
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTHRUST: translesion synthesis-driven hierarchical regulation using a template-activator construct for Cas12a activity†
Lulu
Qin
a,
Wen-Jin
Wang
b,
Xinyi
Xia
a,
Tongshan
Zuo
a,
Yilin
Cai
d,
Guanhong
Xu
a,
Fangdi
Wei
a,
Suling
Wang
a,
Qin
Hu
 a,
Zheng
Zhao
b,
Fan
Zhang
*d,
Ben Zhong
Tang
*b and
Yao
Cen
*ac
a,
Zheng
Zhao
b,
Fan
Zhang
*d,
Ben Zhong
Tang
*b and
Yao
Cen
*ac
aSchool of Pharmacy, Nanjing Medical University, Nanjing, Jiangsu 211166, China
bClinical Translational Research Center of Aggregation-Induced Emission, School of Medicine, The Second Affiliated Hospital, School of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of Hong Kong, Shenzhen (CUHK-Shenzhen), Guangdong 518172, China
cClinical Medical Laboratory Center, Department of Neurology, The Affiliated Taizhou People's Hospital of Nanjing Medical University, Taizhou School of Clinical Medicine, Nanjing Medical University, Taizhou, Jiangsu 225300, China
dState Key Laboratory of Natural Medicines and Jiangsu Provincial Key Laboratory for TCM Evaluation and Translational Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, Jiangsu 211198, China
First published on 16th June 2025
Abstract
The CRISPR/Cas12a system has demonstrated extraordinary capabilities in biosensing and molecular diagnostics, owing to its precise recognition and efficient trans-cleavage ability. However, current crRNA-based Cas12a regulation is relatively crude, requiring costly modifications and multiple components, increasing system complexity. Here, we develop translesion synthesis-driven hierarchical regulation using a template-activator construct for Cas12a activity (THRUST), a powerful and economical Cas12a regulation strategy. This strategy utilizes a bifunctional template-activator construct (TAC) that simultaneously functions as a transcriptional template for T7 RNA polymerase (T7 RNAP) and an activator for Cas12a. T7 RNAP skips the deoxyuridine (dU) lesion while being blocked by the apurinic/apyrimidinic (AP) site. Strategic positioning of transcriptional regulatory units on the TAC allows precise control of crRNA length and simultaneously regulates Cas12a activation at the activator level, thereby achieving hierarchical regulation of Cas12a. Through the construction of “Dim down” and “Light up” biosensing platforms and an aggregation-induced emission lateral flow test, THRUST enriches the CRISPR/Cas12a regulatory toolbox for molecular diagnostics.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins are part of the adaptive immune system of prokaryotes, enabling defense against foreign genetic materials introduced by bacteriophages or plasmids.1 In the field of biosensing, Cas12a is one of the most commonly used Cas proteins. Guided by CRISPR RNA (crRNA, transcribed from the CRISPR array) composed of a scaffold and a spacer,2,3 Cas12a is directed to target double-stranded DNA (dsDNA) containing a protospacer adjacent motif (PAM) sequence or target single-stranded DNA (ssDNA) without PAM. Once activated, Cas12a not only exhibits cis-cleavage activity on the target DNA but also displays efficient non-specific cleavage activity on any ssDNA, that is, multiple turnover trans-cleavage activity,3 allowing Cas12a to provide a novel signal amplification mode in molecular diagnostics. With the above characteristics and its high programmability, a variety of Cas12a-based biosensing platforms have been developed, such as SHELOCK,4 DETECTOR5 and FINDER.6 However, the activity regulation of the Cas12a system remains unidimensional, making it difficult to meet the diverse demands of different detection scenarios for activity intensity. To overcome this limitation, it is crucial to establish an efficient, flexible and hierarchical regulation strategy to precisely modulate the trans-cleavage activity of Cas12a and expand its applications in various fields.The CRISPR/Cas12a system consists of three main regulatory modules. Beyond direct genetic modification of the Cas12a protein,7 current research focuses on the programmable modification of the activator and crRNA. On the one hand, by designing an activator containing a toehold and using a toehold-mediated strand displacement reaction,8,9 a DNA–crRNA complex could be formed and Cas12a could be activated or the activator could be split to co-activate Cas12a,10,11 demonstrating the fine-tuning ability of Cas12a. However, most of these methods rely on the additional provision of an activator, which makes the reaction system more complicated and, to some extent, limits its compatibility with the upstream reaction network, thereby restricting the programmability and flexibility of the CRISPR/Cas system. On the other hand, recent studies have explored the modification of crRNA by various approaches, including elongation-caged crRNA, circular crRNA, light-controlled crRNA, extending DNA/RNA/G-quadruplex (G4) at the end of crRNA, and chemical modification of crRNA (N6-methyladenosine, 2′-O-methyl) to achieve signal transduction switching and activity regulation of Cas12a.12–19 However, optimizing crRNA design for varying sequences often requires multiple trials, significantly reducing cost-effectiveness. More importantly, the “one-touch” activation mechanism, where the activator and crRNA directly trigger Cas12a activity, combined with its ultra-high cleavage efficiency of Cas12a, makes its adjustable range very narrow.20 Furthermore, uncontrolled high cleavage efficiency may interfere with upstream amplification reactions, generate background noise, reduce assay sensitivity, require external activation devices and cause poor compatibility with complex biological samples. In view of the above problems, it is particularly important to develop new regulatory mechanisms, especially to regulate Cas12a activity between hierarchical levels, to improve the sensitivity and specificity of detection in complex biological samples while also simplifying operational steps and reducing economic costs.
T7 RNA polymerase (T7 RNAP) exhibits robust transcriptional activity and strict promoter specificity and is often used as a powerful tool in the preparation of crRNA.21,22 Moreover, T7 RNAP has the ability to translesion synthesis and act as a sensor of DNA lesions during transcription-coupled repair.23 There are many types of DNA lesions, of which deoxyuridine (dU) and apurinic/apyrimidinic (AP) sites are the two most common types. dU is a pyrimidine base that is chemically similar to thymine and can form base pairs with deoxyadenine. However, most DNA polymerases cannot accurately distinguish between thymine and dU, which results in the occasional misincorporation of dU into the DNA chain during DNA replication, replacing normal deoxythymidine.24 The AP site is the cleavage caused by the hydrolysis of the glycosidic linkage connecting deoxyribose to a nucleobase.25 The cleavage can occur spontaneously or be induced by base alkylation or oxidation.26 Both types of DNA lesions can be repaired by excision through DNA glycosylases in a process called base excision repair (BER). Depending on the type of DNA lesions, the impact of lesions on T7 RNAP can be categorized into the following three scenarios: (i) impediment of template base access to the active site, (ii) interference with nucleotide binding and subsequent incorporation into RNA, and (iii) affecting the translocation function of T7 RNAP moving along the DNA template, including the correct elongation of RNA and the positioning of the next round of base pairs. These effects may cause T7 RNAP to correctly bypass the lesions, induce transcriptional mutations, or result in transcriptional arrest.27 Previous studies have shown that building on nucleic acid interactions, the integration of dU can establish base pairs with a geometry more closely resembling Watson–Crick pairing, allowing T7 RNAP to effectively bypass dU.28 In contrast, the AP site introduces a helical distortion that prevents T7 RNAP from loading into the correct active site, thereby rendering it unable to perform translesion synthesis.29 However, existing methods for combining T7 RNAP with Cas12a activity regulation have mainly focused on the direct activation of Cas12a using crRNA generated by T7 RNAP. These methods ignore the potential fine regulation of T7 RNAP on specific behaviors at lesion sites, and face problems such as insufficient regulation accuracy, complex operations and difficulty in extending functional applications to more application scenarios. Therefore, it is crucial to establish an efficient, precise and hierarchical regulation strategy to regulate Cas12a activity.
