
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural cyclodextrins and their derivatives for polymer synthesis
Magdalena A.
Przybyla
 ,
Gokhan
Yilmaz
and
C. Remzi
Becer
*
,
Gokhan
Yilmaz
and
C. Remzi
Becer
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: remzi.becer@warwick.ac.uk; Web: http://www.becergroup.com Web: http://www.twitter.com/remzibecer
First published on 12th November 2020
Abstract
Cyclodextrins (CDs) are a family of cyclic oligosaccharides with a hydrophilic exterior surface and a nonpolar cavity interior, therefore, CDs can form inclusion complexes through noncovalent interactions with a broad range of hydrophobic guests. CD derivatives and CD-based polymers find important uses in different fields such as pharmacy, cosmetics, biomedicine, textiles, and food domain due to their unique properties during the past decade. Hence, in this review, functionalised CDs and CD-based polymers are classified and discussed according to their synthetic approaches comprehensively to help polymer chemists for the development of new CD-based materials for different types of applications.
1 Introduction
Cyclodextrin (CD) is a cyclic oligosaccharide with a bucket-like shape. The hydrophobic cavity of cyclodextrin can encapsulate small hydrophobic molecules and can be utilised in the design of supramolecular structures.1 Cyclodextrin is a cheap, widely available, biocompatible and biodegradable material. Valued for encapsulation properties, cyclodextrin found applications in many fields such as pharmacy,2–5 personal care products,6,7 biomedicine,8–10 food,11–13 molecular recognition14,15 and supramolecular chemistry.16–20 Cyclodextrin also proved to be an extremely versatile molecule for polymer science.16,21,22 It is particularly valued for easy functionalisation, including attachment of initiator moieties as well as unsymmetrical modification.23 Cyclodextrin is compatible with a range of polymerisation techniques such as ATRP (atom transfer radical polymerisation),23–25 ROP (ring opening polymerisation),26,27 and RAFT (reversible addition–fragmentation chain-transfer) polymerisation.28,29 Its water solubility and biodegradability make it a perfect candidate for ‘green polymerisation’.30,31 Hydroxyl groups not directly involved in the polymerisation can be later utilised in post-polymerisation modifications to tailor the product for advanced application. If chains are grown from just one face, the other face can be conjugated with a drug or targeting molecule forming hybrid structures that can be applied in cancer therapy and bioimaging.32 Cyclodextrin can also play a ‘supportive’ role in polymer synthesis, for example, cyclodextrin form inclusion with monomer thus improving its solubility and control over polymerisation reaction.33,34 In emulsion polymerisation, cyclodextrin found application as a phase transfer agent35 and in troublesome aqueous RAFT polymerisation, it can solubilise chain transfer agent.36There are several comprehensive reviews on cyclodextrin-based polymer materials22,37–40 including materials based on host/guest interactions41 and polyrotaxanes.42 A huge amount of work has been done in the field of cyclodextrin-polymer conjugates and this can be overwhelming for beginners. This review is aimed at the newcomers who would like to get an understanding of the opportunities that cyclodextrin brings but also its shortcomings and common pitfalls. The review has four sections: Introduction, Functionalisation of CD, CD-polymer covalent conjugates and Applications. More than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 derivatives of α-, β- and γ-CD have been reported43 which vastly exceeds possibilities of this review. The scope of CD derivatives has been narrowed down to the toolbox of the most common functionalities in the field of CD such as tosylate, halogens or alkyl groups. Particular attention is paid to the derivatives useful for specific applications in polymer chemistry such as CD-initiators or monomers. For more in-depth review on derivatives of cyclodextrins, the reader is directed elsewhere.43,44 The section on covalent conjugates will give an overview of how different polymerisation techniques can be utilised for the synthesis of polymers with certain architectures. The last section will cover representative applications of CD-polymer covalent conjugates in various fields such as preparation of nanoparticles, gene delivery and hydrogels.
000 derivatives of α-, β- and γ-CD have been reported43 which vastly exceeds possibilities of this review. The scope of CD derivatives has been narrowed down to the toolbox of the most common functionalities in the field of CD such as tosylate, halogens or alkyl groups. Particular attention is paid to the derivatives useful for specific applications in polymer chemistry such as CD-initiators or monomers. For more in-depth review on derivatives of cyclodextrins, the reader is directed elsewhere.43,44 The section on covalent conjugates will give an overview of how different polymerisation techniques can be utilised for the synthesis of polymers with certain architectures. The last section will cover representative applications of CD-polymer covalent conjugates in various fields such as preparation of nanoparticles, gene delivery and hydrogels.
There are three cyclodextrins commonly used in polymer chemistry: α-CD, β-CD and γ-CD which are built of six, seven and eight glucopyranose units accordingly, connected via α-1,4-glycosidic bonds (Fig. 1).45 Hydroxyl groups are located on the edges of the cyclodextrin bucket which is in turn made of the sugar backbone and glycosidic oxygen bridges. By convention, primary hydroxyls are at the top and secondary hydroxyls at the bottom of the structure. While secondary hydroxyls form strong hydrogen bonds rigidifying the bottom of the cyclodextrin, primary hydroxyls are free to rotate thus reducing the diameter of the top face and giving cyclodextrin truncated cone-like shape.45 β-CD is the most applied cyclodextrin due to its availability however it has also been reported to exert more potent cytotoxicity which is an important factor for potential bio applications.46 β-CD is rather rigid and forms a complete set of six intramolecular H-bonds, making it the least water soluble out of all known cyclodextrins.45 To increase its water solubility, which is particularly important for encapsulation of hydrophobic molecules in aqueous solvent, some of the hydroxyl groups can be converted to sulphate groups.47 Bigger cyclodextrins such as γ- or δ-CD are no longer symmetrical and as they collapse upon themselves, the cavities become smaller rather than bigger and the complexing capacity of various guest is diminished.
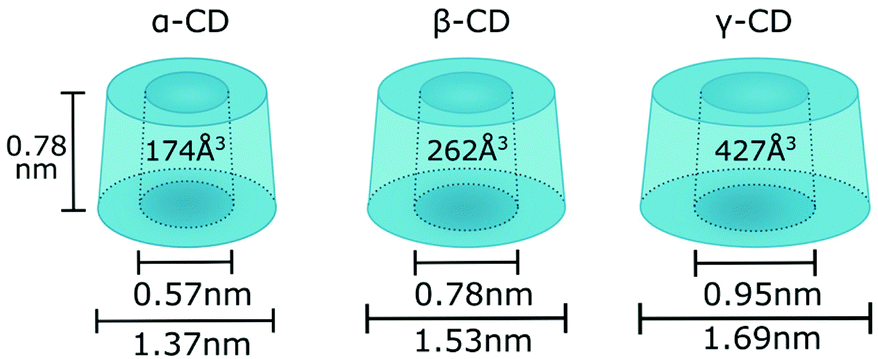 | ||
| Fig. 1 Geometric dimensions of α-, β- and γ-CD. | ||
There are three hydroxyl environments with distinct reactivity, seven hydroxyls each, giving twenty-one in total. Primary hydroxyls at C6 position are most basic and nucleophilic, and hence most reactive.44 Secondary hydroxyls at C2 position are most acidic and those at C3 position are most inaccessible due to steric hindrance and hydrogen bonding and hence least reactive (Fig. 2).43,48
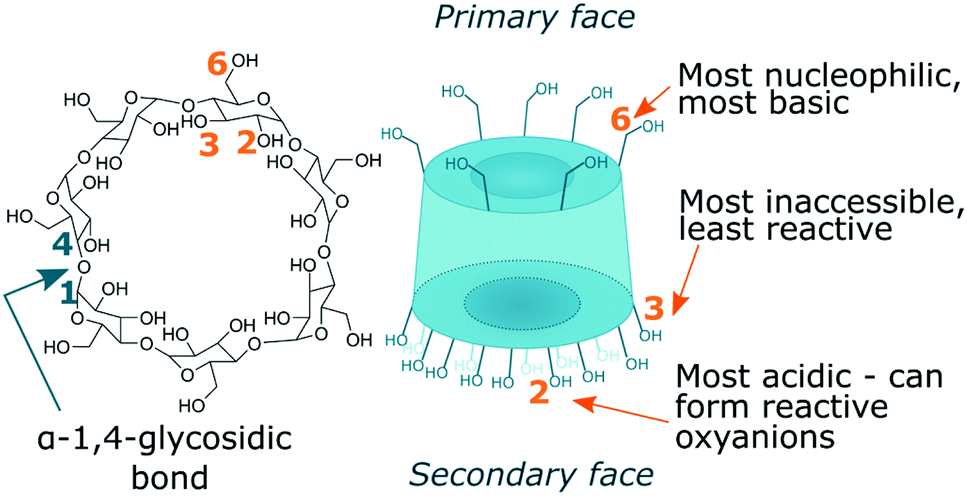 | ||
| Fig. 2 Structure of B-CD with highlighted three hydroxyl groups and their reactivity. | ||
2 Functionalisation of CD
Preparation of CD-polymer conjugates starts with the functionalisation of cyclodextrin. The choice of functionality and degree of substitution depends on the polymerisation technique and desired polymer architecture. Substitution of all hydroxyl groups can give a multiarm initiator for the synthesis of star polymers. Monosubstituted cyclodextrin can be utilised in the preparation of CD-functionalised monomers which upon polymerisation will give polymers with cyclodextrin side chain functionality. Primary and secondary faces can be modified selectively thus enabling for the formation of asymmetric multiarm constructs. Restricting the extent of the reaction to just one face of cyclodextrin is possible by leveraging the aforementioned differences in the reactivity of three hydroxyl groups. However, one needs to take an account on other factors too, such as complexing capabilities of cyclodextrin. Encapsulation of solvent or chemical reagent can have a profound influence on reaction rate and selectivity. The solvent can even switch selectivity as it has been reported for monotosylation.49 Complexation implies certain orientation of the guest molecule and can give access to hindered C3 hydroxyls, which are difficult to modify selectively. Such an approach led to the successful attachment of cinnamyl functionality.50 Further, the chemistry of by-products of reaction needs to be considered as cyclodextrins are unstable under acidic conditions and a base such as pyridine or imidazole is often required to scavenge protons.1,51According to D'Souza et al. protocols for modification of cyclodextrin can be classified into one of the three categories: (a) clever reactions exploiting aforementioned differences in reactivity; (b) long methods involving several protection–deprotection steps; (c) ‘sledgehammer’ indiscriminate reactions with lengthy purification procedure.44 Sometimes no clever methods are available and indiscriminate reactions with lengthy purification procedures have to be followed. There is ongoing research on more efficient and greener synthetic methods such as mechanochemical synthesis under solvent-free conditions using a planetary ball mill.52 Many protocols were established before modern state-of-art analytical facilities were available and selectivity of reported reactions as well as purity of the final product were subsequently questioned or even disproved.53
Reactive electrophiles will attack both rims of the cyclodextrin bucket indiscriminately. Nevertheless, persubstitution usually requires an excess of the reagent and often a mixture of products with varying degrees of substitution is obtained. C-2 and C-3 substituted derivatives and complicated substitutions patterns are often not achievable without protecting groups. Any standard alcohol protecting group can be used as long as it does not require a high concentration of strong acid for deprotection (Table 1).
| Position | Protecting group | Method of introduction | Cleavage | Ref. |
|---|---|---|---|---|
| a Requires prior silylation and subsequent desilylation of the primary face. | ||||
| C6 | Silyl ether | TBDMS | BF3 or TBAF | 54 and 55 |
| C6 | Benzyl | Benzyl chloride | Hydrogenolysis | 56 and 57 |
| C2&C3 | Methyla | Methyl iodide | NaOMe/MeOH | 58 |
| C2&C3 | Acetyla | Acyl chloride | NaOMe/MeOH | 28 and 59 |
tert-Butyldimethylsilyl (TBDMS) is an excellent protecting group for 6-position and can be cleaved with BF3 in tetrahydrofuran55 or tetra-n-butylammonium fluoride.54 Alternatively, primary hydroxyls can be benzylated with benzyl chloride57 and deprotected via palladium catalysed hydrogenolysis.56 The remaining hydroxyl groups can be acetylated prior to the polymerisation to increases the solubility of cyclodextrin derivative in common organic solvent.28 After polymerisation, acetates can be cleaved with NaOMe/MeOH.59
In a similar way, cyclodextrin can be methylated to improve its water solubility – per-O-methyl-β-CD is 10-fold more soluble than native CD.45,60 Such approach is of particular importance in aqueous polymerisation and methylated-CD has been for example used as part of a supramolecular pH-sensitive photoinitiator.61 Similarly, water soluble copolymer with methylated CD and proline has been used as a nanoreactor to mimic enzymes and provide hydrophobic environment for the reaction yet without affecting homogeneity of the reaction solution.62 Random hydroxypropylation also improves water solubility of CD. This comes as a result of disruption of the tight hydrogen bonding network formed by cyclodextrin hydroxyl group.60
Although there is literature available concerning water-soluble CD derivatives,60 no systematic study was performed to assess solubility in common organic solvents which are usually more suitable for polymerisation than water. In general, adding alkyl, silyl and acetyl groups improve solubility cyclodextrin in organic solvents.44 Information on solubility of common CD derivatives is presented in Table 2.
