
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blocky bromination of poly(ether ketone ketone) as a means to preserve crystallizability and rapid crystallization kinetics†
Michelle E.
Pomatto
 ab,
Erin R.
Crater
a,
Garrett F.
Godshall
ab and
Robert B.
Moore
*ab
ab,
Erin R.
Crater
a,
Garrett F.
Godshall
ab and
Robert B.
Moore
*ab
aDepartment of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: rbmoore3@vt.edu
bMacromolecules Innovation Institute, Virginia Tech, Blacksburg, Virginia 24061, USA
First published on 20th January 2024
Abstract
In this work, the post-polymerization bromination of poly(ether ketone ketone) (PEKK, KEPSTAN 8001, T/I = 80/20) with N-bromosuccinimide is reported for the first time. Two microstructures are synthesized – a random microstructure through functionalization in the homogeneous solution state and a blocky microstructure through functionalization in the heterogeneous gel state from a benign solvent, diphenyl acetone. The resultant gel exhibits a nanoscale fibrillar-network morphology with porosity and surface areas of 85.5% ± 3 and 152 ± 12 m2 g−1, respectively. Degrees of bromination between 12–62 mol% were obtained for both the random and blocky brominated PEKK. At similar degrees of bromination, blocky brominated PEKK exhibited increased glass transition temperatures, greater crystallizability, higher melting temperatures and faster crystallization kinetics as compared to the randomly brominated analogs. The crystallization kinetics were analyzed over a range of isothermal crystallization temperatures (210 °C–290 °C) utilizing fast scanning calorimetry. For the random microstructure, the isothermal crystallization temperature resulting in the fastest crystallization kinetics decreased to lower temperatures with increasing mol% bromination, attributed to a comonomer confinement effect. In contrast, the temperature of fastest crystallization for the blocky microstructures remained relatively constant with increasing mol% bromination and similar to that of pure, unfunctionalized PEKK. This behavior was attributed to the longer runs of pristine PEKK units between brominated monomers compared to the random analogs, resulting in a negligible confinement effect. SAXS/WAXS analysis showed that the electron density of the amorphous phase is affected by the copolymer microstructure. It is shown that blocky functionalization of PEKK in the gel state preserves crystallizability and high melting temperatures with minor decreases in crystallization rates. This finding further demonstrates blocky functionalization in the gel state is a facile way to effectively block up functionality while preserving long runs of crystallizable segments along the chains.
Introduction
Poly(ether ketone ketone) (PEKK) is an aromatic high-performance thermoplastic widely used in industrial sectors including aerospace, automotive, and oil and gas industries due to its excellent mechanical performance and chemical resistance properties.1–4 PEKK is a linear semi-crystalline thermoplastic gaining increased interest in these sectors due to excellent properties including high impact strength, chemical resistance, high glass transition temperature and melting temperatures, low flammability and good processibility.3,5,6 While generally referred to as PEKK, commonly utilized grades of PEKK (KEPSTAN 8000, 7000, 6000) are copolymers of terephthalate and isophthalate units and commonly categorized by their “T/I” ratio (Fig. 1). The PEKK copolymer is of particular interest in industry due to the ability to tune the melting and glass transition temperature by tuning the ratio of terephthalate to isophthalate moieties when synthesized.7 It is well understood that the isophthalate moieties act as defects within the crystal structure, with increasing isophthalate content leading to an increase in chain mobility and reduction in the glass transition (Tg), melting temperature (Tm), and crystallization rates.3,7 | ||
| Fig. 1 The molecular structure of poly(ether ketone ketone) copolymer repeat unit comprising of terephthalic, “T” and isophthalic, “I” monomers. X = 80 and Y = 20 for PEKK 80/20. | ||
In addition to the property control imparted by the T/I ratio, it may be advantageous to chemically modify the structure of PEKK by post-polymerization functionalization to further tune Tg, Tm, crystallization kinetics, chemical compatibility, solubility, and other physical and chemical properties. However, due to limited solubility of PEKK in solvents conventionally used for functionalization chemistry, it is not surprising that little attention has been given to the post-polymerization modification of PEKK and subsequent analysis of the effect of functionality on thermo-physical properties. Functionalization chemistries that have been explored are generally limited to reactions that facilitate dissolution by chemically modifying the polymer backbone (e.g., sulfonation for membrane applications,8–10 biocompatibility11 and improved mechanical properties12 as well as reversible dithiolation13 to improve solubility for fiber fabrication). Clearly, new solvents for PEKK must be explored to realize the potential advantages of functionalized PEKK.
Due to the high melting temperature of PEKK (Tm = 300 °C to 360 °C, depending on T/I), high boiling point solvents must be used in order to break down the semi-crystalline structure of PEKK during dissolution. To date, solvents reported for PEKK include: dichloroacetic acid (DCA),14 4-phenylphenol,15–17 and 9-fluorenone.17 These solvents require dissolution temperatures ranging from 180 °C for DCA to over 230 °C for 4-phenylphenol and 9-fluorenone. While these solvents are all generally hazardous, they do provide some valuable attributes. For example, we have found that DCA is an ideal solvent for quantitative NMR analysis of pure PEKK.14 Moreover, solutions of PEKK in 4-phenylphenol and 9-fluorenone have been shown to form semi-crystalline foams or gels upon cooling via a thermally-induced phase separation (TIPS) process.17 These semi-crystalline gels are defined as substantially dilute continuous polymer networks that exhibit solid-like behavior while physically retaining liquids within the amorphous domains of the 3D macromolecular framework.18 These attributes are particularly relevant to our current approach to synthesize and characterize blocky, semicrystalline copolymers formed by post-polymerization functionalization in the heterogeneous gel state.19–22
Halogenation is a particularly useful functionalization of poly(aryl ether ketones) that provides a synthetic pathway for further functionalization and structure modifications including nitration, amination, oxidation, and a variety of other coupling reactions. Using our new solvents for PEKK (i.e., DCA and DPA), we are now able to synthesize random and blocky brominated PEKK with accurate characterization of the degree of bromination by NMR.
In this work, we demonstrate for the first time the efficient post-polymerization bromination of PEKK by two methods (1) halogenation performed in the heterogeneous gel state to create blocky microstructures, and (2) halogenation in the homogeneous solution state to create random copolymers. The purpose of this work is to quantitatively compare matched sets of blocky and random brominated PEKK to assess the influence of microstructure on a range of thermal properties including: the glass transition temperature, Tg, melting behavior, crystallinity, and crystallization kinetics. Additionally, a novel NMR technique to analyze functionalized PEKK is presented due to the discovery of a solvent that allows for room temperature NMR analysis. Finally, in depth analysis of the crystallization kinetics of the BrPEKK copolymers is probed by fast scanning calorimetry (FSC) over a wide range of crystallization temperatures.
Experimental
Materials
Poly(ether ketone ketone) pellets, KEPSTAN® 8001 (vendor reported T/I = 80/20, Tm = 358 °C, Tg = 165 °C, MFR at 380 °C/5 kg = 15 cm3/10 min) were obtained from Arkema and referenced hereon as PEKK. Pellets were washed with acetone and deionized water and dried at 80 °C in a vacuum oven for 8 hours prior to use. Dichloroacetic acid (DCA) and dichloromethane (DCM) were purchased from Sigma-Aldrich and used as received. N-bromosuccinimide (NBS) was purchased from Sigma-Aldrich and purified by recrystallization in water before use. Diphenyl acetone (DPA) was purchased from Oakwood Chemical and used as received.Random bromination of PEKK
To synthesize randomly brominated PEKK (rBrPEKK), PEKK (2.0 g) was dissolved in DCA (20 mL) at 185 °C under constant argon purge. Once fully dissolved, the temperature of the homogeneous solution was lowered to 80 °C and equilibrated for a minimum of 1 hour. A solution of N-bromosuccinimide (NBS) was prepared by dissolving in 5 mL of DCA at room temperature. The degree of substitution was controlled by controlling the molar ratio of NBS to PEKK repeat units. The DCA/NBS solution was introduced to the homogeneous PEKK solution dropwise with vigorous stirring. The reaction proceeded for a minimum of 24 hours under constant argon purge at 80 °C. The reaction was terminated by precipitating into methanol. The white precipitate was washed repeatedly with water and then washed with methanol by Soxhlet extraction for a minimum of 24 hours. The product was dried at 80 °C in a vacuum oven for a minimum of 12 hours before analysis.Blocky bromination of PEKK
To synthesize blocky brominated PEKK (bBrPEKK), a PEKK gel was prepared prior to brominating. To form the PEKK gel, PEKK (2 g) was dissolved in diphenyl acetone (DPA) at 320 °C under constant argon purge to a concentration of 15 w/v%. The homogeneous solution was allowed to cool to room temperature. Within 1–3 minutes, the solution formed an opaque solid indicating the formation of a semi-crystalline polymer gel. The gel was equilibrated at room temperature for 24 hours and then manually broken into small (∼1–2 mm) pieces and transferred to Soxhlet to solvent exchange the DPA with ethanol. After washing with ethanol by Soxhlet extraction for a minimum of 48 hours, the gel was then solvent exchanged to DCM by Soxhlet extraction for a minimum of 24 hours. The DCM swollen gels were then suspended in 95/5 DCA/DCM at 80 °C under reflux and constant argon purge. The addition of the nonsolvent DCM ensured that the PEKK gel did not dissolve during the bromination process. A solution of N-bromosuccinimide (NBS) was prepared by dissolving in 5 mL of DCA at room temperature. The degree of substitution was controlled by controlling the molar ratio of NBS to PEKK repeat unit. The DCA/NBS solution was introduced to the gel suspension dropwise with vigorous stirring. The reaction was allowed to proceed for a minimum of 24 hours under constant argon purge at 80 °C. The reaction was terminated by precipitating into methanol. The brominated gel pieces were then washed repeatedly with water and washed with methanol by Soxhlet extraction for a minimum of 24 hours. The product was dried at 80 °C in a vacuum oven for a minimum of 12 hours.NMR spectroscopy
