAS
- M. Arslan, G. Acik and M. A. Tasdelen, Polym. Chem., 2019, 10, 3806–3821 RSC
- N. Cengiz, T. N. Gevrek, R. Sanyal and A. Sanyal, Chem. Commun., 2017, 53, 8894–8897 RSC
- Y. N. Yuksekdag, T. N. Gevrek and A. Sanyal, ACS Macro Lett., 2017, 6, 415–420 CrossRef CAS
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS
- K. S. Anseth and H. A. Klok, Biomacromolecules, 2016, 17, 1–3 CrossRef CAS
- Z. P. Tolstyka, W. Richardson, E. Bat, C. J. Stevens, D. P. Parra, J. K. Dozier, M. D. Distefano, B. Dunn and H. D. Maynard, ChemBioChem, 2013, 14, 2464–2471 CrossRef CAS
- V. N. Rao, S. Mane, A. Kishore, J. Das Sarma and R. Shunmugam, Biomacromolecules, 2012, 13, 221–230 CrossRef
- E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254–1263 CrossRef CAS
- D. Aydin, M. Arslan, A. Sanyal and R. Sanyal, Bioconjugate Chem., 2017, 28, 1443–1451 CrossRef CAS
- L. Chambre, A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2018, 29, 1885–1896 CrossRef CAS
- A. Castonguay, E. Wilson, N. Al-Hajaj, L. Petitjean, J. Paoletti, D. Maysinger and A. Kakkar, Chem. Commun., 2011, 47, 12146–12148 RSC
- B. Sui, C. Cheng and P. Xu, Adv. Ther., 2019, 2, 1900062 CrossRef CAS
- J. K. Oh, Polym. Chem., 2019, 10, 1554–1568 RSC
- B. Saha, S. Bhattacharyya, S. Mete, A. Mukherjee and P. De, ACS Appl. Polym. Mater., 2019, 1, 2503–2515 CrossRef CAS
- Z. Deng, J. Hu and S. Liu, Macromol. Rapid Commun., 2020, 41, 1900531 CrossRef CAS
- M. Ejaz, H. Yu, Y. Yan, D. A. Blake, R. S. Ayyala and S. M. Grayson, Polymer, 2011, 52, 5262–5270 CrossRef CAS
- Z. Gao, B. Golland, G. Tronci and P. D. Thornton, J. Mater. Chem. B, 2019, 7, 7494–7501 RSC
- L. T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Polym. Chem., 2013, 4, 5527–5536 RSC
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC
- O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 3558–3572 CrossRef CAS
- U. S. Gunay, M. Cetin, O. Daglar, G. Hizal, U. Tunca and H. Durmaz, Polym. Chem., 2018, 9, 3037–3054 RSC
- S. Agar, E. Baysak, G. Hizal, U. Tunca and H. Durmaz, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1181–1198 CrossRef CAS
- G. Delaittre and L. Barner, Polym. Chem., 2018, 9, 2679–2684 RSC
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132 RSC
- N. Cengiz, J. Rao, A. Sanyal and A. Khan, Chem. Commun., 2013, 49, 11191–11193 RSC
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C. H. Lim, M. K. McBride, C. B. Musgrave, R. Shoemaker and C. N. Bowman, Polym. Chem., 2018, 9, 4523–4534 RSC
- C. Ghobril, K. Charoen, E. K. Rodriguez, A. Nazarian and M. W. Grinstaff, Angew. Chem., Int. Ed., 2013, 52, 14070–14074 CrossRef CAS
- M. D. Konieczynska, J. C. Villa-Camacho, C. Ghobril, M. Perez-Viloria, K. M. Tevis, W. A. Blessing, A. Nazarian, E. K. Rodriguez and M. W. Grinstaff, Angew. Chem., Int. Ed., 2016, 55, 9984–9987 CrossRef CAS
- C. Boyer, A. H. Soeriyadi, P. J. Roth, M. R. Whittaker and T. P. Davis, Chem. Commun., 2011, 47, 1318–1320 RSC
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef CAS
- M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 1763–1773 RSC