| Derivative | Solubility |
|---|---|
| Native β-CD | Moderate water solubility45 |
| Soluble in DMF, DMSO1 | |
| Low solubility in NMP1 | |
| Methylated β-CD (various substitution patterns) | Good water solubility (maximum solubility for 13–14 methoxy per CD63 |
| Hydroxypropylated β-CD (various substitution patterns) | Good water solubility60 |
| Sulfated β-CD (various substitution patterns) | Good water solubility60 |
| Peralkylated β-CD | Soluble in common organic solvents44 |
| Per-6-halogeno-per-6-deoxy-β-CD | Soluble in polar organic solvents (DMF, DMSO)44 |
| Perallylated β-CD | Soluble in common organic solvents64 |
| Per-6-thio-per-6-deoxy-β-CD | Poor solubility in acetone, MeOH, chloroform, tetrahydrofuran |
| Good solubility in polar organic solvents – DMF, DMSO27 | |
| Heptakis[2,3,6-tri-O-(2-bromo-2-methylpropionyl]-β-CD | Soluble in common organic solvents such as ether, toluene, dichloromethane except aliphatic hydrocarbons, low water solubility23,65 |
2.1 Substitution of primary hydroxyls
The common functionalisation starting points are halogen and tosylate cyclodextrin derivatives (Fig. 3) Both functional groups can be smoothly displaced via nucleophilic substitution. Whereas mono-tosylation renders an excellent precursor for CD-based monomer, pertosylation of the primary face is a poor choice as the reaction suffers from the formation of 6-anhydro product.53 One of the first reported methods for monotosylation used p-toluenesulfonyl chloride to give 6-O-mono-6-(p-toluenesulfonyl)-6-deoxy-β-CD but required chromatographic purification.66 Reaction with p-toluenesulfonic anhydride, on the other hand, did not require purification by chromatography and gave the product in 61% yield; unreacted anhydride was conveniently removed by filtration.67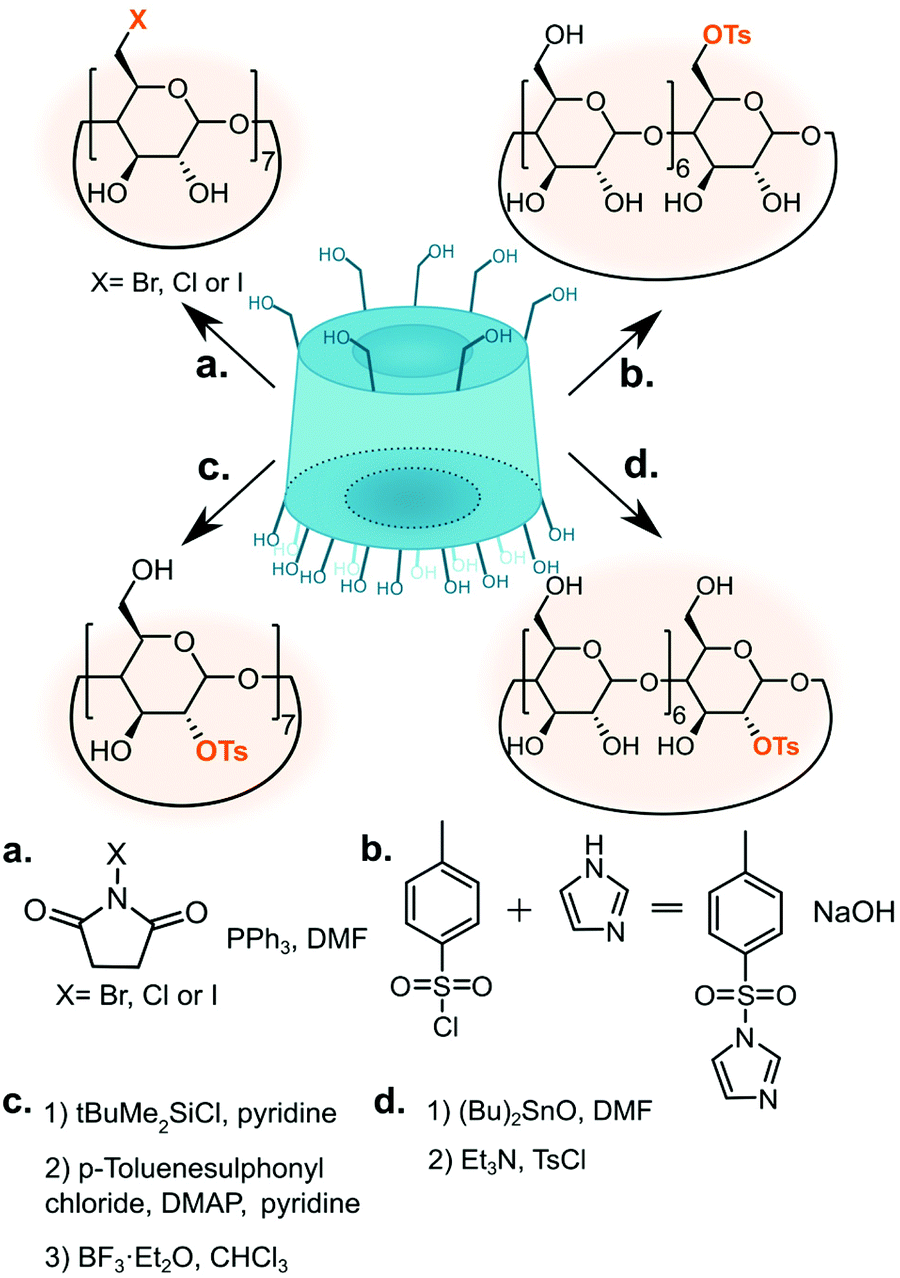 | ||
| Fig. 3 Methods of substituting cyclodextrin hydroxyl groups with tosylate or halogen to give common synthetic intermediates. | ||
More recently, preactivation of tosylate with imidazole was found to improve selectivity towards 6-O-mono-p-toluenesulfonyl-6-deoxy-β-CD product although other substitution patterns are still observed.68 The first protocols for halogenation used methanesulfonyl halides or triphenyl phosphine with iodine gas.57,69,70 Later, more convenient route using N-halosuccinimides was developed.71 This approach is currently favoured as it uses a stable reagent and works for different halogens. Heptakis(6-deoxy-6-halogeno)-CDs are soluble in polar solvents such as pyridine, DMF (dimethylformamide) or DMSO (dimethyl sulphoxide) but their solubility in nonpolar solvent can be increased by esterification (for example acetylation) of the remaining secondary hydroxyls.44
Both halogen and tosylate can be displaced by a nucleophile (Fig. 4). Reaction with excess of sodium or lithium azide provides mono-6-deoxy-6-azido-CD, an excellent partner for click chemistry, in quantitative yield.68,72–74 Alternatively, azido-CD can be prepared via Vilsmeier–Haack type reaction directly from native cyclodextrin with lithium azide, triphenylphosphine and carbon tetrabromide.75 This bypasses the longsome tosylation. Similarly, halogen can be displaced by thiourea to give thiol-CD, useful in thiol–ene reaction. All reported protocols require large excess of thiourea and prolong reflux.76–79 A different method developed by Marsura et al. is based on Mitsunobo reaction where thio-CD can be obtained directly from native cyclodextrin by reaction with a thiol, diisopropyl azodicarboxylate and triphenylphosphine.80
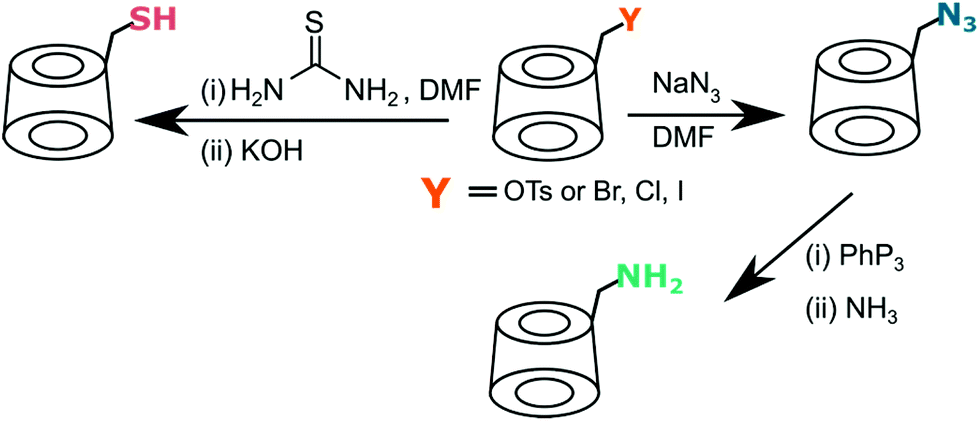 | ||
| Fig. 4 Both tosylate and halogen can be easily displaced by nucleophiles to give synthetically useful cyclodextrin derivatives. | ||
Azide group can be also reduced with triphenylphosphine75 or palladium black under hydrogen gas66 to give amino-CD. For other amines, the direct substitution of tosylate with liquid amine is suitable.81,82 More recently, microwave assisted method was developed by Milton et al. that significantly shortened reaction time compared to standard procedure for tris(2-aminoethyl)amine from 48 h to just half an hour.83 Amine derivatives found its use in the preparation of more complicated CD-based compounds via amine–carboxylic acid coupling. With the aid of DCC (N,N′-dicyclohexylcarbodiimide), TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) derivative of benzoic acid was coupled with heptakis(6-deoxy-6-amino)-β-CD to yield TEMPO-CD for controlled polymerisation.84
2.2 Substitution at C-2 and C-3
Substitutions at 2- and 3-position are less common due to the cumbersome synthesis hindered by the high reactivity of C6 hydroxyls. Selective substitution without protecting groups is often impossible or requires tedious purification. To selectively functionalise C2 hydroxyls, they should be first deprotonated to form 2-alkoxides that are suitable for nucleophilic attack.75,85 Extra care must be taken when choosing a solvent for such reaction as solvent can strongly influence nucleophilicity of oxyanions. Even though C2 hydroxyls are the most acidic, equilibration time is required to avoid deprotonation at other positions. Such approach allowed Jurczak et al. to prepare cyclodextrin with an allyl group.86 After deprotonation with lithium hydride and 24 h of stirring, propargyl bromide was added to yield mono-2-propargyl-β-CD at 38% yield which is very satisfactory for substitution at 2-position (Fig. 5). Monotosylation at C2 was also reported. Ueno and Breslow used 3-nitrophenyl toluenesulfonate as sulfonating agent but the product was isolated at only 10% yield.87 Formation of alkyltin alkoxide C2 prior to tosylation with p-toluene-sulfonyl chloride improved the yield to 29%.88 Both approaches require purification on column chromatography. Once sulfonate is incorporated it can be displaced by nucleophiles in an analogous way to mono-6-deoxy-6-tosylate-CD e.g. with sodium azide.89 To pertosylate C2 hydroxyls, the primary group has to be protected, for example with t-butyI dimethylsilyl groups which can be cleaved with boron trifluoride.90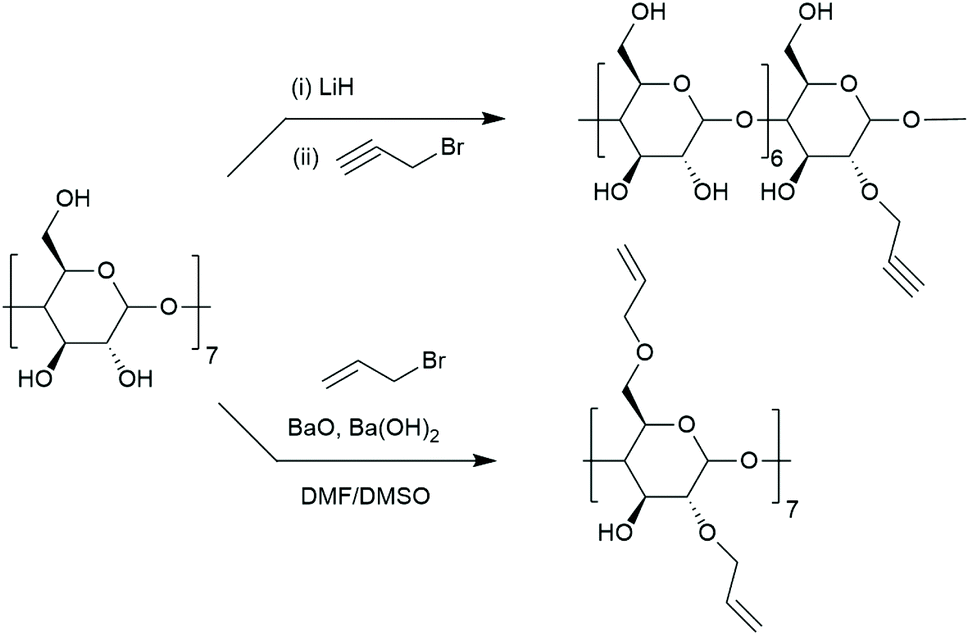 | ||
| Fig. 5 Incorporation of propargyl and allyl group at C-2 position. | ||
C3-position is extremely difficult to modify selectively. Necessary protection of both 2- and 6-positions leads to such a steric crowding that accessibility of 3-positions is too low to react. A promising approach is selective deprotection such as monodesilylation.91
2.3 Persubstitution
Persubstitution i.e. substitution of each hydroxyl group by the same functional group is achievable for a limited set of reagents. Even reactive reagents can struggle as the steric hindrance increases with successive substitutions. In a presence of a base, reactions with alkyl or allyl halogenide proceed smoothly, such as reaction of β-CD with methyl iodide to give per-O-methylated-CD with 74% yield.63Achieving good yields for allylation is more difficult and the first attempt on the synthesis of heptakis(2,3,6-tri-O-allyl)-β-CD used allyl iodide but the reported yield was only 32%.92
Recently, perallylated α-, β- and γ-CDs were obtained by reaction with allyl bromide and sodium hydroxide under argon atmosphere in 4 h in quantitative yield and required only minimal purification.64 As the alkylation reaction is exothermic, low temperature (0 °C) of the reaction is crucial to ensure good yield and avoid incomplete allylation.93 Similarly, cyclodextrin can be easily peracetylated with acetic anhydride although this intermediate has little use in polymer chemistry.94 Peracetylation can be catalysed by base95 or acid.96 Persubstitution by esterification is of particular interest for polymer chemists as it allows for the incorporation of ATRP initiator moiety.23 Whereas some esterification such as perbenzoylation with benzoyl chloride in pyridine are reliable giving almost quantitative yield,97 other result in various degrees of substitution64 as discussed in detail in later.
Another strategy to obtain fully functionalised cyclodextrin is to modify two faces separately, with different reagents. Liu and co-workers reported asymmetrically functionalised cyclodextrin with azide functionality on the top face and ATRP initiator at the bottom.98 Haddleton et al., however, following a similar protocol obtained only partial esterification giving (N3)7-β-CD-(Br)10 as confirmed by MALDI-TOF MS (matrix-assisted laser desorption/ionization-time of flight mass spectrometry).99
Persubstitution can also take place at a specific group (C6, C2, C3) or combination of thereof. As highlighted in section 2.1 primary hydroxyls can be substituted with halogens providing heptakis(6-deoxy-6-halogeno)-CD – useful synthetic intermediates. Primary hydroxyls cannot be selectively alkylated or esterified directly. Instead, a sequence of protection–deprotection steps must follow: the primary face can be silylated, then the secondary face esterified. After deprotection of the primary face it can be reacted with a respective alkyl halide under basic conditions.100 Analogous path was designed for esterification–pivaloylation.101 Similarly, peralkylation of C2 position cannot proceed directly and requires protection of the primary face.100 BaO allows for selective allylation of position 2 and 6 giving heptakis(2,6-O-diallyl)-β-CD (Fig. 5).102 Simple protection of primary face will lead to perallylated cyclodextrin at position 2 upon reaction with allyl halogenide in the presence of BaO.
2.4 CD-functionalised monomers
CD-based monomers can be directly polymerised to obtain polymers with a high density of pendent cyclodextrin groups. Monomer has to be prepared in a well-controlled manner or otherwise extensive purification is required to avoid multivinyl-CD monomer. Such impurity could lead to the formation of branched or crosslinked polymers.The first-reported CD-monomer was based on acrylate and was prepared from the native cyclodextrin with m-nitrophenyl acrylate.103 To avoid side reactions, mild conditions and short reaction times were applied, thus limiting the yield of the reaction to 20%. Selectivity was increased by the inclusion of m-nitrophenyl ester in cyclodextrin cavity. Similarly, other acrylate-based monomers were synthesised from native cyclodextrin.104 Such transesterification reactions are likely to lead to undesired di- and higher substitution products. To achieve higher synthetic precision, Liu et al. used monosubstituted cyclodextrin. Ethylenediamine-modified cyclodextrin was prepared from mono-6-(p-toluenesulfonyl)-6-deoxy-β-CD and reacted with glycidyl methacrylate (Fig. 6).105 This approach is often used nowadays and works well with different amine-CD such as piperazine-modified cyclodextrin.106
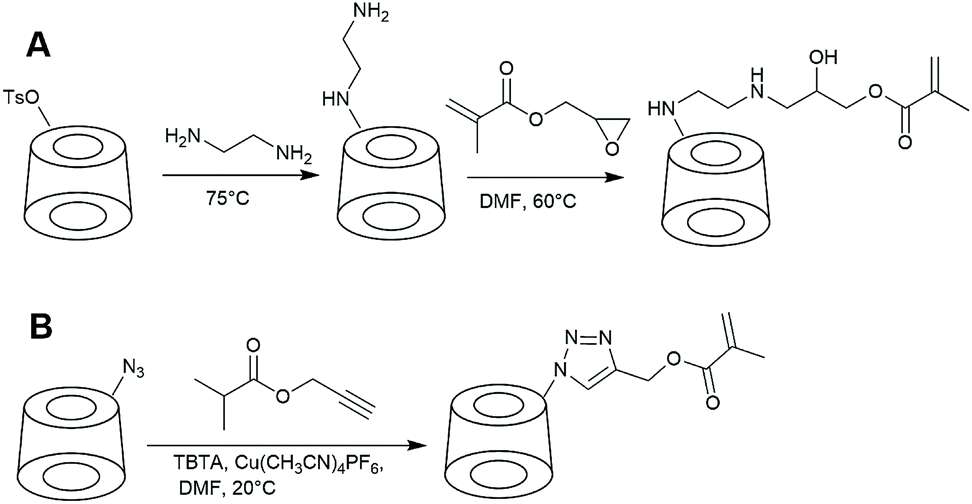 | ||
| Fig. 6 The two most common approaches in preparation of CD-based monomers. A: Amine-CD is reacted with glycidyl-methacrylate. B: Copper mediated azide–alkyne coupling. | ||
To increase coupling efficiency, ‘click’ chemistry can be employed such as CuAAC (copper-catalyzed azide–alkyne cycloaddition) coupling. Recently, methacrylate modified CD (mono-(1H-1,2,3-triazol-4-yl)(methyl)2-methylacryl-β-cyclodextrin) was synthesised by means of azide–alkyne cycloadditions of propargyl methacrylate and mono-6-azido-6-deoxy-β-cyclodextrin. Reaction proceeded with full conversion.107 If microwave radiation is used, the reaction time is reduced from 24 h to 30 min.108
2.5 Incorporation of initiator groups for multiarm polymers
Since the development of controlled polymerisation techniques, the precise engineering of polymer architectures became possible, among them multiarm polymers. Multiarm star-shaped polymer consists of linear arms branching from a core. Such polymers attracted special attention of both academia and industry due to their low viscosity, high arm density and degree of surface functionality. Cyclodextrin is suitable for both core-first and grafting-to approach (Fig. 7). As cyclodextrin can be asymmetrically functionalised with orthogonal initiating functionalities, miktoarms can be prepared.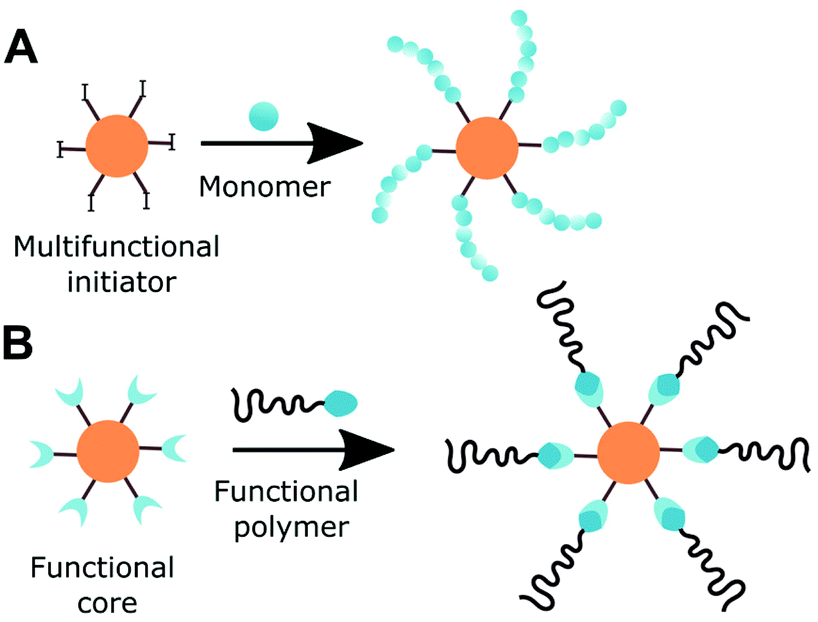 | ||
| Fig. 7 Two applicable to cyclodextrin approaches for the synthesis of multiarm polymers. A: Core-first and B: grafting-to approach. | ||
Cyclodextrin allows for unusually high grafting densities as for typical well-defined multiarm initiators. Primarily, 7,58,109 1458,110 or 2123,64 sites are accessible but intermediate numbers were also reported.111,112
Arms not directly involved in the polymerisation can be further modified to tailor the solubility of the initiator or attach various pendants such as sugars.28 To leverage synthetic precision of controlled polymerisation methods, the initiator has to be well defined and number of functional sites known. Knowledge of the exact degree of substitution is needed to correctly calculate the ratio of reagents for the subsequent polymerisation reaction. The proper characterisation is therefore crucial and MALDI-TOF MS analysis should be carried out to support the structure of the synthesised initiator.