The degrees of bromination of rBrPEKK and bBrPEKK were determined by 1H NMR spectroscopy at room temperature using a unique solvent system14 comprised of DCA and deuterated chloroform (CDCl3) with 0.05% v/v TMS on a Bruker Avance 400 MHz spectrometer. Spectra were collected as the average of 128 scans with a relaxation delay of 10 seconds. The samples were dissolved in DCA at elevated temperature to a concentration of 10% w/v. Once dissolved, the solution was cooled to room temperature and diluted with CDCl3 with 0.05% v/v TMS to a concentration of 5% w/v. The 1H NMR splitting patterns are designated as follows: s (singlet), d (doublet), and m (multiplet). Qualitative 13C NMR spectra were measured using a Bruker Avance 500 MHz spectrometer. The samples were prepared following the same procedure as described above to a final concentration of 10% w/v. Spectra were collected as the average of 256 scans with relaxation delay of 2 seconds. 2D heteronuclear correlation spectroscopy (HSQC) was collected on an Agilent U4-DD2 400 MHz spectrometer. The samples were prepared following the procedure described above.Scanning electron microscopy
The internal surface morphology of PEKK aerogels formed from DPA solutions were characterized using a LEO (Zeiss) 1550 scanning electron microscope (SEM) with an accelerating voltage of 10 kV. The gel samples were solvent exchanged from DPA to ethanol, from ethanol to water, frozen for a minimum of 12 hours, and lyophilized for 24 hours to produce a PEKK aerogel. The aerogel was freeze-fractured utilizing liquid nitrogen and coated with iridium (5 nm) prior to SEM imaging.Surface area analysis
The surface area of the PEKK aerogel was characterized using a Micrometrics 3 Flex Adsorption Analyzer (Micromeritics Instrument Corporation, USA) using nitrogen adsorption. The solvent extracted aerogel sample was prepared following the same procedure described for SEM analysis. The surface area was determined utilizing the Brunauer–Emmett–Teller (BET) method.23Porosity analysis
The porosity of the PEKK aerogel was calculated according to ASTM D6226-15 using an AccuPyc 1340 helium pycnometer (Micromeritics Instrument Corporation, USA) with a 1 cm3 insert and a helium equilibration rate of 0.05 psig min−1. The solvent extracted aerogel sample was prepared following the same procedure described for SEM analysis. The porosity (Π) was calculated as: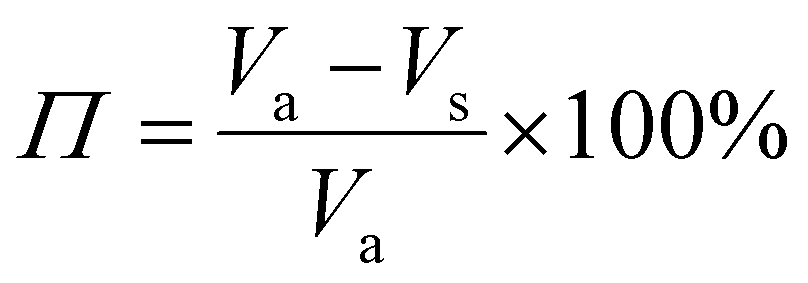 | (1) |
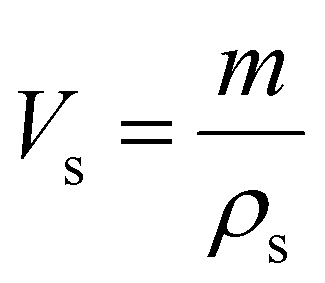 | (2) |
Thermogravimetric analysis
A TA Instruments TGA Q550 thermogravimetric analyzer was used to determine the thermal stability of PEKK and BrPEKK copolymers. Samples were prepared by vacuum drying overnight at 80 °C. All analyses were performed with a ramp rate of 20 °C min−1 in a nitrogen atmosphere. The temperature at which the weight loss of the samples reached 5% (i.e., TD5%) was determined using Trios software (TA Instruments).Differential scanning calorimetry
A TA instruments Q2000 differential scanning calorimeter was used to probe the thermal transitions, degree of crystallinity, and crystallizability of PEKK and BrPEKK copolymers. Samples (4–8 mg) were heated from 30 °C to 400 °C at 10 °C min−1 in nitrogen and isothermally held at 400 °C for 3 minutes to remove all thermal history. Samples were then cooled from 400 °C to 30 °C at 10 °C min−1 to observe changes in heat flow associated with crystallization upon cooling. Samples were then reheated to 400 °C at 10 °C min−1 to observe changes in heat flow associated with the crystallization and melting of crystals which crystallized upon cooling from the melt or cold crystallization upon heating. The glass transition temperatures were determined from fully amorphous PEKK and BrPEKK copolymer samples. Samples were prepared by melt-pressing at 400 °C for 3 minutes to remove thermal history before immediately quenching in ice water to obtain amorphous samples. Samples were dried at 100 °C in a vacuum oven for a minimum of 12 hours before analyzing the Tg from first heat by standard DSC at 10 °C min−1 in nitrogen. The glass transition temperature (Tg), crystallization temperature (Tc), cold crystallization temperature (Tcc) and melting temperature (Tm) were analyzed using Trios software (TA Instruments).The crystallization kinetics of rBrPEKK40 were analyzed by standard differential scanning calorimetry (DSC). Isothermal crystallization experiments were performed by heating rBrPEKK40 samples at 10 °C min−1 to 400 °C, isothermally holding for 3 minutes, rapid cooling (100 °C min−1) to the desired Tc (210 °C–280 °C, Δ 10 °C), isothermally holding at the desired Tc for 30–120 minutes, rapid cooling (100 °C min−1) to 30 °C, and finally heating to 400 °C at 10 °C min−1.
Fast scanning chip calorimetry
A Mettler Toledo Flash DSC 1 with a Huber intra cooler TC100 was employed to analyze the crystallization kinetics of the synthesized BrPEKK copolymers. The crystallization kinetics of PEKK were previously determined14 utilizing the same procedure described here. Prior to evaluation, the chip sensors were individually conditioned, and temperature corrected utilizing Mettler Toledo supplied calibration data and following Mettler Toledo instrument specifications. The temperature was experimentally calibrated by an indium standard. The onset of indium melting versus heating rate was used to determine the corrected horizontal shift of the experimental melting profiles. All experiments were conducted in an ultra-high purity N2 gas environment with a flow rate of 60 mL min−1 to limit oxidation and moisture in the sample chamber. Homogeneous samples were prepared by dissolving 2–3 mg of PEKK or BrPEKK copolymer in DCA and precipitating into methanol, washing excessively with water, and drying at 80 °C under vacuum overnight. Thin (2 μm thickness) samples were cut by microtome from the precipitated PEKK or BrPEKK copolymers and laterally cut to size by scalpel under a microscope. A thin layer of Wacker AK 60000 silicone oil was spread on the sensor prior to sample placement to improve thermal contact and allow for removal of the sample after testing. All samples were pre-melted at 5 K s−1 to 400 °C for 1 s to establish good thermal contact. The sample mass was estimated from the change in heat capacity at the glass transition temperature from an amorphous sample with all samples measured between 1 μg–3 μg. Experiments were repeated over multiple samples and multiple chip sensors to ensure reproducibility.Our previous work demonstrated that the critical cooling rate needed to prevent crystallization upon cooling from the melt for PEKK 80/20 is 10 K s−1.14 Cooling rates of 500 K s−1 were utilized for all experiments. Previous work also determined the time in the melt to completely remove thermal history for PEKK. The minimum time required to remove thermal history at 400 °C is 0.1 s for PEKK.14 Thus, a reasonable melt time of 1 s was utilized for all experiments.
The crystallization kinetics for rBrPEKK32, bBrPEKK31, bBrPEKK40, rBrPEKK52 and bBrPEKK52 were analyzed utilizing an interrupted isothermal crystallization (IIC) method.14,24,25 The time temperature profile for the IIC method is shown in Fig. 2. Select isothermal crystallization temperatures (Tiso) between 200 °C to 310 °C were analyzed by this method. After removing the thermal history at 400 °C for a minimum 1 s, the sample was quenched at 500 K s−1 to the respective Tiso and held for varying amounts of time (0.1 s–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s). The sample was quenched at −500 K s−1 to below the Tg to arrest any further crystallization. The partially crystallized sample was then heated to 400 °C at a heating rate of 500 K s−1 to measure degree of crystallinity as observed by the integration of the melting endotherm upon heating. The maximum isothermal crystallization time (tiso) was determined when the melting enthalpy remained at a constant value with increasing tiso. The maximum isothermal crystallization time was dependent on Tiso. The observed melting enthalpy versus tiso was used to calculate fractional crystallinity and the crystallization half-time (t1/2) over a range of isothermal crystallization temperatures.
000 s). The sample was quenched at −500 K s−1 to below the Tg to arrest any further crystallization. The partially crystallized sample was then heated to 400 °C at a heating rate of 500 K s−1 to measure degree of crystallinity as observed by the integration of the melting endotherm upon heating. The maximum isothermal crystallization time (tiso) was determined when the melting enthalpy remained at a constant value with increasing tiso. The maximum isothermal crystallization time was dependent on Tiso. The observed melting enthalpy versus tiso was used to calculate fractional crystallinity and the crystallization half-time (t1/2) over a range of isothermal crystallization temperatures.
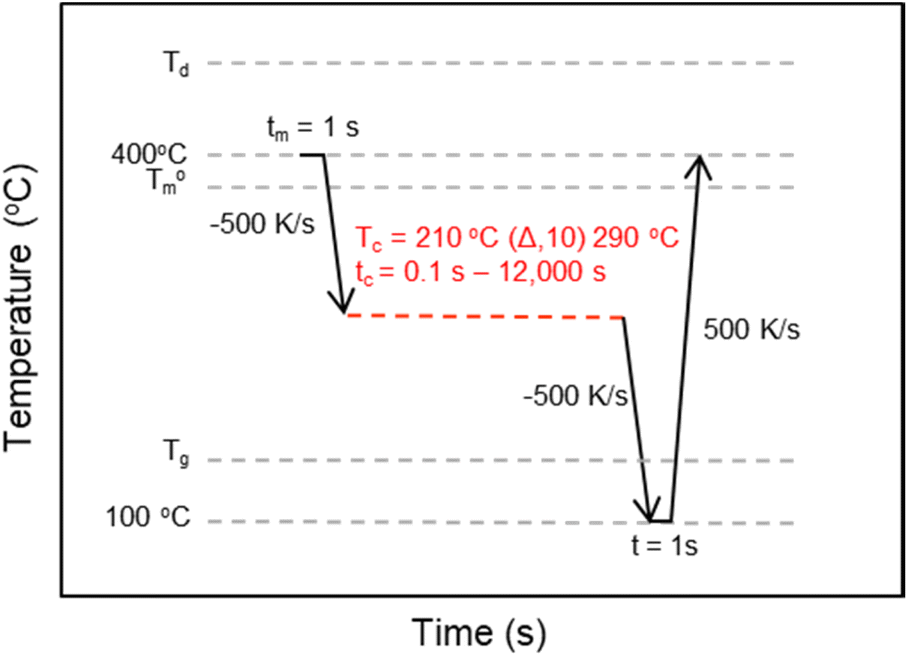 | ||
| Fig. 2 The time–temperature profile for the Flash DSC interrupted isothermal crystallization (IIC) method used to determine the t1/2 of BrPEKK copolymers at a range of Tiso. | ||
SAXS/MAXS/WAXD of PEKK and BrPEKK copolymers
Samples for small, mid, and wide-angle X-ray scattering/diffraction (SAXS, MAXS, WAXS/D) analysis were prepared as follows. The PEKK, rBrPEKK, and bBrPEKK copolymer samples were prepared utilizing a DSC. Samples were loaded into a DSC pan, heated to 400 °C at 10 °C min−1 in nitrogen and isothermally held at 400 °C for 3 minutes to remove all thermal history. Samples were then rapidly cooled at 100 °C min−1 to 280 °C and held for 30 minutes to allow for crystallization to occur. After 30 minutes, the samples were cooled at 10 °C min−1 to room temperature. The samples were then removed from the pan and utilized for SAXS/MAXS/WAXS/D analysis.The SAXS, MAXS, WAXS/D experiments were performed on the solid samples using a Xenocs Xeuss 3.0 SAXS/WAXS system equipped with a GeniX 3D Cu HFVLF microfocus X-ray source utilizing Cu K-α radiation (λ = 0.154 nm). The sample-to-detector distance was 900 mm for SAXS, 370 mm for MAXS, and 43 mm for WAXD. The q-range was calibrated using a silver behenate standard for SAXS and MAXS, and a lanthanum hexaboride standard for WAXD. The exposure times were 2 h, 1 h, and 30 min for SAXS, MAXS, and WAXD, respectively. The two dimensional scattering patterns were collected using a Dectris EIGER 4M detector in an evacuated chamber and reduced using the XSACT software to obtain azimuthally integrated intensity versus scattering vector q profiles, where q = (4π/λ)sin(θ), θ is one half of the scattering angle and λ is the X-ray wavelength. The data were normalized for background, thickness, and absolute intensity, and the SAXS/MAXS/WAXD profiles were merged into a single dataset for visualization purposes.