- M. Arslan, Eur. Polym. J., 2020, 126, 109543 CrossRef CAS
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS
- C. Resetco, B. Hendriks, N. Badi and F. Du Prez, Mater. Horiz., 2017, 4, 1041–1053 RSC
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS
- W. W. Cleland, Biochemistry, 1964, 3, 480–482 CrossRef CAS
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS
- R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 1190–1197 CrossRef CAS
-
R. Singh and G. M. Whitesides, in Sulphur-Containing Functional Groups (1993), John Wiley & Sons, Inc., Chichester, UK, 2010, pp. 633–658 Search PubMed
- D. R. Grassetti and J. F. Murray, Arch. Biochem. Biophys., 1967, 119, 41–49 CrossRef CAS
- N. J. Kavimandan, E. Losi, J. J. Wilson, J. S. Brodbelt and N. A. Peppas, Bioconjugate Chem., 2006, 17, 1376–1384 CrossRef CAS
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS
- J. D. Thomas and T. R. Burke, Tetrahedron Lett., 2011, 52, 4316–4319 CrossRef CAS
- Y. Akiyama, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2004, 15, 424–427 CrossRef CAS
- T. Ishii, M. Yamada, T. Hirase and Y. Nagasaki, Polym. J., 2005, 37, 221–228 CrossRef CAS
- R. Mahou and C. Wandrey, Polymers, 2012, 4, 561–589 CrossRef
- C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830 CrossRef CAS
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef CAS
- G. H. Hsiue, H. Z. Chiang, C. H. Wang and T. M. Juang, Bioconjugate Chem., 2006, 17, 781–786 CrossRef CAS
- N. M. Matsumoto, P. Prabhakaran, L. H. Rome and H. D. Maynard, ACS Nano, 2013, 7, 867–874 CrossRef CAS
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699–2707 CrossRef CAS
- T. Fuoco, A. Finne-Wistrand and D. Pappalardo, Biomacromolecules, 2016, 17, 1383–1394 CrossRef CAS
- H. Tan, L. Zhao, W. Liu, L. Ren, S. Xu, L. Chen and W. Li, RSC Adv., 2014, 4, 60413–60420 RSC
- M. Talelli and M. J. Vicent, Biomacromolecules, 2014, 15, 4168–4177 CrossRef CAS
- J. Li, C. Tian, Y. Yuan, Z. Yang, C. Yin, R. Jiang, W. Song, X. Li, X. Lu, L. Zhang, Q. Fan and W. Huang, Macromolecules, 2015, 48, 1017–1025 CrossRef CAS
- H. S. Han, T. Thambi, K. Y. Choi, S. Son, H. Ko, M. C. Lee, D. G. Jo, Y. S. Chae, Y. M. Kang, J. Y. Lee and J. H. Park, Biomacromolecules, 2015, 16, 447–456 CrossRef CAS
- M. Andersson, K. Elihn, K. Fromell and K. D. Caldwell, Colloids Surf., B, 2004, 34, 165–171 CrossRef CAS
- J. T. Li, J. Carlsson, J. N. Lin and K. D. Caldwell, Bioconjugate Chem., 1996, 7, 592–599 CrossRef CAS
- Y. Ju, M. Zhang and H. Zhao, Polym. Chem., 2017, 8, 5415–5426 RSC
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS
- V. Vázquez-Dorbatt, Z. P. Tolstyka, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207–2212 CrossRef
-
A. P. Daniselson, M. L. Dougherty, R. Falatach, T. A. Wright, E. E. Clark, A. Craig, I. D. Sahu, J. A. Berberich, R. C. Page, G. A. Lorigan and D. Konkolewicz, Biocatalytic Polymerization, Bioinspired Surfactants, and Bioconjugates Using RAFT Polymerization, in ACS Symposium Series, American Chemical Society, 2018, vol. 1285, pp. 219–232 Search PubMed