In theory, the extent of esterification can be tuned by varying reagent ratio to limit the extent of esterification, as summarised in Table 3. Nevertheless, the protocols are often not reproducible and insufficient characterisation of the product is given; without proper mass spectrometry analysis, the degree of substitution remains questionable. Hence, using protecting groups seems to be a more reliable approach. The primary face can be protected as silylates110 and secondary face as methyl ethers58 or acetylates.55,101 Often, particularly for silyl groups, acid scavenger such as imidazole is needed to avoid deprotection of hydroxyls by acidic by-product of esterification. Even though protection–deprotection requires additional steps, it facilitates purification and can speed up the synthesis.
| Degree of substitution | Yield [%] | Reagent and stoichiometry wrt. OH | Wrt to CD | Solvent; other reagents | Reaction conditions | Characterisation | Ref. |
|---|---|---|---|---|---|---|---|
| a Dialysis purification; RT = room temperature, TEA = trimethylamine, DMAP = 4-dimethylaminopyridine, FTIR = Fourier transform infrared spectroscopy. | |||||||
| 1 | N/A |
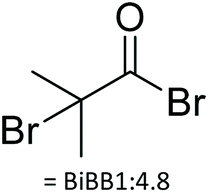
|
4.4 | DMF | (1) 2 h at 0 °C, | NMR | 116 |
| (2) 12 h at RT | |||||||
| N2 atmosphere | |||||||
| 3 | N/A | BiBB 1:5.25 |
4 | DMF | (1) 2 h at 0 °C, | NMR | 117 |
| (2) 12 h at RT | |||||||
| N2 atmosphere | |||||||
| 5 | 78.8 | BiBB 1:3 |
7 | Pyridine, chloroform | 24 h at RT | NMR + FTIR + elemental analysis | 118 |
| N2 atmosphere | |||||||
| 6 | 48 | BiBB 1:3.4 |
16.2 | NMP | 0 °C–>RT, 12 h | NMR | 119 |
| 7 | N/A | BiBB 1:3 |
7 | DMF | (1) 4 h, 0 °C | NMR, FTIR | 120 |
| (2) 30 h, 40 °C | |||||||
| 8 | 82.64 | BiBB 1:2.6 |
8 | NMP; TEA, DMAP | (1) 2 h at 0 °C | NMR | 112 |
| (2) 24 h at RT | |||||||
| N2 atmosphere | |||||||
| 10 | 93 | BiBB 1:2 |
10 | NMP; TEA, DMAP | (1) 2 h at 0 °C, | NMR | 121 |
| (2) 24 h at RT | |||||||
| N2 atmosphere | |||||||
| 14 | 75 | BiBB 2:1 |
41.4 | NMP | (1) 2 h at 0 °C, | NMR + MALDI-TOF MS | 113 |
| (2) 22 h at RT | |||||||
| 16 | 68.5 | BiBB 3.7:1 |
78.3 | NMP | (1) 2 h at 0 °C, | NMR + MALDI-TOF MS | 99 |
| (2) 56 h at RT | |||||||
| 18 | N/A | BiBB 1:1 |
21 | NMP | (1) 2 h at 0 °C | NMR | 122 |
| (2) 22 h at RT | |||||||
| N2 atmosphere | |||||||
| 21 | 17 |
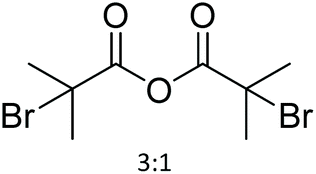
|
64 | Pyridine, DMAP | 96 h at RT | NMR + FTIR + elemental analysis | 23 |
| 21 | 66.8 or 89.5a | BiBB 2:1 |
42 | NMP | (1) 2 h at 0 °C, | NMR + FTIR | 65 |
| (2) 18 h at RT | |||||||
| 21 | 21 |
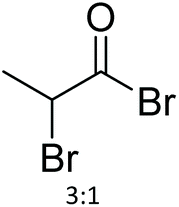
|
64 | NMP | 96 h at RT | NMR + MALDI-TOF MS | 74 |
Although bromine is the predominant halogen in the literature, chloro-CD initiators can also be successfully prepared. Analogously to bromo derivative, chloro initiator was prepared via esterification with 2-chloropropionyl chloride.114 Iodine-CD-based initiator was synthesised for iron-based ATRP. As the direct esterification with iodoisobutyryl bromide was unsuccessful, halogen exchange of octadeca-O-(isobutyryl bromide)-α-CD was implemented.115
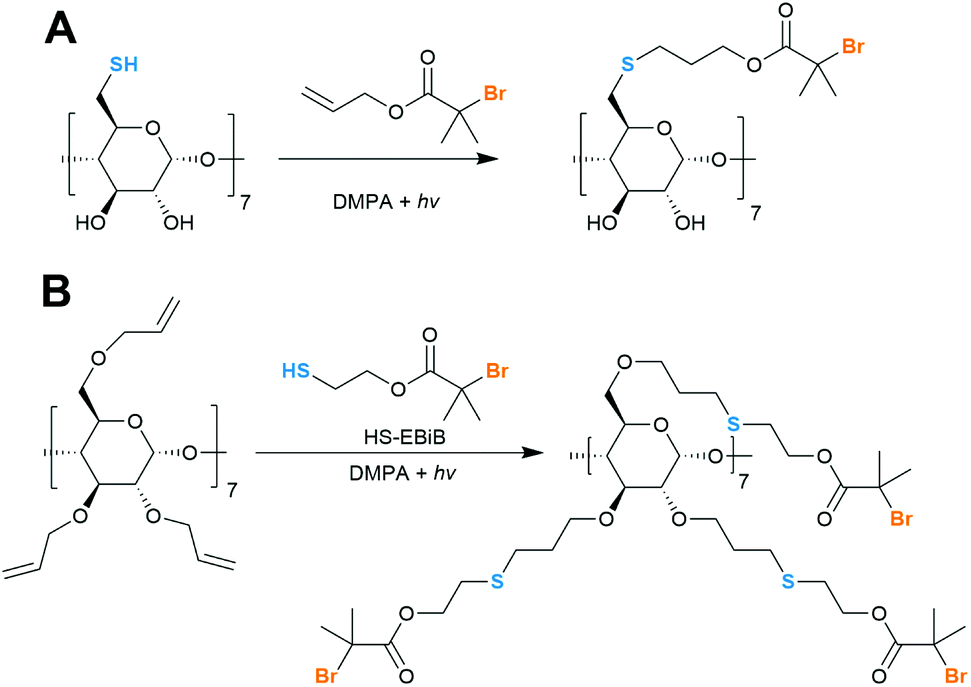 | ||
| Fig. 8 Thiol–ene reaction works for both thio-CD (A, Becer et al.) and allyl-CD (B, Yi et al.) but the first approach requires reduction of cyclodextrin derivative with DTT immediately before the reaction. | ||
Increased solubility of allyl-CD as compared to native CD allowed for reaction to be run in common organic solvent rather than NMP which is difficult to remove. This thiol–ene reaction gave heptakis(2,3,6-tri-O-(6-(3-thiahexyl)-2-bromo-2-methylpropanoate)-β-CD (21-Br-S-β-CD) in 90% yield and the product was duly analysed with MALDI-TOF. To ease data analysis and remove unnecessary isotopic patterns, bromine atoms were removed with tributyltin hydride. 7 and 14-active site species should also be available although this would require a protecting group, for example, benzylation of the secondary face.
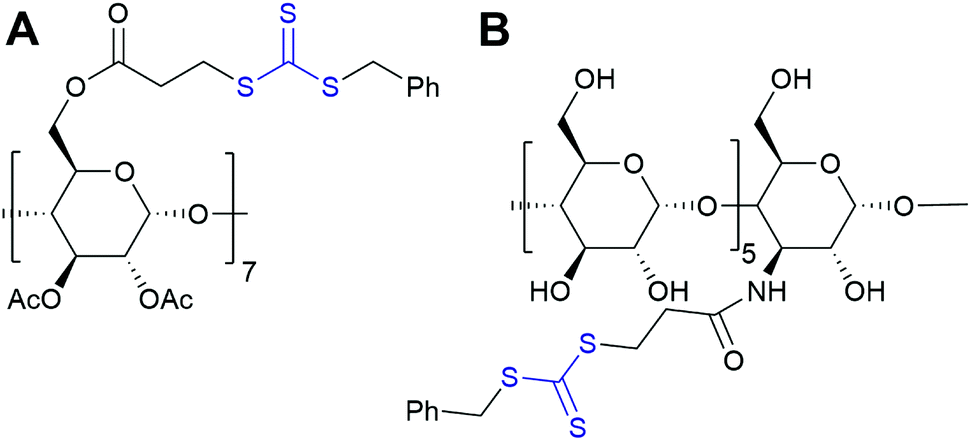 | ||
| Fig. 9 Structures of RAFT agents based on A: β-CD and B: α-CD. | ||
In summary, cyclodextrin can be modified with tosylate group or halogens to give robust synthons which can be further utilised in the synthesis of initiators or used as a place of attachment for polymer chains. TEMPO, ATRP initiator and RAFT agent have been successfully prepared starting from either pre-functionalised or native cyclodextrin. The primary or secondary hydroxyl group can be utilised selectively either by exploiting specific reactions or incorporation of standard protecting groups.
3 CD-polymer covalent conjugates
There are three basic architectures of covalent cyclodextrin-polymer conjugates: multiarm CD-centred polymers, CD-pendant polymers, and CD-capped polymers (Fig. 10). All three can self-assemble to form sophisticated structures. In this section, different polymerisation methods and their compatibility with cyclodextrin will be discussed. Native cyclodextrin has not been reported to interfere with polymerisation as such however cyclodextrin should be thoroughly dried prior to any reaction. As ionic polymerisation is nucleophile-sensitive, naked cyclodextrin face which is not involved as initiators should be protected.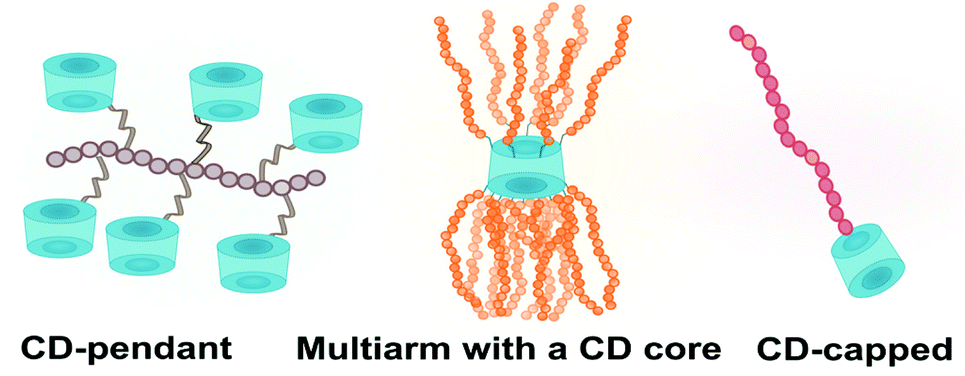 | ||
| Fig. 10 Three basic architectures of covalent cyclodextrin-polymer conjugates. | ||
3.1 Multiarm polymers cyclodextrin-centred polymers
Haddleton et al. reported the first polymerisation from a cyclodextrin core with attached ATRP initiators.23 heptakis[2,3,6-tri-O-(2-bromo-2-methylpropionyl]-β-CD (21-Br-β-CD) was used to polymerise methyl methacrylate (MMA), styrene and to prepare block copolymers by sequential addition. The reactions were run in toluene using standard ATRP conditions, with reagents ratio: [I]:[Cu(I)]:[L] 1:2:4, where L is a bidentate ligand: n-propyl-2-pyridylmethanimine and I is CD-initiator. Although polymerisation of MMA was successful with Đ (dispersity) < 1.15, styrene proved to be problematic due to autoinitiaton and star–star coupling at high temperatures which did not improve upon decreasing concentration of the initiator. SEC analysis showed that multiarm polymers which have more round shape have lower hydrodynamic radii than their linear analogues due to calibration of SEC with linear standards. Hence, polymers were hydrolysed to cleave the arms and give linear polymers for the SEC analysis to avoid bias. Although this approach provides reliable SEC results, hydrolysis can even take up to a week128 and hence slows down the analysis massively. Alternatively, light scattering method can be used.
Chain–chain coupling scales with the number of propagating chain ends and hence is difficult to eliminate during the synthesis of multiarm polymers. It intensifies at high monomer conversion. Hence, to minimise star to star coupling, Reynaud et al. polymerised tert-butyl acrylate in solution rather than bulk with just [1]:[1] molar ratio of [I]:[Cu(I)Br].125 Such polymers can be hydrolysed in TFA to give anionic polymers with potential biomedical applications.129 It was showed that the same polymerisation conditions can be applied to both α and β-CD bromo/chloroproprionyl initiator. Although the obtained dispersity was narrow, detection of linear chains suggests that some leftover 2-bromopropionyl bromide (BPB) from esterification was trapped in the cyclodextrin cavity and initiated growth of unbound chains. This could have been caused by a simplified purification procedure as opposed to chromatographic purification included in similar protocols.23,129 Another study showed that the choice of halogen can have a significant impact on polymerisation. 2-Chloropropionate-CD was showed to work better than 2-bromoisobutyrate-CD for ATRP polymerisation of N-isopropylacrylamide (NIPAM) in polar organic solvents, such as acetonitrile.58
CD-initiator was also employed in aqueous ATRP. A cationic monomer – methyl chloride-quaternised 2-(dimethylamine ethyl) methyl methacrylate was polymerised.25 Various results were reported for reactions run at different temperatures due to the effect of temperature on the solubility of the CD-based initiator – 21Br-β-CD initiator was found only moderately soluble in water. At a higher temperature, lower conversions were obtained. To potentially improve polymerisation parameters, fewer initiator moieties per cyclodextrin could be used and remaining hydroxyls could be modified to increase initiator solubility.
SET-LRP, which employs both Cu(0) and Cu(II), was also employed for formation of CD-based polymers.109 A pre-activated copper wire (washed with HCl) and wrapped around a stirrer bar was the source of Cu(II). The addition of Cu(II) preserved chain end fidelity and suppressed star coupling. This effect was enhanced by running polymerisation in DMSO – solvent promoting disproportionation. The full conversion was achieved while maintaining living ends which allowed for chain extension to form tri-block-copolymer.
The methods reported so far require relatively big quantities of copper, usually >4000 ppm. High content of copper can be problematic, particularly for medical applications, as copper is toxic and difficult to remove completely from the final product.130 Matyjaszewski et al. applied simplified eATRP to prepare star block copolymers with a cyclodextrin core with as little as 50 ppm of Cu complex.24 In eATRP by applying potential, Cu(I)/Cu(II) ratio can be controlled and so the reaction rates and termination. Similarly, ARGET-ATRP was applied for the preparation of (meth)acrylate block copolymers.127 ARGET-ATRP is based on continuous regeneration of active metal species – namely reduction of Cu(II) which arises from irreversible radical termination. Additionally, the reducing agent such as ascorbic acid or Sn(EH)2 can scavenge oxygen and other radical inhibitors improving livingness of the polymerisation.
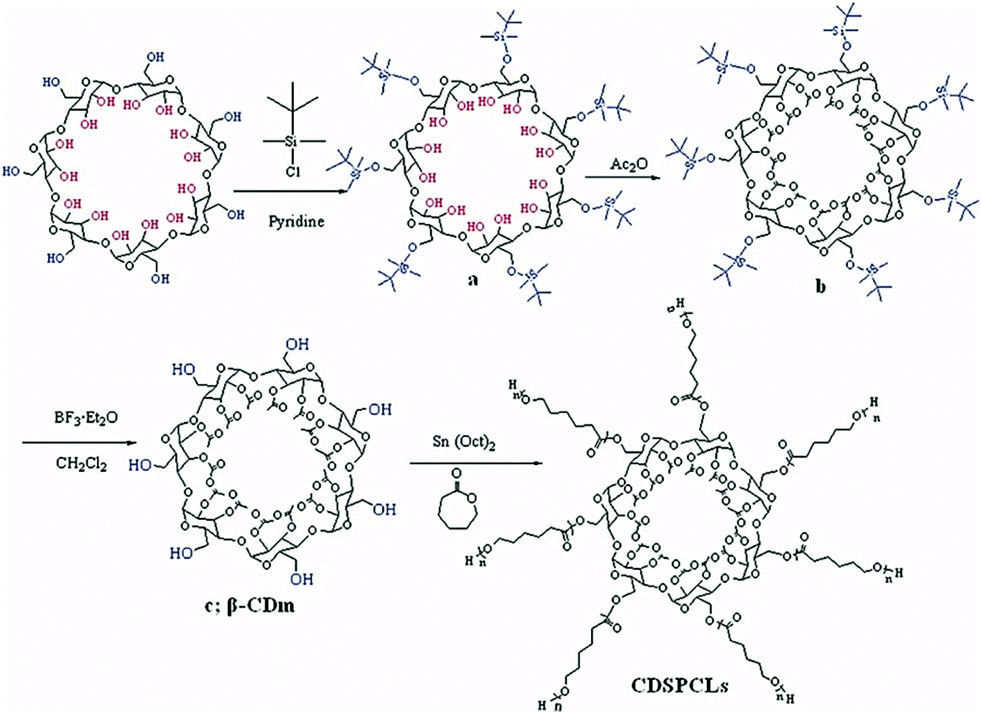 | ||
| Fig. 11 Synthetic route for the preparation of multiarm polymers via ROP. Reprinted with permission from John Wiley and Sons Inc., Copyright 2008.134 | ||
Sn(Oct)2 catalyst ensured fast and simultaneous initiation of all hydroxyl groups. Poly(ε-caprolactone) arms were subsequently coupled with DCC to carboxyl-terminated poly(ethylene glycol) (PEG) to form amphiphilic copolymer that could undergo micellisation.