Herein, we have developed a novel strategy to accurately regulate Cas12a activity, termed translesion synthesis-driven hierarchical regulation using a template-activator construct for Cas12a activity (THRUST), which utilizes the unique translesion synthesis ability of T7 RNAP to bypass dU and be blocked by the AP site. First, a bifunctional template-activator construct (TAC) was constructed, including the T7 promoter and its complementary sequence that initiated transcription of T7 RNAP, and the CRISPR array as a transcription template, with or without modification of the transcription regulatory units (dU or AP site). The THRUST strategy achieved dual functions through the TAC: the TAC served directly as an activator for Cas12a, eliminating the need for an additional activator and simplifying experimental procedures. Simultaneously, the TAC could be used as a template to transcribe crRNA in real time, and by strategically adjusting the types and modification positions of transcription units, hierarchical regulation of Cas12a activity was achieved, including dynamic regulation from “full” to “partial” and then to “blocked” (Fig. 1). Furthermore, uracil DNA glycosylase (UDG) acts as a bridge between dU and the AP site, specifically recognizes and initiates the BER, removes dU from DNA and converts it into the AP site. We constructed two signal output platforms, namely “Dim down” and “Light up”, which provided more diverse strategies and meant that UDG could be sensitively detected. Specifically, we used the peroxidase-like activity of G4/hemin to catalyze the oxidation of Amplex Red (AR) to obtain the oxidation product resorufin, which exhibited typical aggregation-induced emission (AIE) properties. The “Light up” platform effectively reduced background noise, offered an intuitive observation effect and stable signal output, therefore enhancing detection sensitivity and avoiding the false positive problem commonly associated with the “Dim down” platform. The visual AIE lateral flow strip platform was additionally designed to realize the portable, highly sensitive and selective fluorescence detection of analytes. Overall, the THRUST strategy utilized on the bifunctional TAC to provide a system with no extra activator required, streamlining experimental procedures. By exploiting natural mechanisms and lesion design, hierarchical regulation of Cas12a was achieved, significantly improving accuracy and flexibility of regulation, overcoming the limitations of the traditional “ON–OFF” single regulatory mode. Compared with direct modification of crRNA to regulate Cas12a activity, the TAC modification method was not only simpler and economical but also offered high sensitivity and precise regulation of crRNA generation. The approach bridged gaps in accurate activity modulation while presenting great opportunities for integrating Cas12a into sensing and molecular switch platforms, optimizing its performance in various fields.
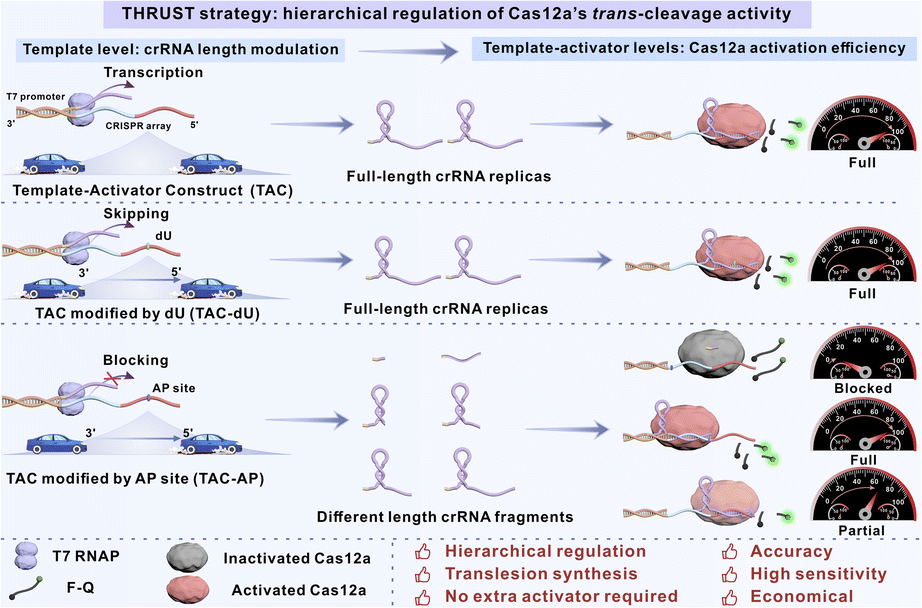 | ||
| Fig. 1 Schematic diagram illustration of the THRUST system. | ||
Results and discussion
Activator level modifications for Cas12a hierarchical regulation
As shown in Fig. 2A, TACs (TAC/TAC-dU/TAC-AP) were constructed using the T7 promoter, its complementary sequence and the CRISPR array (fragments consisting of the blue T1 region and the red T2 region), with or without transcriptional regulatory unit (dU or AP site) in different sites. Specifically, the CRISPR array could be used as a template for T7 RNAP transcription: the T1 region was transcribed to generate a scaffold of crRNA, and the T2 region was transcribed to generate a spacer of crRNA. At the same time, since the generated RNA fragments were complementary to the CRISPR array, which could be used as activators to activate Cas12a, providing a basis for subsequent regulation of Cas12a activity. When TACs were used only as activators, the direct effects of modifying the transcriptional regulatory unit at the activator level on the trans-cleavage activity of Cas12a were initially examined (Fig. 2B). We pre-purchased complete crRNA complementary to the T2 region of TACs. As shown in Fig. 2C and D, we modified the transcriptional regulatory units (dU or AP site) in the T1 and T2 region of the CRISPR array, named TAC-dU2 (a TAC that replaced the second deoxythymidine base in the CRISPR array sequence with dU) to dU39 or TAC-AP2 to AP39 respectively, and observed the fluorescence signal intensity produced by Cas12a trans-cleavage of the F-Q probe to examine the activation tolerance of Cas12a to the modified transcriptional regulatory units in different sites. | ||
| Fig. 2 Activator level modifications and direct activation effects of a series of transcriptional regulatory units for Cas12a hierarchical regulation. (A) TAC consisted of two modules: the T7 promoter with its complementary sequence and the CRISPR array, including T1 and T2 regions. (B) Schematic diagram of the direct effects of Cas12a activation mediated by transcriptional regulatory units. Locations of (C) dU and (D) the AP site in the CRISPR array. Cas12a activation rate when transcriptional regulatory units were modified on (E) a TAC and (F) a double-stranded TAC (dsTAC), which consisted of a TAC and the complementary DNA sequence of the CRISPR array. | ||
Obviously, although dU was a lesion base, it would hardly affect the activation of Cas12a (Fig. 2E). However, the Cas12a system showed different tolerance effects on TAC-AP with AP sites modified at different positions. Specifically, the proximity of the AP site to the center of the TAC was negatively correlated with the tolerance of Cas12a to the TAC. As shown in Table S5, when the AP site was located in the T1 region of the CRISPR array, the Gibbs free energy (ΔG) of TAC-AP2/crRNA and TAC-AP12/crRNA did not change, and thus had no significant effect on the activation efficiency of Cas12a. Although the ΔG of TAC-AP20/crRNA duplexes did not change, there was still a slight inhibitory effect on the activation of Cas12a. We speculated that the AP site in TAC-AP20 was close to the T2 region, it may affect the structure and stability of DNA, resulting in the inability of the REC domain to accurately recognize it, thereby interfering with the effective binding of the REC domain to the TAC and slightly interfering with the trans-cleavage activity of Cas12a.30 As the AP site gradually approached the T2 region bound to the spacer of crRNA, especially the closer it was to the middle part, Cas12a became less tolerant of it. At TAC-AP27, the AP site was located in the complement of the seed region that was still extremely sensitive to base mutations/deletions and was critical for target affinity and specificity.31 This sensitivity would significantly weaken the initial binding efficiency of crRNA and the TAC, thereby inhibiting the trans-cleavage activity of Cas12a. The AP site in TAC-AP33 was located in the binding position in the middle region of spacer's complementary part, resulting in a significant decrease in ΔG. The binding stability of TAC-AP33/crRNA was too weak and did not favor the formation of the R-loop, significantly inhibiting the trans-cleavage activity of Cas12a. Previous studies have shown that the trans-cleavage activity is positively correlated with the length of the activator in the range of 14 to 20 nucleotide (nt), and that 18 nt activators can form a stable complex with Cas12a/crRNA.32 Therefore, when the AP site modification was migrated to the 5′ end of the TAC (TAC-AP39), it had no notable impact on the activation of Cas12a.