Results and discussion
Preparation and characterization of PEKK gel for blocky functionalization template
In our previous work, semicrystalline gels from syndiotactic polystyrene (sPS)21 and poly(ether ether ketone) (PEEK)19,20,22 have been demonstrated to act as effective templates for blocky functionalization, where the polymer chains are organized into a network of tightly packed chain segments in lamellar crystallites and solvent swollen amorphous chains. The tightly packed crystallites sterically exclude the functionalization reagent thus the functionalization reagent reacts only with monomer units that are accessible in the solvent-swollen amorphous component. The resultant copolymer consists of randomly functionalized blocks and unfunctionalized blocks. The unfunctionalized blocks originate from the monomer units that were isolated within the crystalline domains of the gel, thus crystallizability is retained. The schematic in Fig. S1† details the gelation procedure. In this work, PEKK was gelled from a newly discovered, benign gelation solvent, DPA. The microstructure of the 15 wt% PEKK/DPA gel collected by SEM is shown in Fig. 3. The microstructure shows a porous gel with a highly interconnected fibrillar morphology. The crystalline domains and amorphous domains exist within the polymer phase consisting of the interconnected axialitic struts. The surface area determined by nitrogen adsorption is 152 ± 12 m2 g−1, indicating high amounts of surface area available for functionalization. The porosity (Π) of the PEKK/DPA aerogel was calculated utilizing eqn (1) and (2) as 83.4% ± 0.4, a value that is higher than previously reported (71–81%)17 PEKK gels with similar nanoscale morphologies but from different gelation solvents with different wt% and higher densities. Additionally, the porosity was also calculated utilizing the degree of crystallinity and the following relationship: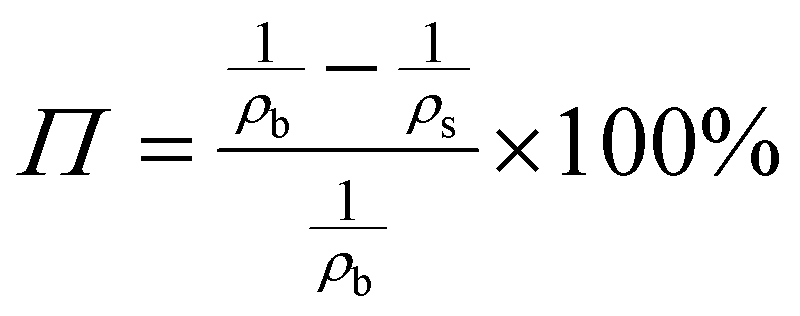 | (3) |
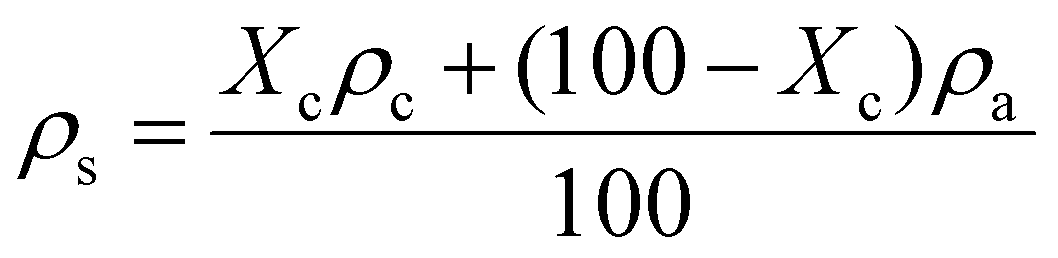 | (4) |
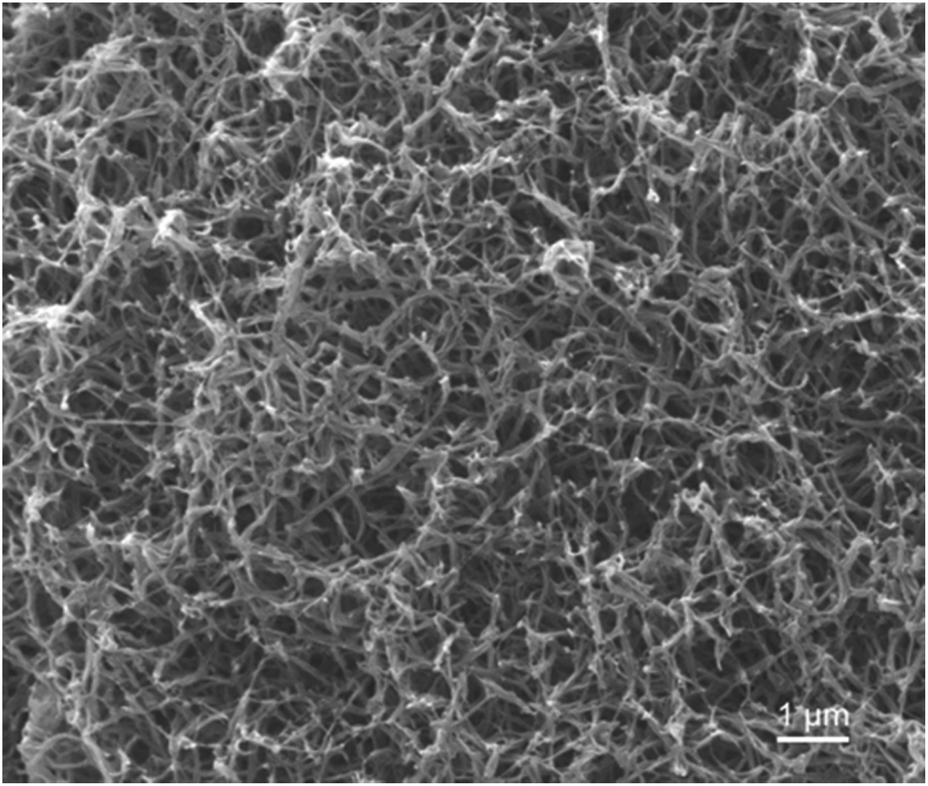 | ||
| Fig. 3 SEM image of 15 wt% PEKK aerogel gelled from DPA. | ||
Bromination of PEKK
Semi-crystalline polymer gels act as a template to produce blocky copolymers, as shown in previous work19–22 where the crystalline regions exclude the functionalization reagent and allow for functionalization to the amorphous region only. It was discovered that PEKK gels from 1,3-diphenylacetone (DPA), a nonhazardous, food grade additive27 resulting in a high porosity, high surface area, nanoscale fibrillar morphology gel. The gel state functionalization process reported here results in a blocky microstructure with runs of pristine crystallizable PEKK retained within the crystalline domains, and amorphous domains containing the functionalized PEKK monomers. The preservation of the pristine crystalline domains allows for significant crystallization even at high degrees of functionalization, similar to that demonstrated with sPS21 and PEEK.19,20Random and blocky microstructures were synthesized by bromination of PEKK in the random and gel state as shown in the reaction scheme in Fig. 4. The bromination of PEKK utilized N-bromosuccinimide (NBS) dissolved in dichloroacetic acid. This reagent system is discussed in detail in previous blocky functionalization work.19 When combined with strong acids such as dichloroacetic acid, NBS is protonated, which promotes the release of a bromonium ion,28 thus becoming a strong brominating reagent.29 This bromonium ion is highly electrophilic, facilitating efficient electrophilic aromatic substitution (EAS) reactions on the deactivated aromatic rings of PEKK that are proximal to the ether linkage, thus dictating the regiochemistry of the bromination reaction. There are 4 available sites for the EAS reaction to occur on the PEKK backbone, including the protons adjacent to the ether linkage in both the isophthalate and terephthalate monomer, as shown in Fig. 4. Due to these multiple reaction sites available for bromination, degrees of bromination greater than 100 mol% are possible. This allows for high degrees of bromination while retaining blocks of unfunctionalized monomers in the gel state bromination process.