A judicious choice of functionality of cyclodextrin and monomers allows for growth of two different polymers at the bottom and the top face of the cyclodextrin without protecting groups. Adeli et al. used biocompatible materials to grow poly(lactic acid) arms from primary hydroxyls and poly(2-ethyl-2-oxazoline) from tosylate secondary hydroxyls.136 The reverse order of polymerisation yielded green solution due to the complexation of Sn(Oct)2 by nitrogen atoms of poly(oxazoline). The chain end group allowed to tune solubility of the final product. Similarly, Shen et al. showed that ring opening polymerisation and ATRP can be coupled to independently functionalise two faces of cyclodextrin.137 Such mixed approaches truly leverage the versatility of CD as an initiator.
γ-CD proved to be an excellent core for the synthesis for a diblock eight-arm star copolymers via ring-opening metathesis polymerisation (ROMP) (Fig. 12).135 As ROMP is functional group-tolerant, hydroxyls not directly involved in the polymerisation did not require any special treatment. Octakis-(6-amino-6-deoxy)-γ-CD was first functionalised with norbornene (Nb) via reaction with N-hydroxysuccinimide precursor (Nb-NHS). After forming an intermediate with Grubbs’ 3rd generation catalyst, cyclodextrin was reacted with norbornene-functionalised hexaethylene glycol. Subsequently, the polymer was coupled with Nb-PEG and quenched with excess ethyl vinyl ether.
 | ||
| Fig. 12 (a) Chemical structures and corresponding cartoon representations of norbornene functionalised γ-CD (γ-CD-Nb8) and polymer chains (Nb-HEG, Nb-PEG). (b) Core-first/graft-from synthesis of a diblock eight-arm star copolymers via ring-opening metathesis polymerisation (ROMP). Reprinted with permission from Royal Society of Chemistry, Copyright 2020.135 | ||
Whereas chain extension with hexaethylene glycol was successful, the second chain extension with PEG was less efficient and homo-arm star polymer were observed in SEC. Higher efficiencies were achievable for lower molecular-weight blocks. Nevertheless, multiarm polymers were characterised with narrow dispersity (Đ = 1.12–1.19).
Among coupling reactions, CuAAC coupling between azide and alkyne group is predominant.139 For more insights and applications of CuAAC coupling to cyclodextrin not only in polymer chemistry, the reader is directed to a detailed review.140 CuAAC coupling can be easily monitored by 1H NMR by the appearance of a triazol proton signal around 8 ppm or disappearance of azide peak in FT-IR spectrum. Unreacted alkyne-polymer chains can be removed with the aid of azido-functionalised resin. As described earlier, azido-CD can be prepared via nucleophilic displacement of halogens. This, however, is limited to the primary face of cyclodextrin. To leverage all hydroxyl groups, a hybrid approach is used: the primary face is modified via click reaction whereas the secondary face is used in ATRP or ROP polymerisation. Such heteroarm star copolymers were afforded by coupling alkynyl-PDEA30 (poly(2-(diethylamino)ethyl methacrylate) to (N3)7-CD-(PNIPAM)14, prepared via ATRP (Fig. 13).98 Alternatively, bromide can be incorporated via esterification with 2-bromopropionic bromide and then subsequently displaced with sodium azide to give β-CD-(N3)21 (heptakis[2,3,6-tri-O-(2-azidopro-pionyl)]-b-cyclodextrin) (Fig. 14).74
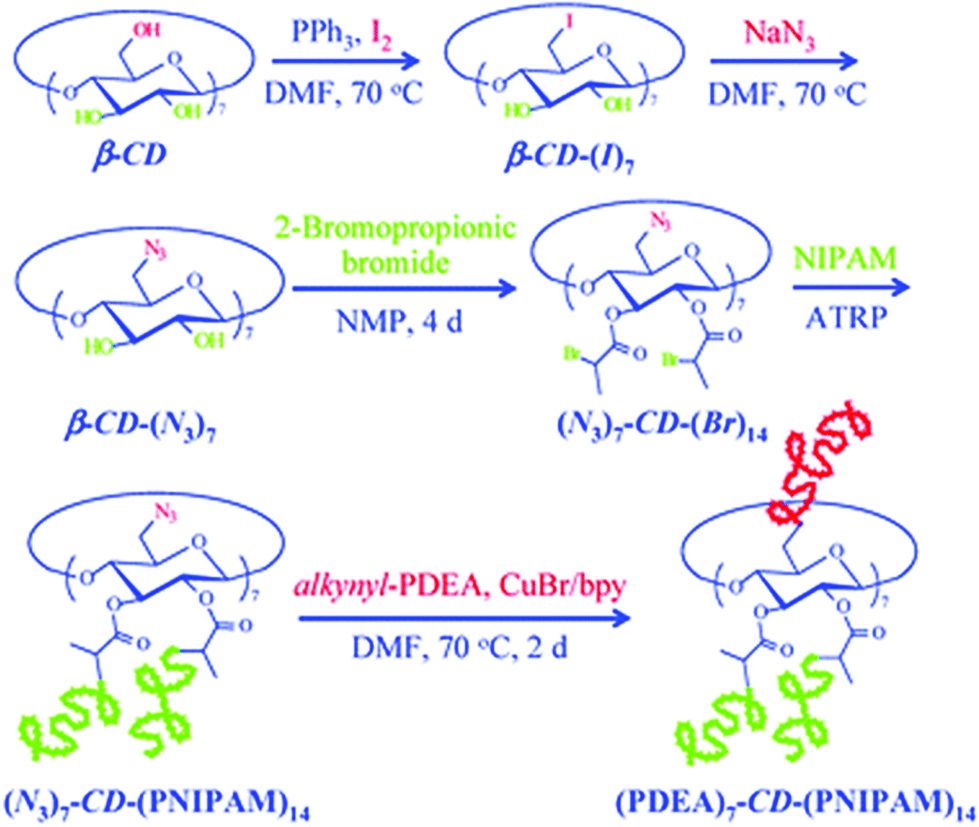 | ||
| Fig. 13 Synthetic scheme for stimuli-responsive double hydrophilic Janus-type A7B14 star copolymers, (PDEA)7-CD-(PNIPAM)14, based on β-CD derivative via the combination of ATRP and click reaction. Reprinted with permission from Royal Society of Chemistry, Copyright 2009.98 | ||
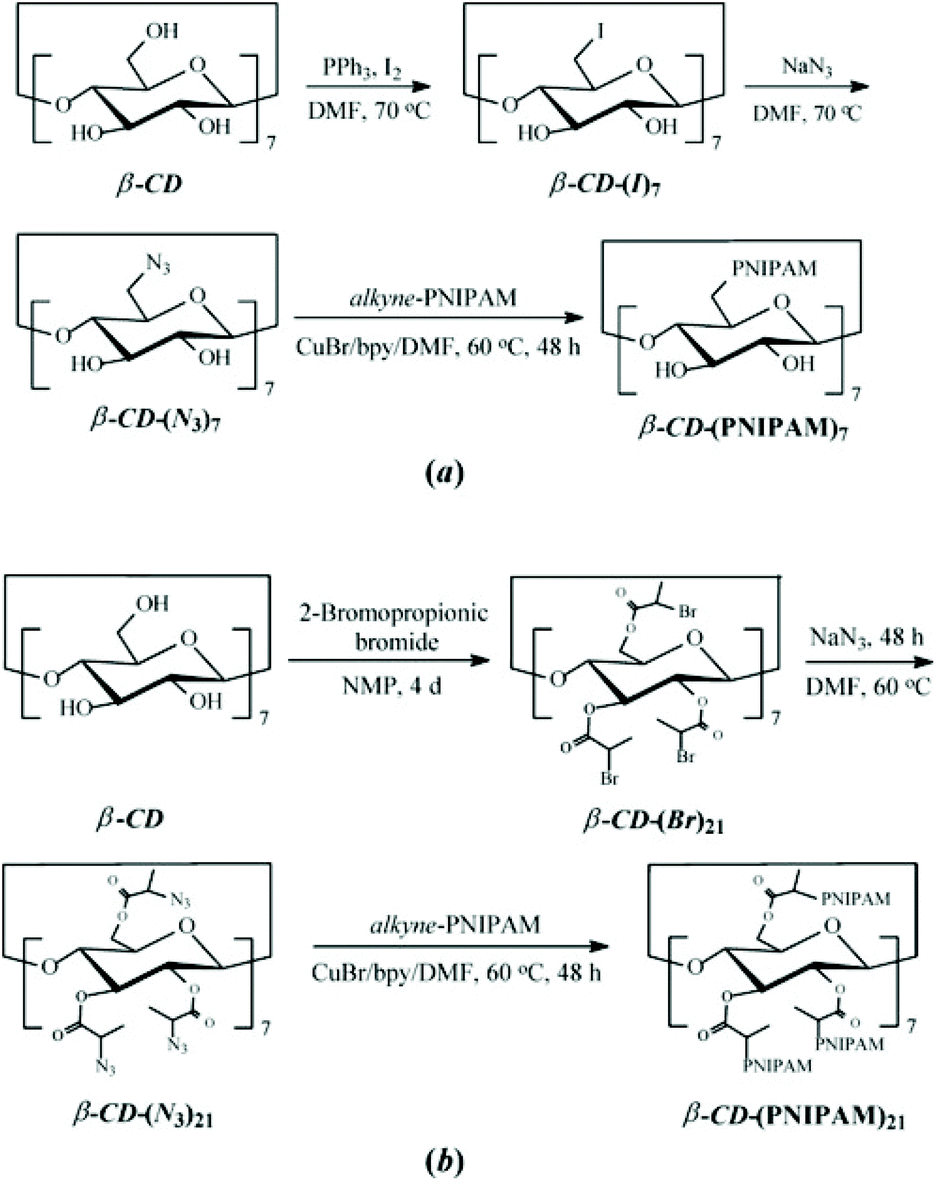 | ||
| Fig. 14 Synthetic routes for the preparation of well-defined (a) 7-arm (b) 21-arm PNIPAM star polymers via “click” reactions of alkyne-PNIPAM azido-β-CD. Reprinted with permission from Wiley Periodicals, Inc., Copyright 2008.74 | ||
Reineke et al. successfully carried out 1,3-dipolar cycloaddition of heptakis-(6-azido-6-deoxy-2,3-di-O-acetyl)-β-CD with alkyne dendrons built of ethyleneamine units.141 The reaction was catalysed by Cu(I) using copper sulfate/sodium ascorbate and only fully-substituted click-clusters were obtained as confirmed by ESI-MS. A different study compared CD-PEG conjugates for biological applications.
Different lengths, namely Mn 550, 2000 and 5000, of alkyne-functionalised PEG methyl ester were coupled to heptakis-(6-deoxy- 6-azido)-β-CD.142 All reactions were run at 70 °C as below this temperature reactions gave low yields. Although coupling reactions of PEG2000 and PEG5000 gave high yields: 99% and 88% accordingly, mass spectrometry analysis revealed the presence of hexasubstituted product, apart from the desired heptasubstituted one. On the other hand, the coupling of limitation of grafting-to approach which becomes less efficient as steric hindrance increases for longer polymer chains. PEG550 was less efficient giving only 55% yield both only heptasubstituted cyclodextrin was observed. Other coupling reactions were also implemented such as thiol–ene base catalysed Michael addition.27 Thiol-functionalised cyclodextrin was reacted with various monomers and vinyl-terminated polymers prepared via catalytic chain transfer polymerisation (CCTP) in the presence of dimethylphenylphosphine or hexylamine.
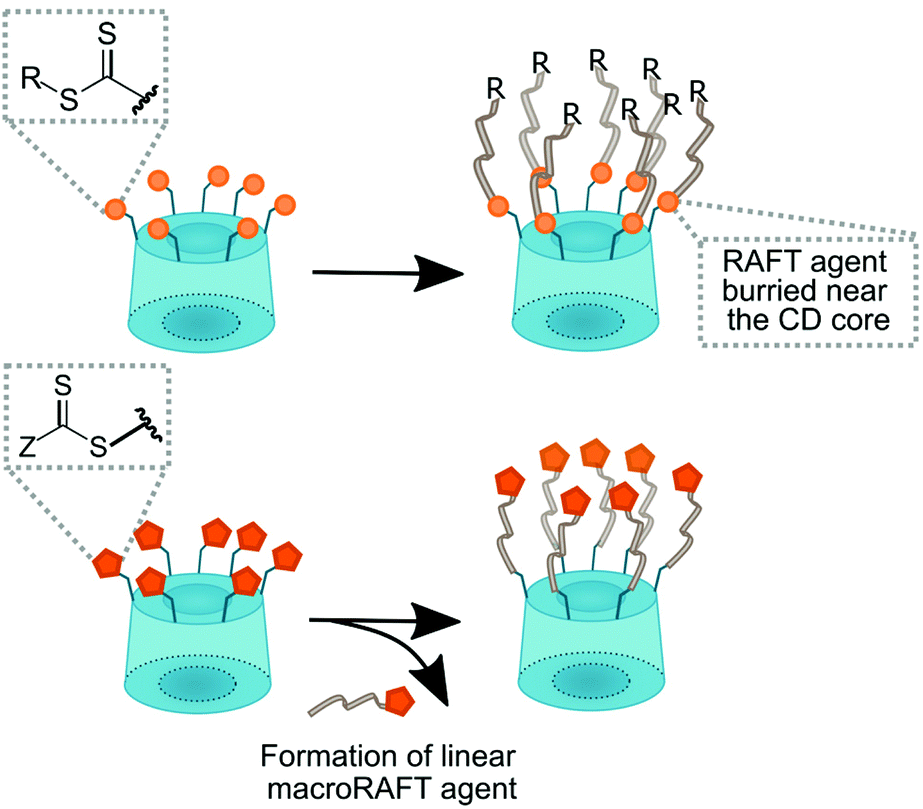 | ||
| Fig. 15 In a CD-RAFT agent, cyclodextrin can be either part of the Z or the R group. | ||
3.2 CD-pendant polymers
Polymers with cyclodextrin side groups can have interesting applications due to the exposure of dangling cyclodextrins that are free to encapsulate small molecules. They can assemble with chains with complementary pendant groups such as adamantane or cholic acid. Pendent cyclodextrins can be further utilised by turning into ATRP initiators via esterification.Such macro initiators give unusually high grafting densities brush polymers (Fig. 16).144 There are two basic approaches to linear polymers with pendent cyclodextrin groups: homo/copolymerisation of a monovinyl CD-based monomer or conjugation of cyclodextrin onto polymer with a complementary functional side chain (Fig. 17).