- K. L. Heredia, T. H. Nguyen, C. W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 28, 3245–3247 RSC
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef CAS
- J. Liu, H. Liu, V. Bulmus, L. E. I. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399–1405 CrossRef CAS
- J. Liu, V. Bulmus, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Macromol. Rapid Commun., 2007, 28, 305–314 CrossRef CAS
- K. Gunasekaran, T. H. Nguyen, H. D. Maynard, T. P. Davis and V. Bulmus, Macromol. Rapid Commun., 2011, 32, 654–659 CrossRef CAS
- C. L. Duvall, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Mol. Pharm., 2010, 7, 468–476 CrossRef CAS
- C. W. Chang, T. H. Nguyen and H. D. Maynard, Macromol. Rapid Commun., 2010, 31, 1691–1695 CrossRef CAS
- Y. Koda, T. Terashima, M. Sawamoto and H. D. Maynard, Polym. Chem., 2015, 6, 240–247 RSC
- T. A. Wright, R. C. Page and D. Konkolewicz, Polym. Chem., 2019, 10, 434–454 RSC
- I. Mukherjee, S. K. Sinha, S. Datta and P. De, Biomacromolecules, 2018, 19, 2286–2293 CrossRef CAS
- R. J. Mancini, J. Lee and H. D. Maynard, J. Am. Chem. Soc., 2012, 134, 8474–8479 CrossRef CAS
- N. Vanparijs, S. Maji, B. Louage, L. Voorhaar, D. Laplace, Q. Zhang, Y. Shi, W. E. Hennink, R. Hoogenboom and B. G. De Geest, Polym. Chem., 2015, 6, 5602–5614 RSC
- N. M. Matsumoto, G. W. Buchman, L. H. Rome and H. D. Maynard, Eur. Polym. J., 2015, 69, 532–539 CrossRef CAS
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS
- M. R. Molla and S. Ghosh, Macromolecules, 2012, 45, 8561–8570 CrossRef CAS
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS
- O. Gok, P. Erturk, B. Sumer Bolu, T. N. Gevrek, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 2463–2477 CrossRef CAS
- J. J. Glass, Y. Li, R. De Rose, A. P. R. Johnston, E. I. Czuba, S. Y. Khor, J. F. Quinn, M. R. Whittaker, T. P. Davis and S. J. Kent, ACS Appl. Mater. Interfaces, 2017, 9, 12182–12194 CrossRef CAS
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518–8520 CrossRef CAS
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun., 2013, 49, 4223–4225 RSC
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054–1056 RSC
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS
- E. W. Lin and H. D. Maynard, Macromolecules, 2015, 48, 5640–5647 CrossRef CAS
- B. Remant, B. Thapa and P. Xu, Mol. Pharm., 2012, 9, 2719–2729 CrossRef
- B. Lou, S. De Koker, C. Y. J. Lau, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2019, 30, 461–475 CrossRef CAS
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2005, 101, 47–58 CrossRef CAS
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106–3113 CrossRef CAS
- Z. Song, Y. Xu, W. Yang, L. Cui, J. Zhang and J. Liu, Eur. Polym. J., 2015, 69, 559–572 CrossRef CAS
- S. Keller, J. T. Wilson, G. I. Patilea, H. B. Kern, A. J. Convertine and P. S. Stayton, J. Controlled Release, 2014, 191, 24–33 CrossRef CAS
- W. Chen, Y. Zou, J. Jia, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2013, 46, 699–707 CrossRef CAS
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. B. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726–12734 CrossRef CAS