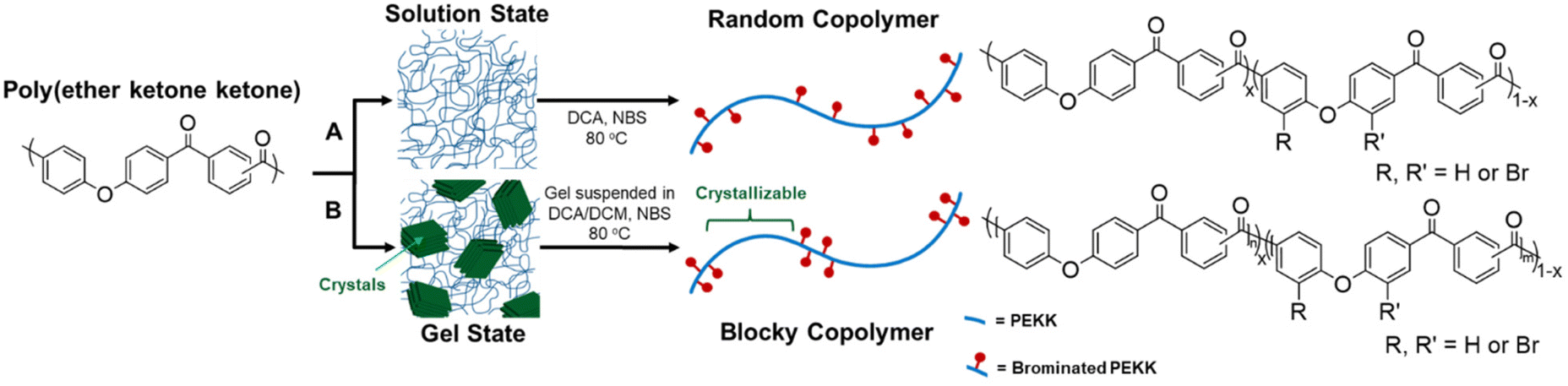 | ||
| Fig. 4 Schematic demonstrating synthesis of (A) random versus (B) blocky brominated microstructure of PEKK. | ||
Degree of bromination determined by NMR spectroscopy
Due to the limited solubility of PEKK, comprehensive NMR characterization of PEKK and functionalized PEKK has been limited. However, we recently discovered that DCA is a good solvent for NMR analysis of PAEKs30 and previous work14 established the 1H and 13C resonances for PEKK 80/20 and PEKK 70/30 from a solvent system of DCA and deuterated chloroform. The aromatic region of the 1H NMR spectra, peak assignments, and integrations for PEKK are summarized in Fig. S2.† PEKK exhibits proton resonances at 7.19–7.22 (m), 7.68–7.72 (t), 7.90–7.94 (m), 8.04–8.06 (m), and 8.16 (s) ppm associated with the aromatic protons of the terephthalic and isophthalic moieties. Fig. 5 shows the 1H spectra of the random and blocky brominated PEKK up to 62% bromination, and the molecular structure of (a) unsubstituted, (b) monosubstituted, and (c) disubstituted BrPEKK with their corresponding proton resonance assignments. The degree of bromination was determined through comparison of the ratio of the integration of resonance peaks associated with the aromatic protons adjacent to the ether linkage (H1a–d) to the remaining protons (H2a–d, H3a–b, H4a, H5a). Protons 2, 3, 4 and 5 are not capable of undergoing an EAS reaction and so the total sum of protons 2, 3, 4 and 5 will remain constant regardless of the degree of bromination. When setting the integration of 7.65 ppm to 8.25 ppm equal to 80, then: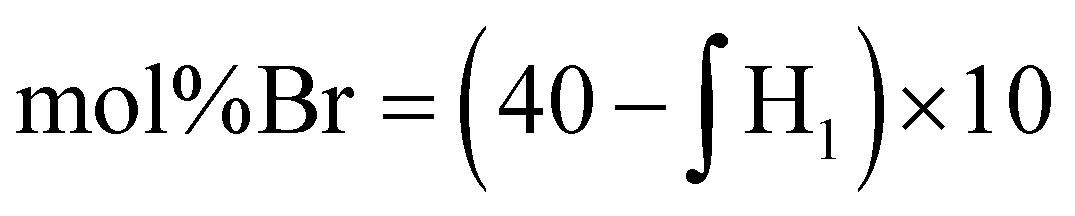 | (5) |
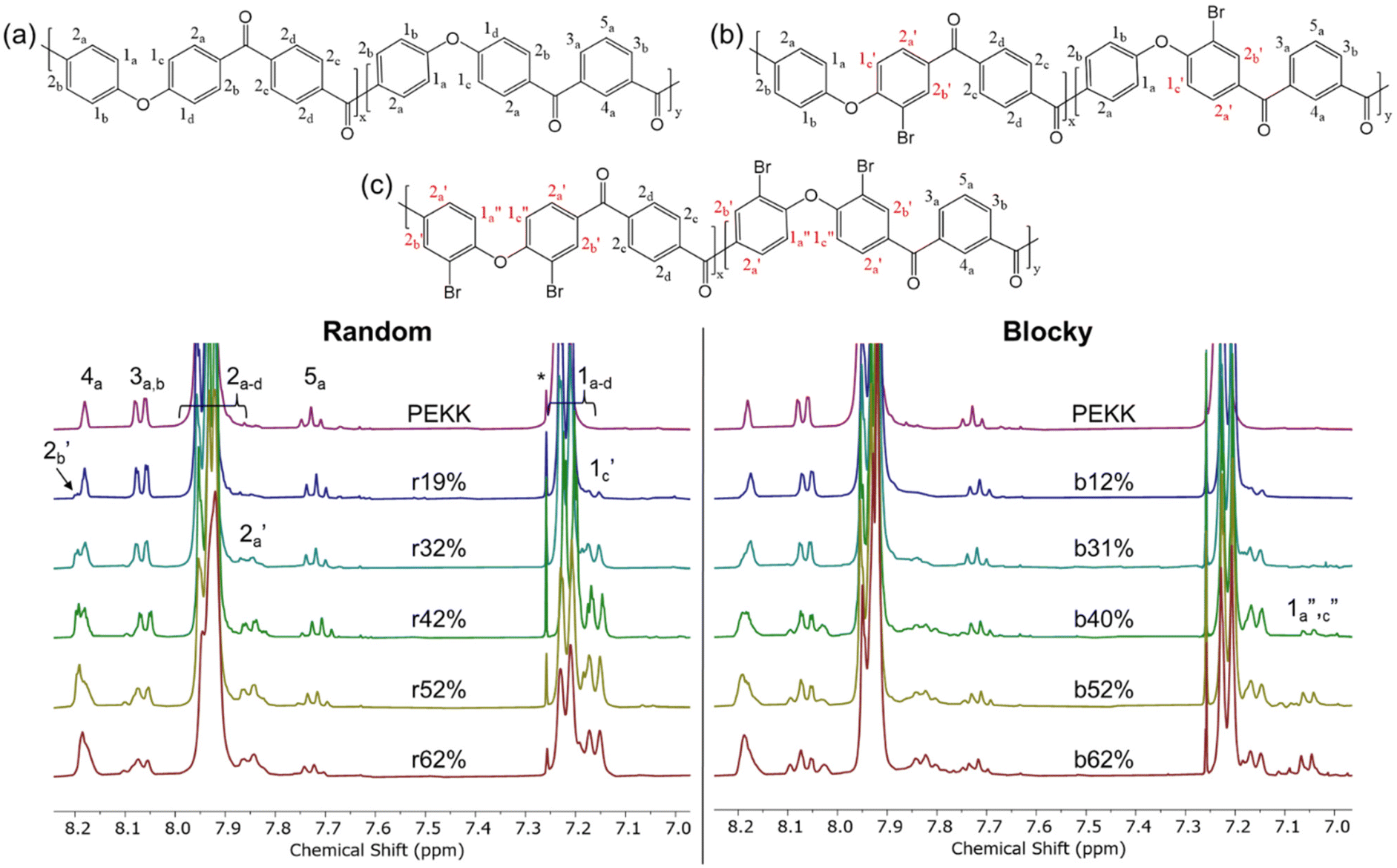 | ||
| Fig. 5 1H NMR of random (left) and blocky (right) brominated PEKK up to 62 mol% bromination and the corresponding chemical structures of (a) pristine PEKK, (b) monosubstituted BrPEKK, and (c) disubstituted BrPEKK. Assignments are further verified by 2D NMR in the ESI.† Spectra are referenced to CDCl3. The asterisk (*) refers to reference solvent resonance. | ||
Using eqn (5), the range of calculated mol% Br of the random and blocky microstructures are shown in Table 1. Compared to previous work19 with PEEK, the maximum degree of bromination is much lower (62% vs. 175%). This is because PEKK has a maximum of two bromine functionalities per monomer, while PEEK has a maximum of four bromine functionalities per monomer, resulting in a higher achievable mol% bromination.
| Calculated mol% bromination | |
|---|---|
| Random | Blocky |
| 19% | 12% |
| 32% | 31% |
| 42% | 40% |
| 52% | 52% |
| 62% | 62% |
Upon bromination, new resonance peaks appear at approximately 7.05 (d), 7.16 (d), 7.85 (m), and 8.20 (m). The peaks at 7.19 ppm to 7.22 ppm, associated with H1a–d, decrease in intensity with increasing degree of bromination due to replacement of H1a–b by the bromine functionality. The new resonance peaks at 7.16 ppm and 7.05 ppm are attributed to shifting of H1a–d protons of monosubstituted (H1c′) and disubstituted (H1a′′,c′′) PEKK monomers, respectively. The systematic shifting upfield is due to increased shielding from the electron dense bromine functionality. The peaks appearing at 7.85 ppm and 8.20 ppm are associated with changes in chemical environment of protons H2a–d where protons H2a′, which are ortho to the bromine functionality, shift upfield due to shielding, and protons H2b′, which are para to the bromine functionality, shift downfield, due to the electronegativity of bromine pulling electron density away from the aromatic ring. Further verification of the 1H and 13C assignments, peak correlations, and monosubstituted versus disubstituted peak assignments of PEKK and BrPEKK are provided by heteronuclear single quantum coherence (HSQC) correlation experiments (Fig. S3†).
From Fig. 5, the amount of unsubstituted (a), monosubstituted (b), and disubstituted (c) PEKK monomers can be determined from analysis of the H1a–d resonance peaks. As previously discussed, H1a–d resonance peaks shift upfield upon monosubstitution (H1c′) or disubstitution (H1a′′,c′′) of the terephthalate or isophthalate monomer. The integration values of the H1a–d, H1c′ and H1a′′,c′′ resonance peaks for PEKK, rBrPEKK and bBrPEKK are shown in Fig. 6a and b. As can be seen in Fig. 6a, the integration value for H1c′ is larger for the random versus the blocky microstructure and H1a′′,c′′ remains at an integration value of 0 for the random but is non-zero for the blocky microstructure. These results can be explained by the confinement effect of blocky functionalization in which the fraction of unfunctionalized monomers is preserved in the crystal regions of the gel. Additionally, the lower number of accessible functionalizable monomers in the amorphous region leads to higher frequency of multiple substitutions. In Fig. 6b the H1a–d peak integration is lower for the random microstructure versus the blocky microstructure indicating more protons are being substituted from pristine monomers at the same degree of functionalization for the random microstructure versus the blocky microstructure.