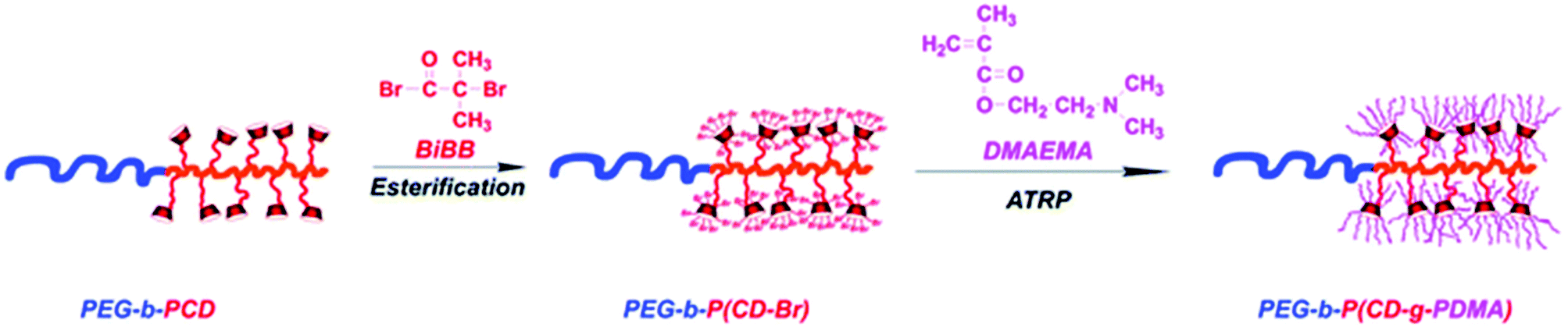 | ||
| Fig. 16 Synthesis of the polycationic brushes via ATRP. Reprinted with permission from The Royal Society of Chemistry, Copyright 2014.144 | ||
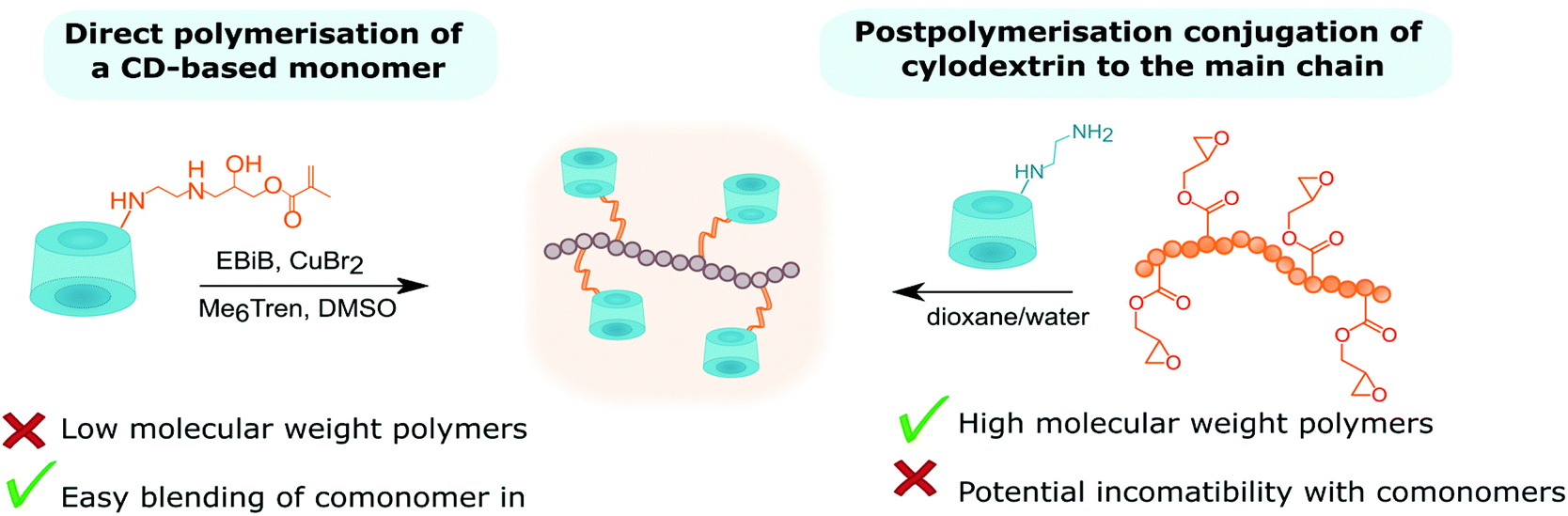 | ||
| Fig. 17 Comparison of two approaches towards CD-pendant polymers. | ||
Attempts to directly polymerise CD-based monomers often lead to low molecular weight polymers due to the build-up of steric hindrance.107,108 Perhaps extending the linker (longer amine) could allow for higher polymerisation degrees. The alternative approach is based on postpolymerisation modification (Table 4). Mono-(6-amino-6-deoxy)-β-CD was coupled to poly(acrylic acid) chains with the aid of a peptide coupling agent – benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), achieving 4% side chain modification.145 Another common approach starts with the synthesis of poly(glycidyl methacrylate). Analogously to the acid group, the epoxy group can be coupled to mono-(6-amino-6-deoxy)-β-CD.146 This approach allows for the synthesis of high molecular weight polymers. One of the shortcomings of postpolymerisation modification is its potential incompatibility with other monomers incorporated into the chain. Incompatibility can be resolved by ‘click chemistry’ which is characterised with high specificity and fidelity in the presence of various functional groups. Copolyesters with pendant alkyne groups were synthesised and subsequently coupled via CuAAC cycloaddition with mono-(6-azido-6-deoxy)-β-CD giving β-CD-functionalised copolyester.147 Copolyesters were made via ring-opening polymerisation of propargyl-modified lactones and ε-caprolactone in the presence of Sn(Oct)2 as a catalyst and adipic acid as co-initiator.
| CD precursor | Polymer precursor | Preparation of CD-pendant polymer | Ref. |
|---|---|---|---|
| Mono-(6-amino-6-deoxy)-β-CD | Poly(acrylic acid) | PyBOP coupling | 145 |
| Mono-(6-amino-6-deoxy)-β-CD | Poly(glycidyl methacrylate) | Amine-epoxy reaction | 146 |
| Mono-(6-azido-6-deoxy)-β-CD | Propargyl-functionalised copolyesters | CuAAC cycloaddition | 147 |
Propargyl-modified lactones were prepared in two steps: the nucleophilic substitution of cyclohexanone with propargyl bromide and Baeyer–Villiger oxidation with an excess of m-chloroperoxy-benzoic acid. To suppress formation of polyrotaxanes during CD-copolyester coupling, polyester chains were capped with bulky 2,2-diphenylethanamine.
Polymers with alkyne side-chain functionality can be also prepared via ATRP. However, polymerisation of propargyl methacrylate resulted in broad dispersity values and cross-linked networks at high monomer conversion.148 This may be ascribed to the coordination of alkyne groups of the monomer to the copper catalyst. The polymerisation of 3-azidopropyl methacrylate, on the other hand, gave well-defined polymers that were coupled to various propargyl-functionalised molecules. Reactions proceeded with nearly full conversion in less than 2 h. Further, it was observed that some coupling reactions were more efficient for azido-polymer chains than analogous azido-monomer. This autocatalytic effect can be explained by complexation of copper complex by triazole formed along the polymer backbone.149
3.3 CD-capped polymers
CD-capped polymers are polymers with cyclodextrin chain end group. They can be prepared by polymerisation from a functional initiator bearing one cyclodextrin moiety or by modification of an existing chain end (Fig. 18). CD-capped polymers often serve as a host in preparation of polymers such as amphiphilic block-copolymers via host–guest interactions.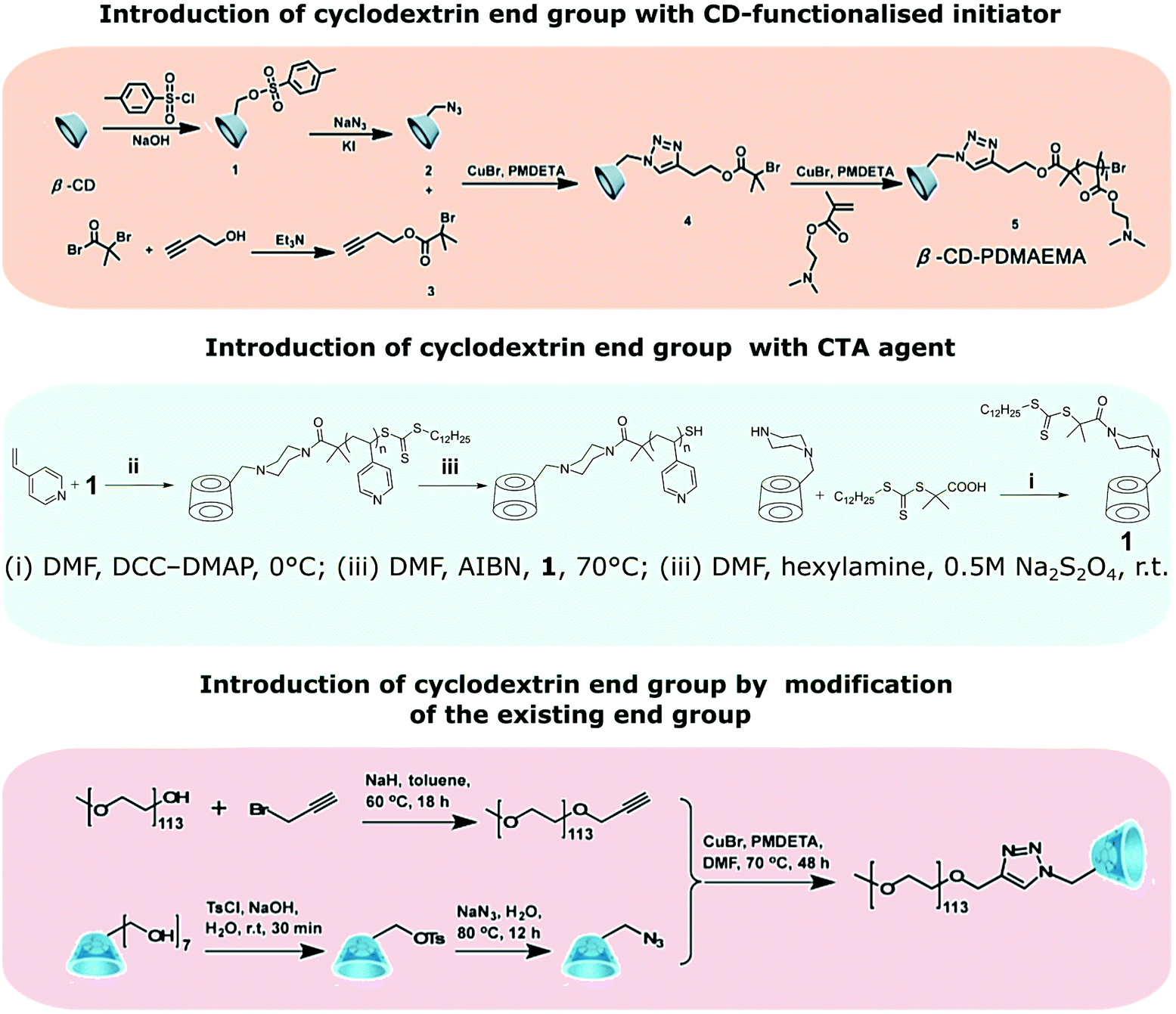 | ||
| Fig. 18 Strategies towards the synthesis of CD-capped polymers. Reprinted with permission from American Chemical Society, Copyright 2014;151 from The Royal Society of Chemistry, Copyright 2008;152 from American Chemical Society, Copyright 2017.153 | ||
Analogously to multiarm initiators, esterification with 2-bromoisobutyryl bromide can provide cyclodextrin ATRP initiator with a single initiating moiety.150 This approach, however, is not free from pitfalls such as formation of undesired multisubstituted cyclodextrin. CuAAC coupling of mono-azido-CD with but-3-ynyl-2-bromo-2-methylpropanoate allows for precise synthesis.151 RAFT polymerisation provides another facile method of introducing cyclodextrin as chain end group by using CD-functionalised CTA agent.152 Alternatively, many chain end groups can be converted to alkyne and then coupled to azido-CD.153
Not only linear polymers can be capped with cyclodextrin. Tian et al. polymerised NIPAM from a cyclodextrin core and used it as a macroinitiator in ATRP polymerisation of monovinyl-CD monomer (Fig. 19).114 This created a polymer with merged encapsulation properties of cyclodextrin and thermoresponsive behaviour of PNIPAM. Not all the arms chain extended, suggesting either loss of chlorine atoms or low initiation efficiency. The analogous reaction for linear polymers was successful hence it was concluded that the monomer was not reactive enough to overcome the steric hindrance of the multiarm polymer. A single polymer chain can be also grown directly from native cyclodextrin via ROP of lactone and other cyclic esters. If the polymerisation is run in bulk there is no competition from the solvent and monomer – lactone gets trapped in the cyclodextrin cavity. This activates lactone towards polymerisation and leads to a polyester-tethered cyclodextrin.154 As different cyclodextrins have different cavity sizes, their initiating efficiency varies: β-CD worked best for lactone whereas α-CD failed completely. The essential role of complexation was confirmed in a later study by inhibiting polymerisation with adamantane which was forms strong inclusion with cyclodextrin (Fig. 20).26
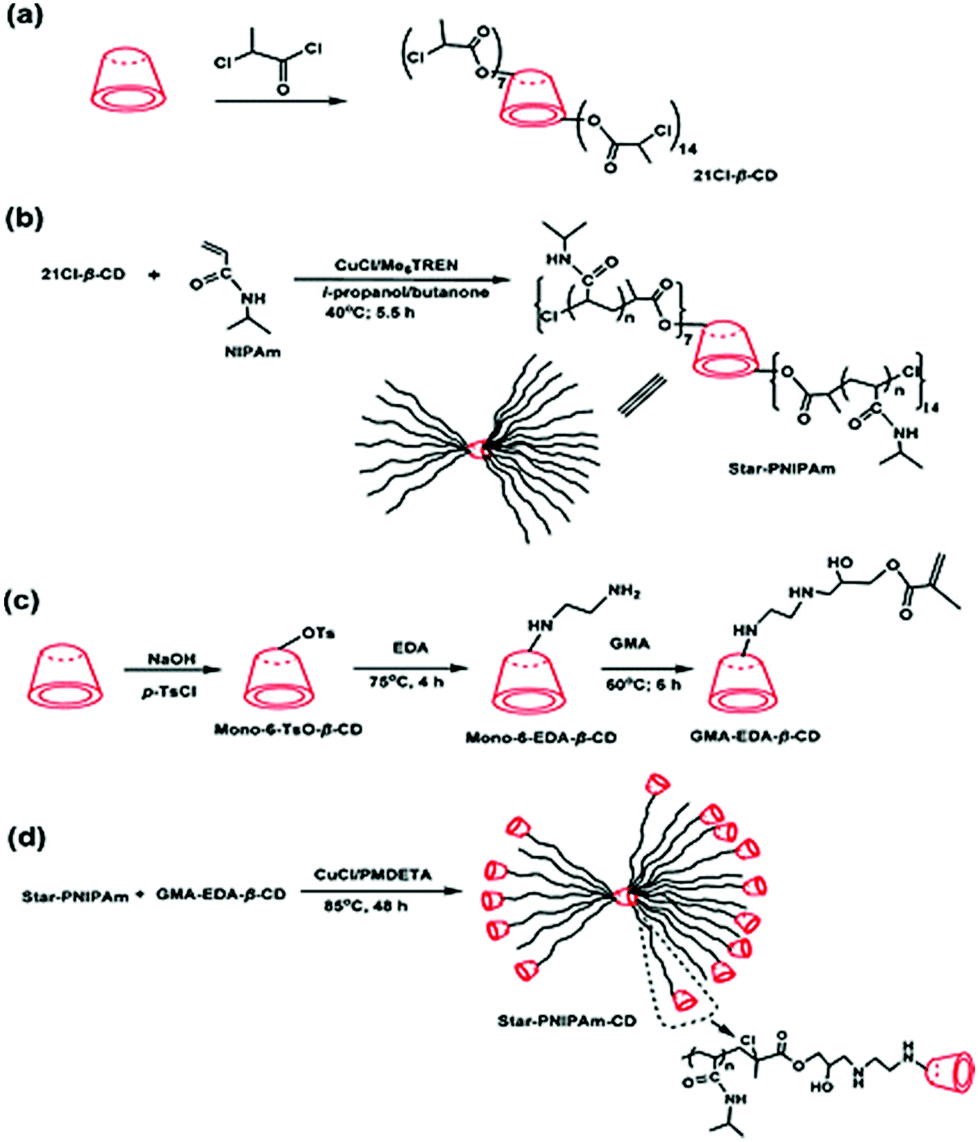 | ||
| Fig. 19 Schematic Illustration of the synthesis routes of (a) heptakis[2,3,6-tri-O-(2-chloropropionyl)]-β-CD, (b) star-PNIPAm, (c) CD-functionalised monomer and (d) star-PNIPAm-CD. Reprinted with permission from American Chemical Society, Copyright 2010.114 | ||
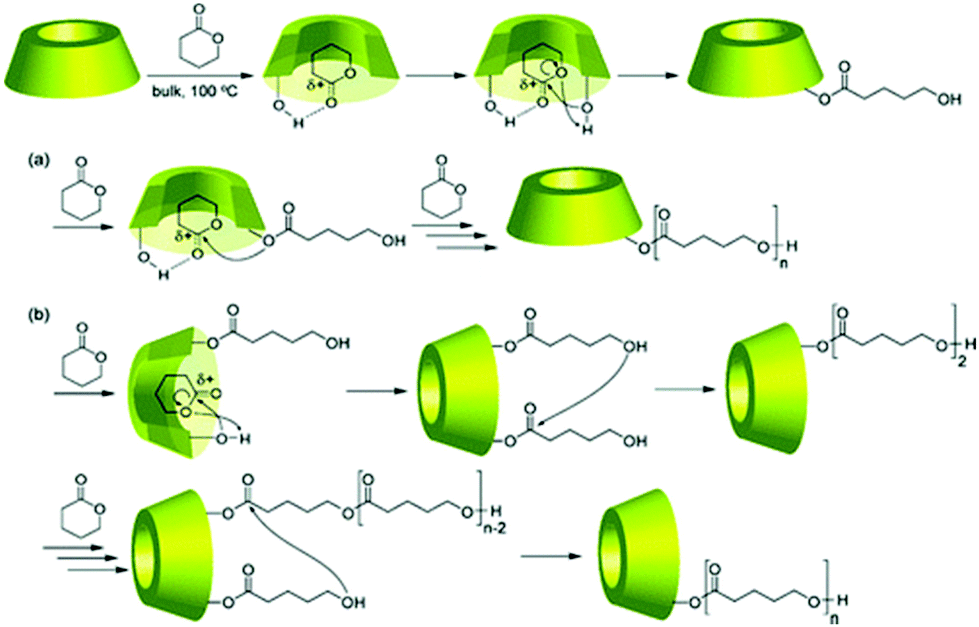 | ||
| Fig. 20 Proposed mechanism of the polymerisation of δ-VL by CD in bulk. Reprinted with permission from American Chemical Society, Copyright 2010.26 | ||
This further study based on solid-state NMR also gave some mechanistic insights into the polymerisation. Namely, it was suggested that it is not the hydroxyl polymer chain end that attacks another lactone but the new monomer. This was confirmed by using mono-2-O-(5-benzyloxypentanoyl)-beta-CD which despite lack of hydroxyl chain end, initiated polymerisation. The monomer, after being activated by formation of a hydrogen bond between lactone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and cyclodextrin OH as observed by FT-IR, gets attached to CD to give disubstituted CD. Then the monomer hydroxyl groups attack carbonyl of the polyester chain and get inserted between CD and polymer chain.