We also added the complementary sequence of the CRISPR array that made up the double-stranded template-activator construct (dsTAC), and further investigated whether the dsTAC would produce similar results in the same circumstances (Fig. S1†). It is widely accepted that the PAM is required to trigger dsDNA unwinding and enable crRNA–DNA hybridization. Although the dsTAC constructed in this work did not contain the classical PAM sequence, there was a nick structure between its CRISPR array complementary sequence and the T7 promoter, which significantly increased the flexibility and structural distortion of DNA, interrupted the continuity of the double helix and reduced the structural stability of the CRISPR array region. In addition, its proximity to the upstream of the crRNA recognition region and its disruption of spatial conformation helped to provide an “invasion portal” for crRNA hybridization, acting as a PAM-like recognition. This structural advantage facilitated the spontaneous formation of RNA–DNA hybrids, allowing the Cas12a/crRNA complex to bypass the dependence on the PAM, and efficiently achieved recognition of dsTAC and activation of Cas12a.33,34 As a result, the specific modulation of Cas12a activity by dsTAC-AP closely resembled that of the TAC (Fig. 2F). In conclusion, it was demonstrated that the trans-cleavage activity of Cas12a in the THRUST system was highly tolerant to dU modified at any site on the TAC, the AP site in the T1 region and the 5′ end of the T2 region, but could not tolerate the AP site modified in the seed region and the middle position of the T2 region.
Template–activator level modifications for Cas12a hierarchical regulation
The TAC was also used as a template for T7 RNAP due to its bifunctional properties. Modifying different transcriptional regulatory units at different positions in the TAC affects the process of T7 RNAP translesion synthesis. First of all, we selected four sites in the CRISPR array for agarose gel electrophoresis experiments (Fig. S2†). The results showed that no matter where the dU modification was located, T7 RNAP could efficiently bypass it and successfully generate complete crRNA replicas. However, as the AP site modification position gradually approached the 5′ end of the CRISPR array, the length of the crRNA fragments generated by transcription gradually increased accordingly. This verified that T7 RNAP was blocked at the AP site and could not complete transcription. This difference could be explained by the distinct interactions of dU and the AP site with the transcription machinery. Studies have shown that the incorporation of dU can form base pairs with a geometry resembling Watson–Crick pairing, stabilizing the nucleic acid duplex and reducing the barrier.28 As a result, T7 RNAP can effectively bypass dU and synthesize full-length crRNA replicas despite the lesion. In contrast, the AP site lacks stacking and complementary interactions with the template, preventing proper entry into the active site of T7 RNAP. This disruption interferes with its movement to the next base position, causing T7 RNAP to remain in an “intermediate stalled state”, slowing down RNA extension and initiating a transcription pause.29 It was confirmed that the AP site had a blocking effect on T7 RNAP transcription, which affected the length of the transcription product. Therefore, by modifying the AP site at different positions on the TAC, it was possible to regulate the length of the transcribed crRNA fragments, thereby regulating the trans-cleavage activity of Cas12a at the template level.Considering the effects of modified transcriptional regulatory units at both the activator and template levels on the Cas12a′s trans-cleavage activity in TACs, we investigated the transcription-coupled activation effects of transcriptional regulatory units at the above specific sites on the transcriptional efficiency and Cas12a′s trans-cleavage activity by monitoring the activation rate of Cas12a (Fig. 3A). The results were consistent with expectations: T7 RNAP was able to efficiently bypass dU modification, successfully transcribe to generate complete crRNA, and fully activate the trans-cleavage activity of Cas12a. However, the AP site is an empty position in the DNA strand where a base is missing, so T7 RNAP loses its normal pairing signal when it encounters the AP site, which usually results in synthesis stopping before it reaches the site. At the same time, the transcription initiation mechanism of T7 RNAP typically results in the 5′ end of the crRNA transcription product extending several nucleotides. So, these result in increasing the length of the product at the AP site by 2 nt compared to the ideal transcript (the RNA product generated according to the CRISPR array on the template). The modest increase in nucleotide extension at the 5′ end of crRNA does not adversely affect the trans-cleavage activity of Cas12a.18 Consequently, the activity of the crRNA transcribed by TAC-dU in this work was not affected by the additional extension sequence. When the AP site was modified close to the T7 promoter complementary sequence (TAC-AP2 and TAC-AP12), only shorter crRNA fragments could be transcribed, and Cas12a was completely inactivated. Interestingly, although TAC-AP20 exhibited the capacity to transcribe a 22 nt crRNA fragment (of which 19 nt was the conserved scaffold sequence), it nevertheless succeeded in activating Cas12a, thereby achieving the desired fluorescence. It was hypothesized that the 19 nt length of the scaffold of the mature crRNA and the 3 nt extension of the 5′ end would not affect the cleavage activity.16,18,32 Due to the complete complementarity of the sequence between the T1 region of TAC-AP and the 22 nt crRNA fragment, the strong force of DNA–RNA base pairing may cause some crRNA fragments to act as spacer, while some were still folded to form the scaffold, and the two formed split crRNA. Therefore, when TAC-AP20 was used, the T1 region acted as an activator to bind to the generated split crRNA, which still achieved the activation of Cas12a′s trans-cleavage activity. Likewise, there was a similar possibility that TAC-AP27 transcribed 29 nt crRNA fragments, forming split crRNA. In this case, the T1 region together with the 5 nt in the T2 region still acted as an activator to trigger the activation of Cas12a. However, when the 29 nt crRNA fragment was used as a scaffold, the 5 nt at its 3′ end formed an overhanging structure, and the resulting steric hindrance prevented the spacer and TAC-AP27 from entering the RuvC domain of Cas12a, leading to a decrease in Cas12a trans-cleavage activity.32,34 TAC-AP33 was transcribed to produce a 35 nt crRNA fragment that was ineffective in activating Cas12a in either case. On the one hand, when the T2 region served as the activator, this fragment could not directly activate Cas12a due to the presence of only an 11 nt spacer. On the other hand, when the T1 region served as the activator, the backbone in the generated split crRNA formed an 11 nt overhang structure at its 3′ end. Compared to the 5 nt overhang of TAC-AP27, the 11 nt overhang caused a more pronounced steric hindrance effect, which significantly reduced the activation of Cas12a. As the position of the AP site modification gradually approached the 5′ end of the CRISPR array, the length of the transcribed crRNA gradually increased to 41 nt, and the corresponding spacer was 17 nt. At this point, the T2 region acted as an activator of the system, but the binding of the 17 nt spacer to the target DNA was not stable enough,32 resulting in insufficient activation of the trans-cleavage activity of Cas12a.
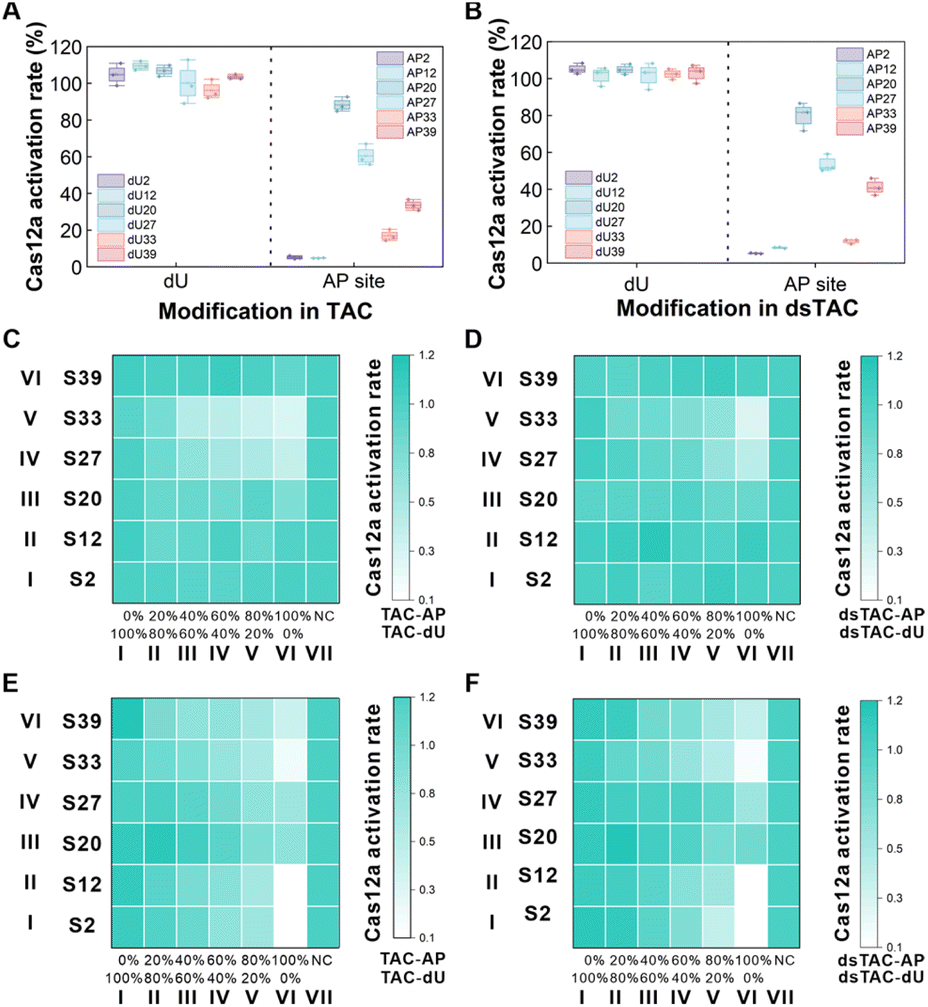 | ||
| Fig. 3 The transcription-coupled activation and direct activation effects of a series of transcriptional regulatory units for Cas12a hierarchical regulation. The transcription-coupled activation effects of transcriptional regulatory units were modified on (A) a TAC and (B) a dsTAC as assessed by Cas12a activation rate. The Cas12a activation rate affected by different ratios of (C) TAC-dU/-AP and (D) dsTAC-dU/-AP at the activator level. The horizontal axis represented the change in the activator AP site modification ratio from 0% to 100%, and the vertical axis represented a mixture of TAC-dU and TAC-AP at the different modified positions mentioned above (S2 to S39). NC was a 100% unmodified TAC/dsTAC in the system. The transcription-coupled activation effects at different ratios of (E) TAC-dU/-AP and (F) dsTAC-dU/-AP on Cas12a′s activity at the template-activator levels. | ||
Given that T7 RNAP transcription also normally occurs on a double-stranded template, we further explored the performance of the dsTAC under the same conditions (Fig. 3B). The results showed that the transcriptional regulatory behaviour of the modified dU and AP site on T7 RNAP was similar to that of the TAC at the template level. At the same time, based on the nick structure-assisted PAM-free activation mechanism, the modified dU and AP sites in the dsTAC further fine-tuned Cas12a activity at the activator level, eventually achieving a hierarchical regulation effect of Cas12a activity at the template-activator levels. In short, without requiring an additional activator, the THRUST system modified the AP site at different positions on the TAC, making it impossible for T7 RNAP to perform translesion synthesis, thereby obtaining different lengths of crRNA fragments/split crRNA products in a controllable manner and further achieving hierarchical and accurate regulation of the trans-cleavage activity of Cas12a.
Subsequently, the mixtures of different modified positions of TAC-dU and TAC-AP were named S2 to S39; for example, S2 represents a mixture of TAC-dU2 and TAC-AP2. Using TAC-dUs and TAC-APs at different ratios at the above positions, we investigated the activation effects on the hierarchical regulation of the THRUST strategy at the activator level. As demonstrated in Fig. 3C and D, when a TAC or dsTAC acted as an activator, only dU modification at any site did not impact the trans-cleavage activity of Cas12a (rows I–VI of column I). In the case of the AP site being introduced at positions S2, S12, S20, and S39, no significant effect on the activity of Cas12a was observed (rows I, II, III and VI). However, when the AP site was introduced at positions S27 and S33 (rows IV and V), its ability to inhibit Cas12a activation was positively correlated with the percentage of AP site concentration. In accordance with the results presented in Fig. 1E and F, this further substantiated the finding that dU modification on the activator exerts no effect on Cas12a trans-cleavage activity. However, when the AP site modification was introduced, its inhibitory effect on Cas12a trans-cleavage activity was significantly enhanced as the modification position gradually approached the middle part of the T2 region.
We further verified the transcription-coupled activation effects of the transcriptional regulatory units on hierarchical regulation in the TAC and dsTAC at the template-activator levels. As shown in Fig. 3E and F, T7 RNAP could effectively bypass dU, thereby successfully transcribing across the dU base, obtaining complete crRNA replicas, activating Cas12a to cleave the F-Q probe and generate fluorescence (rows I–VI of column I). However, increasing the concentration of TAC-AP at S2, S12, and S33 significantly weakened the activation effect of Cas12a (rows I, II and V). Under the condition of 100% TAC-AP, Cas12a activity was almost completely inhibited. In contrast, the S20 and S27 sites (rows III and IV) effectively activated Cas12a by forming split crRNA. And S39 partially activated Cas12a by generating a crRNA product with a 17 nt spacer (row VI of column VI). Consistent with the results in Fig. 2A and B, T7 RNAP could effectively bypass the dU base and transcribe smoothly. The AP site had a significant blocking effect on T7 RNAP and prevented transcription. In short, the THRUST strategy used the bifunctional TAC to simplify the system components without the need to add an additional activator and to achieve precise hierarchical regulation of Cas12a trans-cleavage activity.
Detection of the BER enzyme by the THRUST strategy
Various exogenous and endogenous factors, such as radiation and drugs, can cause alkylation and oxidation of bases, thereby damaging the integrity and functionality of DNA and resulting in DNA lesions and gene mutations. Among these DNA lesions, dU is particularly common. In the presence of UDG, a key enzyme in the BER process, DNA sequences with dU base lesions would produce an AP site. Abnormal UDG activity usually precedes the onset of cancer, making it a potential early diagnostic marker and prognostic indicator for cancer.35 Consequently, the development of a simple, efficient, highly specific and sensitive method for analyzing UDG activity holds significant clinical value. Since conversion of the dU and AP site at S2 had the greatest difference in effect on the activity of the THRUST system and both could serve as activators of Cas12a, we chose this site to construct the “Dim down” signal output platform (Fig. S3†). As shown in Fig. S4,† when UDG was absent, T7 RNAP could successfully transcribe complete crRNA, which activated Cas12a to cleave the F-Q probe under the activation of TAC-dU2 with no additional provision of the activator, and produced fluorescence. In the presence of UDG, the enzyme recognized the U/T base pair and cleaved the N-glucosidic bond linking the sugar and the damaged base, creating an AP site. T7 RNAP could not bypass it and only obtained very short RNA fragments. Cas12a remained inactive, and no fluorescence signal was generated. Under optimized experimental conditions (Fig. S5†), the performance of the sensing platform was explored (Fig. S6†). The fluorescence intensity exhibited a linear decrease as the UDG concentration ranged from 0.0001 to 0.0020 U mL−1. Based on the 3σ rule, the limit of detection (LOD) was determined to be 0.0000249 U mL−1. When the dsTAC was used as the bifunctional template, the UDG concentration decreased linearly from 0.0004 to 0.010 U mL−1, and the LOD was calculated to be 0.0000932 U mL−1. Consistent with previous speculation, as the dsTAC lacked the classic PAM sequence, its sensitivity in binding to Cas12a was lower than that of the TAC, and therefore its detection sensitivity was also lower than that of the TAC. However, the “Dim down” signal output platform was easily affected by the background signal and was difficult to observe, resulting in false positive results, which limited its application in the field of biological diagnosis.In order to overcome the limitations of the above problems, we utilized G4 as a signal probe to develop a “Light up” signal output platform (Fig. 4A). When UDG was present, it recognized TAC-dU2 and created an AP site, which blocked T7 RNAP transcription and further inhibited Cas12a activation, preventing the cleavage of G4. The peroxidase-like activity of G4/hemin was utilized.36 AR was oxidized to generate resorufin under the catalysis of H2O2 and generated red fluorescence (Fig. S7†). As shown in Fig. S8,† in HEPES/DMSO mixed solutions with varying proportions, the emission intensity of resorufin significantly enhanced as aggregation progressed, ultimately reaching 135 times the original emission intensity, showing typical AIE characteristics, which was consistent with the reported literature.37 Conversely, when UDG was absent, TAC-dU2 transcribed complete crRNA under the action of T7 RNAP and activated Cas12a, which indiscriminately trans-cleaved G4. The system lacked intact G4 to bind with hemin to oxidize AR, resulting in no significant red fluorescence. Under the optimized conditions (Fig. S9†), we explored the performance of the proposed “Light up” platform by monitoring the fluorescence signal changes in response to varying UDG concentrations. The UDG concentration increased linearly from 0.00005 to 0.0040 U mL−1, and the LOD was calculated to be 0.0000108 U mL−1 (Fig. 4B and C). Benefiting from the rapid aggregation characteristics and high sensitivity of resorufin, the THRUST system achieved a balance between detection time and sensitivity: the detection time was short while maintaining a low LOD, showing better overall performance compared to other methods (Fig. 4D).38–46 To assess the selectivity of the sensing strategy, various DNA enzymes were chosen as interfering substances (Fig. 4E). The fluorescence signal could be restored when the target UDG was present. In a word, this method demonstrated excellent selectivity and anti-interference properties, making it a promising candidate for detecting BER enzymes in complex environments. Furthermore, we evaluated whether the THRUST strategy could be implemented in a one-pot assay. However, the signal recovery level of the one-pot method was significantly lower than that of the step-by-step method (Fig. S10†). We speculated that it was due to the difficulty of achieving optimal compatibility for both T7 RNAP and Cas12a activities within a single reaction environment. Additionally, the unique design of the TAC in the THRUST system allowed it to perform the dual role of a T7 RNAP transcription template and an activator of Cas12a. As the transcription reaction of T7 RNAP proceeded, most of the TACs may have been used by T7 RNAP, resulting in a significant reduction in the number of TACs that could freely activate Cas12a, making it impossible to efficiently activate Cas12a at the same time.
 | ||
| Fig. 4 Transcriptional switch-based detection of UDG activity and its application in biosensing. (A) Schematic diagram of the “Light up” signal output platform. (B) Fluorescence intensity of different concentrations of UDG at 598 nm and (C) the linear relationship between fluorescence intensity and UDG concentration. (D) Comparison of detection time and LOD between the THRUST system and other systems. (E) Selectivity and anti-interference of the THRUST system for UDG among flap endonuclease 1 (FEN1), human 8-oxoguanine DNA glycosylase (hOGG1), human alkyladenine DNA glycosylase (hAAG), T4 polynucleotide kinase (T4 PNK), apurinic–apyrimidinic endonuclease I (APE1), terminal deoxynucleotidyl transferase (TdT), nicking endonuclease (Nt.BbvCI), and exonuclease I (Exo I). (F) Relative activity of UDG in response to different concentrations of uracil glycosylase inhibitor (UGI). (G) Fluorescence intensity induced by human cervical cancer cell lines (HeLa cells), human lung adenocarcinoma cell lines (A549 cells), HeLa-heated cells, A549-heated cells, HeLa cells + UGI, A549 cells + UGI, normal human bronchial epithelium (BEAS-2B cells) and cell lysis buffer. | ||
UDG is a vital player in the BER process, and its inhibitors can dissociate the UDG-DNA interaction or obstruct access to the UDG active site, resulting in UDG inactivation. UGI can inhibit UDG activity by forming a highly stable and irreversible complex at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio.38 As illustrated in Fig. 4F, the relative activity of UDG declined progressively with increasing UGI concentration and showed a dose-dependent manner. By calculation, we obtained the IC50 value of UGI for inhibiting UDG activity, which was 0.0431 U mL−1. The results showed that this method not only could screen UDG inhibitors, but also evaluate their inhibitory effects, and has the potential for wide application in UDG-related drug development and disease diagnosis.
:1 stoichiometric ratio.38 As illustrated in Fig. 4F, the relative activity of UDG declined progressively with increasing UGI concentration and showed a dose-dependent manner. By calculation, we obtained the IC50 value of UGI for inhibiting UDG activity, which was 0.0431 U mL−1. The results showed that this method not only could screen UDG inhibitors, but also evaluate their inhibitory effects, and has the potential for wide application in UDG-related drug development and disease diagnosis.
To evaluate the feasibility of the THRUST system for clinical diagnosis, we monitored UDG activity in HeLa cells, A549 cells, and BEAS-2B cells (Fig. 4G). As UDG was overexpressed in HeLa and A549 cells, it facilitated the conversion of TAC-dU2 to TAC-AP2, inactivating Cas12a and maintaining the integrity of the G4 sequence, thereby oxidizing AR to resorufin, which produced a strong red fluorescence signal in their lysates by the “Light up” signal output system. To verify whether the signal enhancement in cell lysates was caused by active UDG, we designed a UGI inhibition experiment and compared it with inactivated HeLa and A549 cells. The results showed that UGI could effectively inhibit UDG activity, confirming that the detected fluorescence signal increase was specifically attributed to active UDG in HeLa and A549 cell lysates.
Further studies showed (Fig. S11†) that the fluorescence intensity of resorufin increased with the increase in HeLa cell number, from 5 cells to 200 cells, with a LOD of 3 cells. Similarly, in A549 cells, the resorufin fluorescence intensity also increased with the increase in cell number, from 25 to 500 cells, with a LOD of 8 cells. These results further demonstrated the high sensitivity and scalability of the “Light up” platform in detection. With its unique sensitivity and efficient regulation of Cas12a activity, the THRUST strategy had a universal range of application potential in precision diagnosis and molecular detection.
Broadening the scope of the THRUST system in practical application
Based on the “Light up” fluorescent sensing platform, we further developed the AIE lateral flow strip platform (Fig. 5A). The nucleic acid detection system was built around two main components: the Cas12a cleavage reaction and lateral flow platform detection. The test strip mainly consisted of three parts: a sample pad, nitrocellulose (NC) membrane and absorbent pad.47 Streptavidin (SA) and biotin-modified G4 (bio-G4) pre-conjugated to SA (SA-bio-G4) were applied to the NC membrane to create the control line (C line) and the test line (T line). As for the C line, it showed red fluorescence in any case because bio-G4 was pre-attached to the strip via SA. In the absence of UDG, TAC-dU2 activated Cas12a, which cleaved bio-G4, preventing the cleaved bio-G4 fragments from binding to hemin. When the reactant was applied to the test paper, the liquid migrated to the absorption pad by capillary action. Since bio-G4 had been cleaved, the T line lacked an intact G4/hemin structure, resulting in no obvious red fluorescence after the AR/H2O2 mixture was dripped. In contrast, in the presence of UDG, T7 RNAP could not transcribe complete crRNA due to the produced TAC-AP, inactivating Cas12a and leaving bio-G4 intact. The SA on the T line captured the complete bio-G4, and the peroxidase-like activity of G4/hemin catalyzed the production of resorufin after the addition of the AR/H2O2 mixture, and produced red fluorescence. | ||
| Fig. 5 Rapid detection of UDG using the AIE lateral flow test strip. (A) Schematic diagram of the THRUST system for the practical application of the AIE lateral flow test strip. (B) Actual detection under a 365 nm UV flashlight. (C) Photographs of the fluorescence signal of UDG content in HeLa cells. | ||
To verify the ability of the THRUST system to analyze biological samples, we first used cell lysates with different UDG contents for detection (Fig. 5B). In the control with no UDG and BEAS-2B cells with low UDG expression, T7 RNAP transcribed TAC-dU2 to obtain complete crRNA, and after activating Cas12a to cleave bio-G4, the T line had no fluorescence. In contrast, only in A549 cells and HeLa cells with high UDG expression, T7 RNAP transcribed very short crRNA fragments, which could not activate Cas12a, thus ensuring the integrity of bio-G4 and making the T line show red fluorescence. Building on the qualitative analysis of UDG achieved by this AIE lateral flow strip platform, we further attempted to perform actual detection in cell lysates with different numbers of HeLa cells. As depicted in Fig. 5C, with the rise in the number of HeLa cells, indicating a gradual increase in UDG concentration, the red fluorescence on the T line gradually deepened. This work verified the effectiveness of the THRUST system in analysing biological samples. Through the qualitative detection of UDG activity, the significant advantages of the “Light up” platform combined with AIE characteristics were demonstrated. Firstly, the platform could effectively reduce background noise and provide stable signal output, significantly improving detection sensitivity. Secondly, the convenient lateral flow strip detection achieved efficient monitoring of UDG concentration in HeLa cells, and the intuitive readout method showed extremely high ease of use and rapid detection capabilities. These results demonstrated that this strategy was not only suitable for the analysis of UDG activity but also had a wide range of potential applications for the sensitive and efficient detection of complex biological samples.