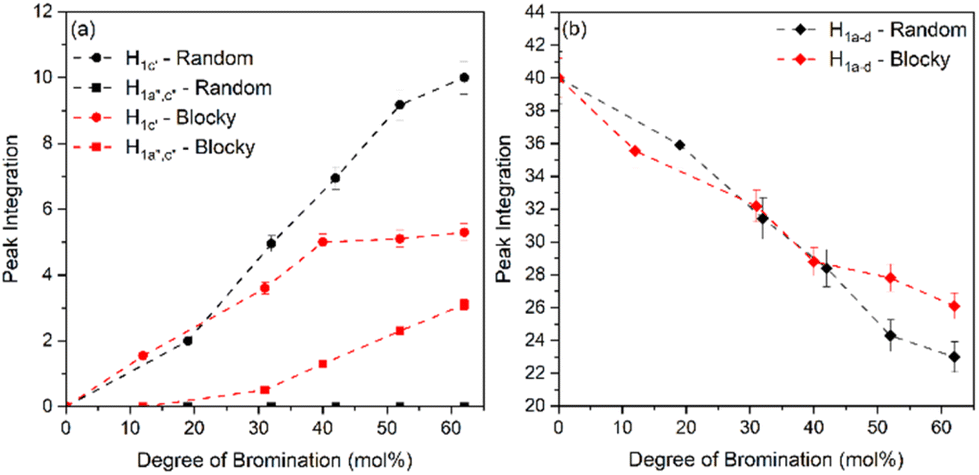 | ||
| Fig. 6 Peak integration versus degree of bromination of (a) monosubstituted, H1a′,c′ (circles), disubstituted, H1a′′,c′′ (squares) and (b) unsubstituted, H1a–d (diamonds) random (black) and blocky (red) BrPEKK. The blocky microstructure shows an increase in disubstituted monomers due to the chemical confinement effect of gel state functionalization. | ||
The integration value of H1a–d can be thought of as the fraction of crystallizable units of the polymer where the blocky microstructure retains a higher fraction of crystallizable units compared to the random microstructure. It is acknowledged that the ratio of the bromination of the isophthalate unit to the terephthalate unit is higher for the blocky versus the random due to the preference of terephthalate units to crystallize and minimal inclusion of the isophthalate units in the crystal structure.7 When taking into account the crystallizability of the terephthalate monomer compared to the isophthalate monomer and the amount of crystallizable terephthalate monomers preserved in the blocky microstructure, the fraction of crystallizable units for blocky BrPEKK is assumed to be higher than what was calculated from NMR and shown in Fig. 6. Future work will explore utilizing 2D NMR to determine the number of functionalized terephthalate versus isophthalate units to show differences in the comparative amounts of bromination of these units between the random and blocky microstructures.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was utilized to probe the thermal properties of the brominated PEKK before further thermal analysis by DSC and FSC. The degradation profiles for PEKK and BrPEKK are shown in Fig. S4.† The thermal stability decreases with an increase in mol% bromination from a TD5% of 600 °C to 506 °C for pure PEKK and rBrPEKK62, respectively. However, there is no significant difference in thermal stability when comparing random and blocky microstructures of the same degree of functionality. The TGA analysis indicates all materials are thermally stable at temperatures utilized for further thermal analysis (up to 400 °C). The Td5% for all materials is tabulated in Table S1.†Differential scanning calorimetry
Differential scanning calorimetry (DSC) was utilized to determine the influence of random versus blocky microstructure on the thermal properties of rBrPEKK and bBrPEKK. The measured values of crystallization temperature upon cooling (Tc), crystallization upon heating (i.e., cold crystallization, Tcc), melting temperature (Tm), degree of crystallinity (Xc), and glass transition temperature (Tg) are summarized in Table 2. The degree of crystallinity for each sample was determined using the following equation: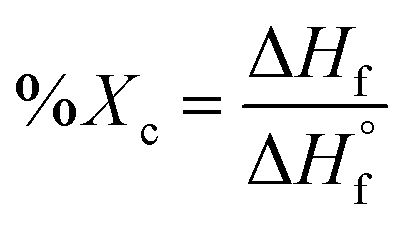 | (6) |
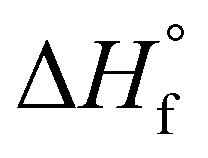 is the enthalpy of fusion of 100% crystalline PEKK. The
is the enthalpy of fusion of 100% crystalline PEKK. The 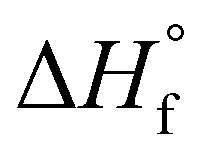 of KEPSTAN 8001 has not been experimentally determined. The
of KEPSTAN 8001 has not been experimentally determined. The 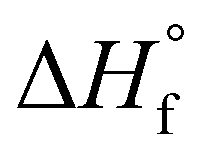 of KEPSTAN 6002 (T/I ratio of 60/40) was calculated from molecular dynamics simulations (150 J g−1)31 and WAXS and DSC experiments (202 J g−1).32 However, the most often referenced value used for %Xc calculations of PEKK is 130 J g−1,33 and this value was utilized for this work.
of KEPSTAN 6002 (T/I ratio of 60/40) was calculated from molecular dynamics simulations (150 J g−1)31 and WAXS and DSC experiments (202 J g−1).32 However, the most often referenced value used for %Xc calculations of PEKK is 130 J g−1,33 and this value was utilized for this work.
| Sample | T c (°C) | T cc (°C) | T m (°C) | X c (%) | T g (°C) |
|---|---|---|---|---|---|
| a %Xc determined from first heat for the aerogel. %Xc determined for rBrPEKK and bBrPEKK from second heat after cooling from 400 °C at 10 °C min−1. b T g by midpoint of amorphous samples. | |||||
| PEKK | 310 | — | 352 | 53 | 163.5 |
| rBrPEKK19 | 290 | — | 345 | 26 | 163.9 |
| bBrPEKK12 | 292 | — | 351 | 38.7 | 164.5 |
| rBrPEKK31 | 250 | — | 333 | 22 | 164.7 |
| bBrPEKK32 | 280 | — | 345 | 28 | 164.9 |
| rBrPEKK42 | 220 | 241 | 316 | 12.4 | 165.8 |
| bBrPEKK40 | 281 | — | 343 | 22 | 166.4 |
| rBrPEKK52 | — | 261 | 309 | 1 | 166.4 |
| bBrPEKK52 | 271 | — | 341 | 18 | 167.2 |
| rBrPEKK62 | — | — | — | 0 | 166.6 |
| bBrPEKK62 | 266 | — | 338 | 11 | 169.7 |
All samples were heated into the melt, cooled, and subsequently heated to determine the effect of microstructure on Tm, Tc, Tcc, %Xc, Tg and crystallizability. After removal of thermal history, the cooling and second heat thermograms are shown in Fig. 7a and b, respectively. Upon cooling (Fig. 7a), all blocky materials exhibit a pronounced crystallization exotherm and the Tc decreases with increasing degree of bromination. In contrast, the crystallization exotherm for the random materials diminishes profoundly above 31 mol% bromination and is totally absent above 52 mol%. Relative to the blocky brominated samples, a significantly greater depression in Tc is observed for the random samples. As discussed previously, this difference in crystallization behavior upon cooling is attributed to a retention of long runs of pristine crystallizable PEKK units during heterogeneous bromination in the gel state. Since bromination in the random state yields a statistically shorter average run length of pristine crystallizable PEKK units, the probability of a crystallizable segment approaching the crystal growth surface diminishes (relative to the blocky microstructure), which consequently decreases crystallizability and the rate of crystallization.
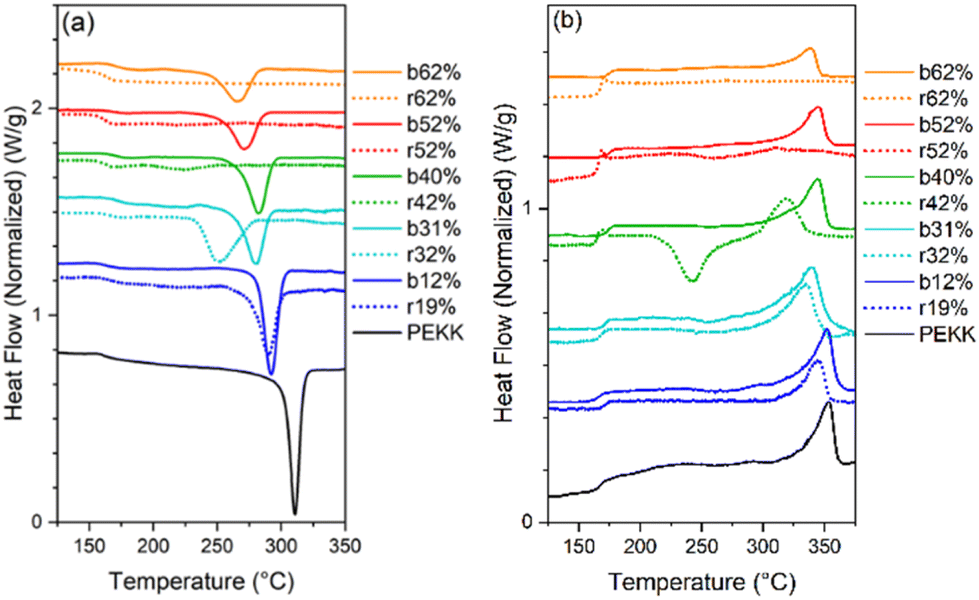 | ||
| Fig. 7 Standard DSC scans of blocky (solid lines) and random (dashed lines) BrPEKK (a) first cool after removal of thermal history and (b) second heat. All DSC scans were performed at 10 °C min−1 in a nitrogen atmosphere. | ||
Upon heating (Fig. 7b), the blocky materials all exhibit distinct melting endotherms with a minimal melting point depression. In contrast, the melting endotherms for the random materials show a decrease in intensity, profound broadening at degrees of bromination greater than 31 mol%, and a significantly greater melting point depression. While the blocky bBrPEKK62% remains crystallizable, the rBrPEKK62% sample remains amorphous. It is also observed that cold crystallization occurs in the rBrPEKK42% and rBrPEKK52% materials, indicating a significant decrease in crystallization kinetics compared to the blocky analogs. No cold crystallization is observed for the blocky materials indicating that all crystallization occurred quickly (during the cooling cycle) for the blocky microstructures and slowed significantly for the random microstructures. This behavior is again attributed to the long runs of pristine PEKK in the blocky microstructure facilitating the ability to quickly form thicker crystals with higher melting temperatures.
The Tc, Tm and Xc determined from the data in Fig. 7 are plotted in Fig. 8a–c as a function of mol% bromination. The random and blocky materials exhibit a decrease in Tc, Tm, and Xc as compared to pristine PEKK due to the introduction of the bromine functionality acting as a defect along crystallizable segments of PEKK. The introduction of these defects shortens the runs of pristine PEKK monomers between brominated units, resulting in slower crystallization kinetics, thinner crystallites, and decreased crystallizability. However, the random copolymers exhibit a larger decrease in Tc, Tm, and Xc with increasing mol% bromination compared to the blocky microstructure. Fig. 8a compares the Tc upon cooling for the blocky and random analogs. For the blocky samples, the Tc decreases from 310 °C for pristine PEKK to 271 °C for bBrPEKK62. For the random samples, the Tc decreases more rapidly from 310 °C for pristine PEKK to 220 °C for rBrPEKK42, with no crystallization upon cooling observed for rBrPEKK52 or rBrPEKK62. Fig. 8b compares the Tm upon cooling for the blocky and random analogs. For the blocky samples, the Tm decreases from 352 °C for pristine PEKK to 338 °C for bBrPEKK62. For the random samples, the Tm decreases more rapidly from 352 °C for pristine PEKK to 309 °C for rBrPEKK52 with no melting observed upon heating for rBrPEKK62. Additionally, Fig. 8c compares the Xc of the blocky and random analogs. For the blocky samples, the Xc decreases from 53% for pristine PEKK to 11% for bBrPEKK62. For the random samples, the Xc decreases more rapidly from 53% for pristine PEKK to only 1% for rBrPEK52 with no crystallinity observed for rBrPEKK62.