O and cyclodextrin OH as observed by FT-IR, gets attached to CD to give disubstituted CD. Then the monomer hydroxyl groups attack carbonyl of the polyester chain and get inserted between CD and polymer chain.
In summary, cyclodextrin is compatible with various polymerisation methods, nonetheless, it is mainly found in ATRP protocols. Out of controlled polymerisation techniques, RAFT and nitroxide mediated polymerisation seem to be the least versatile for the preparation of cyclodextrin-polymer conjugates and there is little literature available concerning those two techniques and cyclodextrins. These techniques are particularly inefficient for the preparation of multiarm polymers. This is as cyclodextrin is a relatively small core and steric hindrance leads to increased termination events. Nonetheless, cyclodextrin has been successfully applied as ATRP and ROP initiator, and CD-functionalised monomers were polymerised in controlled manner. Cyclodextrin is particularly promising for the preparation of multiarm polymers as cyclodextrin can overcome common problems such as low arm density and can be applied in the synthesis of miktoarm polymers as well. There are several synthetic strategies towards cyclodextrin-pendant and capped polymers. Such materials can find applications in building more complex structures via self-assembly.
4 Utilisation of CD-polymer conjugates
Since the 1930s when systematic studies on cyclodextrin have started, cyclodextrin has found applications in various fields. As the strongest features of cyclodextrin are encapsulation of small hydrophobic molecules and availability to form non-covalent interactions it is not surprising that cyclodextrin became popular in pharmaceutical155–157 and food industry12 as well as analytical chemistry.158,159 Allying cyclodextrin with controlled living polymerisation has also proved to be fruitful. It is outside the scope of this article to comprehensively review all fields that CD-polymer conjugates have been applied in. For a comprehensive review on cyclodextrin applications in drug delivery,4,160,161 cancer therapy2,8,162 and hydrogels20,163 the reader is directed elsewhere. Here, we aim to provide some of the pioneering examples as well as recent studies in those fields to show the newcomers the scope of applications of cyclodextrin-polymer conjugates.4.1 Gene and drug delivery
Cyclodextrin can be asymmetrically functionalised allowing for combining multiple functions. Introduction of azide moieties on the primary rim and alpha-bromopropionate functionalities on the secondary rim led to the construction of gene delivery vector with magnetic resonance imaging contrast (Fig. 21).32N,N-Dimethylaminoethyl methacrylate (DMA) cationic chains grown with ATRP can complex anionic plasmid deoxyribonucleic acid (DNA) whereas the azide handle allows for attachment of functionalised gadolinium complex. Self-assembly led to micellar nanoparticles with entrapped DNA – theranostic nanocarriers. Multiarm polymer enhanced relaxation of the magnetic contrast compared to commercial standards. Analogously, drug rather than gene carriers can be designed.164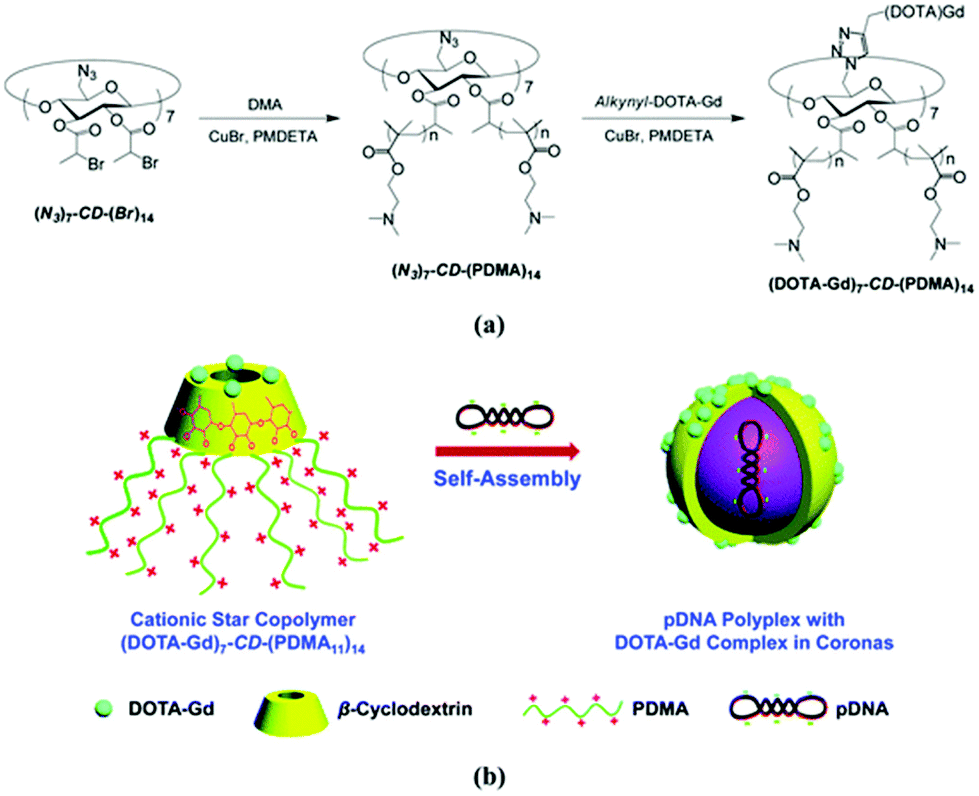 | ||
| Fig. 21 (a) Synthetic route for the preparation of well-defined star copolymers, (DOTA-Gd)7-CD-(PDMA)14. (b) Schematic illustration of (DOTA-Gd)7-CD-(PDMA)14 star copolymers dually acting as pDNA delivery vectors and MR imaging contrast agents. Reprinted with permission from The Royal Society of Chemistry, Copyright 2014.32 | ||
Recently, Becer et al. combined two features of cyclodextrin: encapsulation and selective functionalisation to prepare supramolecular cationic glycosylated polymers (Fig. 22).165 Glycosylation is a common tool for improving bioavailability and targeting properties of such polymers.166 Cationic polymer arms were grown from the primary face of cyclodextrin leaving access to the hydrophobic cavity from the bottom face for assembly with a glycosylated copolymer containing adamantane. This allowed for better structural optimisation than combining sugar and cationic groups on a single polymer. Increasing length of PDMAEMA improved the transfection efficiency up to an upper limit where increased steric hindrance excluded DNA plasmids. Pendant sugar groups enhanced cellular uptake of DNA in human skin explants and increased expression of delivered DNA in leukocytes as compared to non-glycosylated equivalents.
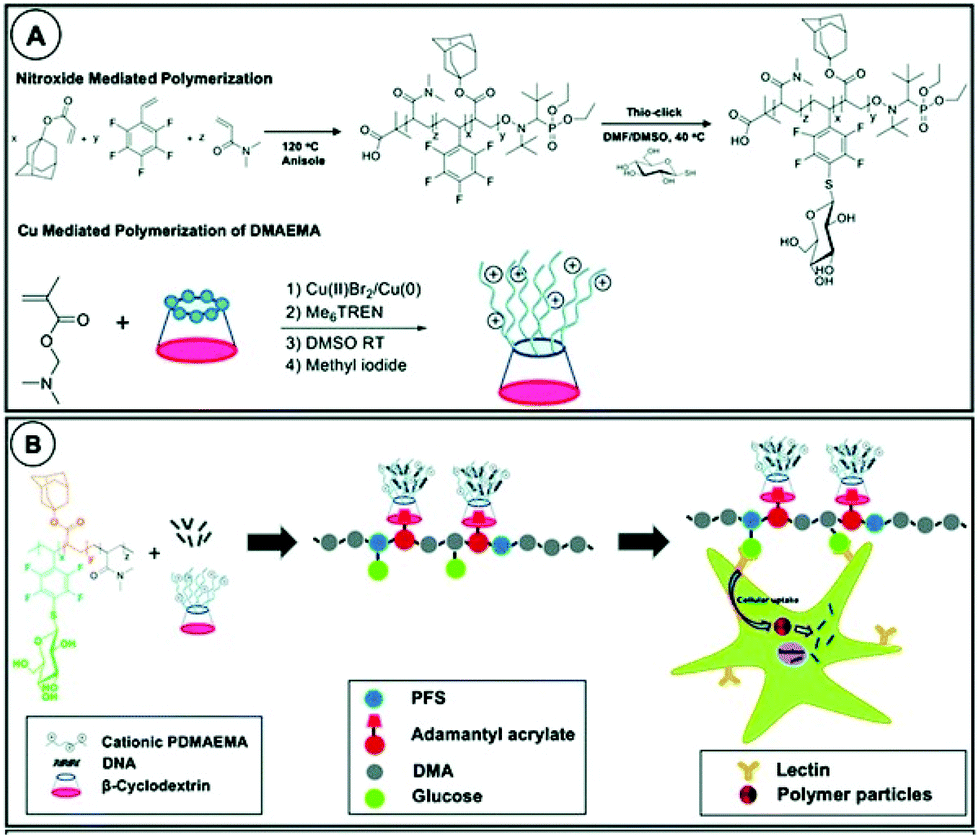 | ||
| Fig. 22 A: Polymerisation of the glycopolymer complexation groups; B: schematic presentation of the complexation. Reprinted with permission from The Royal Society of Chemistry, Copyright 2020.165 | ||
Multiarm polymers with CD core can also self-assemble and encapsulate drugs. pH-Responsive statistical PDPA-POEGMA (2-(diisopropylamino) ethyl methacrylate – oligo(ethylene glycol)-methylether-methacrylate) copolymers were grown from a cyclodextrin core for chemo/photothermal cancer therapy.167 Hydrophobic PDPA segment encapsulates hydrophobic doxorubicin (DOX) and releases the drug through swelling in an acidic environment as the segment becomes hydrophilic. Drug release is hence enhanced in the acidic environment of a tumour but leaching to the bloodstream (pH 7.4) is limited. DOX encapsulation efficiency was the highest (40.20%) for copolymer with equimolar content of DPA and OEGMA, providing a balance between water solubility in water and available volume of the hydrophobic domain for drug encapsulation. Co-loading of micelles with photothermal agent enhanced drug activity. However, only 52% of drug was released in in vitro test suggesting micelles might be too stable or drug is bound too strongly.
4.2 Functional nanoparticles
Multiarm polymers with a cyclodextrin core can be utilised in preparation of functional nanoparticles. Such polymers can form unimolecular micelles i.e. micelles built of a single molecule. Unimolecular micelles are characterised with increased stability, particularly against dilution.168 Lin et al. established a synthetic protocol for ferroelectric (PbTiO3) magnetic (Fe3O4) multifuncitonal nanoparticle.169 A triblock copolymer CD-P4VPb-PtBA-b-PS was prepared via ATRP from BiBB-functionalised cyclodextrin core and used to direct aggregation of inorganic compounds via interaction of metallic precursor with a specific polymer block (Fig. 23). It was found that appropriate solvent mixture was crucial for obtaining well defined, uniform core–shell nanoparticles. This strategy is versatile, allowing for the incorporation of different metals and tuning size by varying lengths of polymer blocks. The external, not bound to metal block can alter the solubility of the particle – PS outer chains aid solubility in common organic solvents and PEO in water. Recently, fluorescent unimolecular micelles were developed for non-invasive optical fluorescence imaging: organelle labelling and tumour localisation.170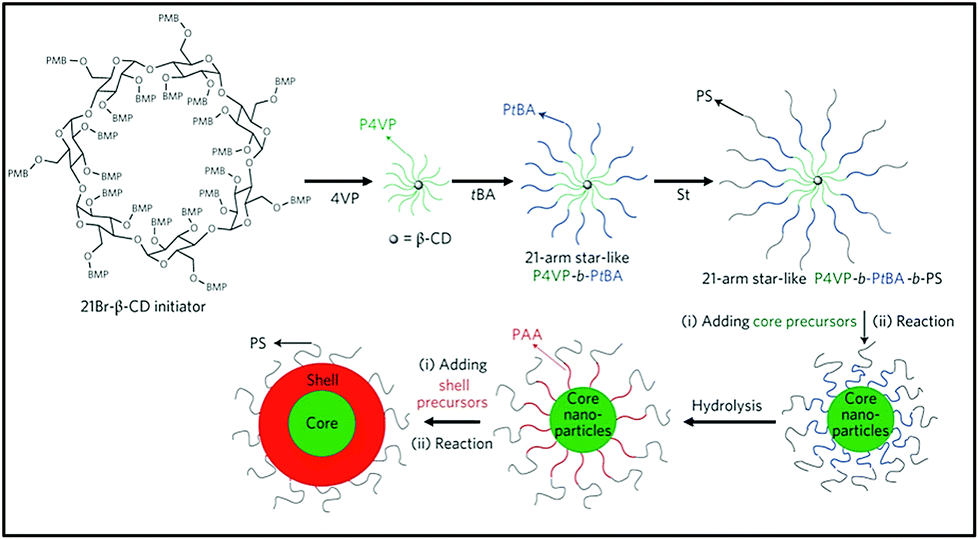 | ||
| Fig. 23 Schematic representation of synthetic strategies for functional nanoparticles. | ||
The ultra-small size of nanoparticles led to enhanced accumulation in tumours. Incorporation of biocompatible CD-core and hydrophilic POEGMA chains bypassed common problems encountered for fluorophore-based materials, namely toxicity and poor biodegradability. Encapsulation of a dye in polymer improved its photostability, compared to stand-alone dyes. The ultra-small size of nanoparticles led to the enhanced accumulation in tumours.
Functional nanoparticles can be also prepared from metallic nanoparticles by attaching cyclodextrin-pendant polymers to the particle surface. Iron oxide particles were coated with silica that provided amino functional end groups that were suitable for further modifications. This allowed for an easy attachment of 2-bromopropionyl bromide that initiated polymerisation of glycidyl methacrylate on the particle surface. Epoxy group of the methacrylate was utilised in appending cyclodextrins. Magnetic nanoparticles were proved to catalyse substrate-selective oxidation of alcohols. They also work as adsorbent, removing impurities such as bisphenol A. Purification was aided by magnetic separation of nanoparticles from clean liquid after complexation (Fig. 24).171
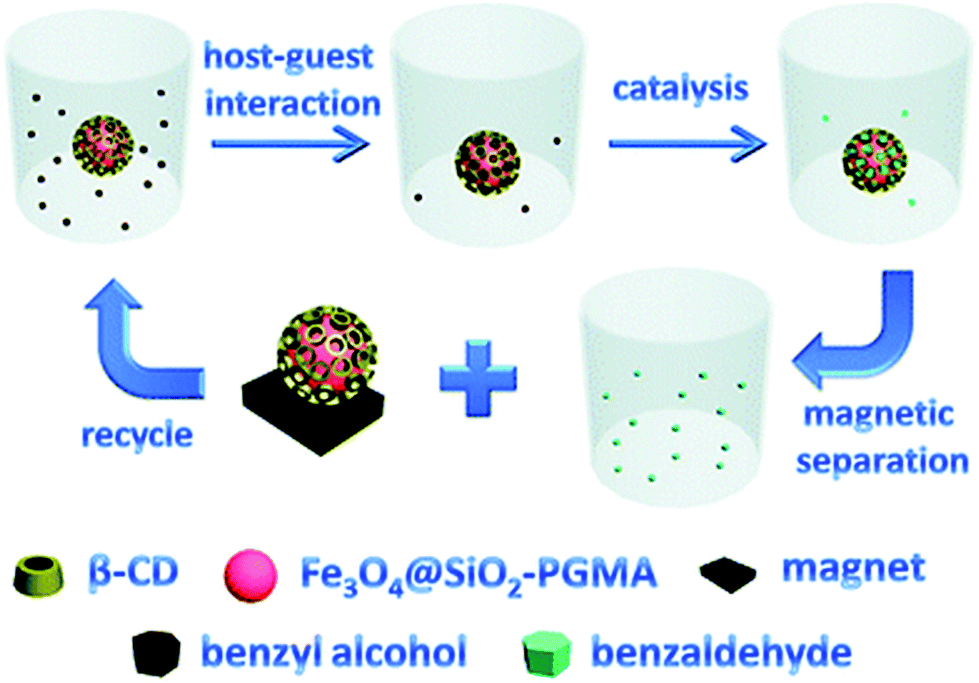 | ||
| Fig. 24 The schematic representation of the substrate-selective catalysis and recycling of the immobilized catalyst. Reprinted with permission from The Royal Society of Chemistry, Copyright 2011.171 | ||
4.3 Hydrogels
Cyclodextrin is a very promising building block for flexible hydrogels. Although covalently crosslinked hydrogels can be responsive, hydrogels based on supramolecular interactions are more tuneable, responsive to external stimuli such as light172 or redox145 stimuli and capable of self-healing.20,172 By combining polymer chains bearing CD moieties with polymer chains bearing complementary guest molecules, supramolecular interactions are created that provide durability and improved mechanical properties. Recently, a photoregulated photochromic hydrogel formed of spiropyran functionalised polyacrylamide (PAM) chain, decorated with azobenzene and β-CD pendant groups that could self-heal within 10 s was reported.173 Toughness of the hydrogel was controlled by dynamic host–guest interaction between cyclodextrin and azobenzene as azobenzene isomerises upon irradiation with light (Fig. 25). Depending on the wavelength of the light, toughness could be weakened or strengthened. By varying the content of pendant groups on PAM chains, tensile and rupture strain can be tuned, thus allowing for optimisation of mechanical properties of the gel. This is, however, limited by the water solubility of azobenzene functionalised PAM, capping the maximum azobenzene content in the gel.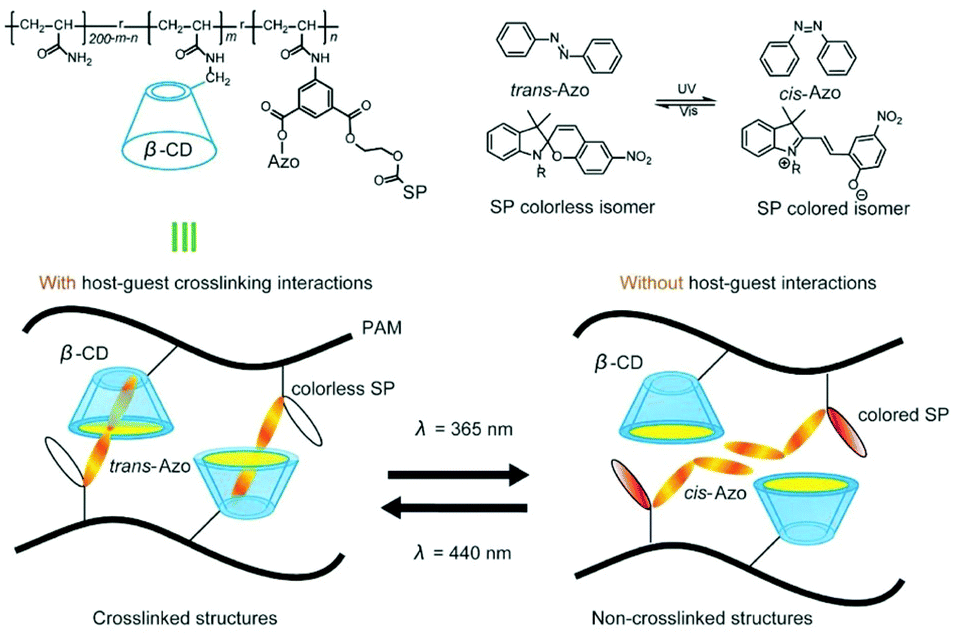 | ||
| Fig. 25 The polymer structure and schematics of supramolecular interactions. β-CD and Azo groups were introduced to provide the polymer with host–guest interactions. In the polymer structure, m and n represented the molar ratio of the host (β-CD) and guest (Azo) moieties, respectively. Reprinted with permission from John Wiley and Sons Inc., Copyright 2018.173 | ||
Easy functionalisation combined with inclusion properties of cyclodextrin allows to prepare covalent/sliding-ring network via one-pot synthesis (Fig. 26).174 Azido-α-CD was used not only as a sliding cross-linker but also as a chain end group to prevent rotaxanes from dissociation. Azido functionality on CD was used to covalently attach dialkyne PEG chains forming the network via CuAAC. Resulting hydrogels are very elastic yet strong. Curing temperature and wt% (weight percentage) of reagents have an impact on the degree of covalency which affects the swelling capability of the network. Further, by precomplexing cyclodextrin with sodium 4-(phenylazo)benzoate, almost purely covalent network can be obtained. The sliding ring networks were characterised with higher equilibrium swelling ration values as the dynamic cross-linking points allow the network to stretch more than static crosslinking points in covalent analogues. Interestingly, increasing the crosslinking density of the sliding ring network did not result in a linear trend in increasing overall stiffness of the material. This can be explained by the fact that with increasing number of cross-linking points, the movement of the crosslinks become restricted. Thus, sliding ring hydrogels cured at high weight% of reagents would behave similarly to covalently cross-linked networks. Positive preliminary cytotoxicity revealed potential of such tuneable hydrogels in tissue engineering applications particularly if further modified via remaining azido-groups on cyclodextrin to attach biologically relevant pendants.