Conclusion
In summary, we have developed a THRUST strategy based on a bifunctional TAC and achieved hierarchical and precise regulation of Cas12a trans-cleavage activity. Firstly, this strategy used the template as the activator with no extra activator required, simplifying the addition of experimental components. Secondly, the strategy utilized T7 RNAP-mediated translesion synthesis to control the length of crRNA, enabling “full-partial-blocked” hierarchical and precise regulation of the CRISPR/Cas12a system. Compared with direct crRNA modification, THRUST simplified the design process by chemically modifying the activator, providing a more economical and flexible approach to system regulation. Furthermore, by utilizing the generation mechanism of the AP site, we developed a biosensor capable of detecting UDG with high sensitivity and specificity. Based on the conventional “Dim down” fluorescence signal output platform, the “Light up” fluorescence platform was constructed by combining with signal output molecules with AIE characteristics, which significantly improved detection sensitivity. Additionally, an AIE lateral flow strip platform was further developed, featuring high sensitivity, portability, and ease of use, making it suitable for practical applications. Overall, this work not only offers a novel and hierarchical strategy for the activation and inhibition of the CRISPR/Cas12a system but also establishes a versatile platform for advancing Cas12a-based biosensor design. By combining precise molecular control with sensitive detection capabilities, this method provides new ideas for innovative applications in areas such as molecular diagnostics and enzyme activity monitoring, further narrowing the gap between basic research and practical applications in the field of point-of-care testing.Experimental
Materials and reagents
All of the materials, reagents, apparatus and oligonucleotide sequences (Tables S1–S4†) used in the experiment are listed in the ESI.†The construction and assembly of TACs
1 μM T7 promoter and 1 μM template (with or without transcriptional regulatory units) were mixed in hybridization buffer. Deionized water was added to make the volume reach 50 μL. The mixture was incubated in a 95 °C water bath for 10 min, slowly cooled to room temperature to allow annealing and hybridization to form TACs and stored at −20 °C for use.Fluorescence process of Cas12a′s trans-cleavage activity with TACs
0.5 nM TACs were mixed with 10 nM crRNA, and 10 nM Cas12a, 100 nM reporter FAM-TTTATT-BHQ1 (F-Q), Buffer II and deionized water were added to make the total volume reach 100 μL. The reaction was carried out at 37 °C for 1 h, and the fluorescence intensity at 528 nm was measured with an excitation wavelength of 488 nm.Fluorescence detection of the THRUST system
0.5 nM TACs were mixed with 30 U T7 RNAP, 0.1 mM ribonucleoside triphosphates (rNTPs), and 1 × T7 RNAP reaction buffer to make a 50 μL system and incubated at 37 °C for 1 h. Finally, 10 nM Cas12a, 100 nM reporter FQ, Buffer II, and deionized water were added to make the total volume reach 100 μL. The reaction was carried out at 37 °C for 1 h, and the fluorescence intensity at 528 nm was measured with an excitation wavelength of 488 nm.Detection of UDG in the “Dim down” platform
0.5 nM TAC-dU2 and different concentrations of UDG were prepared in a 20 μL reaction solution containing 1 × UDG buffer and reacted at 37 °C for 1 h. Then, 30 U T7 RNAP, 0.1 mM rNTPs, and 1 × T7 RNAP buffer were added to make the system volume 50 μL and incubated at 37 °C for 1 h. Finally, 10 nM Cas12a, 100 nM fluorescent probe F-Q, Buffer II, and deionized water were used to make the total volume 100 μL. The reaction was carried out at 37 °C for 1 h, and the fluorescence intensity at 528 nm was measured under 488 nm excitation.Detection of UDG in the “Light up” platform
2 nM TAC-dU2 and different concentrations of UDG were prepared into a 20 μL reaction solution containing 1 × UDG buffer and reacted at 37 °C for 1 h. Then 30 U T7 RNAP, 0.1 mM rNTPs, and 1 × T7 RNAP buffer were added to a volume of 30 μL, and allowed to react at 37 °C for 1 h. Finally, 10 nM Cas12a, 0.4 μM G4, and Buffer II were added to 50 μL, and allowed to react at 37 °C for 0.5 h. Finally, 30 μM AR, 200 nM hemin, 1 mM H2O2, HEPES buffer (20 mM HEPES, 10 mM KCl, 100 mM NaCl, pH 7.3) and deionized water were added to reach a total volume of 100 μL. The reaction was carried out at room temperature for 10 min, and the fluorescence intensity was measured at 598 nm under 490 nm excitation.Detection of UDG using a G4-based test trip
To detect UDG fluorescence using a test strip, the cleavage product of bio-G4 in the “Light up” platform was incubated with AR and hemin in HEPES buffer. This solution was then applied to the sample well of the test strip. Following this, a combination of resorufin and 1 mM of H2O2 was added to the test zone and allowed to incubate at room temperature for 5 min. Images of the T and C lines on the test strip were then captured using a camera.Data availability
The data supporting this article are included in the ESI.†Author contributions
Y. C., F. Z., and B. Z. T. conceived and supervised the project and provided financial support. L. Q. performed the experiments and contributed to the data analysis. W. J. W., X. X., T. Z., Y. C., G. X., F. W., S. W., Q. H., and Z. Z. participated in the experiments and discussed the results. L. Q. wrote the manuscript, and Y. C., F. Z., and B. Z. T. revised the manuscript. All authors reviewed and approved the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21705080, 31900905), Natural Science Foundation of Jiangsu Province (No. BK20221304), “Blue Project” Foundation of the Higher Education Institutions of Jiangsu Province, Shenzhen Key Laboratory of Functional Aggregate Materials (ZDSYS20211021111400001), and the Science Technology and Innovation Commission of Shenzhen Municipality (KQTD20210811090142053).Notes and references
- P. Horvath and R. Barrangou, Science, 2010, 327, 167–170 CrossRef CAS PubMed.
- G. Gasiunas, R. Barrangou, P. Horvath and V. Siksnys, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2579–2586 CrossRef PubMed.
- S. Y. Li, Q. X. Cheng, J. K. Liu, X. Q. Nie, G. P. Zhao and J. Wang, Cell Res., 2018, 28, 491–493 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. D. Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- Y. Yin, W. Xie, M. Xiong, Y. Gao, Q. Liu, D. Han, G. Ke and X. B. Zhang, Angew. Chem., Int. Ed., 2023, 62, e20230983 Search PubMed.
- W. Kang, F. Xiao, X. Zhu, X. Ling, S. Xie, R. Li, P. Yu, L. Cao, C. Lei, Y. Qiu, T. Liu and Z. Nie, Angew. Chem., Int. Ed., 2024, 63, e202400599 CrossRef CAS PubMed.
- N. Bagheri, A. Chamorro, A. Idili and A. Porchetta, Angew. Chem., Int. Ed., 2024, 63, e202319677 CrossRef CAS PubMed.
- Y. Wu, W. Luo, Z. Weng, Y. Guo, H. Yu, R. Zhao, L. Zhang, J. Zhao, D. Bai, X. Zhou, L. Song, K. Chen, J. Li, Y. Yang and G. Xie, Nucleic Acids Res., 2022, 50, 11727–11737 CrossRef CAS PubMed.
- S. R. Rananaware, E. K. Vesco, G. M. Shoemaker, S. S. Anekar, L. S. W. Sandoval, K. S. Meister, N. C. Macaluso, L. T. Nguyen and P. K. Jain, Nat. Commun., 2023, 14, 5409 CrossRef CAS PubMed.
- S. Zhao, Q. Zhang, R. Luo, J. Sun, C. Zhu, D. Zhou and X. Gong, Chem. Sci., 2024, 15, 18347–18354 RSC.
- X. Fei, C. Lei, W. Ren, X. Liu and C. Liu, Anal. Chem., 2023, 95, 12169–12176 CrossRef CAS PubMed.
- Y. Wu, D. Chang, Y. Chang, Q. Zhang, Y. Liu, J. D. Brennan, Y. Li and M. Liu, Small, 2023, 19, 2303007 CrossRef CAS PubMed.
- M. Hu, Z. Qiu, Z. Bi, T. Tian, Y. Jiang and X. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2092933177 Search PubMed.
- Z. Hu, A. Sun, J. Yang, G. Naz, G. Sun, Z. Li, J. Gogo Liu, S. Zhang and X. Zhang, Chem. Sci., 2023, 14, 5945–5955 RSC.
- W. Zhang, Y. Zhong, J. Wang, G. Zou, Q. Chen and C. Liu, Nucleic Acids Res., 2025, 53, gkaf040 CrossRef PubMed.
- Y. Li, Y. Wu, R. Xu, J. Guo, F. Quan, Y. Zhang, D. Huang, Y. Pei, H. Gao, W. Liu, J. Liu, Z. Zhang, R. Deng, J. Shi and K. Zhang, Nat. Commun., 2023, 14, 7722 CrossRef CAS PubMed.
- L. T. Nguyen, B. M. Smith and P. K. Jain, Nat. Commun., 2020, 11, 4906 CrossRef CAS PubMed.
- W. Huang, J. Wang, C. Wang, Y. Liu, W. Li, Q. Chen, J. Zhai, Z. Xiang and C. Liu, Adv. Sci., 2025, 12, e2411305 CrossRef PubMed.
- R. Zhao, W. Luo, Y. Wu, L. Zhang, X. Liu, J. Li, Y. Yang, L. Wang, L. Wang, X. Han, Z. Wang, J. Zhang, K. Lv, T. Chen and G. Xie, Nucleic Acids Res., 2023, 51, 10795–10807 CrossRef CAS PubMed.
- J. Xu, L. Cao, S. Yang, Y. Jian, Y. Liu, Z. Shen, Q. Liu, X. Chen, M. Li, S. Li, X. Zuo, M. Li and H. Wang, Aggregate, 2024, 6, e663 CrossRef.
- G. Wang, W. Tian, X. Liu, W. Ren and C. Liu, Anal. Chem., 2020, 92, 6702–6708 CrossRef CAS PubMed.
- Y. H. Chen and D. F. Bogenhagen, J. Biol. Chem., 1993, 268, 5849–5855 CrossRef CAS PubMed.
- T. Ji, N. Xie, J. Ding, M. Wang, X. Guo, Y. Chen, S. Yu, Y. Feng and B. Yuan, Anal. Chem., 2023, 95, 8384–8392 CrossRef CAS PubMed.
- Z. J. Liu, S. Martinez Cuesta, P. Van Delft and S. Balasubramanian, Nat. Chem., 2019, 11, 629–637 CrossRef CAS PubMed.
- C. Liu, B. H. Le, W. Xu, C. Yang, Y. H. Chen and L. Zhao, Nucleic Acids Res., 2023, 51, e73 CrossRef CAS PubMed.
- A. Agapov, A. Olina and A. Kulbachinskiy, Nucleic Acids Res., 2022, 50, 3018–3041 CrossRef CAS PubMed.
- M. Tanasova, S. Goeldi, F. Meyer, P. C. Hanawalt, G. Spivak and S. J. Sturla, Chembiochem, 2015, 16, 1212–1218 CrossRef CAS PubMed.
- S. Tornaletti, L. S. Maeda and P. C. Hanawalt, Chem. Res. Toxicol., 2006, 19, 1215–1220 Search PubMed.
- E. Worle, A. Newman, J. D Silva, G. Burgio and D. Grohmann, Nucleic Acids Res., 2022, 50, 10153–10168 CrossRef PubMed.
- I. Strohkendl, F. A. Saifuddin, J. R. Rybarski, I. J. Finkelstein and R. Russell, Mol. Cell, 2018, 71, 816–824 CrossRef CAS PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- J. C. Cofsky, D. Karandur, C. J. Huang, I. P. Witte, J. Kuriyan and J. A. Doudna, elife, 2020, 9, e55143 CrossRef CAS PubMed.
- Y. Jeon, Y. H. Choi, Y. Jang, J. Yu, J. Goo, G. Lee, Y. K. Jeong, S. H. Lee, I. Kim, J. Kim, C. Jeong, S. Lee and S. Bae, Nat. Commun., 2018, 9, 2777 CrossRef PubMed.
- K. Fugger, I. Bajrami, D. S. M. Silva, S. J. Young, S. Kunzelmann, G. Kelly, G. Hewitt, H. Patel, R. Goldstone, T. Carell, S. J. Boulton, J. Macrae, I. A. Taylor and S. C. West, Science, 2021, 372, 156–165 CrossRef CAS PubMed.
- C. Li, X. Li, S. Wei and T. Wu, Anal. Chem., 2025, 97, 3680–3686 CrossRef CAS PubMed.
- L. Xiong, J. Wang, F. Yang, B. Z. Tang and X. He, Anal. Chem., 2024, 96, 9244–9253 CrossRef CAS PubMed.
- J. Tao, H. Zhang, M. Weinfeld and X. C. Le, ACS Meas. Sci. Au, 2024, 4, 459–466 CrossRef CAS PubMed.
- W. Zeng, Z. Chen, Y. Lei, W. Liang, Y. Chai, R. Yuan and Y. Zhuo, Anal. Chem., 2024, 96, 15915–15923 CrossRef CAS PubMed.
- Y. Du, Y. Cui, X. Li, G. Sun, Y. Zhang, A. Tang, K. Kim and D. Kong, Anal. Chem., 2018, 90, 8629–8634 CrossRef CAS PubMed.
- Y. Du, H. Jiang, Y. Huo, G. Han and D. Kong, Biosens. Bioelectron., 2016, 77, 971–977 CrossRef CAS PubMed.
- X. Yuan, Y. Lei, Y. Li, X. Zhou, X. Yang, Y. Chai, R. Yuan and Y. Zhuo, Anal. Chem., 2024, 96, 17013–17020 CrossRef CAS PubMed.
- T. Liu, Z. Li, M. Chen, H. Zhao, Z. Zheng, L. Cui and X. Zhang, Biosens. Bioelectron., 2021, 194, 113607 CrossRef CAS PubMed.
- K. Dong, W. Shu, J. Zhang, S. Cheng, J. Zhang, R. Zhao, T. Hua, W. Zhang and H. Wang, Biosens. Bioelectron., 2023, 226, 115118 CrossRef CAS PubMed.
- X. Song, Q. Ding, J. Zhang, R. Sun, L. Yin, W. Wei, Y. Pu and S. Liu, Anal. Chem., 2021, 93, 13687–13693 CrossRef CAS PubMed.
- Q. Zhang, C. Li, F. Ma, X. Luo and C. Zhang, Biosens. Bioelectron., 2022, 213, 114447 CrossRef CAS PubMed.
- M. Chao, S. Tai, M. Mao, W. Cao, C. Peng, W. Ma, Y. Feng and Z. Wang, Aggregate, 2024, 6, e644 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02575c |
| This journal is © The Royal Society of Chemistry 2025 |