- D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid Commun., 2014, 35, 1340–1344 CrossRef CAS
- R. Bej, J. Sarkar and S. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 194–202 CrossRef CAS
- D. Basak, R. Bej and S. Ghosh, Polym. Chem., 2015, 6, 6465–6474 RSC
- R. Bej, P. Rajdev, R. Barman and S. Ghosh, Polym. Chem., 2020, 11, 990–1000 RSC
- X. Liu, J. He, D. Hu, Y. Niu, X. Xiaa and Y. Lu, RSC Adv., 2014, 4, 48897–48900 RSC
- J. Zhuang, B. Zhao and S. Thayumanavan, ACS Macro Lett., 2019, 8, 245–249 CrossRef CAS
- A. J. Van Der Vlies, C. P. Oneil, U. Hasegawa, N. Hammond and J. A. Hubbell, Bioconjugate Chem., 2010, 21, 653–662 CrossRef CAS
- A. W. Jackson and D. A. Fulton, Macromolecules, 2012, 45, 2699–2708 CrossRef CAS
- S. Zhu, Z. W. Li and H. Zhao, Langmuir, 2015, 31, 4129–4136 CrossRef CAS
- X. Yang, D. Chen and H. Zhao, Acta Biomater., 2016, 29, 446–454 CrossRef CAS
- Y. Z. You, K. K. Kalebaila, S. L. Brock and D. Oupicky, Chem. Mater., 2008, 20, 3354–3359 CrossRef CAS
- Z. Jiang and S. Thayumanavan, Isr. J. Chem., 2020, 60, 132–139 CrossRef CAS
- D. C. González-Toro and S. Thayumanavan, Eur. Polym. J., 2013, 49, 2906–2918 CrossRef
- J. H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS
- D. C. González-Toro, J. H. Ryu, R. T. Chacko, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2012, 134, 6964–6967 CrossRef
- J. H. Ryu, S. Bickerton, J. Zhuang and S. Thayumanavan, Biomacromolecules, 2012, 13, 1515–1522 CrossRef CAS
- J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS
- N. M. Matsumoto, D. C. González-Toro, R. T. Chacko, H. D. Maynard and S. Thayumanavan, Polym. Chem., 2013, 4, 2464–2469 RSC
- B. Liu and S. Thayumanavan, Chem, 2019, 5, 3166–3183 CAS
- X. Ji, J. Liu, L. Liu and H. Zhao, Colloids Surf., B, 2016, 148, 41–48 CrossRef CAS
- N. Boehnke, J. K. Kammeyer, R. Damoiseaux and H. D. Maynard, Adv. Funct. Mater., 2018, 28, 1–11 CrossRef
- Q. Yu, X. Ma, Y. Liu and H. Zhao, Chem. – Eur. J., 2019, 25, 16712–16717 CrossRef CAS
- N. Cengiz, Eur. Polym. J., 2020, 123, 109441 CrossRef CAS
- I. Altinbasak, R. Sanyal and A. Sanyal, RSC Adv., 2016, 6, 74757–74764 RSC
- S. Y. Choh, D. Cross and C. Wang, Biomacromolecules, 2011, 12, 1126–1136 CrossRef CAS
- Y. Zhang, P. Heher, J. Hilborn, H. Redl and D. A. Ossipov, Acta Biomater., 2016, 38, 23–32 CrossRef CAS
- M. M. Perera and N. Ayres, Polym. Chem., 2017, 8, 6741–6749 RSC
- H. W. Chien, X. Xu, J. R. Ella-Menye, W. B. Tsai and S. Jiang, Langmuir, 2012, 28, 17778–17784 CrossRef CAS
- R. Iwata, R. Satoh, Y. Iwasaki and K. Akiyoshi, Colloids Surf., B, 2008, 62, 288–298 CrossRef CAS
- T. N. Gevrek, M. Cosar, D. Aydin, E. Kaga, M. Arslan, R. Sanyal and A. Sanyal, ACS Appl. Mater. Interfaces, 2018, 10, 14399–14409 CrossRef CAS
- H. Peng, K. Rübsam, X. Huang, F. Jakob, M. Karperien, U. Schwaneberg and A. Pich, Macromolecules, 2016, 49, 7141–7154 CrossRef CAS
- A. M. Akimoto, T. Takarada and M. Maeda, Colloids Surf., B, 2013, 103, 360–365 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2020 |