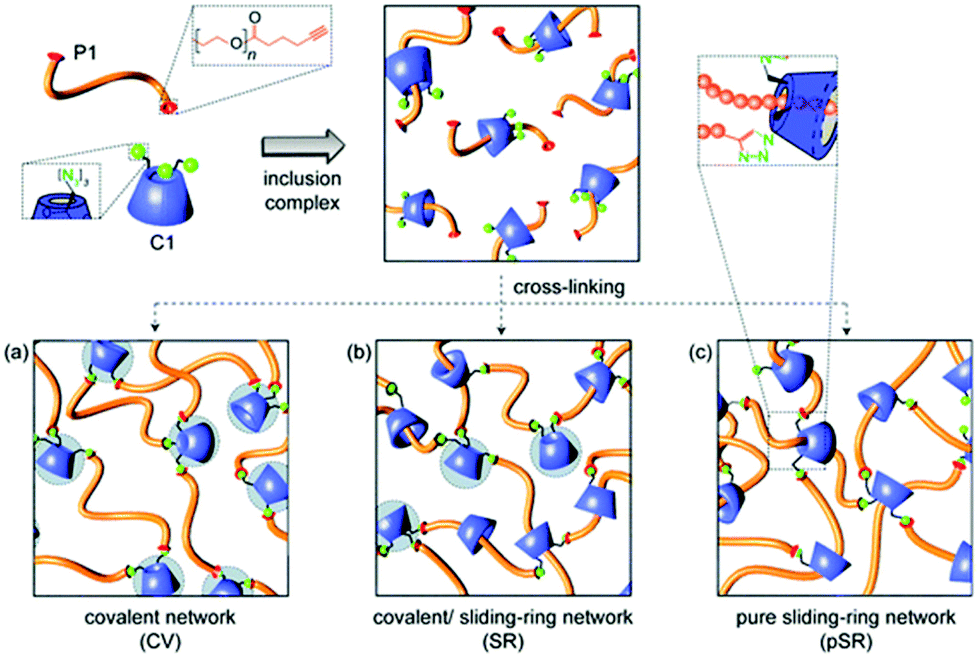 | ||
| Fig. 26 Different hydrogel networks based on cyclodextrins arising from a one-pot synthesis. Reprinted with permission from The Royal Society of Chemistry, Copyright 2013.174 | ||
Incorporation of cyclodextrin into acrylic hydrogels by postpolymerisation modification of glycidyl methacrylate led to preparation of soft contact lenses ocular drug delivery.175 It was found that cyclodextrin did not affect majority of hydrogel physical properties such as swelling degree and viscoelasticity but decreased friction coefficient. Cyclodextrin was capable of complexing diclofenac – a nonsteroidal anti-inflammatory drug. Incorporation of drugs into contact lenses improves drug ocular bioavailability. Pendant cyclodextrins increased loading capacity of the hydrogel by 1300%. Attachment of cyclodextrin to polymer chains slowed down drug release and prevented leakage of the drug to contact lenses storage solution. Leveraging hydrophobic cavity of cyclodextrin allows for the formation of self-healing or thermoresponsive hydrogels. Dynamic and reversible complexation provides good mechanical properties while crosslinking prevents complete dissolution. The choice of a suitable guest is crucial as the binding affinity will greatly affect the dynamicity of the hydrogel. Polymers with cholic acid are promising due to biocompatibility of cholic acid. Such polymers can self-assemble with polymer chains earing pendant cyclodextrins.176 Choosing a guest that can isomerise opens up possibilities of synthesising photosensitive hydrogels. As photoirradiation leads to isomerisation of azobenzene connection of alpha-CD bearing and azobenzene bearing chains is controlled by light.172 However, it was noted that morphological changes in the hydrogel structure were relatively difficult to achieve likely due to the low association constant.
4.4 Miscellaneous applications
Cyclodextrin-centred polymers also found application in surface science. Multiarm polymers were grafted onto a membrane to enhance pore selectivity for filtration. Ulbricht et al. showed that both molecular weight and number of polymer arms, particularly their flexibility, can influence size-selective ultrafiltration.177 7 and 21-arms CD-block copolymers made of 2-dimethylamino(ethyl) and propargyl methacrylates were ‘clicked’ onto azide-modified membrane surface. All polymers enhanced size selectivity. Less densely grafted stars with longer arms had the smallest impact on the liquid flux due to their increased flexibility. This is ascribed to the higher flexibility of longer arm chains and decreased repulsion between neighbouring chains. Sample to sample variation was observed which can likely be attributed to wide dispersity of grafted polymers (1.2 < D > 2.7); potentially optimisation of polymerisation conditions should yield well-defined polymers and hence more uniform functionalisation of the membrane.Multiarm polymers grown from a cyclodextrin core were also tested as antimicrobial and antiviral agents. High grafting density and tunability of the number of functional sites allowed Xiao et al. to optimise structure of a cationic multiarm polymer.112 Antimicrobial activity depends primarily on charge density and hence number and length of polymeric arms as well as the shape of the polymeric structure. 8-Arm acrylamide and guanidine-based monomer copolymer was found to have high charge density and a good exposition of charged side chains to generate strong electrostatic interaction with bacterial membrane. In addition, incorporation of polyhexamethylene guanidine hydrochloride made the polymer an effective antiviral agent.
In summary, cyclodextrin-polymer conjugates found applications in fields such as nanoparticle preparation, antimicrobial agents, hydrogels, drug and gene delivery. The versatility of such conjugates comes from alloying unique features of cyclodextrin and polymers at the same time. As cyclodextrin is compatible with controlled polymerisation methods, the precise engineering of such materials is possible. Asymmetric functionalisation of cyclodextrin is particularly promising and resultant hybrid materials are expected to revolutionise fields such as cancer theranostics which combines diagnosis and treatment.
5 Conclusions and outlook
Cyclodextrin is a widely available, biodegradable molecule that allows for the design of novel materials. Two faces of cyclodextrin can be selectively modified thus enabling the preparation of asymmetric structures and providing easy access to multifunctional materials. Cyclodextrin proved to be compatible with many polymerisation techniques such as ATRP and ROP but less so with RAFT and NMP. ‘Click chemistry’ can be used both for grafting polymer arms and attachment of initiator moiety. Facile reactions such as CuAAC coupling allow for precise engineering of desired materials. Cyclodextrin-polymer conjugates have already found applications in drug delivery, cancer therapy, hydrogels, preparation of inorganic materials and more.In order to fully harness the potential of cyclodextrin polymers more attention has to be paid to the proper characterisation and purification of prefunctionalised cyclodextrin precursors. Researchers in the field of polymer chemistry tend to use protocols from the early stages of the development of cyclodextrin derivatives. Nevertheless, the synthesis is often not optimal and in-depth characterisation of the product is missing. At the same time, organic chemists have been developing new protocols with improved selectivity and yields. Particularly, more attention has to be paid to greener and faster methods such as microwave assisted reactions. There is unrealised potential in asymmetrical functionalisation of cyclodextrins and merging different polymerisation techniques to access materials that are difficult to prepare otherwise. ‘Click chemistry’ has been also mainly limited to CuAAC coupling and other efficient coupling chemistries are waiting to be explored. Advances in the synthesis should lead to the emergence of new applications in the fields that CD-polymer conjugates have already been successfully applied as well as new ones.
Conflicts of interest
There are no conflicts to declare.References
- W. J. Shieh and A. R. Hedges, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 673–683 CrossRef.
- B. Tian, S. Hua and J. Liu, Carbohydr. Polym., 2020, 232, 115805 CrossRef CAS.
- P. Jansook, N. Ogawa and T. Loftsson, Int. J. Pharm., 2018, 535, 272–284 CrossRef CAS.
- J. Zhou and H. Ritter, Polym. Chem., 2010, 1, 1552–1559 RSC.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS.
- F. G. Hougeir and L. Kircik, Dermatol. Ther., 2012, 25, 234–237 CrossRef.
- M. Kfoury, L. Auezova, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2019, 17, 129–143 CrossRef CAS.
- H. Wei and C. Y. Yu, Biomater. Sci., 2015, 3, 1050–1060 RSC.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS.
- R. Mejia-Ariza, L. Graña-Suárez, W. Verboom and J. Huskens, J. Mater. Chem. B, 2017, 5, 36–52 RSC.
- B. Tian, D. Xiao, T. Hei, R. Ping, S. Hua and J. Liu, Polym. Int., 2020, 69, 597–603 CrossRef CAS.
- G. Astray, C. Gonzalez-Barreiro, J. C. Mejuto, R. Rial-Otero and J. Simal-Gándara, Food Hydrocolloids, 2009, 23, 1631–1640 CrossRef CAS.
- L. Szente and J. Szejtli, Trends Food Sci. Technol., 2004, 15, 137–142 CrossRef CAS.
- Y. M. Zhang, Q. Y. Xu and Y. Liu, Sci. China: Chem., 2019, 62, 549–560 CrossRef CAS.
- J. Hu and S. Liu, Acc. Chem. Res., 2014, 47, 2084–2095 CrossRef CAS.
- F. Seidi, A. A. Shamsabadi, M. Amini, M. Shabanian and D. Crespy, Polym. Chem., 2019, 10, 3674–3711 RSC.
- D. Gontero, M. Lessard-Viger, D. Brouard, A. G. Bracamonte, D. Boudreau and A. V. Veglia, Microchem. J., 2017, 130, 316–328 CrossRef CAS.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS.
- R. Periasamy, J. Carbohydr. Chem., 2020, 39, 189–216 CrossRef CAS.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS.
- X. Yao, P. Huang and Z. Nie, Prog. Polym. Sci., 2019, 93, 1–35 CrossRef CAS.
- F. Yhaya, A. M. Gregory and M. H. Stenzel, Aust. J. Chem., 2010, 63, 195–210 CrossRef CAS.
- K. Ohno, B. Wong and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2206–2214 CrossRef CAS.
- P. Chmielarz, S. Park, A. Sobkowiak and K. Matyjaszewski, Polymer, 2016, 88, 36–42 CrossRef CAS.
- J. Li, H. Xiao, Y. S. Kim and T. L. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6345–6354 CrossRef CAS.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2007, 40, 3154–3158 CrossRef CAS.
- Q. Zhang, G. Z. Li, C. R. Becer and D. M. Haddleton, Chem. Commun., 2012, 48, 8063–8065 RSC.
- M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4498–4512 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719–727 CrossRef CAS.
- X. Hu, Y. Hu, G. Xu, M. Li, Y. Zhu, L. Jiang, Y. Tu, X. Zhu, X. Xie and A. Li, Environ. Res., 2020, 180, 108796 CrossRef CAS.
- P. Tang, Q. Sun, L. Zhao, Y. Tang, Y. Liu, H. Pu, N. Gan, Y. Liu and H. Li, Chem. Eng. J., 2019, 366, 598–607 CrossRef CAS.
- Y. Li, Y. Qian, T. Liu, G. Zhang, J. Hu and S. Liu, Polym. Chem., 2014, 5, 1743–1750 RSC.
- M. Fischer and H. Ritter, Macromol. Rapid Commun., 2000, 21, 142–145 CrossRef CAS.
- J. Storsberg and H. Ritter, Macromol. Chem. Phys., 2002, 203, 812–818 CrossRef CAS.
- R. J. Leyrer and W. Mächtle, Macromol. Chem. Phys., 2000, 201, 1235–1243 CrossRef CAS.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Macromolecules, 2011, 44, 7220–7232 CrossRef CAS.
- N. Yao, W. Lin, X. Zhang, H. Gu and L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 186–196 CrossRef CAS.
- N. Tarannum, Suhani and D. Kumar, J. Polym. Res., 2020, 27, 89 CrossRef CAS.
- M. A. Fernández, O. F. Silva, R. V. Vico and R. H. de Rossi, Carbohydr. Res., 2019, 480, 12–34 CrossRef.
- F. Van De Manakker, T. Vermonden, C. F. Van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3157–3175 CrossRef CAS.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Prog. Polym. Sci., 2014, 39, 235–249 CrossRef CAS.
- S. Loethen, J. M. Kim and D. H. Thompson, Biomedical applications of cyclodextrin based polyrotaxanes, 2007, vol. 47 Search PubMed.
- M. Řezanka, Environ. Chem. Lett., 2018, 17, 49–63 CrossRef.
- A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977–1996 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS.
- L. Szente, A. Singhal, A. Domokos and B. Song, Molecules, 2018, 23, 1–15 Search PubMed.
- J. Folkman, P. B. Weisz, M. M. Joullié, W. W. Li and W. R. Ewing, Science, 1989, 243, 1490–1493 CrossRef CAS.
- F. Sallas and R. Darcy, Eur. J. Org. Chem., 2008, 957–969 CrossRef CAS.
- K. Fujita, S. Nagamura and T. Imoto, Tetrahedron Lett., 1984, 25, 5673–5676 CrossRef CAS.
- J. Jindřich and I. Tišlerová, J. Org. Chem., 2005, 70, 9054–9055 CrossRef.
- G. Crini, S. Fourmentinn and E. Lichtfouse, Cyclodextrin Fundamentals, Reactivity and Analysis, 2018 Search PubMed.
- L. Jicsinszky, M. Caporaso, K. Martina, E. C. Gaudino and G. Cravotto, Beilstein J. Org. Chem., 2016, 12, 2364–2371 CrossRef CAS.
- P. R. Ashton, P. Ellwood, I. Staton and J. F. Stoddart, J. Org. Chem., 1991, 56, 7274–7280 CrossRef CAS.
- P. Fügedi, Carbohydr. Res., 1989, 192, 366–369 CrossRef.
- P. Zhang, L. Chang-Chun, A. W. Coleman, H. Parrot-Lopez and H. Galons, Tetrahedron Lett., 1991, 32, 2769–2770 CrossRef CAS.