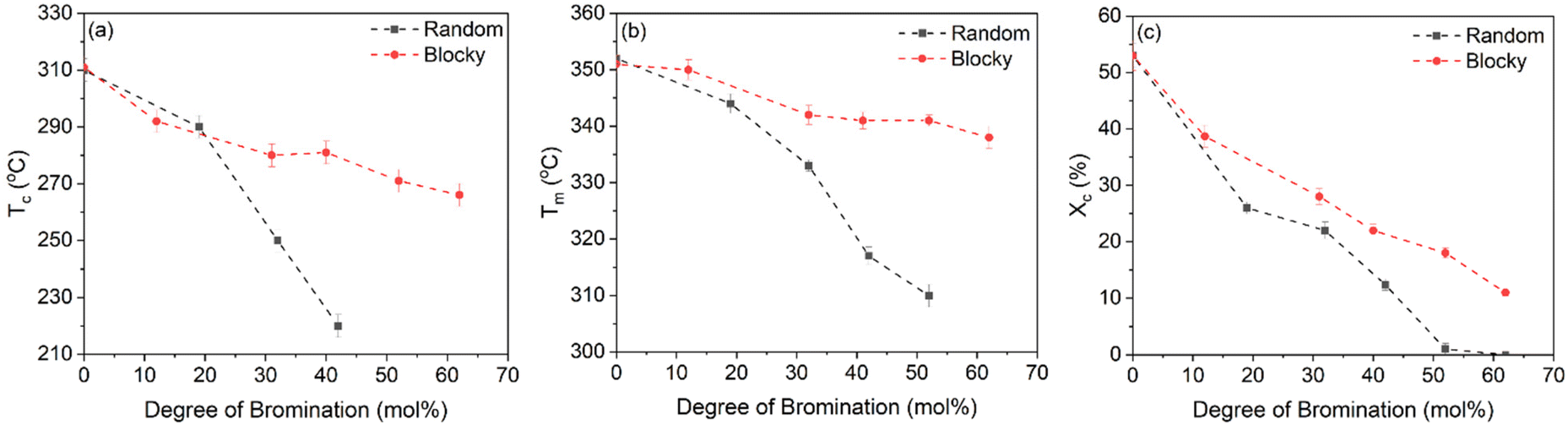 | ||
| Fig. 8 The (a) Tc, (b) Tm, and (c) Xc as a function of degree of bromination for blocky (red, circles) and random (black, squares) BrPEKK. Dashed lines are to guide the eye. | ||
DSC heating scans from melt quenched samples (i.e., 100% amorphous, Fig. S5†) reveal an increase in Tg for both the blocky and random microstructures with increasing degree of bromination, and these Tg data are plotted in Fig. 9. The blocky microstructure exhibits a higher Tg compared to the random microstructure for all degrees of bromination with an increase in Tg from 163.5 °C for pristine PEKK to 169.7 °C for bBrPEKK62 and 166.6 °C for rBrPEKK62. Similar behavior was previously observed in PEEK19 and is attributed to the addition of the bromine functionality on the backbone which increases the Tg due to the steric hindrance of torsional rotation of the phenyl rings due to the bulkiness of the bromine groups.34,35 The blocking up of the bromine functionality leads to an increase in the steric hindrance of the torsional rotation in the blocky microstructure compared to the random microstructure. Additionally, the NMR data above indicates that the blocky microstructures above 40% bromination contain disubstituted monomers while the random microstructure contains only monosubstituted monomers. Thus, it is hypothesized that the increased density of bromine functionalities on the blocky microstructure further increases the barrier to torsional rotation leading to an increase in the Tg for the blocky microstructure as compared to the random microstructure.
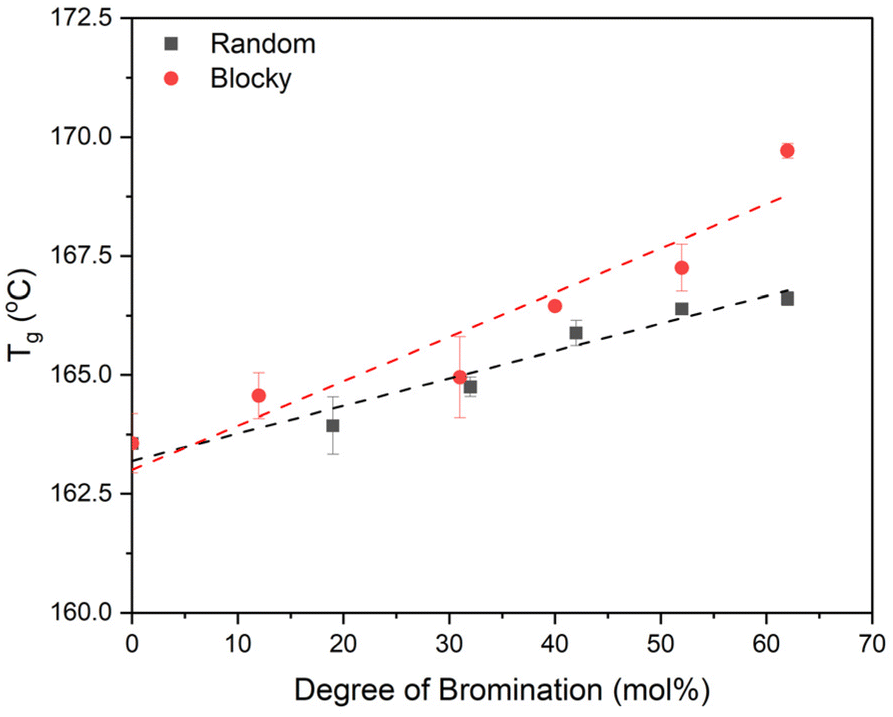 | ||
| Fig. 9 Glass transition temperatures (Tg) of random (black squares) and blocky (red circles) amorphous BrPEKK determined from the midpoint of amorphous samples. | ||
Isothermal crystallization kinetics by FSC
The crystallization kinetics of PEKK 80/20 were recently explored14 utilizing fast scanning calorimetry (FSC) which allows for analysis of crystallization at high degrees of supercooling due to rapid cooling (4000 K s−1) and heating (40000 K s−1) capabilities. Cooling rates during polymer melt processes can reach up to several thousand Kelvin per second.36 Due to such high cooling rates, crystallization commonly occurs at high degrees of supercooling where fast crystallizing materials cannot be evaluated by standard differential scanning calorimetry (DSC). Utilizing FSC, the effect of microstructure on the crystallization kinetics of functionalized PEKK over a wide range of temperature can be explored.
The crystallization kinetics of rBrPEKK32, bBrPEKK31, bBrPEKK40, rBrPEKK52 and bBrPEKK52 are too fast to analyze by standard DSC but too slow for traditional analysis by direct evaluation from the isothermal step by FSC (due to such small sample size). Due to this phenomenon, the crystallization kinetics were analyzed utilizing the previously described interrupted isothermal crystallization (IIC) method. This method utilizes the melting enthalpy upon heating after isothermal crystallization to calculate the degree of crystallinity instead of heat flow during crystallization for traditional isothermal crystallization.24,25 The crystallization kinetics of rBrPEKK40 were slow enough for traditional analysis by direct evaluation from the isothermal step. The degree of crystallinity of rBrPEKK52 was too low for traditional isothermal crystallization by standard DSC and so it was analyzed by the IIC method. The crystallization kinetics of the PEKK homopolymer were previously collected14 utilizing the IIC method.
The crystallization half-time, t1/2, or time when fractional crystallinity reaches 50% of maximum crystallinity during the isothermal crystallization process (i.e., Φ = 0.5), was extracted from the fractional crystallinity versus time data collected from the IIC experiments on the Flash DSC and the heat flow versus time experiments on the standard DSC and plotted in Fig. 10a as a function of isothermal crystallization temperature, Tiso. The t1/2 of PEKK is replotted from previous work and shown in Fig. 10a. The crystallization kinetics of BrPEKK exhibits parabolic behavior due to the balance of nucleation and diffusion-controlled crystallization with limits between the Tg and 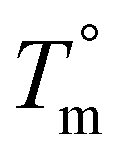 . Compared to pure PEKK, the bulk crystallization rate decreases (i.e., t1/2 increases) for the random and blocky analogs with an increase in mol% bromination. The inclusion of Br-functionalized units (i.e., defects) on the backbone of the crystallizable polymer creates non-crystallizable segments that must be relegated to the amorphous fraction. With an increase in the number of structural defects along the chains, the probability of a non-crystallizable segment approaching the crystal growth surface increases, thus decreasing the rate of crystallization and the overall crystallizability. For all comparable degrees of bromination, the blocky copolymers crystallize significantly faster than the random analogs. The blocky copolymers exhibit kinetics on time scales similar to the pristine PEKK 80/20 polymer, while the random analogs exhibit vastly slower kinetics with the shortest t1/2 ranging from 100 s for rBrPEKK32 up to 1500 s for rBrPEKK52. The difference in kinetics between the blocky and random microstructure as a function of degree of bromination is clearly exhibited in the plot of the t1/2versus degree of bromination at Tiso of 240 °C (Fig. 10b). With increasing degree of bromination, the random microstructure exhibits an exponential increase in t1/2 as compared to the blocky microstructure. The vastly different kinetics are attributed to the microstructure. Due to the semi-crystalline, gel-state functionalization process, the blocky copolymers contain long, uninterrupted segments of crystallizable PEKK units that are preserved in the functionalized microstructure. These defect-free segments facilitate rapid crystallization compared to the random microstructures that contain statistically shorter runs of defect-free segments.
. Compared to pure PEKK, the bulk crystallization rate decreases (i.e., t1/2 increases) for the random and blocky analogs with an increase in mol% bromination. The inclusion of Br-functionalized units (i.e., defects) on the backbone of the crystallizable polymer creates non-crystallizable segments that must be relegated to the amorphous fraction. With an increase in the number of structural defects along the chains, the probability of a non-crystallizable segment approaching the crystal growth surface increases, thus decreasing the rate of crystallization and the overall crystallizability. For all comparable degrees of bromination, the blocky copolymers crystallize significantly faster than the random analogs. The blocky copolymers exhibit kinetics on time scales similar to the pristine PEKK 80/20 polymer, while the random analogs exhibit vastly slower kinetics with the shortest t1/2 ranging from 100 s for rBrPEKK32 up to 1500 s for rBrPEKK52. The difference in kinetics between the blocky and random microstructure as a function of degree of bromination is clearly exhibited in the plot of the t1/2versus degree of bromination at Tiso of 240 °C (Fig. 10b). With increasing degree of bromination, the random microstructure exhibits an exponential increase in t1/2 as compared to the blocky microstructure. The vastly different kinetics are attributed to the microstructure. Due to the semi-crystalline, gel-state functionalization process, the blocky copolymers contain long, uninterrupted segments of crystallizable PEKK units that are preserved in the functionalized microstructure. These defect-free segments facilitate rapid crystallization compared to the random microstructures that contain statistically shorter runs of defect-free segments.
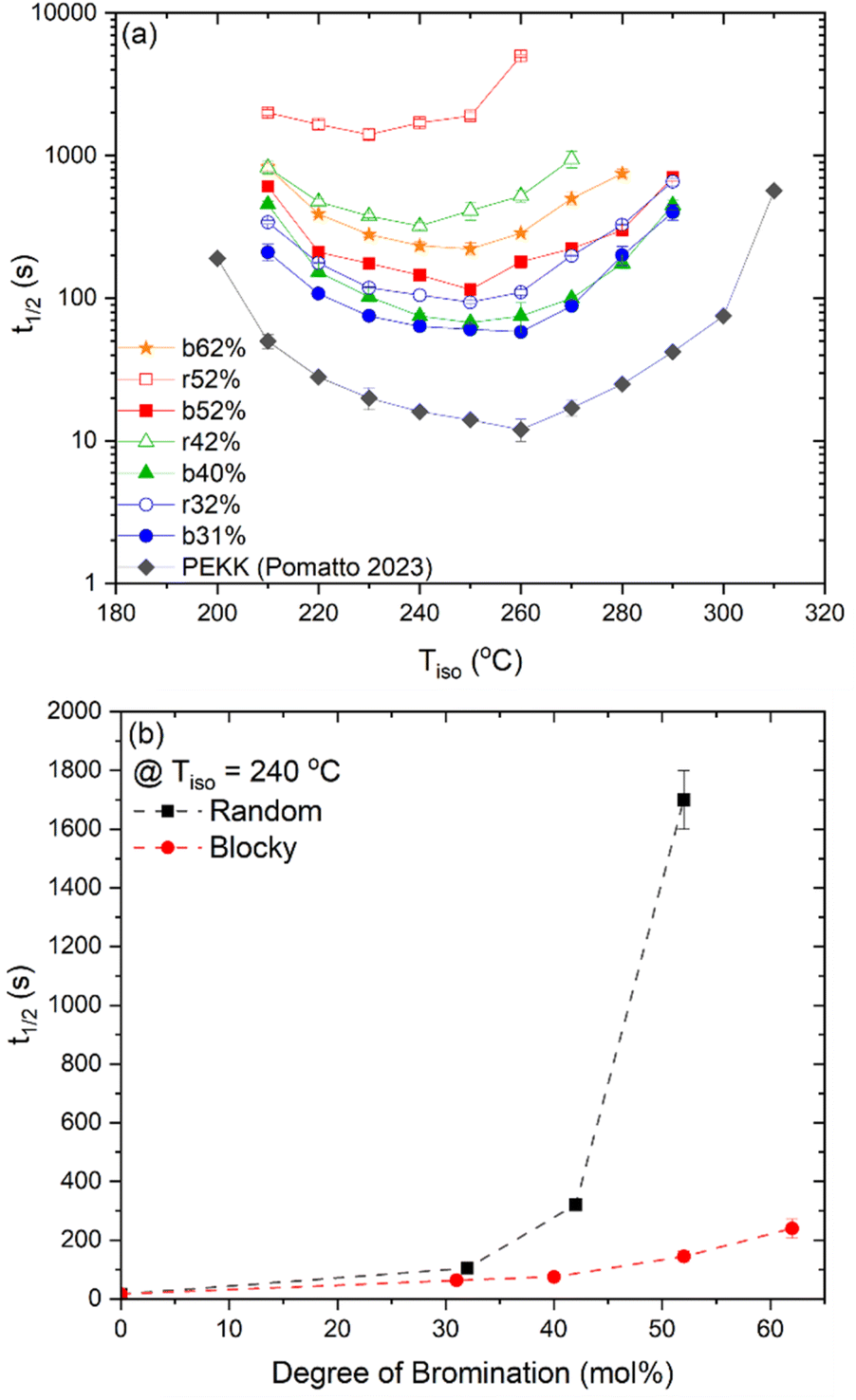 | ||
| Fig. 10 (a) Crystallization half time (t1/2) for PEKK14 (black diamonds), bBrPEKK31 (blue circles), rBrPEKK32 (blue open circles), bBrPEKK40 (green triangles), rBrPEKK42 (green open triangles), bBrPEKK52 (red squares), rBrPEKK52 (red open squares) and bBrPEKK62 (orange stars) over a wide range of temperatures. (b) Crystallization half time (t1/2) versus degree of bromination of random (black squares) and blocky (red circles) BrPEKK isothermally crystallized at 240 °C. Dashed lines are to guide the eye. | ||
Due to the rapid cooling capabilities of the Flash DSC, isothermal crystallization of the BrPEKK copolymers at high degrees of supercooling (i.e., temperatures far below the 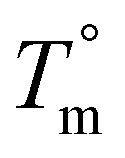 ) is achievable, allowing for comparisons of the parabolic minimum of the t1/2versus Tiso profiles. For each of the brominated copolymers, the Tiso corresponding to the fastest crystallization kinetics (shortest t1/2) shifts to lower temperatures with increasing mol% bromination, compared to pure PEKK. This behavior is more prominent for the random copolymers compared to the blocky copolymers. The copolymer Tiso shifts from 260 °C for pristine PEKK to only 250 °C for bBrPEKK52 but shifts to 230 °C for rBrPEKK52. This shifting of Tiso to lower temperatures is attributed to the comonomer confinement effect also known as the chemical confinement effect. The comonomer confinement effect refers to the phenomenon in which non-crystallizable (e.g., functionalized) comonomers interrupt the chain folding capabilities of the crystallizable segments of the copolymer.37 Consequently, homogeneous nucleation at high degrees of supercooling becomes more difficult and thus heterogeneous nucleation dominates at lower temperatures compared to the homopolymer. Thus, the parabolic minimum of the t1/2versus Tiso shifts to lower Tiso. Similar trends have been observed in other semi-crystalline copolymer systems including isotactic polypropylene-co-1-butene,38 isotactic polypropylene-co-1-octene,39 amide 6-co-amide 6,6,40 and PEKK 60/40 and 70/30.2,41 The random copolymers exhibit a larger shift of the parabolic curve to lower Tiso due to the statistically random distribution of brominated monomers along the polymer chains (i.e., greater confinement of crystallizable PEKK segments). The blocky microstructure has longer uninterrupted runs of pure PEKK, resulting in a reduced influence of the comonomer confinement effect on the t1/2 parabolic minimum.
) is achievable, allowing for comparisons of the parabolic minimum of the t1/2versus Tiso profiles. For each of the brominated copolymers, the Tiso corresponding to the fastest crystallization kinetics (shortest t1/2) shifts to lower temperatures with increasing mol% bromination, compared to pure PEKK. This behavior is more prominent for the random copolymers compared to the blocky copolymers. The copolymer Tiso shifts from 260 °C for pristine PEKK to only 250 °C for bBrPEKK52 but shifts to 230 °C for rBrPEKK52. This shifting of Tiso to lower temperatures is attributed to the comonomer confinement effect also known as the chemical confinement effect. The comonomer confinement effect refers to the phenomenon in which non-crystallizable (e.g., functionalized) comonomers interrupt the chain folding capabilities of the crystallizable segments of the copolymer.37 Consequently, homogeneous nucleation at high degrees of supercooling becomes more difficult and thus heterogeneous nucleation dominates at lower temperatures compared to the homopolymer. Thus, the parabolic minimum of the t1/2versus Tiso shifts to lower Tiso. Similar trends have been observed in other semi-crystalline copolymer systems including isotactic polypropylene-co-1-butene,38 isotactic polypropylene-co-1-octene,39 amide 6-co-amide 6,6,40 and PEKK 60/40 and 70/30.2,41 The random copolymers exhibit a larger shift of the parabolic curve to lower Tiso due to the statistically random distribution of brominated monomers along the polymer chains (i.e., greater confinement of crystallizable PEKK segments). The blocky microstructure has longer uninterrupted runs of pure PEKK, resulting in a reduced influence of the comonomer confinement effect on the t1/2 parabolic minimum.
SAXS/WAXS of PEKK and BrPEKK copolymers
PEKK, rBrPEKK and bBrPEKK copolymers were analyzed by small and wide-angle X-ray scattering (SAXS/WAXS) to further characterize differences between the blocky and random microstructures. The SAXS/WAXS profiles of isothermally crystallized PEKK, rBrPEKK and bBrPEKK are shown in Fig. 11a and b. The sharp peaks in the wide-angle region (q > 8 nm−1) are primarily attributed to form 1 crystalline reflections of pristine PEKK crystallites.7,42 For ease of comparison, offset WAXS data for the rBrPEKK and bBrPEKK are shown in Fig. S6.† Close inspection of the WAXS data for bBrPEKK62% suggests a significant presence of the form II polymorph of PEKK7,42 (as indicated by arrows in Fig. S6†). We observed similar differences in polymorphic composition between random and blocky copolymers of sPS,43 but further studies will be required to probe the origin of this observation. Retention of crystallinity (i.e., sharp WAXS reflections) is evident for all blocky copolymers, in agreement with the DSC data (Fig. 7). In comparison, the random copolymers exhibit crystalline reflections only up to 42 mol%, whereas an amorphous halo is observed for rBrPEKK52 and rBrPEKK62% (Fig. 11a). This lack of crystallinity in the random copolymers at high degrees of bromination is again consistent with the DSC data (Fig. 7).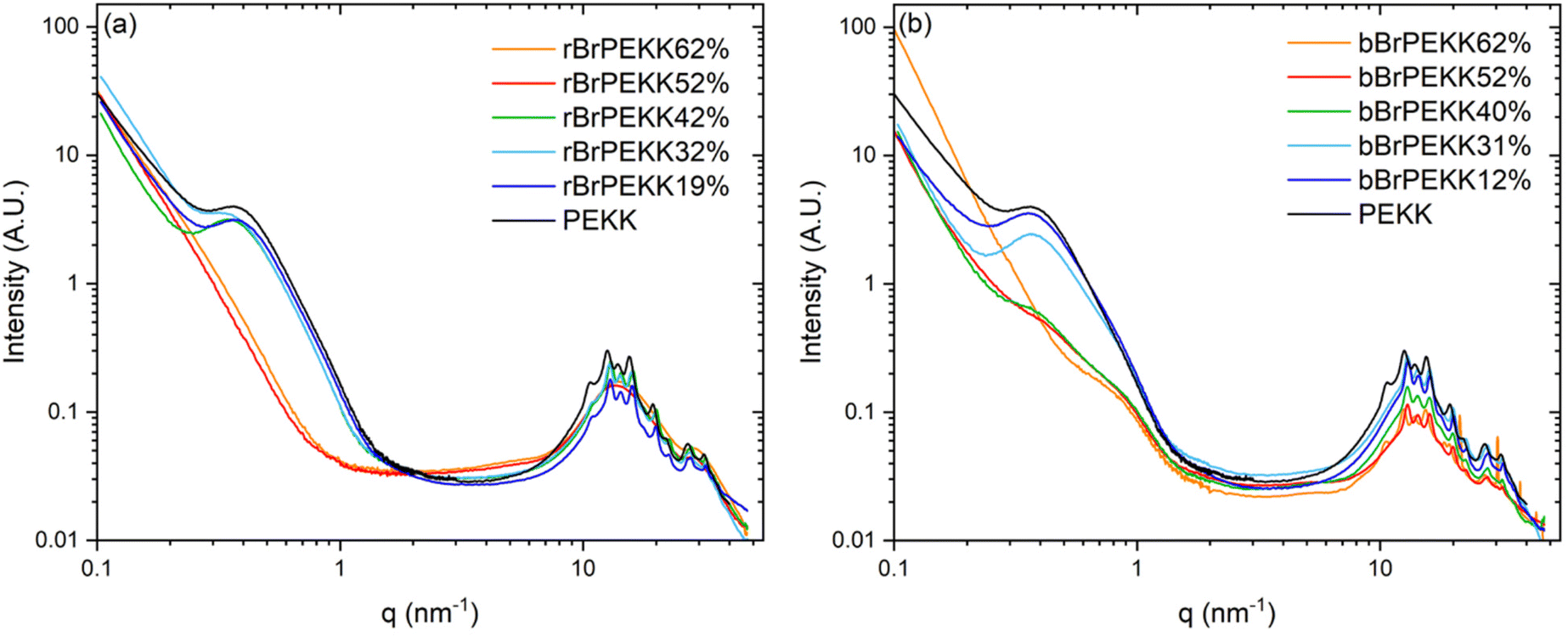 | ||
| Fig. 11 Small, mid, and wide-angle X-ray scattering profiles of (a) random BrPEKK and (b) blocky BrPEKK. All samples were isothermally crystallized from the melt at 280 °C for 30 minutes in a nitrogen atmosphere. | ||
In the small-angle region (0.1 < q < 1 nm−1) of the scattering profiles (Fig. 11), the characteristic interlamellar long period peak is observed in neat PEKK at q = 0.38 nm−1.32 The same feature is observed in random BrPEKK copolymers only up to 42 mol% (Fig. 11a), since this feature is only observed in samples that contain crystallinity. In the SAXS patterns of the blocky BrPEKK copolymers (Fig. 11b), the absolute intensity of the interlamellar long period feature decreases with increasing degree of bromination. This reduction in intensity is attributed to the blocky microstructure afforded by functionalizing in the semicrystalline gel state. Significant crystallization in the blocky brominated PEKK samples effectively concentrates the electron-rich bromine atoms into the amorphous phase, thus increasing the average electron density of the amorphous phase. This results in lower contrast between the crystalline and amorphous phases, leading to a reduction in absolute scattering intensity of the long period peak. Calculated estimations of electron density contrast in these systems support this proposed origin of a reduction in scattering contrast for the blocky microstructures, as detailed in the ESI.†
Conclusions
The bromination of PEKK by acid-catalyzed electrophilic aromatic substitution utilizing NBS in DCA was successful in both the homogeneous solution state and heterogeneous gel state. Diphenyl acetone was discovered as a new gelation solvent for PEKK resulting in nanoscale fibrillar-network morphologies with high porosity and surface area. The heterogeneous gel state functionalization of PEKK resulted in a blocky microstructure attributed to steric exclusion of NBS from the tightly packed crystallites within the gel. Consequently, bromination is restricted to the amorphous segments between crystallites within the gel. The retention of pristine unfunctionalized blocks preserves crystallizability even at high degrees of bromination (62 mol%) while the random analog (62 mol%) is no longer crystallizable. Blocky BrPEKK samples exhibited higher glass transition temperatures, melting temperatures, crystallization rates and crystallizability as compared to the random BrPEKK analogs. The t1/2versus Tiso for blocky and random BrPEKK exhibited parabolic behavior and shifted to longer t1/2 and lower Tiso upon functionalization. The random analogs exhibited larger shifts in the parabolic minimum as compared to the blocky analogs, attributed to the comonomer confinement effect. SAXS and WAXS analysis further discern differences in the microstructures of the random and blocky brominated copolymers as supported by scattering length density estimations. This work demonstrates blocky functionalization of PEKK as a facile method to effectively block up functionality while preserving long runs of crystallizable segments on the backbone resulting in enhanced thermal properties as compared to randomly functionalized analogs.Author contributions
Michelle E. Pomatto: Conceptualization; data curation; formal analysis; investigation; methodology roles/writing – original draft; writing – review & editing. Erin R. Crater: Data curation; formal analysis of SAXS/WAXS; writing – original draft. Garrett F. Godshall: Data curation. Writing – original draft. Robert B. Moore: Conceptualization; project administration; supervision; writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Arkema for providing KEPSTAN PEKK used for this work. We thank Glenn A. Spiering for guidance with the SLD calculations. The authors also thank the Institute for Critical Technology and Applied Science (ICTAS), the Macromolecules Innovation Institute (MII), and the Department of Chemistry for laboratory support. This material is based upon work supported by the National Science Foundation under grant no. DMR-1809291 and DMR-2104856. Purchase of the Xenocs Xeuss 3.0 SAXS/WAXS instrument used to obtain results included in this publication was supported by the National Science Foundation under the award DMR MRI 2018258.References
- R. L. Mazur, P. C. Oliveira, M. C. Rezende and E. C. Botelho, J. Reinf. Plast. Compos., 2014, 33, 749–757 CrossRef.
- T. Choupin, B. Fayolle, G. Régnier, C. Paris, J. Cinquin and B. Brulé, Polymer, 2017, 111, 73–82 CrossRef CAS.
- L. Quiroga Cortés, N. Caussé, E. Dantras, A. Lonjon and C. Lacabanne, J. Appl. Polym. Sci., 2016, 133, 43396 CrossRef.
- E. A. M. Hassan, D. Ge, L. Yang, J. Zhou, M. Liu, M. Yu and S. Zhu, Composites, Part A, 2018, 112, 155–160 CrossRef CAS.
- M. Reitman, D. Jaekel, R. Siskey and S. M. Kurtz, in PEEK Biomaterials Handbook, ed. S. M. Kurtz, William Andrew Publishing, Oxford, 2012, pp. 49–60, DOI:10.1016/B978-1-4377-4463-7.10004-1.
- S. Z. D. Cheng, R.-M. Ho, B. S. Hsiao and K. H. Gardner, Macromol. Chem. Phys., 1996, 197, 185–213 CrossRef CAS.
- K. H. Gardner, B. S. Hsiao, R. R. Matheson and B. A. Wood, Polymer, 1992, 33, 2483–2495 CrossRef CAS.
- S. Swier, Y. S. Chun, J. Gasa, M. T. Shaw and R. A. Weiss, Polym. Eng. Sci., 2005, 45, 1081–1091 CrossRef CAS.
- S. Vetter, B. Ruffmann, I. Buder and S. P. Nunes, J. Membr. Sci., 2005, 260, 181–186 CrossRef CAS.
- J. V. Gasa, S. Boob, R. A. Weiss and M. T. Shaw, J. Membr. Sci., 2006, 269, 177–186 CrossRef CAS.
- B. Yuan, Q. Cheng, R. Zhao, X. Zhu, X. Yang, X. Yang, K. Zhang, Y. Song and X. Zhang, Biomaterials, 2018, 170, 116–126 CrossRef CAS PubMed.
- Z. C. Kennedy, J. F. Christ, M. D. Fenn, L. Zhong, W. Chouyyok, A. M. Arnold, A. C. Denny, A. M. Albrecht, J. A. Silverstein, R. L. Erikson and J. Chun, Addit. Manuf., 2022, 49, 102492 CAS.
- S. L. Aristizábal, L. Upadhyaya, G. Falca, A. Y. Gebreyohannes, M. O. Aijaz, M. R. Karim and S. P. Nunes, J. Membr. Sci., 2022, 660, 120798 CrossRef.
- M. E. Pomatto and R. B. Moore, Polymer, 2023, 125810 CrossRef CAS.
- D. Rusakov, A. Menner and A. Bismarck, Macromol. Rapid Commun., 2020, 41, 2000110 CrossRef CAS PubMed.
- D. Rusakov, A. Menner, F. Spieckermann, H. Wilhelm and A. Bismarck, J. Appl. Polym. Sci., 2022, 139, 51423 CrossRef CAS PubMed.
- D. Rusakov, A. Menner and A. Bismarck, Macromol. Mater. Eng., 2023, 308, 2200704 CrossRef CAS.
- S. J. Talley, X. Yuan and R. B. Moore, ACS Macro Lett., 2017, 6, 262–266 CrossRef CAS PubMed.
- C. R. Kasprzak, L. J. Anderson and R. B. Moore, Polymer, 2022, 251, 124918 CrossRef CAS.
- L. J. Anderson, X. Yuan, G. B. Fahs and R. B. Moore, Macromolecules, 2018, 51, 6226–6237 CrossRef CAS.
- K. F. Noble, A. M. Noble, S. J. Talley and R. B. Moore, Polym. Chem., 2018, 9, 5095–5106 RSC.
- L. J. Anderson and R. B. Moore, Solid State Ionics, 2019, 336, 47–56 CrossRef CAS.
- D. Dollimore, P. Spooner and A. Turner, Surf. Technol., 1976, 4, 121–160 CrossRef CAS.
- J. Seo, A. M. Gohn, O. Dubin, H. Takahashi, H. Hasegawa, R. Sato, A. M. Rhoades, R. P. Schaake and R. H. Colby, Polym. Cryst., 2019, 2, e10055 Search PubMed.
- X. Tardif, B. Pignon, N. Boyard, J. W. P. Schmelzer, V. Sobotka, D. Delaunay and C. Schick, Polym. Test., 2014, 36, 10–19 CrossRef CAS.
- V. V. Josh Kemppainen , Evan Pineda, Gregory Odegard, Thermomechanical Property Prediction of Amorphous and Crystal PEKK via Molecular Dynamics, Report E-20122, NASA, NTRS, 2022.
- Synthetic flavoring substances and adjuvants, US Code Fed. Regul, 1999, 52-59.
- T. R. Bock, Catalytic phosphonation of high performance polymers and POSS, Novel components for polymer blend and nanocomposite fuel cell membranes, Doctoral Dissertation, Albert Ludwigs University, Germany, 2006, p. 20949871 Search PubMed.
- K. Rajesh, M. Somasundaram, R. Saiganesh and K. K. Balasubramanian, J. Org. Chem., 2007, 72, 5867–5869 CrossRef CAS PubMed.
- S. J. Talley, C. L. AndersonSchoepe, C. J. Berger, K. A. Leary, S. A. Snyder and R. B. Moore, Polymer, 2017, 126, 437–445 CrossRef CAS.
- C. Li and A. Strachan, Polymer, 2019, 174, 25–32 CrossRef CAS.
- S. Tencé-Girault, J. Quibel, A. Cherri, S. Roland, B. Fayolle, S. Bizet and I. Iliopoulos, ACS Appl. Polym. Mater., 2021, 3, 1795–1808 CrossRef.
- D. Blundell and B. Osborn, Polymer, 1983, 24, 953–958 CrossRef CAS.
- A. K. Mukherjee and A. Mohan, J. Appl. Polym. Sci., 1992, 44, 773–779 CrossRef CAS.
- M. S. McCaig, E. D. Seo and D. R. Paul, Polymer, 1999, 40, 3367–3382 CrossRef CAS.
- J. E. K. Schawe, J. Therm. Anal. Calorim., 2014, 116, 1165–1173 CrossRef CAS.
- W. Hu, V. B. F. Mathot, R. G. Alamo, H. Gao and X. Chen, Crystallization of statistical copolymers, Springer International Publishing, 2016, pp. 1–43, DOI:10.1007/12_2016_349.
- D. Kalapat, Q. Tang, X. Zhang and W. Hu, J. Therm. Anal. Calorim., 2017, 128, 1859–1866 CrossRef CAS.
- D. Mileva, R. Androsch, D. Cavallo and G. C. Alfonso, Eur. Polym. J., 2012, 48, 1082–1092 CrossRef CAS.
- T. Wang, X. Li, R. Luo, Y. He, S. Maeda, Q. Shen and W. Hu, Thermochim. Acta, 2020, 690, 178667 CrossRef CAS.
- T. Choupin, B. Fayolle, G. Régnier, C. Paris, J. Cinquin and B. Brulé, Polymer, 2018, 155, 109–115 CrossRef CAS.
- K. H. Gardner, B. S. Hsiao and K. L. Faron, Polymer, 1994, 35, 2290–2295 CrossRef CAS.
- G. B. Fahs, S. D. Benson and R. B. Moore, Macromolecules, 2017, 50, 2387–2396 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01338c |
| This journal is © The Royal Society of Chemistry 2024 |