- S. P. Cornes, M. R. Sambrook and P. D. Beer, Chem. Commun., 2017, 53, 3866–3869 RSC.
- M. Bouzitoun, R. Mlika, H. Gam, H. B. Ouada, M. Majdoub and H. Sfihi, Mater. Sci. Eng., C, 2006, 26, 481–485 CrossRef CAS.
- A. Wycisk, A. Döring, M. Schneider, M. Schönhoff and D. Kuckling, Polymers, 2015, 7, 921–938 CrossRef CAS.
- S. Srinivasachari, Y. Liu, G. Zhang, L. Prevette and T. M. Reineke, J. Am. Chem. Soc., 2006, 128, 8176–8184 CrossRef CAS.
- L. Szente and J. Szejtli, Adv. Drug Delivery Rev., 1999, 36, 17–28 CrossRef CAS.
- X. Li, J. Shi, K. Wu, F. Luo, S. Zhang, X. Guan and M. Lu, J. Photochem. Photobiol., A, 2017, 333, 18–25 CrossRef CAS.
- E. G. Doyagüez, J. Rodríguez-Hernández, G. Corrales, A. Fernández-Mayoralas and A. Gallardo, Macromolecules, 2012, 45, 7676–7683 CrossRef.
- J. Szejtli, A. Lipták, I. Jodál, P. Fügedi, P. Nánási and A. Neszmélyi, Starch/Staerke, 1980, 32, 165–169 CrossRef CAS.
- Y. Yi, Polym. Chem., 2020, 11, 659–663 RSC.
- J. Li and H. Xiao, Tetrahedron Lett., 2005, 46, 2227–2229 CrossRef CAS.
- L. D. Melton and K. N. Slessor, Carbohydr. Res., 1971, 18, 29–37 CrossRef CAS.
- N. Zhong, H. S. Byun and R. Bittman, Tetrahedron Lett., 1998, 39, 2919–2920 CrossRef.
- T. T. Nielsen, V. Wintgens, C. Amiel, R. Wimmer and K. L. Larsen, Biomacromolecules, 2010, 11, 1710–1715 CrossRef CAS.
- J. Defaye and A. Gadelle, Angew. Chem., Int. Ed. Engl., 1991, 30, 78–80 CrossRef.
- P. R. Ashton, R. Königer, J. F. Stoddart, D. Alker and V. D. Harding, J. Org. Chem., 1996, 61, 903–908 CrossRef CAS.
- K. Chmurski and J. Defaye, Supramol. Chem., 2000, 12, 221–224 CrossRef CAS.
- K. Tsujihara, H. Kurita and M. Kawazu, Bull. Chem. Soc. Jpn., 1977, 50, 1567–1571 CrossRef CAS.
- H. Parrot-Lopez, C. C. Ling, P. Zhang, A. Baszkin, G. Albrecht, C. de Rango and A. W. Coleman, J. Am. Chem. Soc., 1992, 114, 5479–5480 CrossRef CAS.
- J. Xu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 404–419 CrossRef CAS.
- S. Hanessian, A. Benalil and C. Laferrière, J. Org. Chem., 1995, 60, 4786–4797 CrossRef CAS.
- B. Siegel, J. Inorg. Nucl. Chem., 1979, 41, 609–610 CrossRef CAS.
- K. Fujita, T. Ueda, T. Imoto, I. Tabushi, N. Toh and T. Koga, Bioorg. Chem., 1982, 11, 72–84 CrossRef CAS.
- B. Jing, X. Chen, X. Wang, Y. Zhao and H. Qiu, ChemPhysChem, 2008, 9, 249–252 CrossRef CAS.
- M. T. Rojas, A. E. Kaifer, R. Königer and J. F. Stoddart, J. Am. Chem. Soc., 1995, 117, 336–343 CrossRef CAS.
- F. Sallas, P. Leroy, A. Marsura and A. Nicolas, Tetrahedron Lett., 1994, 35, 6079–6082 CrossRef CAS.
- G. Maccarrone, E. Rizzarelli and G. Vecchio, Polyhedron, 2002, 21, 1531–1536 CrossRef CAS.
- C. A. Haskard, C. J. Easton, B. L. May and S. F. Lincoln, Inorg. Chem., 1996, 35, 1059–1064 CrossRef CAS.
- A. Puglisi, J. Spencer, J. Clarke and J. Milton, J. Inclusion Phenom. Macrocyclic Chem., 2012, 73, 475–478 CrossRef CAS.
- T. Kakuchi, A. Narumi, T. Matsuda, Y. Miura, N. Sugimoto, T. Satoh and H. Kaga, Macromolecules, 2003, 36, 3914–3920 CrossRef CAS.
- D. Rong and V. T. D'Souza, Tetrahedron Lett., 1990, 31, 4275–4278 CrossRef CAS.
- K. Chmurski, P. Stepniak and J. Jurczak, Synthesis, 2015, 47, 1838–1843 CrossRef CAS.
- A. Ueno and R. Breslow, Tetrahedron Lett., 1982, 23, 3451–3454 CrossRef CAS.
- T. Murakami, K. Harata and S. Marimoto, Tetrahedron Lett., 1987, 28, 321–324 CrossRef CAS.
- I. W. Muderawan, T. T. Ong, C. L. Teck, D. J. Young, B. C. Chi and S. C. Ng, Tetrahedron Lett., 2005, 46, 7905–7907 CrossRef CAS.
- A. W. Coleman, P. Zhang, H. Parrot-Lopez, C. C. Ling, M. Miocque and L. Mascrier, Tetrahedron Lett., 1991, 32, 3997–3998 CrossRef CAS.
- J. Gu, T. Chen, Q. Wang, T. Chen and C. C. Ling, Carbohydr. Res., 2015, 410, 36–46 CrossRef CAS.
- A. Leydet, C. Moullet, J. P. Roque, M. Witvrouw, C. Pannecouque, G. Andrei, R. Snoeck, J. Neyts, D. Schols and E. De Clercq, J. Med. Chem., 1998, 41, 4927–4932 CrossRef CAS.
- J. Ni, S. Singh and L. X. Wang, Carbohydr. Res., 2002, 337, 217–220 CrossRef CAS.
- V. K. Potluri, J. Xu, R. Enick, E. Beckman and A. D. Hamilton, Org. Lett., 2002, 4, 2333–2335 CrossRef CAS.
- C. M. Lian, L. P. Jiang and D. L. Liu, Chin. Chem. Lett., 2014, 25, 134–136 CrossRef CAS.
- L. Jicsinszky, K. Martina, M. Caporaso, P. Cintas, A. Zanichelli and G. Cravotto, Phys. Chem. Chem. Phys., 2015, 17, 17380–17390 RSC.
- J. Boger, R. J. Corcoran and J.-M. Lehn, Helv. Chim. Acta, 1978, 61, 2190–2218 CrossRef CAS.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
- Q. Zhang, L. Su, J. Collins, G. Chen, R. Wallis, D. A. Mitchell, D. M. Haddleton and C. R. Becer, J. Am. Chem. Soc., 2014, 136, 4325–4332 CrossRef CAS.
- K. Takeo, H. Mitoh and K. Uemura, Carbohydr. Res., 1989, 187, 203–221 CrossRef CAS.
- F. Santoyo-González, J. Isac-García, A. Vargas-Berenguel, R. Robles-Díaz and F. G. Calvo-Flores, Carbohydr. Res., 1994, 262, 271–282 CrossRef.
- R. J. Bergeron, M. P. Meeley and Y. Machida, Bioorg. Chem., 1976, 5, 121–126 CrossRef CAS.
- M. Furue, H. Akira and S. Nozakura, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 357–360 CrossRef CAS.
- A. Harada, M. Furue and S. Nozakura, Macromolecules, 1976, 9, 701–704 CrossRef CAS.
- Y. Y. Liu, X. D. Fan and L. Gao, Macromol. Biosci., 2003, 3, 715–719 CrossRef CAS.
- M. Zhang, Q. Xiong, W. Shen and Q. Zhang, RSC Adv., 2014, 4, 30566–30572 RSC.
- J. S. Diget, L. W. Städe and T. T. Nielsen, Eur. Polym. J., 2019, 116, 84–90 CrossRef CAS.
- M. Munteanu, S. W. Choi and H. Ritter, Macromolecules, 2008, 41, 9619–9623 CrossRef CAS.
- Y. Abdouni, G. Yilmaz and C. R. Becer, Macromol. Rapid Commun., 2017, 38, 1–5 Search PubMed.
- Z. Guo, X. Chen, X. Zhang, J. Xin, J. Li and H. Xiao, Tetrahedron Lett., 2010, 51, 2351–2353 CrossRef CAS.
- K. M. Xiu, J. J. Yang, N. N. Zhao, J. S. Li and F. J. Xu, Acta Biomater., 2013, 9, 4726–4733 CrossRef CAS.
- Y. Pan, Y. Xue, J. Snow and H. Xiao, Macromol. Chem. Phys., 2015, 216, 511–518 CrossRef CAS.
- F. Li, M. Cao, Y. Feng, R. Liang, X. Fu and M. Zhong, J. Am. Chem. Soc., 2019, 141, 794–799 CrossRef CAS.
- Y. Y. Liu, Y. B. Zhong, J. K. Nan and W. Tian, Macromolecules, 2010, 43, 10221–10230 CrossRef CAS.
- M. H. Stenzel-Rosenbaum, T. P. Davis, V. Chen and A. G. Fane, Macromolecules, 2001, 34, 5433–5438 CrossRef CAS.
- Y. Guo, J. Liu, K. Zhang, H. Zhang, Y. Li and Z. Lei, Biochem. Eng. J., 2017, 121, 188–195 CrossRef CAS.
- C. Gao, M. Liu, S. Lü, X. Zhang and H. Duan, J. Mater. Chem. B, 2014, 2, 6823–6829 RSC.
- Z. Gao, M. Chen, Y. Hu, S. Dong, J. Cui and J. Hao, Polym. Chem., 2017, 8, 2764–2772 RSC.
- W. Lin, G. Ma, J. Wu and S. Chen, Colloids Surf., B, 2016, 146, 888–894 CrossRef CAS.
- Y. Li, H. Guo, J. Zheng, J. Gan, K. Wu and M. Lu, Colloids Surf., A, 2015, 471, 178–189 CrossRef CAS.
- S. Li, M. Xiao, A. Zheng and H. Xiao, Mater. Sci. Eng., C, 2014, 43, 350–358 CrossRef CAS.
- S. P. Rwei, Y. Y. Chuang, T. F. Way, W. Y. Chiang and S. P. Hsu, Colloid Polym. Sci., 2014, 293, 493–503 CrossRef.
- L. Zhang and M. H. Stenzel, Aust. J. Chem., 2009, 62, 813–822 CrossRef CAS.
- K. Koyanagi, Y. Takashima, T. Nakamura, H. Yamaguchi and A. Harada, Beilstein J. Org. Chem., 2016, 12, 2495–2502 CrossRef CAS.
- K. Karaky, S. Reynaud, L. Billon, J. François and Y. Chreim, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5186–5194 CrossRef CAS.
- P. Chmielarz, Chem. Pap., 2017, 71, 161–170 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS.
- D. Neugebauer, B. S. Sumerlin, K. Matyjaszewski, B. Goodhart and S. S. Sheiko, Polymer, 2004, 45, 8173–8179 CrossRef CAS.
- F. A. Plamper, H. Becker, M. Lanzendörfer, M. Patel, A. Wittemann, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- L. Mueller and K. Matyjaszewski, Macromol. React. Eng., 2010, 4, 180–185 CrossRef CAS.
- I. N. Topchieva, P. Mischnick, G. Kühn, V. A. Polyakov, S. V. Elezkaya, G. I. Bystryzky and K. I. Karezin, Bioconjugate Chem., 1998, 9, 676–682 CrossRef CAS.
- C. Huin, Z. Eskandani, N. Badi, A. Farcas, V. Bennevault-Celton and P. Guégan, Carbohydr. Polym., 2013, 94, 323–331 CrossRef CAS.
- X. S. Feng, D. Taton, E. L. Chaikof and Y. Gnanou, J. Am. Chem. Soc., 2005, 127, 10956–10966 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6455–6465 CrossRef CAS.
- R. Li, X. Li, Y. Zhang, A. O. Delawder, N. D. Colley, E. A. Whiting and J. C. Barnes, Polym. Chem., 2020, 11, 541–550 RSC.
- M. Adeli, Z. Zarnegar and R. Kabiri, Eur. Polym. J., 2008, 44, 1921–1930 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2961–2974 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- P. A. Faugeras, B. Boëns, P. H. Elchinger, F. Brouillette, D. Montplaisir, R. Zerrouki and R. Lucas, Eur. J. Org. Chem., 2012, 4087–4105 CrossRef CAS.
- S. Srinivasachari, K. M. Fichter and T. M. Reineke, J. Am. Chem. Soc., 2008, 130, 4618–4627 CrossRef CAS.
- Y. Rojas-Aguirre, M. A. Torres-Mena, L. J. López-Méndez, S. L. Alcaraz-Estrada, P. Guadarrama and J. M. Urucha-Ortíz, Carbohydr. Polym., 2019, 223, 115113 CrossRef CAS.
- M. Jesberger, L. Barner, M. H. Stenzel, E. Malmström, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2003, 3847–3861 CrossRef CAS.
- M. Zhang, Q. Xiong, Y. Wang, Z. Zhang, W. Shen, L. Liu, Q. Wang and Q. Zhang, Polym. Chem., 2014, 5, 4670–4678 RSC.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef.
- Y. Y. Liu, X. D. Fan and Y. B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3516–3524 CrossRef CAS.
- O. Jazkewitsch, A. Mondrzyk, R. Staffel and H. Ritter, Macromolecules, 2011, 44, 1365–1371 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS.
- Y. Zhu, X. Zheng, B. Yu, W. Yang, N. Zhao and F. Xu, Macromol. Biosci., 2014, 14, 1135–1148 CrossRef CAS.
- B. W. Liu, H. Zhou, S. T. Zhou, H. J. Zhang, A. C. Feng, C. M. Jian, J. Hu, W. P. Gao and J. Y. Yuan, Macromolecules, 2014, 47, 2938–2946 CrossRef CAS.
- J. Zeng, K. Shi, Y. Zhang, X. Sun and B. Zhang, Chem. Commun., 2008, 3753–3755 RSC.
- W. Yasen, R. Dong, L. Zhou, J. Wu, C. Cao, A. Aini and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 9006–9014 CrossRef CAS.
- Y. Takashima, M. Osaki and A. Harada, J. Am. Chem. Soc., 2004, 126, 13588–13589 CrossRef CAS.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS.
- T. Loftsson and D. Duchêne, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS.
- J. Cheng, K. T. Khin and M. E. Davis, Mol. Pharmaceutics, 2004, 1, 183–193 CrossRef CAS.
- S. Li and W. C. Purdy, Chem. Rev., 1992, 92, 1457–1470 CrossRef CAS.
- G. K. E. Scriba, J. Chromatogr. A, 2016, 1467, 56–78 CrossRef CAS.
- F. Trotta, M. Zanetti and R. Cavalli, Beilstein J. Org. Chem., 2012, 8, 2091–2099 CrossRef CAS.
- B. Kost, M. Brzezinski, M. Socka, M. Basko and T. Biela, Molecules, 2020, 25, 3404 CrossRef CAS.
- X. Yao, J. Mu, L. Zeng, J. Lin, Z. Nie, X. Jiang and P. Huang, Mater. Horiz., 2019, 6, 846–870 RSC.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Polym. J., 2020, 52, 839–859 CrossRef CAS.
- T. Liu, X. Li, Y. Qian, X. Hu and S. Liu, Biomaterials, 2012, 33, 2521–2531 CrossRef CAS.
- A. K. Blakney, R. Liu, G. Yilmaz, Y. Abdouni, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Polym. Chem., 2020, 11, 3768–3774 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- T. Jia, S. Huang, C. Yang and M. Wang, J. Mater. Chem. B, 2017, 5, 8514–8524 RSC.
- X. Pang, L. Zhao, M. Akinc, J. K. Kim and Z. Lin, Macromolecules, 2011, 44, 3746–3752 CrossRef CAS.
- X. Pang, L. Zhao, W. Han, X. Xin and Z. Lin, Nat. Nanotechnol., 2013, 8, 426–431 CrossRef CAS.
- X. Ma, X. Shi, S. Bai, J. Zhang, M. Hou, T. Zhang, B. S. Li, P. Xue, Y. Kang and Z. Xu, Chem. Commun., 2018, 54, 6252–6255 RSC.
- Y. Kang, L. Zhou, X. Li and J. Yuan, J. Mater. Chem., 2011, 21, 3704–3710 RSC.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS.
- C. Xiong, L. Zhang, M. Xie and R. Sun, Macromol. Rapid Commun., 2018, 39, 2–7 CrossRef.
- S. Tan, A. Blencowe, K. Ladewig and G. G. Qiao, Soft Matter, 2013, 9, 5239–5250 RSC.
- J. F. Rosa dos Santos, C. Alvarez-Lorenzo, M. Silva, L. Balsa, J. Couceiro, J. J. Torres-Labandeira and A. Concheiro, Biomaterials, 2009, 30, 1348–1355 CrossRef CAS.
- Y. G. Jia and X. X. Zhu, Chem. Mater., 2015, 27, 387–393 CrossRef CAS.
- J. Büning, I. Frost, H. Okuyama, L. Lempke and M. Ulbricht, J. Membr. Sci., 2020, 596, 117610 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |