
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom rocks to bioactive compounds: a journey through the global P(V) organophosphorus industry and its sustainability
Sosthène P.-M.
Ung
 * and
Chao-Jun
Li
* and
Chao-Jun
Li
Department of Chemistry and FQRNT Centre for Green Chemistry and Catalysis, McGIll University, 801 Sherbrooke Street West, Montréal, Québec H3A 0B8, Canada. E-mail: sosthene.ung@mail.mcgill.ca
First published on 4th November 2022
Abstract
This tutorial review aims at presenting the usefulness and the diversity of organophosphorus compounds within the context of a more sustainable phosphorus chemistry. Three topics will be emphasized while reflecting on green chemistry: firstly, a short sum-up of the phosphorus resources and their industrial and environmental ties will be given to contextualize the framework of this review. The rich nomenclature, properties, and applications of organophosphorus compounds will then be highlighted, emphasizing P(V) comparatively to P(III) ones. Finally, a brief review of the current synthetic routes towards organophosphorus molecules and their inherent challenges will be described before presenting a few promising alternatives.
 Sosthène P.-M. Ung | Dr Sosthène Pierre-Marie Ung obtained his MSc. in organic chemistry from l'École Nationale Supérieure de Chimie of Montpellier, France in 2013. He then joined McGill University in Montréal, Canada in 2015 for doctoral studies to work under Prof. Chao-Jun Li's supervision to develop sustainable catalytic systems for the construction of C–P(V) bonds and graduated in 2022. |
 Chao-Jun Li | Dr Chao-Jun Li is E. B. Eddy Professor and Canada Research Chair (Tier I) in Green Chemistry at McGill University and is the co-director of the FRQNT Centre for Green Chemistry and Catalysis. He is a Fellow of the Royal Society of Canada, Royal Society of Chemistry, The American Association for the Advancement of Science, American Chemical Society, Chemical Institute of Canada, Chinese Chemical Society, and The World Academy of Sciences (TWAS). He has published >500 research articles, 8 books, 14 patents and has given >475 plenary/keynote/invited talks. |
Sustainability spotlightWith the growing need for the establishment of a greener chemistry, a closer look at the current organophosphorus chemistry field is required. Most synthetic phosphorus compounds are indirectly obtained through the functionalization of extracted phosphate rocks, a finite resource which is asymmetrically spread around the globe. The overreliance on energy-costly processes, on transformations generating toxic by-products, and the use of the same hazardous intermediates within the organophosphorus industry will be described. In line with the sustainable development goals 12 and 15 of the United Nations about sustainable production patterns & industrialization, the proposed tutorial review showcases an accessible and up-to-date description of the various applications of P(V) molecules and their current industrial synthetic routes, before investigating the inherent sustainability challenges of the field and highlighting more sustainable prospective alternatives. |
1. An overview of phosphorus processes
1.1 The universality of phosphorus in life-essential structures
While organic chemistry focuses mainly on the chemical behavior of carbon, hydrogen, and a few period 2 elements such as nitrogen and oxygen, and elements from the halogen group, the chemistry of sulfur and phosphorus remains somewhat marginal despite their abundance in natural patterns. Indeed, phosphorus, the predominant element examined in the present review, is omnipresent in biomolecules and essential to life. Two remarkable examples can be found in biochemistry, thus highlighting its significance: one example sees phosphorus playing a role in the architecture of information encoding biopolymers. The second shows phosphorus as the universal mediator for energy transfer in life processes. Contrary to sulfur which exists directly in the peptide structure, phosphorus is present as phosphates in the skeleton of polynucleotides such as DNA or RNA. Each nucleoside is linked to another unit through a 3′-5′ phosphodiester linkage on the sugar rings (Fig. 1a). Being anionic at pH = 7, phosphates confer the overall biopolymer architecture a good water-solubility and thus physiological compatibility, which would otherwise be lacking due to the relative hydrophobicity of the nitrogenous base.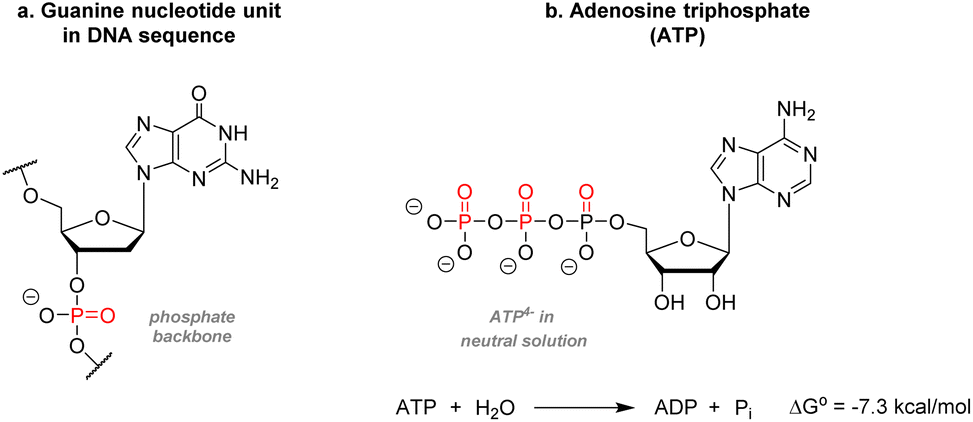 | ||
| Fig. 1 Common phosphate containing biomolecules. (a) Guanine nucleotide unit (b) adenosine triphosphate. | ||
Furthermore, the negative charges allow for specific proteins to bind to the DNA strands easily. After elucidation of the DNA structure by Watson, Crick, Wilkins, and Franklin,1 it was shown that this sugar-phosphate backbone twists around itself to form the well-known double helices structure. Phosphorus is also present in adenosine triphosphate (ATP), the molecular unit of energy present in all known forms of life (Fig. 1b). Synthesized during cellular respiration, photosynthesis, or fermentation, ATP is used to fuel multiple metabolic reactions, transport macromolecules across the cellular membrane, DNA and RNA synthesis, muscle contractions, etc. The chemical energy transferred during those processes relies on the hydrolysis of the terminal phosphate yielding one adenosine diphosphate unit, one free phosphate anion, and 7.3 kcal mol−1 per ATP molecule consumed.2 Phosphorylation of proteins – understood in biochemistry by adding a phosphate unit onto an alcohol functional group – is also the most abundant modification and has a prominent role in activating, deactivating, or modifying the role of enzymes.3 Phosphorus can also be found in cell membranes as phospholipids or inorganic apatite in bones and teeth enamel.
This entire section will give a brief timeline of phosphorus discoveries. A short overlook of the global phosphorus resources available and the industrial processes will then be given. Finally, we will briefly investigate the ties linking phosphorus and the environment.
1.2 A brief history of phosphorus discoveries
Despite its universal role in life processes, elemental phosphorus was isolated in 1669 by German alchemist Hennig Brand during his efforts to find the philosopher's stone.4 Brand obtained a liquid that instantly gave off fumes and burst into flames when distilling urine. Moreover, this substance could solidify and glow in the dark, which is why he coined it phosphorus, from ‘Φωσφόρος’ which means light-bearer in Greek. P4, in the white phosphorus form, was thus well-known from the beginning as a pyrophoric substance, as it readily converts to phosphorus pentoxide P2O5 in the presence of oxygen from the air (Scheme 1).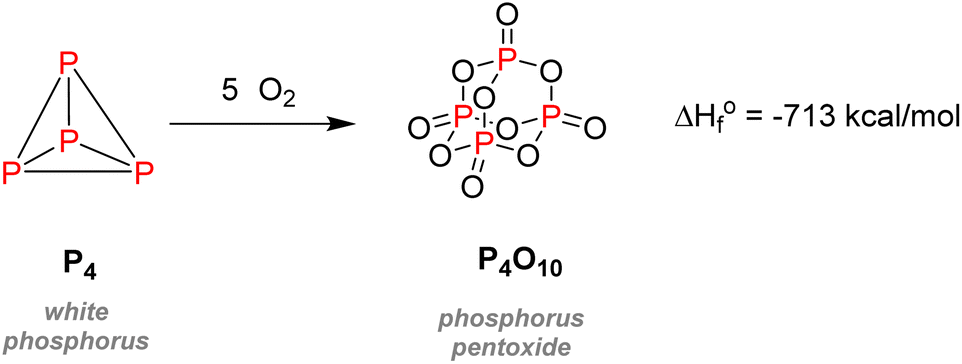 | ||
| Scheme 1 Oxidation of white phosphorus. | ||
After its discovery, phosphorus was soon found to be present in plants, bones, and minerals such as pyromorphite, and by the late 18th century, PH3 was prepared by Gengembre,5 followed by Gay-Lussac and Thénard, who prepared PCl3 and PCl5 in 1808 (Fig. 2).6 By the 1830s, white phosphorus was used in France and England in striking matches, unfortunately leading to many fatalities amongst factory workers.7,8 In 1842, Lawes and Murray patented in Great Britain the first technique for synthesizing P-fertilizer coined ‘superphosphate’ using sulfuric acid and bones.9 However, even though bones and later guano had replaced urine as the primary source of phosphorus, the discovery of phosphorus-rich mineral deposits by the late 19th century changed the face of the blooming phosphorus economy. Those phosphate rocks deposits, such as in Suffolk in England, were readily exploited to satisfy the increasing demand for fertilizers. Later discovery of the wet process to obtain phosphoric acid and the invention of the electric furnace in 1888 by Readman paved the way for more efficient industrialization and commercialization of phosphorus compounds.10 As for metal–phosphorus complexes, the pioneering works of Rose, Hofmann, Cahours, and Gal during the second half of the 19th century foreshadowed the extensive use of transition-metal phosphine complexes in modern catalysis.11–14 Although Lassaigne had already prepared alkyl phosphates in 1820,15 it was both Arbuzov and Michaelis,16,17 now primarily considered as the founders of modern organophosphorus chemistry, who became well-known for developing coupling methodologies to construct C–P bond, thus creating new tools for the introduction of phosphorus moieties in organic synthesis. The diverse use of inorganic phosphates came about gradually from the 1930s until the 1950s, when they became frequently employed for toothpaste, surfactants, water softening and food technology.18 During this era, a German chemist called Gerhard Schrader also patented the first organophosphorus insecticides: hexaethyltetraphosphate (HETP) and tetraethylphosphate (TEPP).19 However, his discoveries are also closely associated with the most historically pernicious use of phosphorus: chemical weaponry. The use of elemental phosphorus as incendiary bullets dates back from World War I. Nevertheless, it was Schrader who developed diverse nerve agents, amongst which the infamous Tabun and Sarin gas,20 which are now classified as weapons of mass destruction and formally banned under the Chemical Weapons Convention of 1993.21 Although elemental phosphorus has now been restricted in the case of incendiary devices, white phosphorus munitions are still in usage to produce smoke and camouflage movement. The effects of white phosphorus munitions remain still heavily dangerous, causing significant burn injuries and respiratory damages.
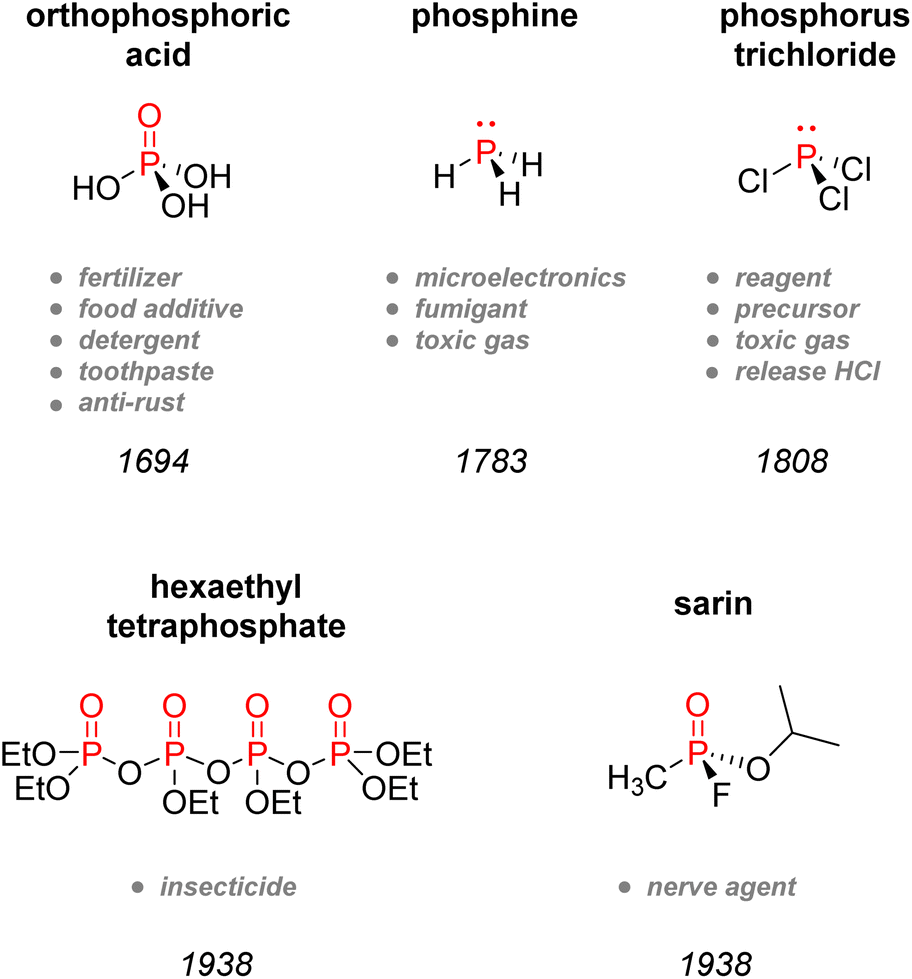 | ||
| Fig. 2 Timeline of the discovery of significant phosphorus-containing compounds. | ||
1.3 Phosphate rocks: a scarce resource
Phosphorus is not a rare element on the surface of the earth. It is thought to make up between 1000 to 1300 ppm by mass of the Earth's continental crust, and as such, is more abundant than chlorine or sulfur, or even carbon.22–25 However, most of the exploitable phosphorus sources reside in sedimentary phosphate rocks, typically fluorapatite Ca5(PO4)3F. The availability of those phosphate rocks is finite, and various estimation of the longevity of the world's reserve varies between complete depletion coming about within the next 70 to 400 years.26 The continuously changing assessment over this controversial issue can be explained by relying on limited but ever-changing new available information and considering the future economic and technological dynamics of exploitation and evaluation.27 The exact magnitude of the scarcity of phosphorus is still far from settled. However, a concept known as peak phosphorus, the point in time when the maximum production rate of phosphorus will be reached, has emerged as an analogy to the concept of peak oil.28 Most of the commercial reserves of phosphate rocks (94%) are split between only 11 countries (Table 1), and 70% of the reserves lies solely within Morocco/Western Sahara. In 2015, the global extraction capacity of phosphate rocks reached 223 × 106 tons, with China, the US, and Morocco representing 70% of the extraction alone.29| Country | Reserves of P-rocks in 2019 (ton × 106) | Country | Production in 2019 (ton × 106) |
|---|---|---|---|
| Morocco | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
China | 110 |
| China | 3200 | Morocco | 36 |
| Algeria | 2200 | USA | 23 |
| Syria | 1800 | Russia | 14 |
| Brazil | 1700 | Jordan | 8 |
| Saudi Arabia | 1400 | Saudi Arabia | 6.2 |
| South Africa | 1400 | Vietnam | 5.5 |
| Egypt | 1300 | Brazil | 5.3 |
| Australia | 1200 | Egypt | 5 |
| USA | 1000 | Peru | 3.7 |
| Finland | 1000 | Israel | 3.5 |
| Jordan | 1000 | Tunisia | 3 |
1.4 The phosphorus industry and processes
It has been estimated that over 90% of the phosphate rock exploited is converted to phosphoric acid according to the ‘wet process’ (Scheme 2). The method consists of the digestion of fluorapatite minerals by concentrated sulfuric acid. Calcium sulfate which is yielded as a by-product (about 4–6 tons produced per ton of phosphoric acid) is later precipitated and filtered out, and the wet phosphoric acid obtained is purified and concentrated.30 | ||
| Scheme 2 Conversion of phosphate rock to orthophosphoric acid through the wet process. | ||
It is estimated that this process accounts for more than half of the global supply of sulfur taking the form of sulfuric acid, thus showing how tightly the phosphorus and sulfur industries are interdependent.31 Most of the phosphoric acid produced is used for the fertilizer industry. The 4 commonly sold phosphorus fertilizers and their production in 2016 are shown above (Fig. 3).32 The remaining part of phosphate rocks can be converted to molecular phosphorus P4 for subsequent reactions. Historically, phosphoric acid was also synthesized through this process by oxidizing the white phosphorus obtained to afford phosphorus pentoxide, the anhydride version of phosphoric acid. However, because the reaction is strongly endothermic, this thermal process requires electric furnaces, which are highly energy intensive. These issues caused the development of the wet process, which circumvented the need to go through a white phosphorus intermediate to obtain phosphoric acid. Nevertheless, this thermal process (coined the Wöhler process) remains the most efficient pathway to produce molecular phosphorus. An electric furnace heating at around 1450 °C is fed the three raw materials: phosphate rocks that are either sintered or pelletized, dried metallurgical coke for the carbon source, and silica deposits such as pebbles, gravel, or coarse sand (Scheme 3).33
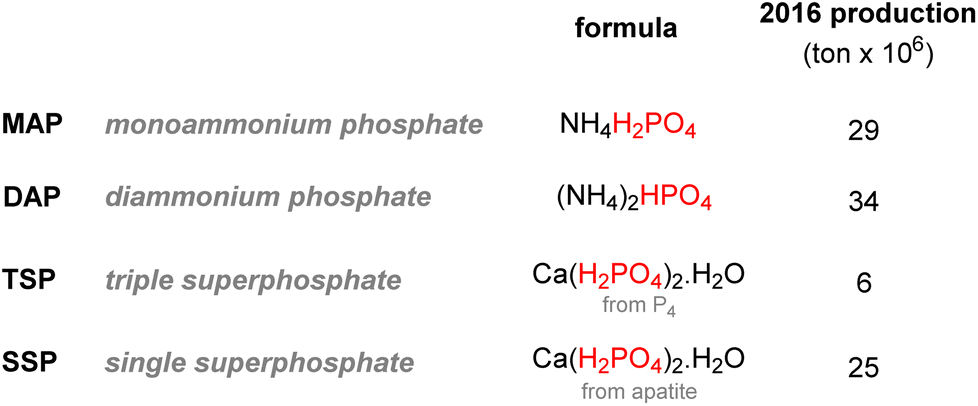 | ||
| Fig. 3 Commonly sold phosphorus-containing fertilizers. | ||
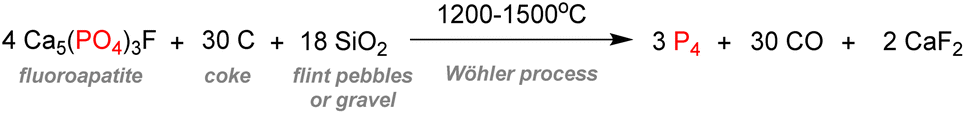 | ||
| Scheme 3 Conversion of phosphate rocks to white phosphorus through the Wöhler process. | ||
The P-gas produced are then condensed and washed with water. The white phosphorus afforded is then stored underwater to prevent it from self-ignition. Even though the wet process mostly supplanted the thermal pathway, around 83% of the P4 obtained is still converted to phosphoric acid through combustion.33 This phosphoric acid has the advantage of being purer than the one obtained through the wet process and can be used accordingly for more sensitive applications. Only about 12% of the white P4 produced is converted into fine chemicals and heavy phosphorus compounds, including PCl3, PCl5, and P4S10 with the remaining being used for all other industrial phosphorus species (Fig. 4).34
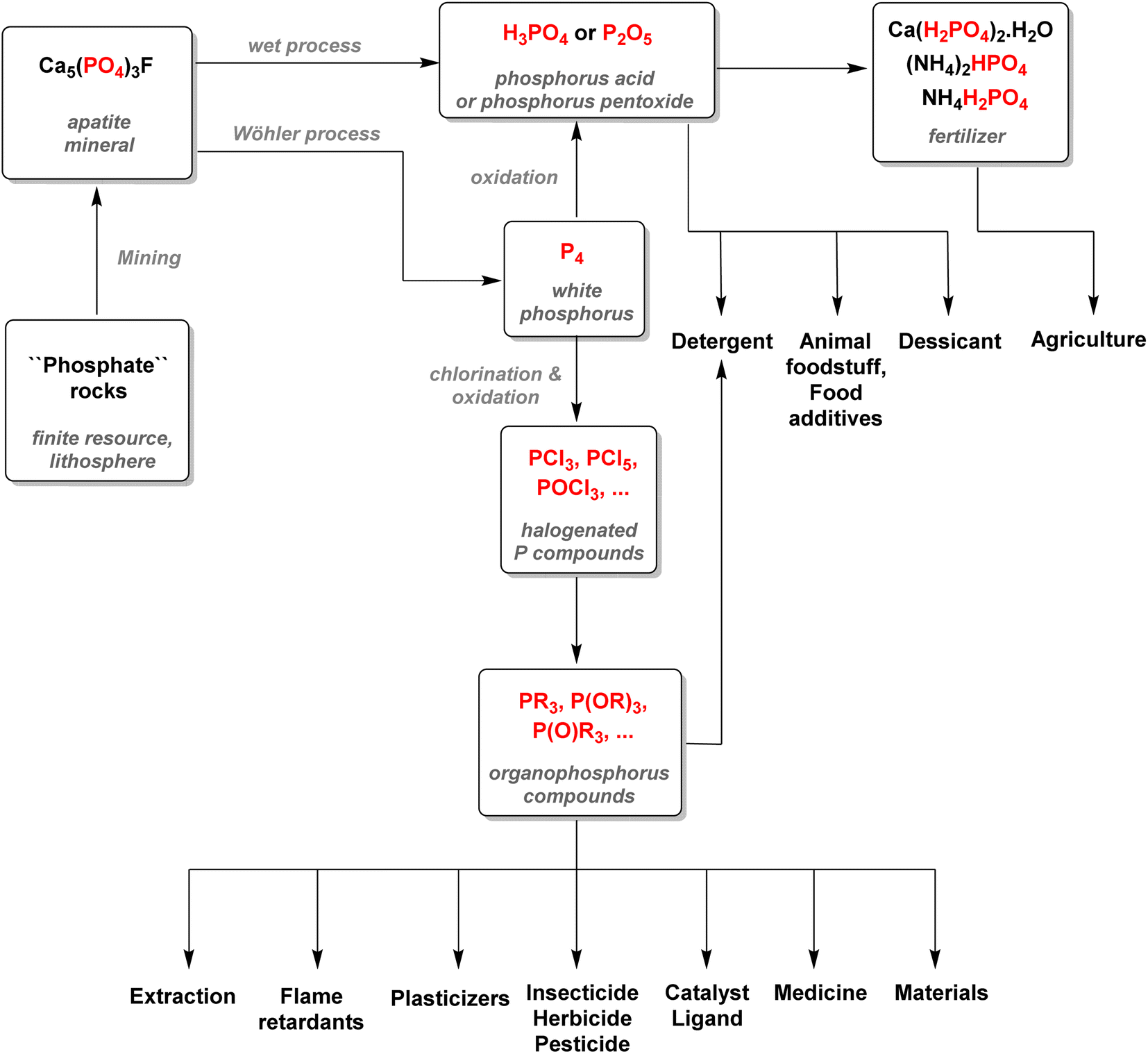 | ||
| Fig. 4 Overview of the phosphorus industry and application. | ||
One of the most important classes of inorganic compounds synthesized from elemental phosphorus is phosphorus halides. Phosphorus trichloride is produced by the combustion of either molten white phosphorus or red phosphorus with gaseous chlorine (Scheme 4a). The high heat afforded by this exothermic reaction keeps the process at a boiling point and allows for phosphorus trichloride to be collected by distillation.35 The world production surpasses 300000 tons per year,36 and it is the most critical building block for the synthesis of many valuable organophosphorus species, as the final section of this review will explain in further detail. Phosphorus pentachloride is another halogenated phosphorus compound of relative importance, and it is produced by reacting phosphorus trichloride with an excess of chlorine gas (Scheme 4b). As a point of comparison, around 10000 tons of PCl5 were produced in 2000 worldwide.37 It is mainly used to synthesize acyl chloride or the chlorination of alcohol as PCl5 is an efficient chlorinating agent thanks to the enhanced electrophilicity of its pentavalent phosphorus center. Finally, phosphoryl chloride, another precursor used to produce phosphate esters, can be synthesized by oxidizing PCl3 with air or pure oxygen (Scheme 4c). Overall, those halogenated phosphorus species are particularly crucial in the bigger picture as they are precursors for the synthesis of organophosphorus molecules.38 Nevertheless, their production and handling are particularly problematic due to their high toxicity and hazardous characteristics.
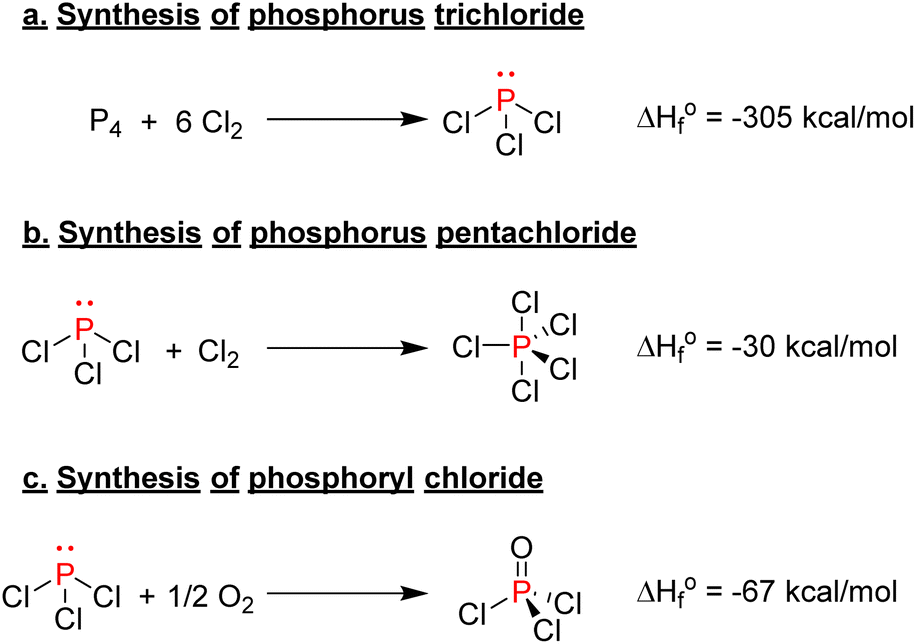 | ||
| Scheme 4 Synthesis of organophosphorus chloride species. | ||
1.5. The phosphorus cycle, human-caused perturbations, and environmental concerns
 | ||
| Fig. 5 Simplified schematic of the natural phosphorus cycle on Earth. | ||
2. Organophosphorus compounds: chemical properties and applications
This section will be detailed on organophosphorus compounds: their nomenclature, chemical properties, and applications in various fields. In addition, an emphasis will be given on sustainability issues linked to the industry, and a focus on pentavalent organophosphorus compounds will be made.2.1. Nomenclature and classification
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or phosphorothioate PS bond arranged within a tetrahedral geometry. Another way to classify organophosphorus compounds is according to the numbers of P–C, P–O, or P–N bonds or according to the phosphorus oxidation state: it goes from −III in the case of phosphines PR3 or +V in the case of phosphates P(O)(OR)3.
O or phosphorothioate PS bond arranged within a tetrahedral geometry. Another way to classify organophosphorus compounds is according to the numbers of P–C, P–O, or P–N bonds or according to the phosphorus oxidation state: it goes from −III in the case of phosphines PR3 or +V in the case of phosphates P(O)(OR)3.
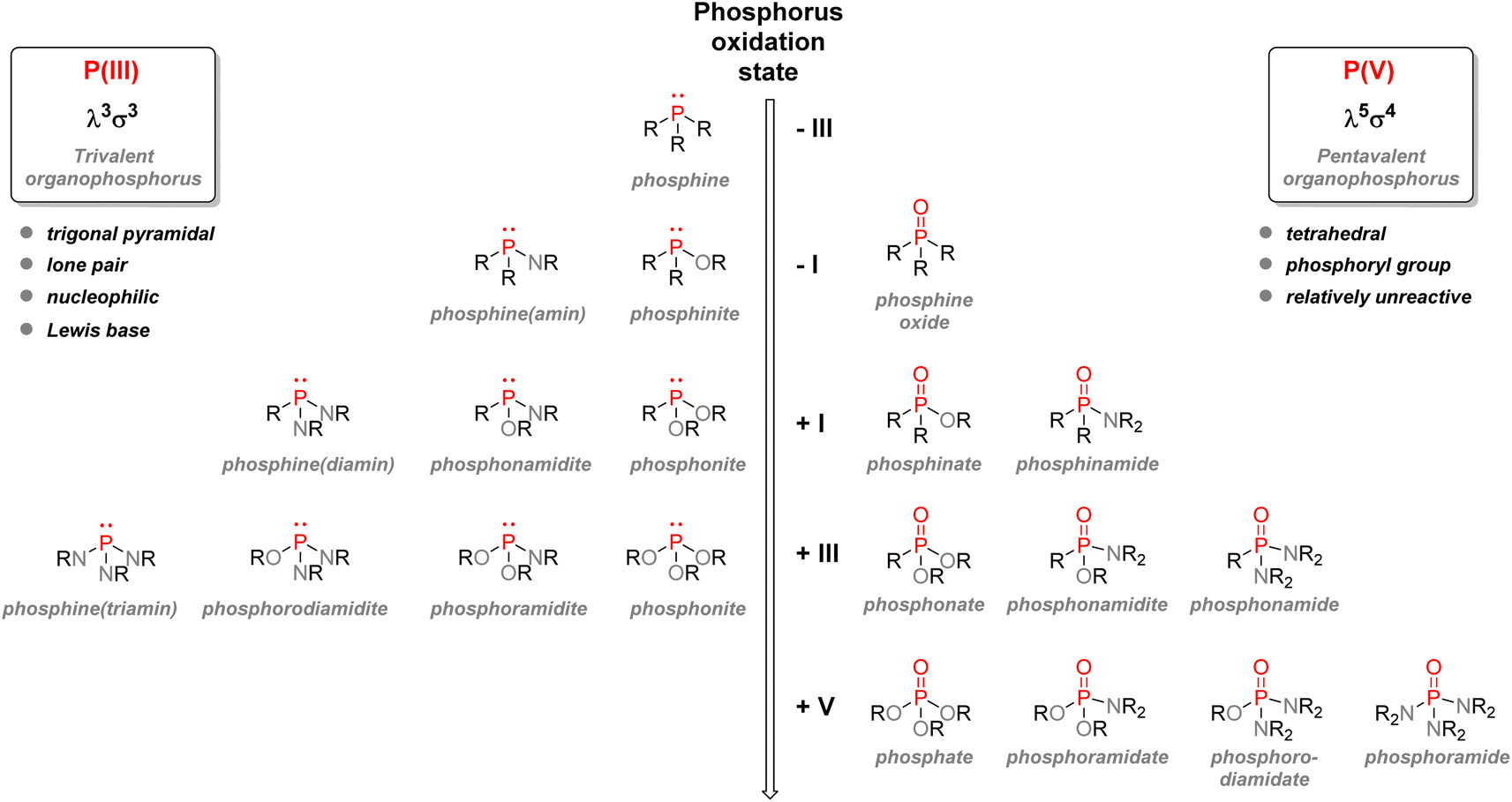 | ||
| Fig. 6 Nomenclature of organophosphorus compounds: trivalent and pentavalent species sorted by oxidation number. | ||
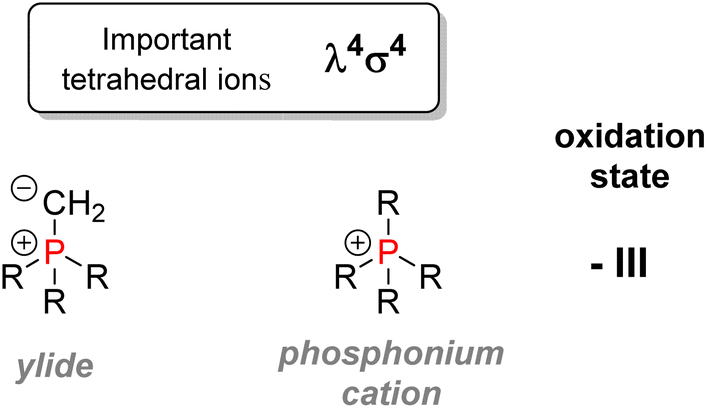 | ||
| Fig. 7 Important tetrahedral organophosphorus ions. | ||
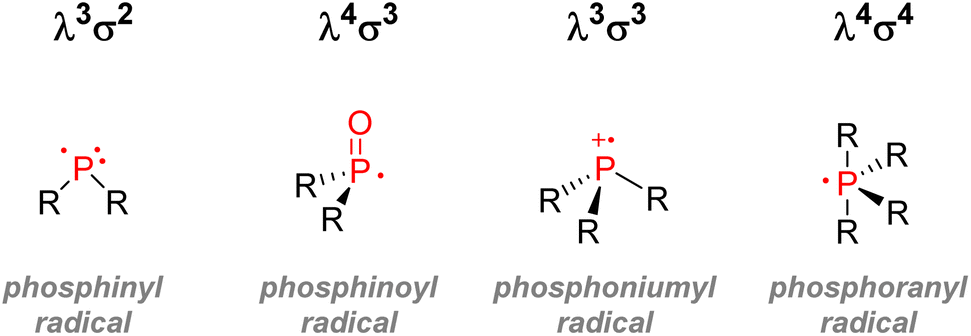 | ||
| Fig. 8 Important organophosphorus radicals. | ||
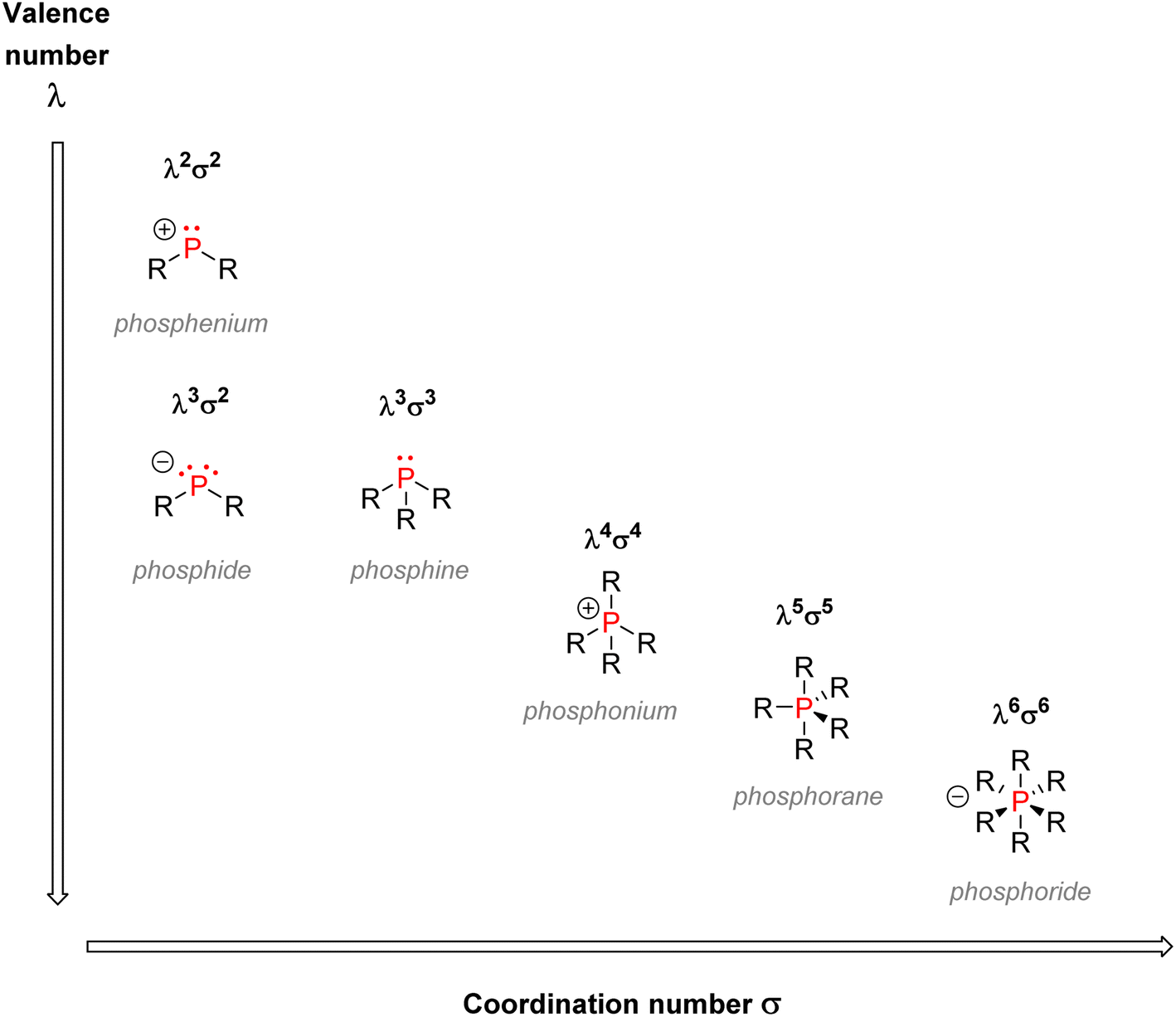 | ||
| Fig. 9 Valence & coordination numbers for organophosphorus species. | ||
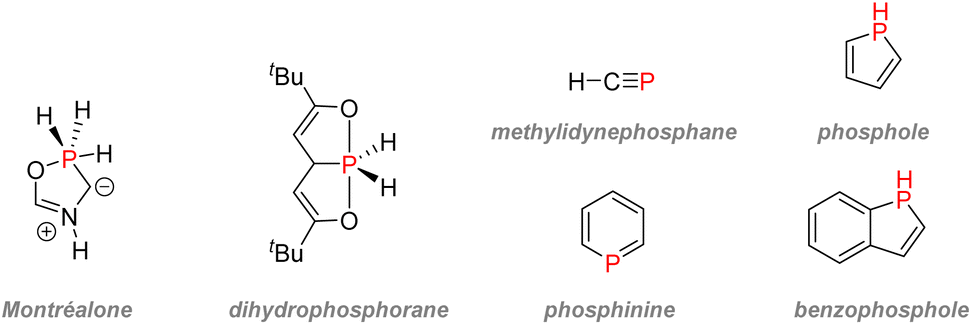 | ||
| Fig. 10 Examples of phosphorus-containing heterocycles. | ||
2.2. Trivalent phosphorus: a versatile lone pair of electrons
In contrast to this back-bonding effect, electron-donating substituents can enhance the σ donation of the phosphorus to the metal; this can be measured through the Tolman electronic parameter.53 Changing substituents can also change geometrical configurations of the organometallic complex: bulky substituents can widen the solid angle – called Tolman angle – between the metal center at the vertex and the cone formed by the phosphine substituents.54 As such, bulky substituents can saturate the metal's coordination sphere and affect its catalytic properties. Chelating diphosphine ligands can also be adjusted according to their bite angle.55 Consequently, phosphine ligands are very popular for catalyst design: they convey facile tunability of electronic and steric parameters thanks to their highly systematic and predictive behavior when playing with the substituents.
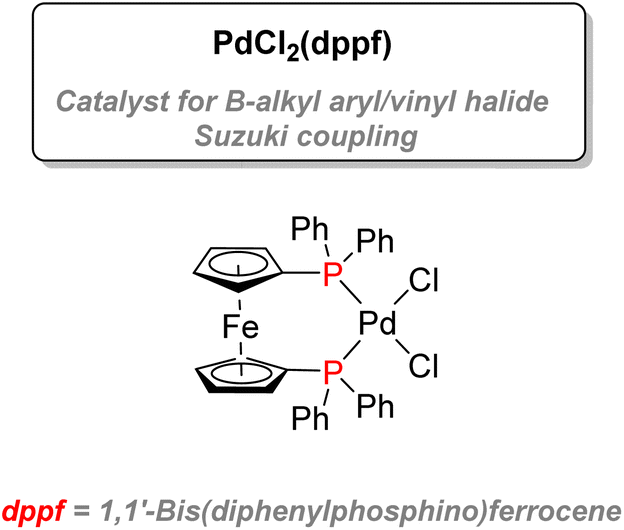 | ||
| Fig. 11 Palladium–diphosphine complex used for Suzuki coupling. | ||
Diphosphine ligands were found to be ineffective in the case of Csp3–Csp3 couplings. Two factors hinder this reaction: slow oxidative addition of the alkyl halide to the palladium center and a quick β-hydride elimination can occur instead of the expected transmetalation step. In this case, bulky electron-rich phosphines such as PCy3 or P(i-Pr)3 were found to promote the cross-coupling product (Scheme 5).58 A better σ-donation of the phosphine increases the electron density at the palladium center and enhances the oxidative addition step by promoting the formation of a coordinatively and electronically unsaturated complex. The steric hindrance of bulky phosphine also disfavors β-hydride elimination olefin byproduct by blocking empty coordination sites.
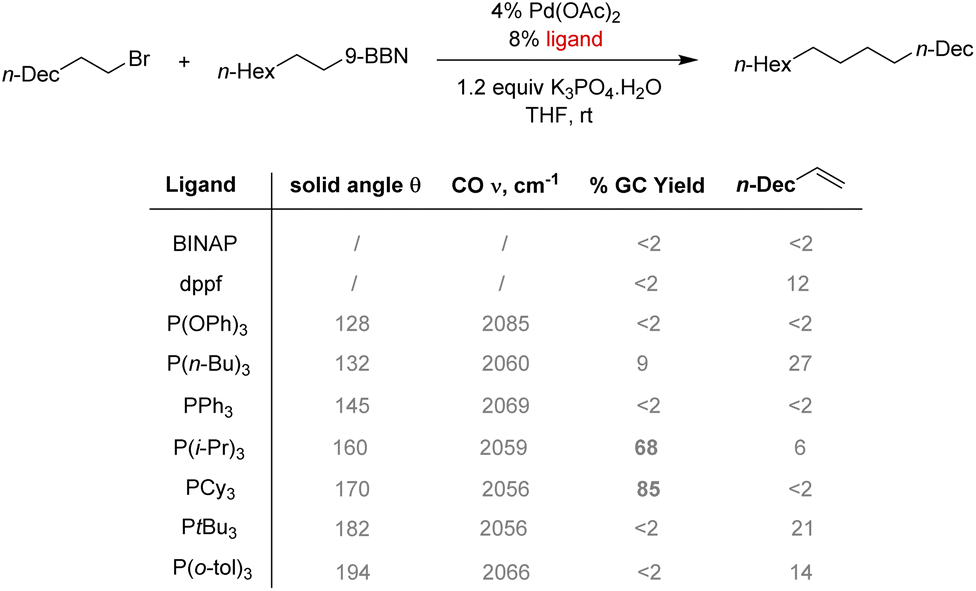 | ||
| Scheme 5 The electronic and steric effects of phosphine ligands on Csp3–Csp3 Suzuki coupling. | ||
In the case of Noyori's system for asymmetric hydrogenation of diketones with BINAP/Ru, bulky and chiral diphosphine BINAP have been used to promote enantioselective transformations (Scheme 6).59 BINAP–ruthenium(II) dicarboxylate complexes were isolated and analyzed by X-ray crystallography, showing that the rigid configuration of the ligand backbone completely blocks off any coordination on one face on the complexes.60 It only allowed for coordination on two quadrants of the remaining face due to the steric hindrance produced by phenyl substituents.61 Thus, the use of chiral phosphines can favor topic discrimination for substrate coordination in a process called dynamic kinetic resolution. Those stereoselective processes can yield highly enantiopure products. This technique was used to synthesize L-DOPA by Knowles for the industrial synthesis of (−)-menthol.62,63
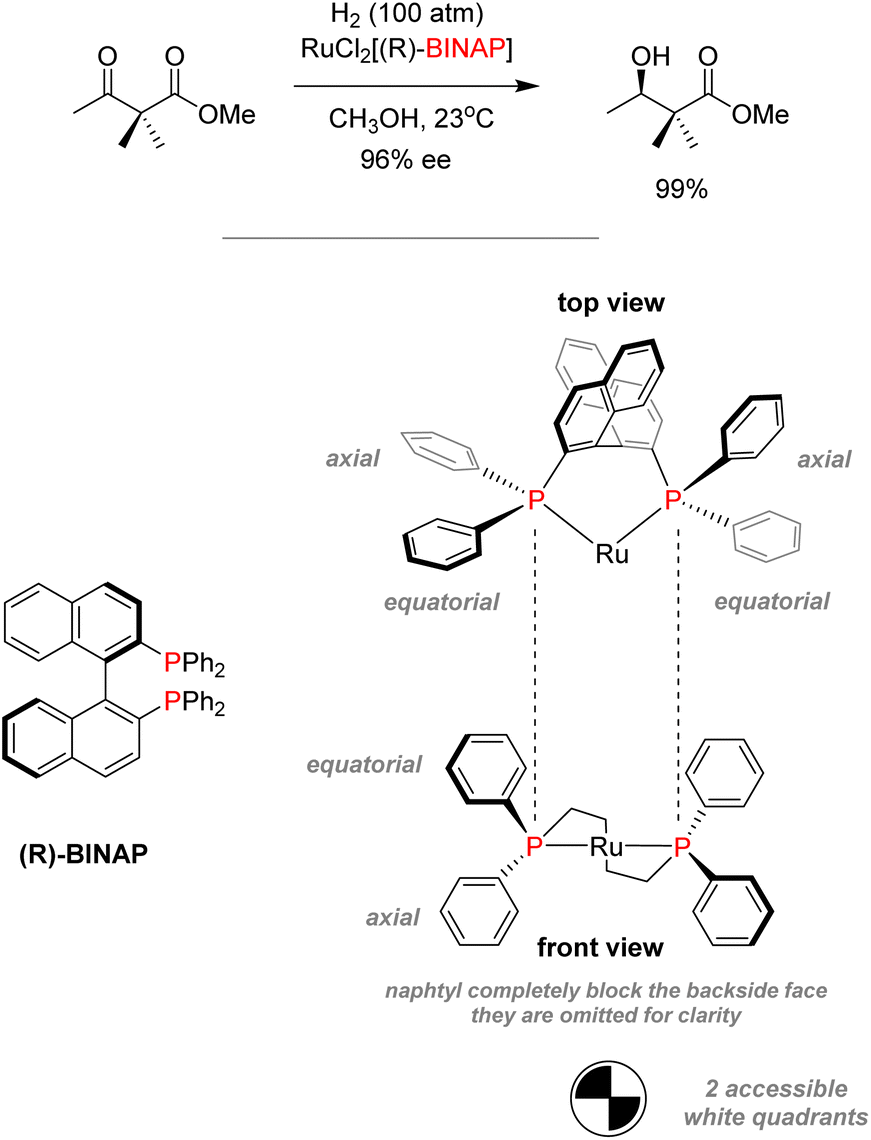 | ||
| Scheme 6 Noyori asymmetric hydrogenation of diketones with ruthenium–BINAP complexes. | ||
In addition to classical diphosphine ligands, another noteworthy class of P(III) ligands discovered in the 1970s has proven particularly interesting and has been the subject of many recent advances in designing efficient catalytic systems.64–68 Pincer ligands are tridentate ligands which coordinate metal centers tightly with a meridional configuration, thus achieving better thermal and air stability. Moreover, the restricted coordination sites of such organometallic complexes often limit the generation of unwanted side products. For P(III) containing pincer ligands, the central chelating atom can either be a carbon for PCP ligands or a nitrogen for PNP ligands. Akin to their simpler diphosphine counterparts, pincer ligands can easily be tuned by selecting appropriate substituents on the phosphorus for electronics & sterics or by designing the central framework and spacers for specific geometries or electronics. Such ligands activated relatively unreactive bonds such as C–C, H–H or C–F bonds. Milstein's direct dehydrogenative synthesis of amides from alcohol and amines in 2007 was a significant milestone.69 Those early discoveries paved the way for the development of various applications using organometallic pincer complexes for more sustainable catalysis: among those are atom-economic hydrogen transfer reactions such as hydrogenation & dehydrogenation of organic molecules, efficient cross-couplings, hydrogen storage materials, greenhouses gas conversion, or plastic depolymerization.70–72 Novel development, including the use of non-symmetrical frameworks or merging metallocenes with pincer ligands has also been reported.73,74
2.3. Pentavalent phosphorus: a stable phosphoryl bond
O bond, and more occasionally, other chalcogens such as PS or PSe. Contrary to amine oxides which necessitate specific oxidizing agents to be formed but can easily be reduced, trivalent phosphines can readily be oxidized under air (or by almost any other oxidant) to form PO bearing phosphine oxides. However, their reduction reveals much more demanding and requires very strong reductants such as silicon hydrides or lithium aluminium hydrides. Phosphoryl bonds are considered as mostly stable and somewhat unreactive. In that aspect, they are not similar in reactivity with CN or CO bonds as PO does not undergo nucleophilic addition easily. However, oxygen can react with strong acids or potent electrophiles. The stability comes from a high bond dissociation energy, varying between 129 to 139 kcal mol−1, which can explain the oxophilicity of trivalent phosphorus and their tendency to form P(V) adducts.79 The formation of PO bonds is usually a decisive factor for many reactions, e.g., the driving force of the Wittig reaction or the Wittig–Horner reaction. The stability of the phosphoryl bond was somewhat of a controversial topic for past chemists: early literature drew PO bond as shown (Fig. 12a), then a later convention was to describe phosphoryl bonds as undergoing a back bonding of sort between a pair of electrons from the oxygen and the empty 3d orbitals on phosphorus as shown by the resonance equation (Fig. 12b). To this date, many phosphorus specialists argue that the zwitterionic form describes phosphoryl bonds the best.80,81
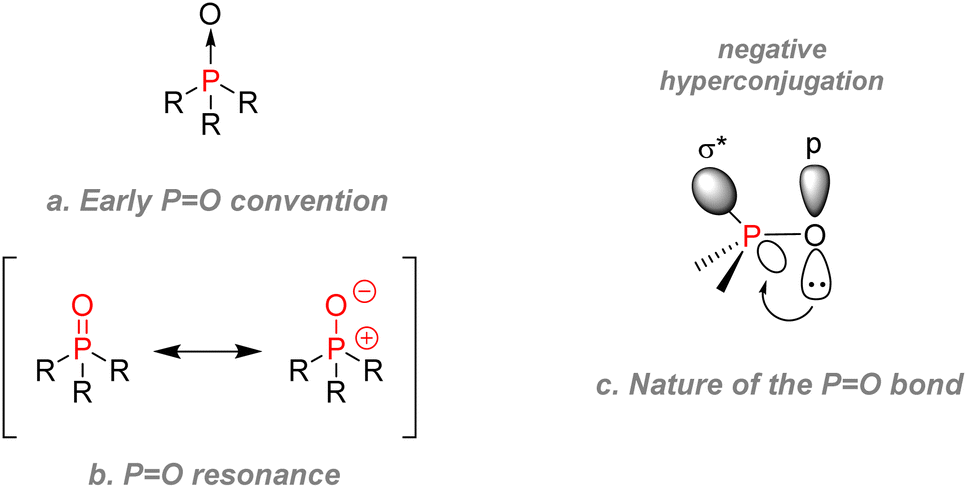 | ||
| Fig. 12 Drawing conventions and nature of phosphoryl PO bond. (a) Early convention (b) resonance (c) nature of the P=O bond. | ||
However, computational chemistry showed that hyperconjugation was probably between the oxygen lone pair electrons and the empty anti-bonding σ* orbitals of the P–R bonds (Fig. 12c).82,83 Overcoming the controversy are several unequivocal metrics: phosphoryl bonds are also short: 1.475 to 1.490 Å compared to 1.60 Å for a typical single P–O bond from a P–O–P unit. Regarding their dielectric constant, phosphoryl bonds are also highly polar (4.51 D in triphenylphosphine oxide).79 The phosphoryl oxygen is thus highly susceptible to forming hydrogen bonds, which can make P(V) species prone to water solubility in some cases. Those characteristics make the 31P NMR of the phosphoryl group shift according to the solvent as it is sensitive to protic environments.84 Finally, we can classify phosphoryl moieties in terms of substituent effects: the inductive donating effect of the phosphinyl group (where R = C for R2P(O)–) is comparable with the methoxy group for a saturated system.
Nevertheless, phosphinyl groups are moderately electron-withdrawing by resonance, making them meta director and deactivator in the case of electrophilic aromatic substitutions.85,86 Alternatively, the strong electron-withdrawing effect of phosphoryl bonds enables them to stabilize carbanions or saturated carbons at the α position. Those properties make the phosphoryl bond a singular one in nature and will explain the bioactivity of many phosphorylated molecules. The following sections will detail several applications of those P(V) containing structures.
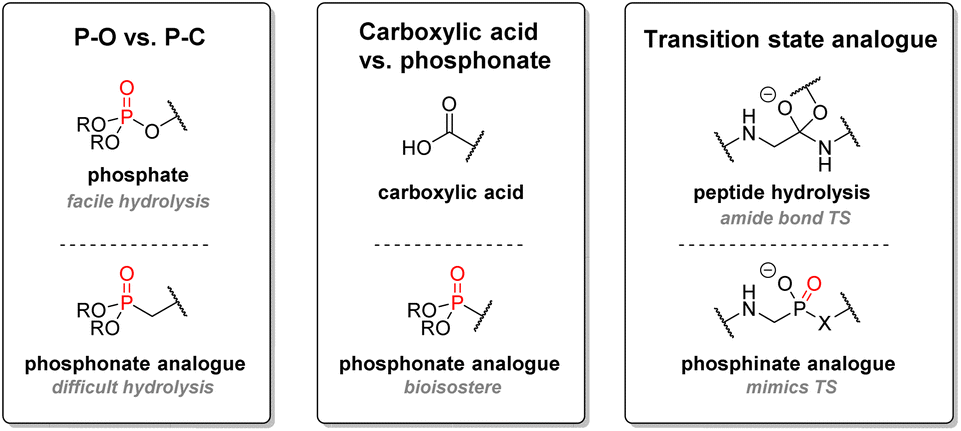 | ||
| Fig. 13 Strategies for P(V) organophosphorus use in medicinal chemistry. | ||
In terms of pharmacokinetics, P(V) structures exhibit high polarity and thus generally good aqueous solubility. They have also been found to have a sufficiently long half-life in human liver microsome conditions, allowing for good bioavailability before metabolism. With a few exceptions, most structures containing P(V) moieties were also usually stable under acidic or basic conditions. Functional groups containing acidic phosphorus such as phosphates, phosphinates, or phosphonates suffer from the significant drawback of turning into charged species under biological conditions, thus decreasing cell permeability and oral bioavailability. One way to circumvent those inconveniences is to use solubility-augmented prodrugs, which will be metabolized into the active therapeutic compound. Phosphine oxide-based drugs are still infrequently used, but recent examples have shown promising results.88
2.3.2.1. Early hits. The cyclic phosphorodiamidate anticancer drug called ‘cyclophosphamide’, discovered in 1958, is used as an immunosuppressor drug to treat cancers, leukemia, and lymphoma (Fig. 14). After metabolism, cyclophosphamide is converted to acrolein and an active chlorinated phosphorodiamidate, which acts as a nitrogen mustard compound.89 Cross-linking with DNA strands will occur, and cell death will ensue. Cyclophosphamide figures in the World Health Organization's list of essential medicines.90 Another entry in this list is Fosfomycin, an epoxide phosphonic acid used as an antibiotic to treat bladder infections.91 Foscarnet, a very simple trisodium phosphonate salt first synthesized in 1924, was found in 1978 to be an effective antiviral agent by inhibiting viral DNA polymerase.92 It is effective against herpes and has been used for salvage therapy treatments against HIV.93
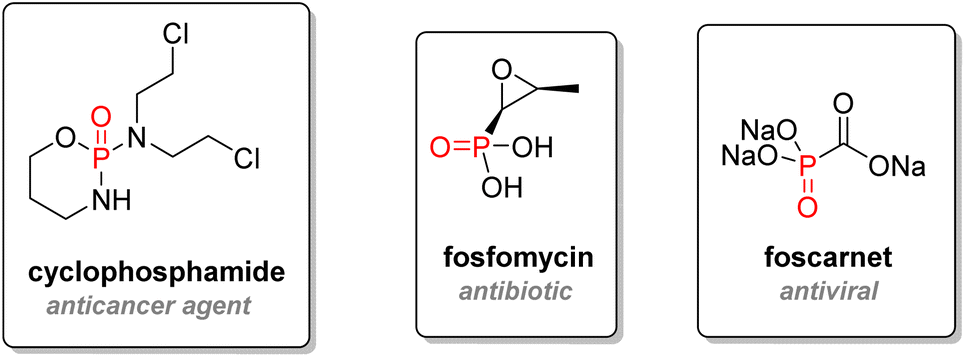 | ||
| Fig. 14 Early organophosphorus therapeutics. | ||
2.3.2.2. Bisphosphonates. Following the rationale of substituting oxygen by carbon between two P(V) to mimic phosphates, the idea of using biphosphonic acid derivatives as analogs of pyrophosphoric acid emerged. Similar to pyrophosphoric acid, bisphosphonates exhibit an intrinsic affinity for calcium ions and accumulate very well in bones, where they inhibit excessive bone resorption (Fig. 15). This class of drugs is used against osteoporosis, Paget's disease of bones, or other bone disorders.94 The first generation of bisphosphonates included derivatives of etidronic or clodronic acids bearing simple R1 and R2 substituents on the central carbon. Over the years, a better structure–activity relationship was established, and the current drugs have a dramatically increased potency, going up to a 10
000-fold upgrade compared with etidronate in the case of zoledronate.95 The current strategy is to use a hydroxyl R1 substituent to better the affinity with calcium or magnesium ions, while using nitrogen in the R2 substituent will increase the hydrogen bonds with the target enzyme. Bisphosphonate moieties have also been cross-linked to other drugs to take advantage of the exceptional binding affinity of these structures to the hydroxyapatite from bones. This method was coined osteoporotic drug delivery system.96
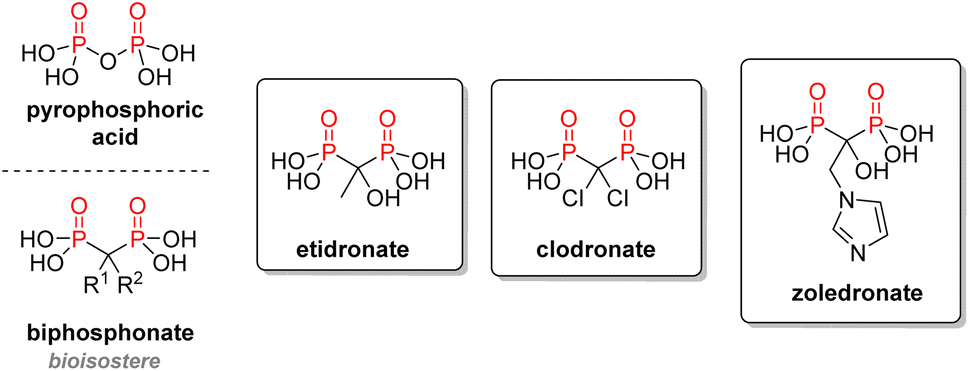 | ||
| Fig. 15 Bisphosphonates as bone resorption inhibitors. | ||
2.3.2.3. Aminophosphonates. Another crucial structural motif of organophosphorus drugs is aminophosphonates: α-aminophosphonic acids are bioisosteres of α-amino acids and resemble the high-energy transition state of the hydrolysis of ester and amide bonds. The simple amino-acid mimetics strategy is advantageous in the case of antibacterial drugs (Fig. 16). For instance, alafosfalin, which is a phosphonodipeptide, releases L-aminoethanephosphonic acid after metabolism. The phosphonate behaves like the natural substrate for L-alanine racemase except for its higher binding affinity to the cofactor, thus inhibiting the enzyme and subsequent cell wall biosynthesis.97 Dehydrophos, a peptide analog produced by Streptomyces luridus, also shows wide-spectrum antibacterial activity.98
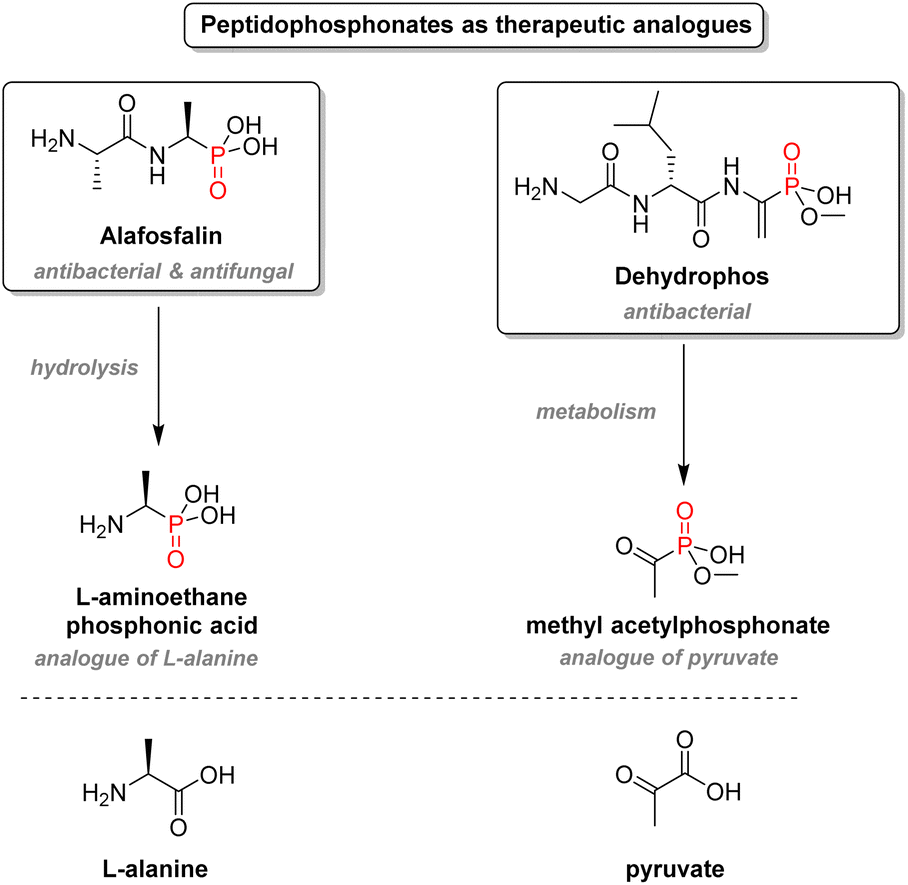 | ||
| Fig. 16 Peptidophosphonates as therapeutic analogs. | ||
Fosinopril is a successful β-aminophosphinate therapeutic used to treat hypertension and is commercialized by Bristol-Myers Squibb. The compound is administered as a prodrug: the phosphinic acid is esterified in order to improve bioavailability, and subsequent metabolism will cleave this P–O bond to release the active compound, Fosinoprilat (Scheme 7). This molecule is designed to be an analog of the transition state emerging from the hydrolysis of the dipeptidic substrate from the Angiotensin-Converting Enzyme (ACE).99
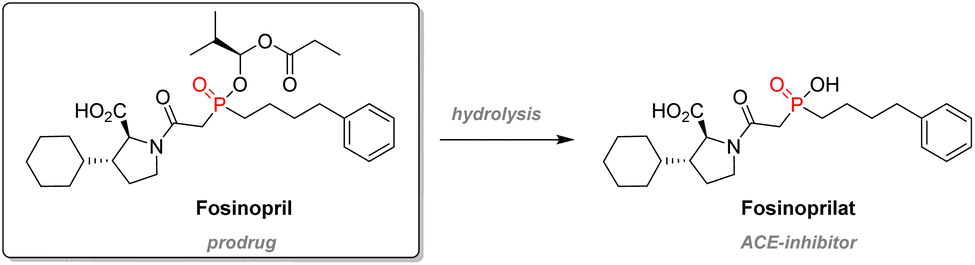 | ||
| Scheme 7 Metabolism of Fosinopril. | ||
Another mechanism of action for organophosphorus drugs is to be used as an irreversible inhibitor. This strategy includes the most nefarious examples of chemical warfare: nerve agents blocking the acetylcholinesterase enzyme. Details about their mode of action will be explained below in the agrochemistry application section. As for therapeutics, diaryl α-aminophosphonates exhibited promising inhibition of serine protease enzymes, which could lead to several applications such as anti-cancer agents, therapeutics against inflammatory disease, infections, or against hypertension.100–102 Finally, aminophosphonates can also be used for treatment against bone disease: incadronate merges both aminophosphonate and bisphosphonate structures and has been used against hypercalcemia and cancer development in vitro (Fig. 17).103,104 Macrocyclic polyaminophosphonates chelating ligands have been used with heavy lanthanide nuclei for either imagery or targeted radiotherapy of bone cancer.102,105,106
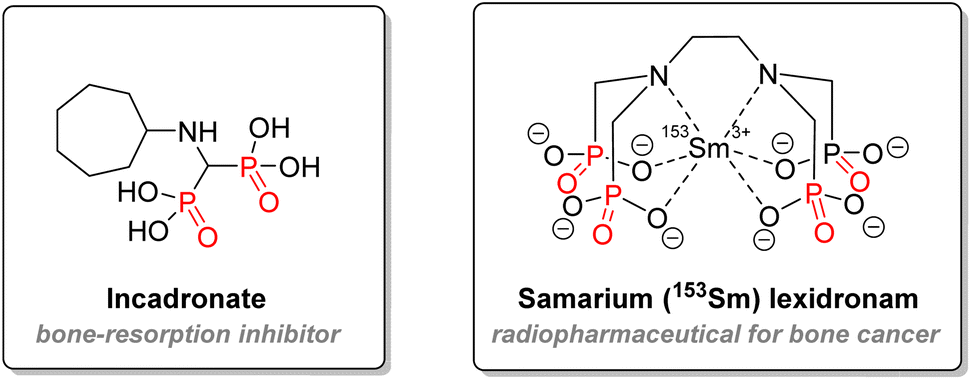 | ||
| Fig. 17 Incadronate and lexidronam. | ||
2.3.2.4. Antiviral nucleotide analogs. Organophosphorus nucleotide analogs are also a prevalent drug design strategy for antivirals. Those analogs behave and compete with natural nucleotides (or deoxynucleotides) to be incorporated into either viral DNA or RNA strands (Fig. 18). After phosphorylation steps, the creation of the consecutive phosphodiester bond with a new nucleotide for the strand is inhibited, either because of a missing 3′-hydroxyl group on the drug or because of steric interactions on the carbohydrate mimic. This mode of action thus blocks the growth of the viral strand and terminates the chain. This approach successfully inhibited viral reverse transcriptase in the case of hepatitis B: adefovir dipivoxil and tenofovir disoproxil are both phosphorylated adenosine derivatives prodrugs, and cleavage of the pivaloyloxymethylated P–O bonds will occur through metabolism, thus releasing the active forms of the drugs.107 Tenofovir can also be used against HIV. Remdesivir is a phosphoramidate prodrug synthesized by Gilead, which metabolite mimics adenosine triphosphate to interfere and block viral RNA polymerase. It was initially designed as a treatment for hepatitis C or Ebola virus but was later tested against SARS-CoV-2.108,109 Similar mechanisms of action are at work with other FDA approved phosphoramidate nucleotide drugs such as Sofosbuvir (blocking RNA polymerase from hepatitis C) or Cidofovir (blocking DNA polymerase from cytomegalovirus).110,111
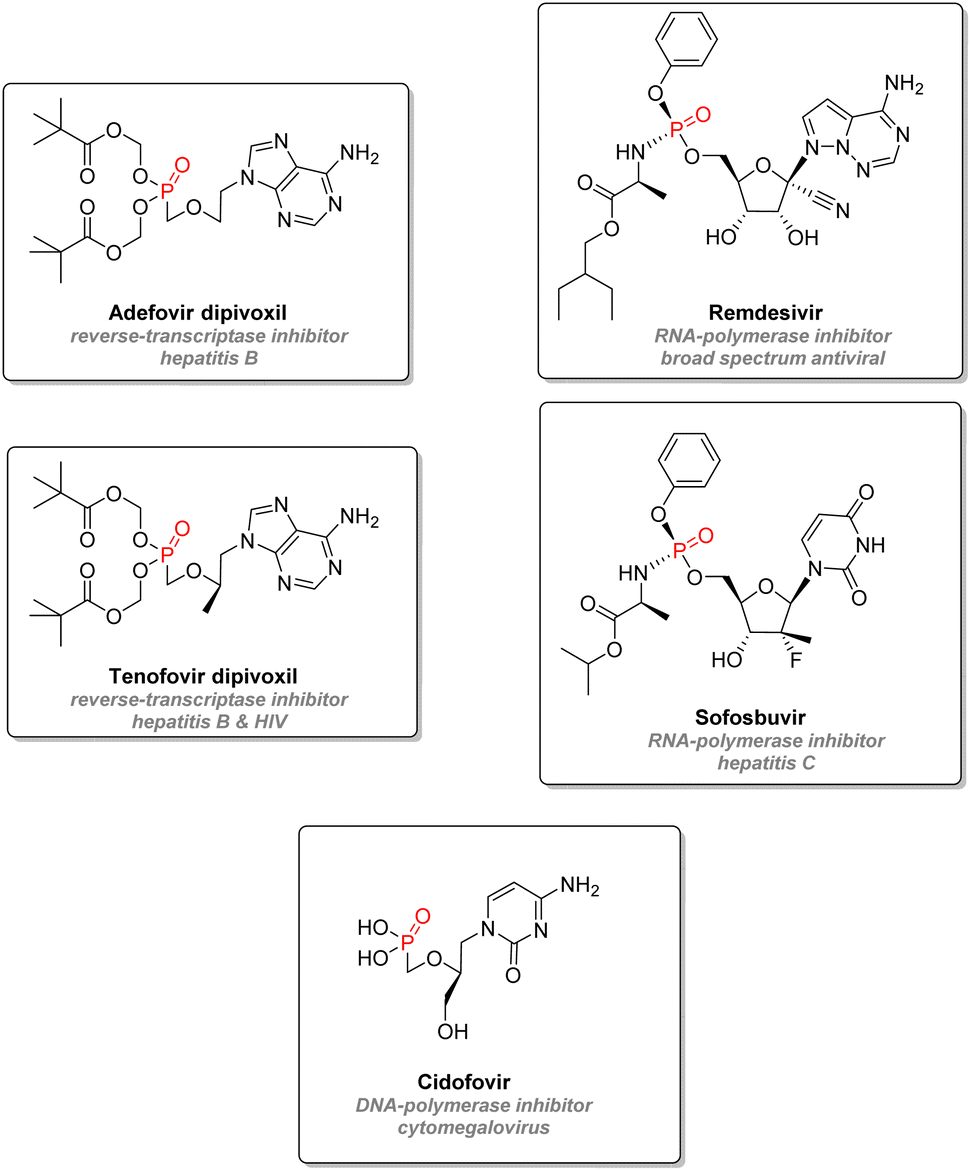 | ||
| Fig. 18 Nucleotide analogue antivirals. | ||
2.3.3.1. Insecticides. The usage of organophosphorus species – often generally coined “organophosphates” – for agrochemical purposes dates back to WWII. As previously stated in Section 1.1.2, Schrader in Germany and Ghosh in the UK independently investigated the use of phosphoryl derivatives as insecticides before the war and discovered that the mammalian toxicity of those compounds was surprisingly exceptionally high.112 Tabun, a clear colorless liquid phosphonamidite, was the first organophosphorus chemical intended as an insecticide which was repurposed as a warfare nerve agent. Regrettably, it paved the way for developing the whole G-series and V-series of lethal organophosphorus chemical weapons. However, proper substituent modification on the organophosphorus structure could reduce mammalian toxicity, thus allowing for a more specific and selective pesticide application.79 All organophosphorus insecticides act as irreversible acetylcholinesterase inhibitors: they all mimic the molecular shape of the neurotransmitter acetylcholine and can dock in the enzymatic active site (Fig. 19). However, instead of having the serine residue hydrolyzing acetylcholine, serine gets phosphorylated, thus impeding the usual regulation of acetylcholine and finally causing muscle paralysis and eventual death of the insect.113
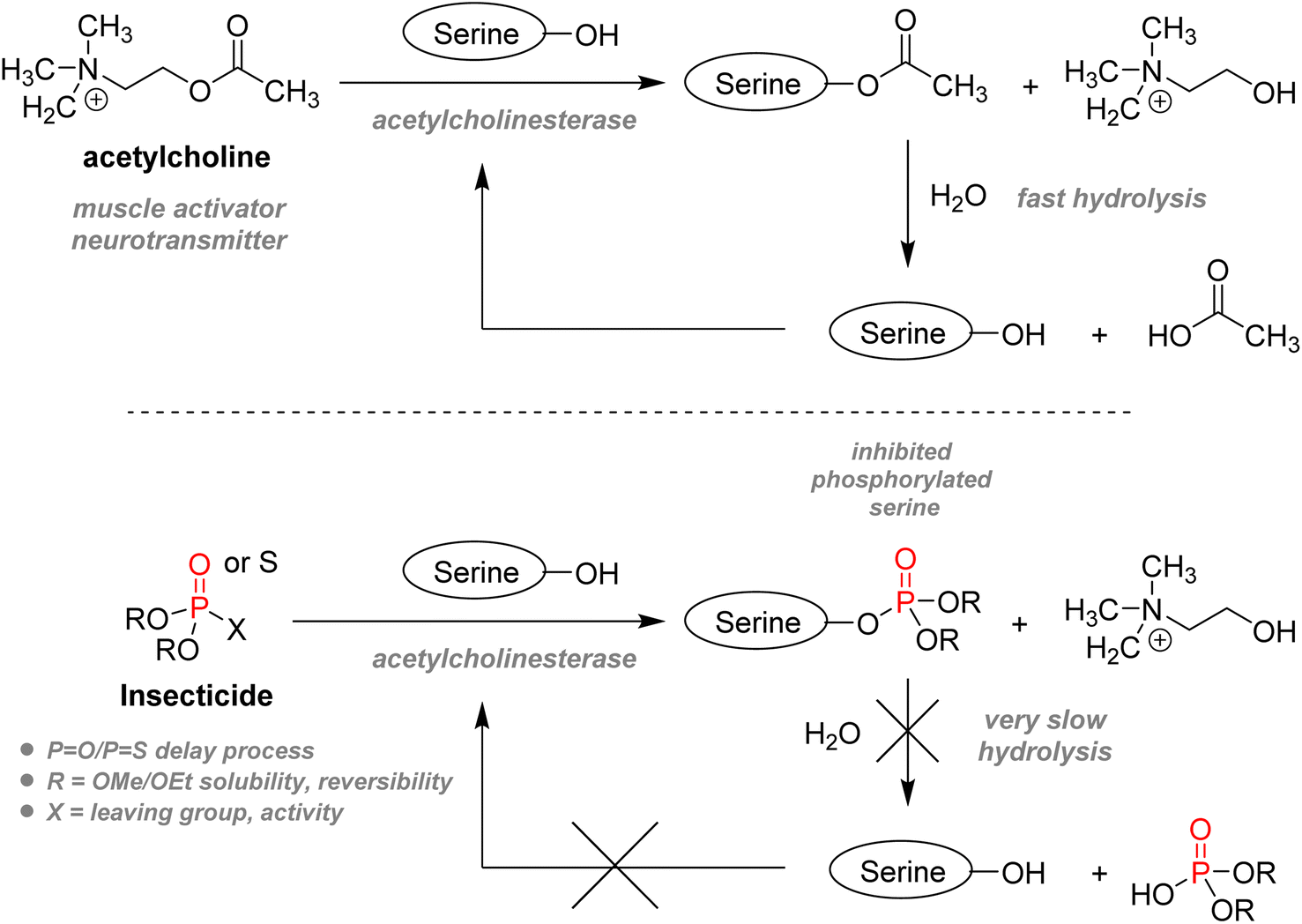 | ||
| Fig. 19 Mode of action of organophosphorus acetylcholinesterase inhibitors. | ||
As electrophilic phosphorylating agents, most insecticides of the organophosphorus family are either phosphorothioate (one sulfur replacing one oxygen atom: PS replacing PO, or P–S replacing P–O), phosphorodithioate, or vinyl phosphates (Fig. 20). Three key features play a role in the design: firstly, usually, two methyl, ethyl esters, or amino residues are present in order to balance the solubility, as free hydroxyl or thiol group would be ionized under physiological conditions, thus impeding a good bioavailability. Substituents also determine the rate at which the phosphorylated acetylcholinesterase serine residue goes back to its active state: hours for methyl esters to days with ethyl esters.114 Secondly, the phosphoryl group can also be replaced by PS. As electrophilicity of the P center is crucial for bioactivity, PS compounds are less reactive towards acetylcholinesterase. However, it will take several hours for in vivo oxidase to convert thions PS to active oxons PO compounds.114 This delay allows for lesser mammalian toxicity as part of the insecticide can be eliminated during this process while still being active against faster-metabolizing insects. Thirdly, the remaining substituent should act as an effective leaving group which can be substituted by serine. Efficiency is increased with electron-withdrawing groups. This explains why the p-nitrophenoxy substituent of parathion makes it highly active and toxic, resulting in its global ban from sales and imports. The emergence of malathion in the 1950s is an important landmark as it bears significantly lower toxicity towards humans. It is suitable for general purposes such as household or garden use but has also been used for public health in the struggle against mosquitoes or flies. Chlorpyrifos, developed in the 1960s, is still currently one of the most widely used agricultural pest control agents and the first organophosphate used in the US in 2012,115 despite increasing studies highlighting long-term exposure health problems. Another subcategory of P(V) insecticides are the vinylphosphates, including the highly volatile and regulated dichlorvos and the less toxic tetrachlorvinphos.
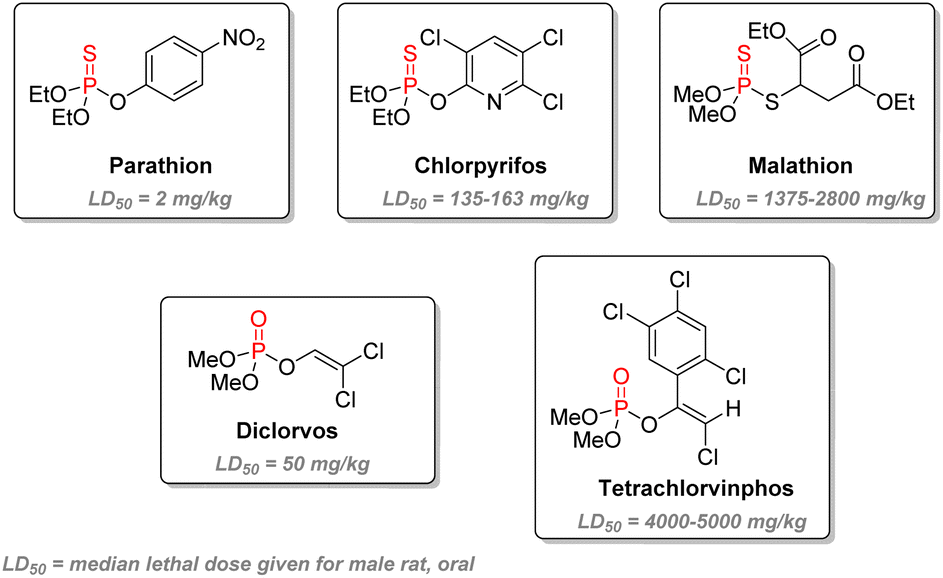 | ||
| Fig. 20 Examples of organophosphorus insecticides and their corresponding LD50. | ||
Organophosphorus insecticides react with air, light, and water and are more easily hydrolyzed and biodegraded than organochlorine insecticides, which significantly impact the environment. For instance, formerly extensively used but now banned organochlorine insecticide DDT has a 2.8 year half-life in soil vs. 1 or 2 weeks for malathion or 3–6 months for chlorpyrifos.116 However, their substantial use has shown contaminations in grains, vegetables, fruits, and milk. The ongoing concern with organophosphorus insecticides is their acute human toxicity: yearly worldwide, organophosphate poisoning has been estimated to be 3 million, causing 200000 deaths.117,118 Detrimental effects on bees, fish, birds, and wildlife have also been reported.119
2.3.3.2. Herbicides. Another important application for P(V) compounds in agricultural chemistry is herbicide (Fig. 21). The most successful herbicide is glyphosate – commercially sold as Roundup – an α-aminophosphonate type systemic herbicide first marketed in 1974. It is not only the world's currently most used herbicide but also pesticide overall by weight. It has been the most used herbicide in the US for agriculture since 2001, for the industry/commercial/government market in 2009–2012, and it was ranked second for home and garden market in the same period.115 The mechanism of action of the phosphonate is to inhibit 5-enolpyruvyl-shikimate-3-phosphate synthase (EPSPS), an enzyme part of the amino-acid biosynthetic pathway from glucose. This enzyme is specific to plants and does not exist in mammals. The efficient disruption of the amino-acid synthesis causes the death of many plants without selectivity, making glyphosate a powerful broad-spectrum herbicide.120 Thousands of compounds have been tested to inhibit EPSPS, but only glyphosate has been commercialized so far. Transgenic glyphosate-resistant crops such as soybean, maize, cotton, or canola breeds are commonly used with the potent herbicide.121 Key features include good uptake through the leaves, excellent translocation (movement of the herbicide inside the plant from the uptake location to the site of action), the absence of natural resistance amongst higher-plants, very low toxicity (LD50 oral for male rats above 5000 mg kg−1),122 and a relatively short half-life in soil (30–174 days).123 However, recent controversies have emerged related to the possible human carcinogenicity and environmental issues linked to the use of glyphosate.124,125 Contradictory studies and changing legal status have shown the need to reassess data about the herbicide standards.126,127
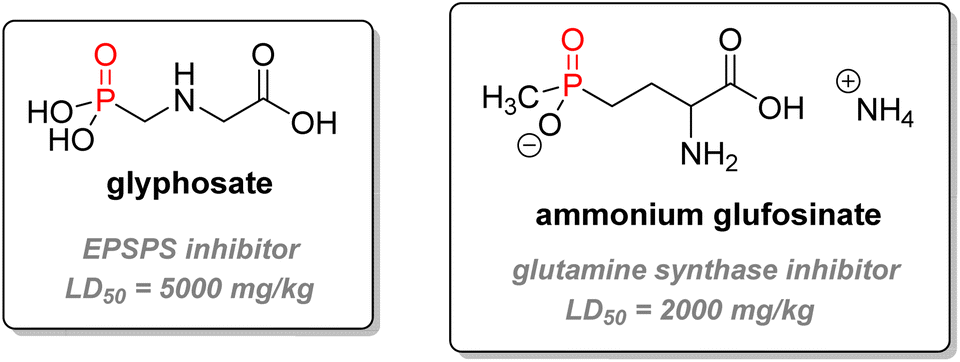 | ||
| Fig. 21 Common organophosphorus herbicides. | ||
Glufosinate is a non-selective phosphinate herbicidal compound of lesser importance, which can also be used as an antibiotic or a fungicide. It is the active metabolite of bialaphos, a natural peptide derivative synthesized by Streptomyces hygroscopicus.128 It acts as a glutamine synthase inhibitor, impeding photosynthesis and causing the death of the plant. Glufosinate ammonium bears a lower LD50 than glyphosate (416–2000 mg kg−1 for rat, oral).120 Other organophosphorus compounds used as agrochemicals include fungicide Fosetyl-Al or plant growth regulators ethephon or glyphosine (Fig. 22).
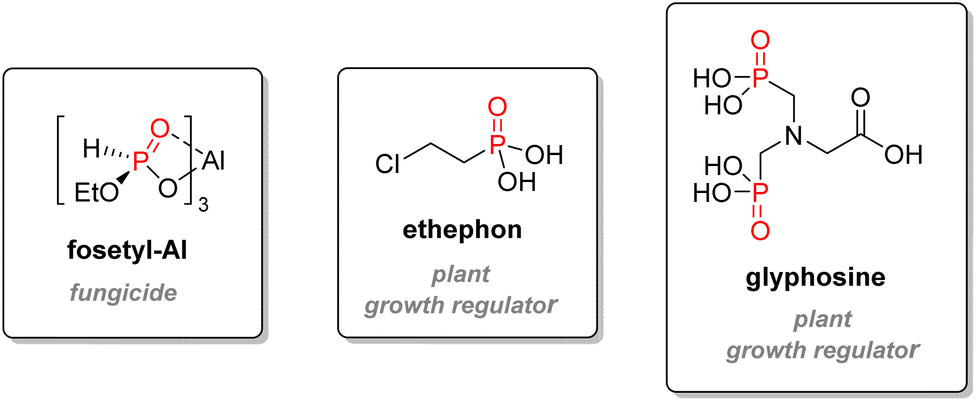 | ||
| Fig. 22 Examples of organophosphorus agrochemicals. | ||
 | ||
| Fig. 23 Common organophosphorus structures for organic electronics. | ||
2.3.4.1. Organic light-emitting diodes. Currently used organic light-emitting diode (OLED) technology has been a significant advance in display and light-source technology. Electrons injected from the cathode will be transported through an electron transport layer to the emissive layer. Similarly, the lack of electrons, known as holes, will be injected by the anode and transported through the hole transport layer to the emissive layer as well. This emissive layer will recombine both electrons and holes into excitons, radiating light according to the dye used in the layer (Fig. 24). However, conventional OLEDs rely on light emitted by fluorescence, limiting the conversion of excitons into light only to the singlet excitons, representing a 25% internal quantum efficiency. Another improved strategy exists with phosphorescent organic light-emitting diodes (PhOLED), which can harness the remaining triplet state excitons and increase efficiency. Nevertheless, phosphorescent dyes (called guests or dopants) inside the emissive layer should be spaced out inside a matrix (host) in order to avoid triplet–triplet quenching between the excited molecules.129
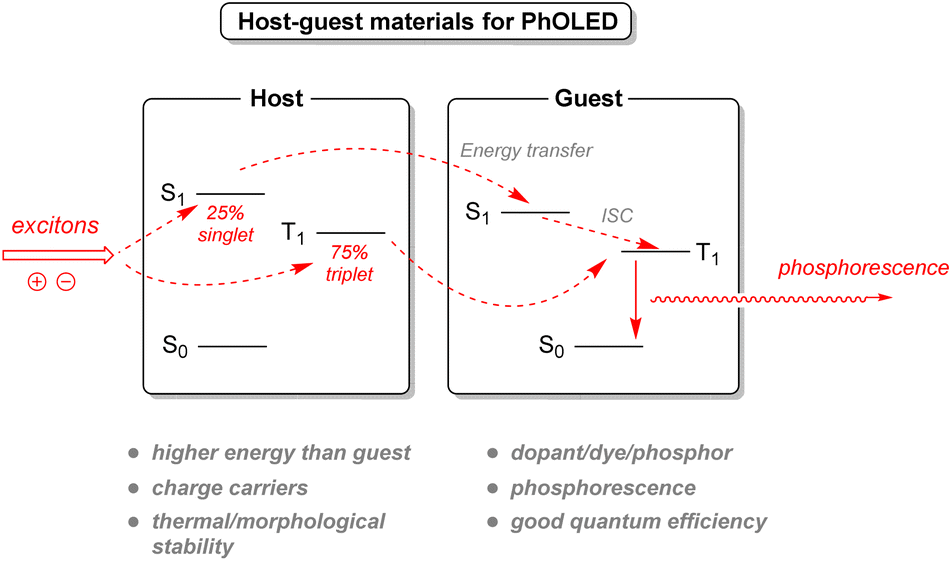 | ||
| Fig. 24 Roles of host–guest materials for PhOLED. | ||
2.3.4.1.1. Phosphine oxides. Phosphine oxides have been used both in the electron transport layer and in the host material: the high polarity and inductively withdrawing effect of the P
O bond makes phosphine oxide especially well-suited for electron transport. Their high triplet state energy can also effectively capture triplet state excitons and redistribute them to the lower energy triplet state of the light-emitting guest molecule through energy transfer. Thanks to their tetrahedral geometry, phosphine oxide can interrupt conjugation and give good thermal and morphological stability with their steric bulk while keeping a necessary high triplet state energy. In order to improve charge transport, some host materials can be ‘bipolar’, and transport both electrons and holes by using both an electron-withdrawing group such as a phosphine oxide moiety and an electron-donating one such as a tertiary amine: for instance (Fig. 25(1)) combines an active fluorophore bifluorene backbone with a diphenylphosphine oxide. It has been used as an electron transport host material for high-efficiency blue electrophosphorescent devices.130 Bipolar host materials using hole-transport moieties such as carbazole and electron transport ones like phosphine oxide have been used to construct deep blue colored PhOLED (Fig. 25(2)).131 Polymeric bipolar hosts combining carbazoles and phosphine oxides have also shown good charge transport capabilities and a high triplet state (Fig. 25(3)).132 Phosphine oxides have also directly been used as light-emitting material (Fig. 25(4)) by fusing them with fluorene or carbazole in the architecture for blue emission,133,134 or by directly incorporating classic guest phosphorescent complexes such as blue FIrPic and yellow [(fbi)2Ir(acac)] inside a fluorinated phosphine oxide polymeric backbone to give an efficient single polymer for white PhOLED.135
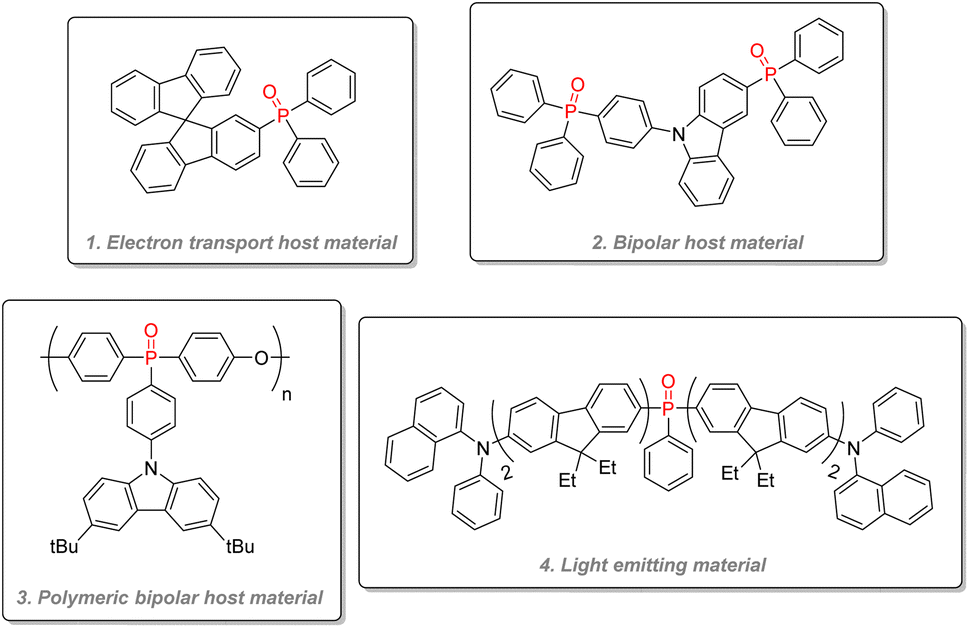 | ||
| Fig. 25 Examples of phosphine oxides as PhOLED materials. | ||
The excellent review from Jeon & Lee compiled many phosphine oxide-containing materials used for PhOLEDs and their physical data.136 Despite the outstanding abilities of phosphine oxide-based PhOLED material in terms of high triplet state energy and electron transport, their stability towards UV or electrical stress is compromised when P–C bond cleavage occurs during excited or charged states.137,138 This could significantly lower the life-time of blue PhOLED and remains an ongoing obstacle to be overcome.
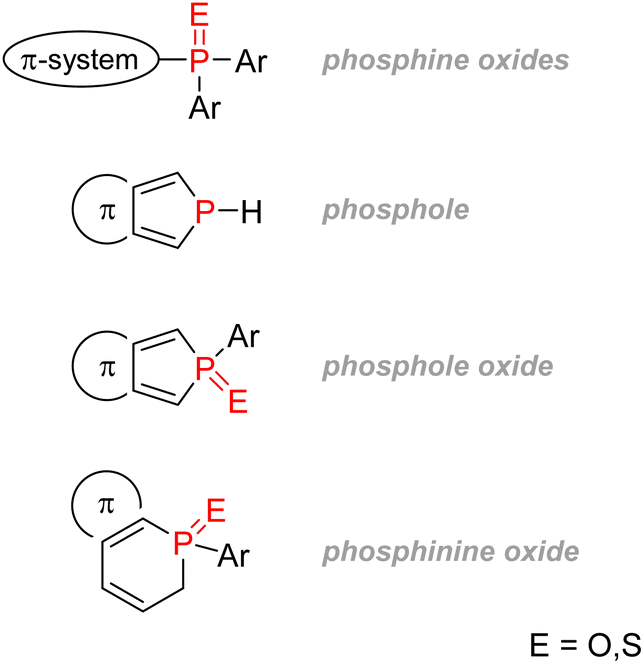
2.3.4.1.2. Phospholes. Phosphorus-containing heterocycles have also found applications in organic electronics. Phosphole, the phosphorus relative of pyrrole and thiophene, is a P(III) 5-membered ring bearing very weak aromaticity: 1H-phosphole has a calculated nucleus-independent chemical shift (NICS)(0) of −5.6 compared with −11.9 with furan or −13.3 for thiophene, which is more aromatic. The reason for such low aromaticity for phosphole and phosphole oxides is that P(III) has a high energy inversion barrier and keeps a pyramidal configuration, disrupting a good overlap of the π electrons.139 Contrary to pyrrole, the phosphorus lone-pair does not participate in the aromaticity, as it is instead an interaction between the diene π electrons and the σ* from the P–R bond that takes place (Fig. 26). Changing the P–R exocyclic substituent can change this very σ*–π interaction and thus tune the aromaticity of the phosphole ring.
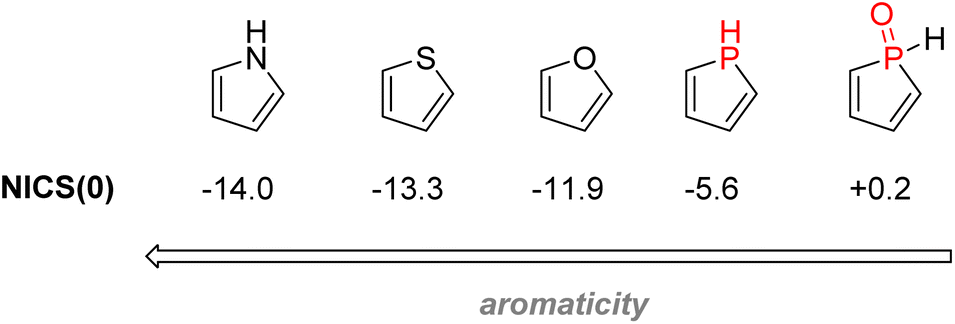 | ||
| Fig. 26 Calculated aromaticity of phospholes and phosphole oxides compared to other electron-rich heteroarenes. | ||
Moreover, phospholes can act as standard platform structures for obtaining diverse modified π-conjugated materials as functionalization on the phosphorus lone pair is easy: oxidation (oxygen, sulfur, or selenium), alkylation, or coordination with a transition metal are all possible. Finally, due to low LUMO energy, phospholes have electron acceptor properties that can also be modified according to the phosphorus's P–R substituents or oxidation state. Those properties make phospholes and phosphole oxides containing π-systems versatile tools for material chemists.
Both dibenzooxophosphole, used as an electron acceptor unit, and carbazole, used as a hole acceptor unit, were integrated into bipolar host material for OLED purposes with good efficiency in combination with Os(II) guest dyes (Fig. 27(1)).140 Highly performant thioxophosphole (Fig. 27(2)) was directly used as a fluorescent dye for OLED and showed the highest quantum efficiency for phosphole emitting layer material.141
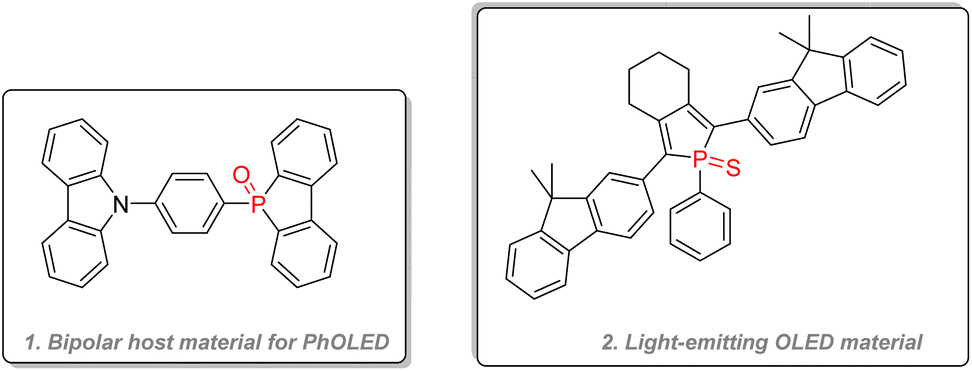 | ||
| Fig. 27 Phospholes as OLED materials. | ||
2.3.4.2. Dye-sensitized solar cells, bioimaging, electrochromic device, and other stimuli-responsive materials.
2.3.4.2.1. Phospholes. Aside from OLEDs, phosphole oxides and sulfides have also been used as dyes for dye-sensitized solar cells (DSSC) (Fig. 28),142,143 for electrochemical cells, and electrochromic devices.144,145 Polyaromatic phospholes and phosphole oxides have also been used in reversible stimuli-responsive color-changing structures, which can be indispensable for smart materials such as display, thermochromic paints, or self-dimming glass. Reus & Baumgartner detailed many examples of those stimuli-responsive chromic organophosphorus architectures in their review, showing among other interesting properties electrochromism (change of color after a redox reaction), photochromism (change of color according to the UV/visible light irradiation), thermochromism (change of color according to the temperature), halochromism (change of color according to the pH) or even mechanochromism (change of color according to the mechanical stress).146
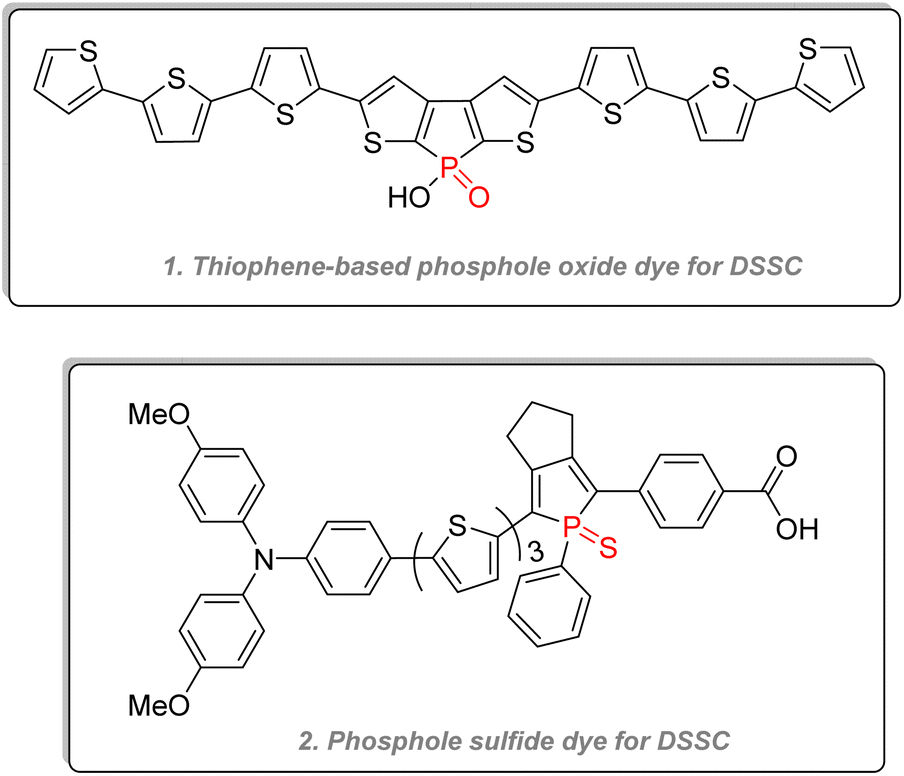 | ||
| Fig. 28 Phospholes used for dye-sensitized solar cells. | ||
2.3.4.2.2. Phosphinine derivatives and P-containing 6 membered rings. Phosphorus-containing six-membered rings such as phosphinine and phosphinine oxide bear unique optoelectronic properties. Jiang reported in 2015 the use of a phosphorus dipyrromethene and its aza counterpart as fluorescent marker dyes for bioimaging (Fig. 29(1)). The compound showed better water solubility than the currently used BODIPY dye.147 Other phosphorus versions of traditional dyes have been synthesized, such as phospha-rhodamine or phospha-fluorescein, and all have shown good quantum efficiency, water solubility, and excellent resistance towards photo-bleaching.148,149 A very ingenious strategy to detect calcium ions in living cells was to combine a far-red fluorescent phospha-fluorescein scaffold with calcium probes (Fig. 29(2)).150 Polyaromatic systems incorporating phosphinine derivatives have recently been studied by Romero-Nieto et al., and the compound (Fig. 29(3)) showed outstanding photochemical properties and good suitability for photoelectrochemical cells.151 An excellent review written by Regulska & Romero-Nieto focuses on the applications of extended π systems based on phosphorus-containing six-membered heterocycles.152
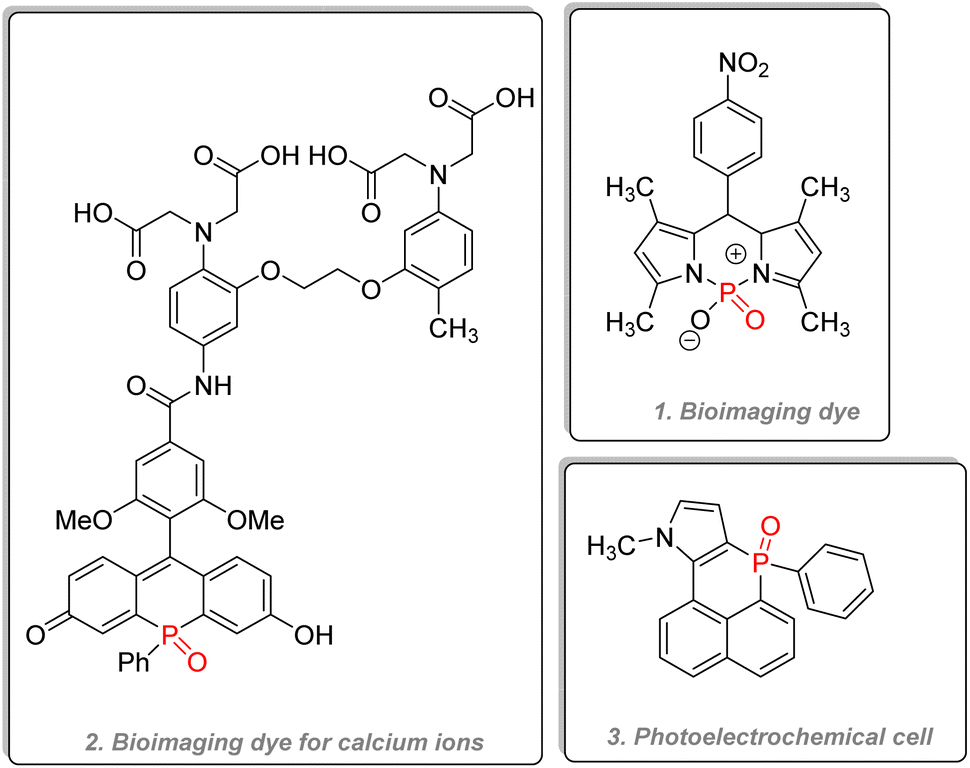 | ||
| Fig. 29 Phosphinines in organic electronics. | ||
Aluminium hydroxide is a mass-produced, and inexpensive flame retardant used widely along with other metal hydrides for cheap commodity materials such as thermoplastic polyolefins. However, Al(OH)3 is often required in very high loadings, which can negatively affect the material properties. More advanced and sensitive materials such as engineering polymers or high-performance ones often necessitate more suitable retardants. The oldest technology used for those materials was based on halogenated flame retardants, once considered the workhorse of this field. Their decomposition into halogen acid gas acts as a radical quencher, thus obstructing the combustion process.154 Despite claims from manufacturers, some popular halogenated flame retardants have proven to have a high bioaccumulation rate and to be toxic for humans and animals, causing harmful neurobehavioral effects and endocrine disruption.155 This led regulatory authorities in Europe and North America to ban several brominated and chlorinated compounds, thus pushing for increased use of phosphorus-based flame retardants.
Inorganic forms of phosphorus such as red phosphorus previously used in nylon 66 or polyethylene have been used as flame retardants, but the hazards linked to handling and coloring have since restricted its use. Cheap and efficient ammonium phosphates, whose use as fertilizer was already covered earlier, can also be used as a flame retardant for cellulosic materials such as textile, paper, and wood. Organophosphorus flame retardants have two main mechanisms of action, one in the condensed (solid) phase and one in the gas phase. First, phosphorus will cross link between the carbon decomposition products occurring on the solid-state of the material and form inorganic glasses like polyphosphate and phosphoric acid, thus inhibiting pyrolysis. Second, in combination with other material, phosphoric acid residues can promote intumescence, insulating the unburnt parts. Volatile phosphorus fragments (PO˙, HPO2˙ and PO2˙) act as radical scavenger and quench reactive H and OH radicals to delay exothermic processes and decrease the supply of flammable gases.156 Organophosphorus flame retardants can be used either as additives or as reactive.
The first category, the most used one, contains most of the classic organophosphorus flame retardants.157 Triphenyl phosphate (Fig. 30(1)) is used in cellulose acetate films and engineering thermoplastics. Tricresyl phosphate is used in rubbers and flexible PVC. Resorcinol bis(diphenyl phosphate) (Fig. 30(2)) is a less volatile and highly successful compound used for engineering thermoplastics, polyamides, vinyls, thermoplastic polyesters, thermosets, and various elastomers. Bisphenol A bis(diphenyl phosphate) (Fig. 30(3)) is a cheaper option than the resorcinol diphosphate, but it requires more loading for similar properties. Phosphonates such as dimethyl methylphosphonate (Fig. 30(4)) were used in polyester resins and ductwork, but they are now more restricted. Aluminium diethyl phosphinate (Fig. 30(5)) is used in thermoplastics such as polyamides, polyesters, ABS.
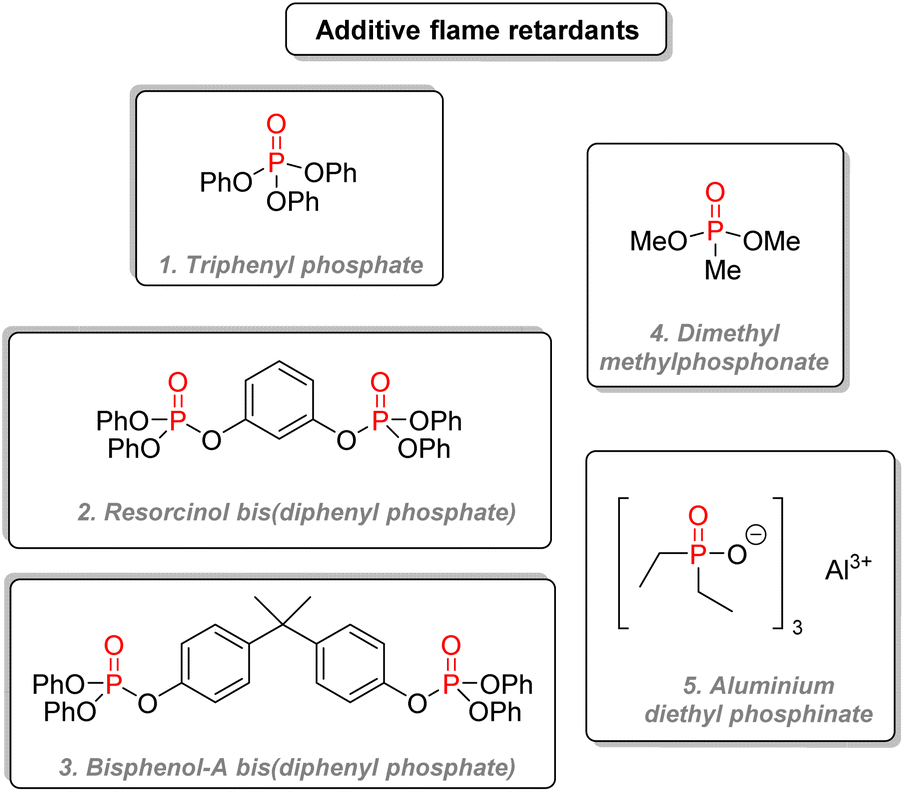 | ||
| Fig. 30 P(V) organophosphorus as additive flame retardants. | ||
Reactive flame retardants bear reactive chemical moieties, which allow them to be directly cross-linked into the material, making them an integral part of them. This often leads to a lesser impact on the physical properties of the overall material and a decreased leaching into the environment. Bis(4-amino-phenyl)phenylphosphine oxide (Fig. 31(1)) was used in polyurethane resins.158 Phosphinate adducts such as 9,10-dihydro-9-oxa-10-phosphaphenanthrene-9-oxide (Fig. 31(2)), which is also used as an additive, can also be added as a reactive flame retardant in epoxy resins while keeping high thermal stability.159 Derivatives of this structure also gave good flame retardant properties for the electrolytes of Li-ion batteries.160,161
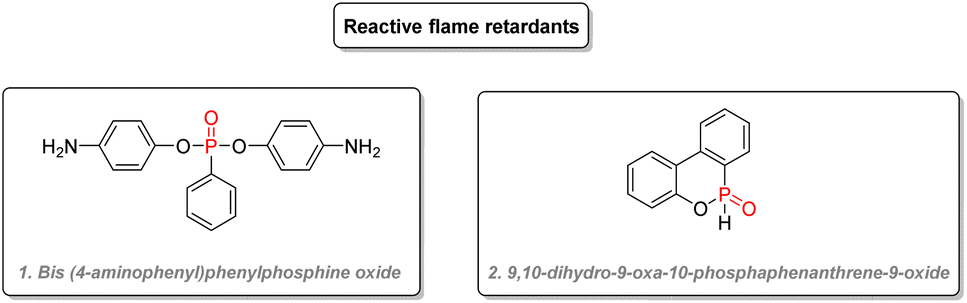 | ||
| Fig. 31 P(V) organophosphorus as reactive flame retardants. | ||
However, growing concern about the ubiquitous presence of organophosphorus flame retardants residues in outdoor and indoor environments, in addition to the rising issue of their potential toxicity, has led reports to question whether the substitution of halogenated compounds by P-flame retardants additives was the right choice.162 One interesting direction to solve this issue would be to use biocompatible and bio-resourced phosphorylated biopolymers such as casein or DNA as flame retardant coating, as a few recent reports showed.163–165
2.3.7.1. Uranium extraction. P(V) compounds are essential in nuclear chemistry as extraction agents. Nuclear power plants typically rely on the fission of heavy actinide isotopes such as 235U to generate power. Uranium is generally obtained by purifying natural uranium ores according to different processes. According to the DAPEX (dialkyl phosphoric extraction) procedure, the uranium ore is treated with a strong acid such as nitric acid and then purified by solvent extraction, using a non-polar solvent such as kerosene and two organophosphorus extraction agents di-(2-ethylhexyl)phosphoric acid (Fig. 32(1)) with a 4% loading and tri-n-butyl phosphate (Fig. 32(2)) in an equivalent amount. Long alkyl substituents give those organophosphorus chemicals good lipophilicity and thus good solubility in inert solvents while coordinating selectively with uranyl ions, thanks to their high polarity. After complexation with di-(2-ethylhexyl)phosphoric acid (ligand L), uranyl complexes such as (UO2)2(NO3)2L2 are formed.171 After separating the impurities by removing the aqueous phase, the uranium-rich kerosene phase is then treated with a strip solution containing either HCl, HF, or carbonate to transfer uranyl ions in the aqueous phase. The role of tributyl phosphate is thought to have a synergistic effect compared to using the phosphoric acid ligand alone: the tributyl phosphate additive is thought to contribute to the stability of (UO2)2(NO3)2L2 complex by hydrogen bonding.172 Uranium dioxide will then be converted to UF6 for isotopic enrichment before use as nuclear fuel.
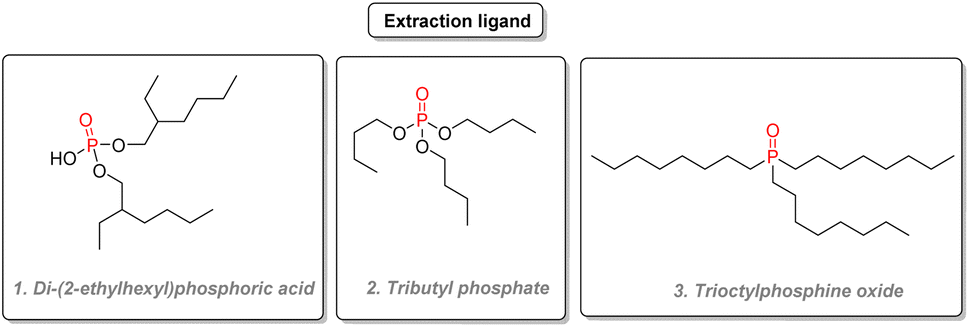 | ||
| Fig. 32 Organophosphorus as extraction ligands. | ||
Other organophosphorus compounds have been used for uranium extraction, such as trioctylphosphine oxide (Fig. 32(3)) and bis(2,4,4-trimethyl pentyl)phosphinic acid. A summary of all the extraction agents used for uranium in acidic solution and their comparison is given in a review written by Yoon and coworkers.173 Phosphonate compounds such as dibutylbutyl phosphonate can be used as extractants as well.174 As an alternative to uranium ores, uranium can also be extracted from phosphate rocks, which can be profitable when the demand is high. Since the 1990s, processes using both di-(2-ethylhexyl)phosphoric acid and trioctylphosphine oxides are considered state of the art concerning uranium extraction from phosphate rocks. A comprehensive review published in 2014 details the different processes and the challenges ahead for this unconventional source.175
2.3.7.2. Recycling nuclear waste. After nuclear fuel usage, nuclear powerplants produce residual materials, which are mixtures of fission products and still valuable uranium and plutonium materials. Recovering those energy-carrier isotopes to recycle them for conversion, enrichment, and fuel usage is the central role of reprocessing plants. This strategy aims at a more economical and sustainable use of actinides in a context where uranium resources are finite. The reprocessing plants use the PUREX (plutonium uranium extraction) process to produce UO2(NO3)2 and Pu(NO3)4, which can either be re-enriched in the case of uranium or used in a mixed uranium–plutonium fuel called mixed oxide fuel (MOX) for plutonium. The PUREX process relies on liquid–liquid extraction to isolate useful actinides and separate them from wasteful fission products. After the dissolution of the actinides in nitric acid, the actinide nitrates are treated with kerosene and 30% tri-n-butyl phosphate, which binds selectively to uranium and plutonium of +IV and +VI oxidation state to form stable complexes.176 The other fission by-products will remain in the aqueous layer. Plutonium complexes can later be separated from the uranium ones by using a reducing agent such as ferrous sulfamate or hydrazine to obtain plutonium species to a +III oxidation state, which would then migrate towards the aqueous phase.
2.3.7.3. Miscellaneous. In addition to uranium extraction, the use of P(V) compounds can be extended to the extraction of valuable rare-earth elements.177–182 Finally, more recent strategies include the design of pillarenes or calixarenes bearing phosphine oxide moieties to extract uranium from ionic liquids.183,184
3. Synthesis of organophosphorus: classical pathways & associated challenges
After examining their different applications, we will explore in this section the industrial synthetic ways for fine organophosphorus chemicals and consider their associated limitations and sustainability issues. For clarity, the reactions shown here are the most used synthesis pathways towards non-nitrogen-containing organophosphorus compounds. Critical alternative pathways are also shown in dashed lines, but many other reactions have been overlooked when less relevant. For instance, the reductions of phosphine oxides to phosphines or phosphates to phosphites performed with lithium aluminium hydride have been disregarded as they show lesser industrial value. Therefore, the aim here is not to present an exhaustive list of all synthetic routes for organophosphorus molecules but rather to show the trends in a simplified manner, as described above (Fig. 33).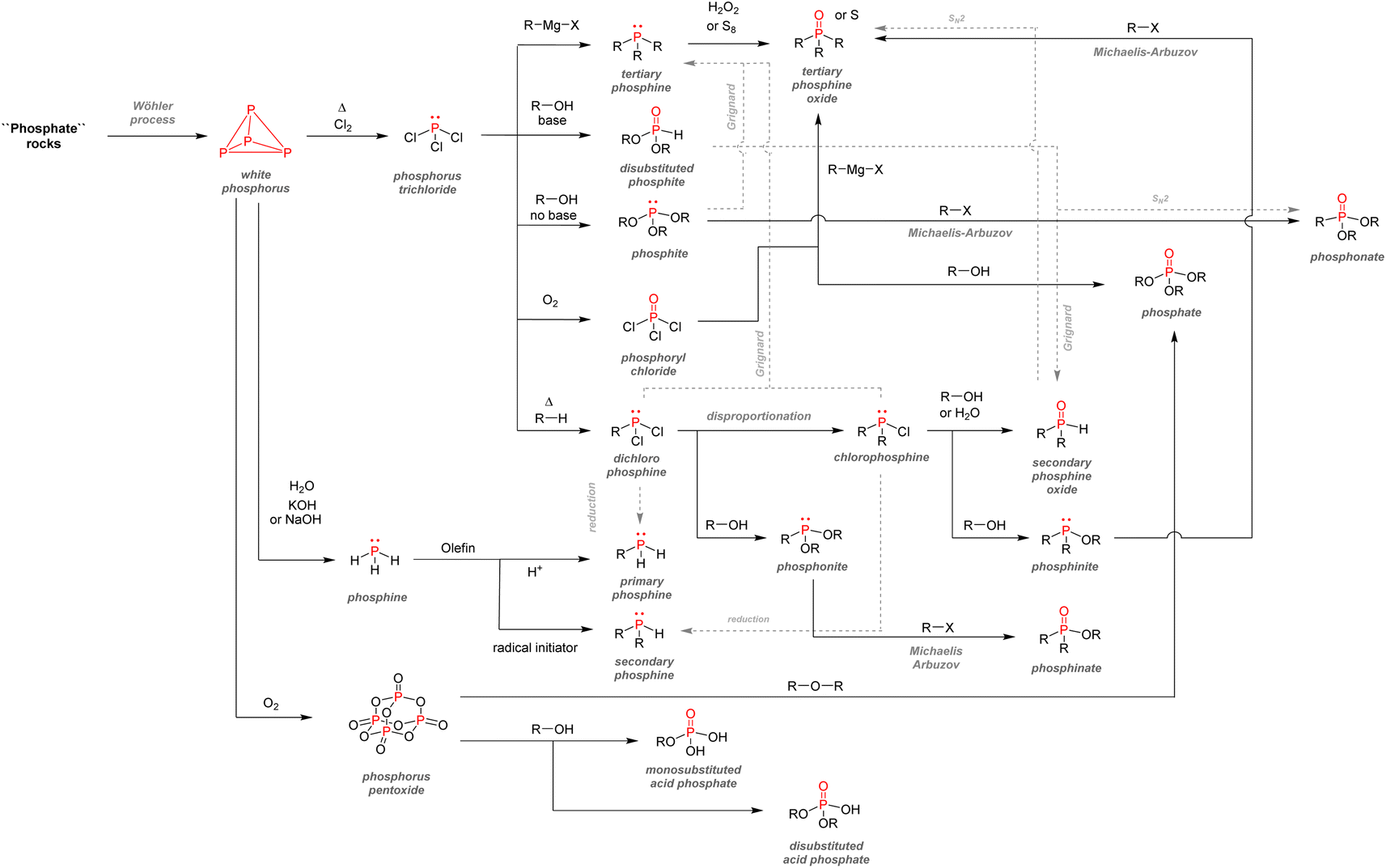 | ||
| Fig. 33 Overview of synthetic routes towards organophosphorus compounds. | ||
All the synthetic pathways to organophosphorus compounds start from the same universal intermediate, which is white phosphorus. By itself, molecular phosphorus does not have direct functionalization pathways used on an industrial scale towards useful organophosphorus. For example, both white phosphorus and PCl3 do not have significant applications other than in the military field. However, white phosphorus can be converted to other intermediates: phosphorus trichloride, phosphine, and to a lesser extent, phosphorus pentoxide (anhydride form of pure orthophosphoric acid). The latter can be formed through the exothermic oxidation of white phosphorus and can subsequentially provide monosubstituted and disubstituted acid phosphates after reaction with alcohols.
Phosphine PH3 can be prepared by treating white phosphorus with water and either sodium or potassium hydroxide. Phosphine is a dangerous and toxic gas by itself, it but can be converted to either primary or secondary phosphines through hydrophosphination of olefins (more details below). In the big picture, phosphorus trichloride remains the lowest common denominator as a ubiquitous intermediate for most organophosphorus. Its preparation necessitates the treatment of white phosphorus with toxic gaseous chlorine under reflux. Phosphorus trichloride can provide access to tertiary phosphines after alkylation through Grignard reagents. Subsequent oxidation by hydrogen peroxide or octasulfur S8 yields tertiary phosphine oxides or sulfides. Treatment of PCl3 with alcohol can yield either disubstituted or trisubstituted phosphites according to the presence or absence of base additives. Monoalkylation of PCl3 occurs through a high-temperature reaction with alkanes, affording dichlorophosphines PRCl2. Consecutive alcohol substitution gives phosphonites, whereas PRCl2 disproportionation produces chlorophosphines PR2Cl. Chlorophosphines also provide access to secondary phosphine oxides after oxidation or phosphinite after alcohol substitution. Finally, the oxidation of phosphorus trichloride can form another common intermediate: phosphoryl chloride P(O)Cl3. Treatment with carbanion equivalent will afford tertiary phosphine oxides while substitution with alcohol will generate trisubstituted phosphates. Considering that P4, PCl3, and POCl3 are ubiquitous P-intermediates, obvious sustainability issues stand out when looking at the current industrial procedures:
-Energy costly processes are necessary, such as the white phosphorus synthesis from apatite or the synthesis of PCl3.
-The use of toxic reagents chlorine gas is also indispensable, and phosphorus chlorides intermediates will provide hazardous halide by-products after functionalization.
-In terms of redox efficiency, the global synthetic route can also be perfected: the preparation of oxidized organophosphorus (oxidation number from +I to +V) such as phosphites, phosphonates, and phosphates require the reduction of phosphorus from naturally occurring apatite (+V) to molecular phosphorus (0) and then re-oxidation.
-In terms of redox economy,185 P-intermediates with a more balanced oxidation number would be highly desirable in order to obtain both phosphines (−III) and phosphates (+V), rather than the currently used PCl3 (+III) and POCl3 (+V).
-Finally, all P-intermediates, namely P4, PCl3, and POCl3, are eminently hazardous and uneasy to handle.
3.1. Towards more sustainable P-building blocks?
Considering the growing need for more sustainable chemical transformations, better alternatives complying with the 12 principles of green chemistry and concepts such as atom economy should be developed for the organophosphorus industry.186,187 Different strategies currently being investigated on a laboratory scale to solve this problem are highlighted in this section. The list of pathways presented below is far from exhaustive, and a more curious reader should refer to the few reviews already published on the subject.188–191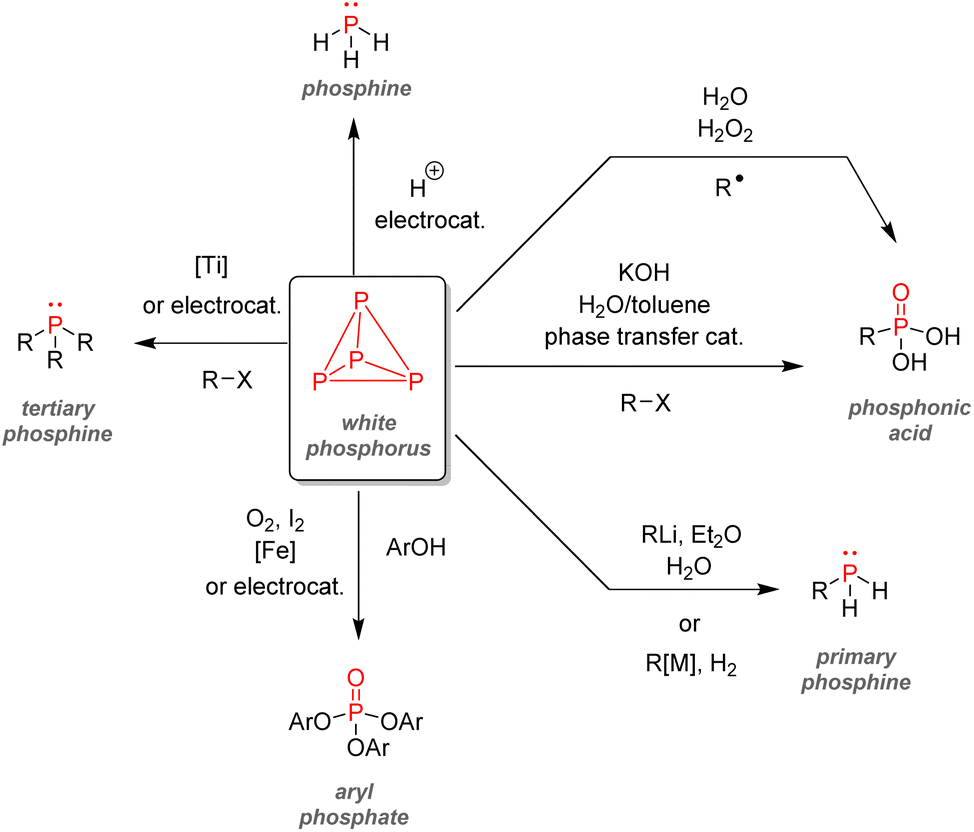 | ||
| Fig. 34 White phosphorus as an intermediate for organophosphorus synthesis. | ||
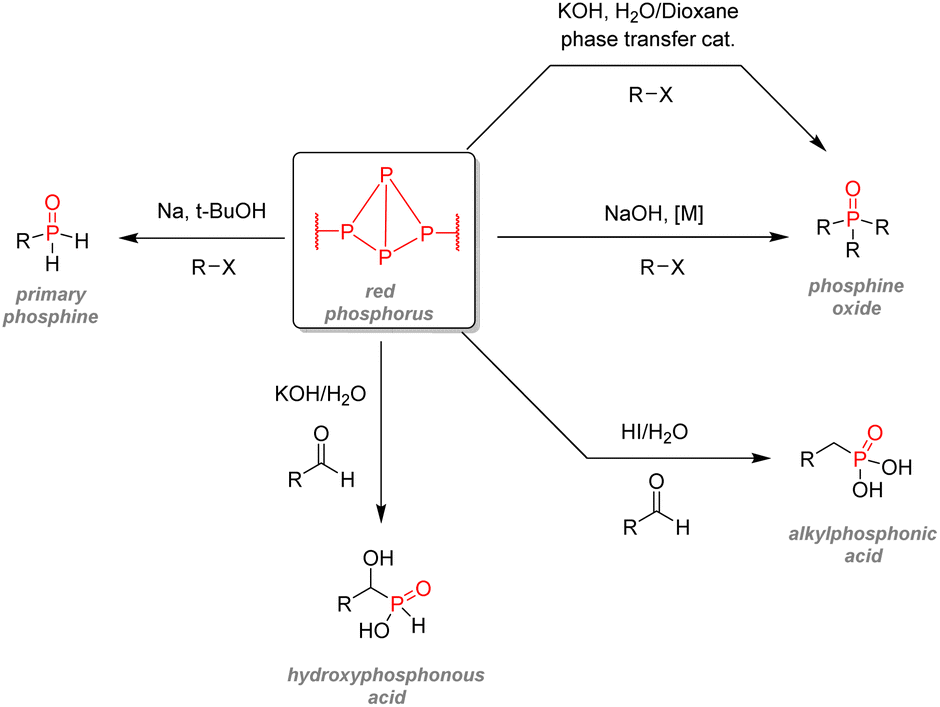 | ||
| Fig. 35 Red phosphorus as an intermediate for organophosphorus synthesis. | ||
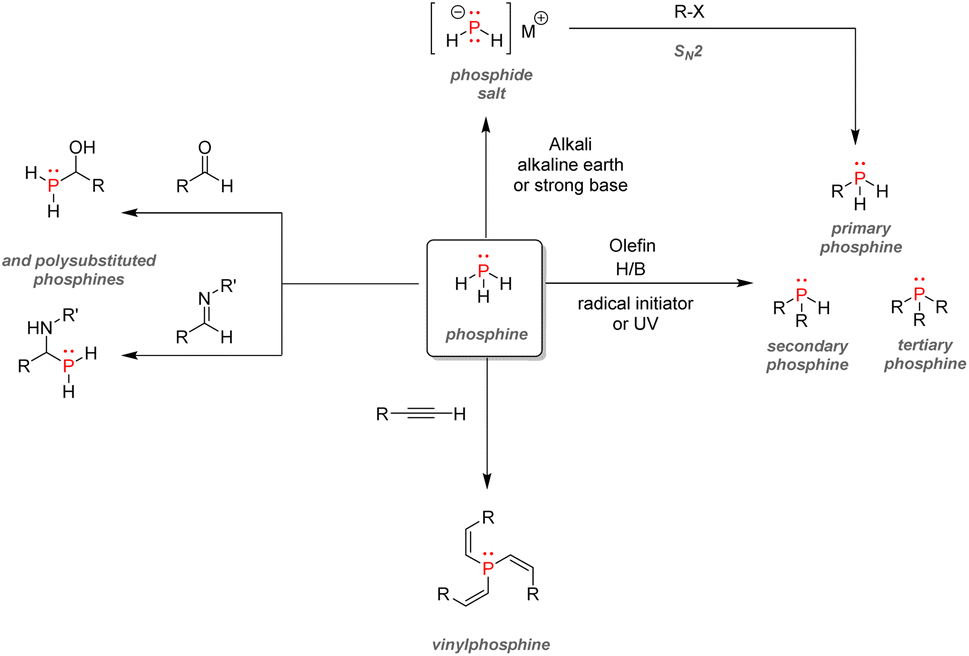 | ||
| Fig. 36 Phosphine as an intermediate for organophosphorus synthesis. | ||
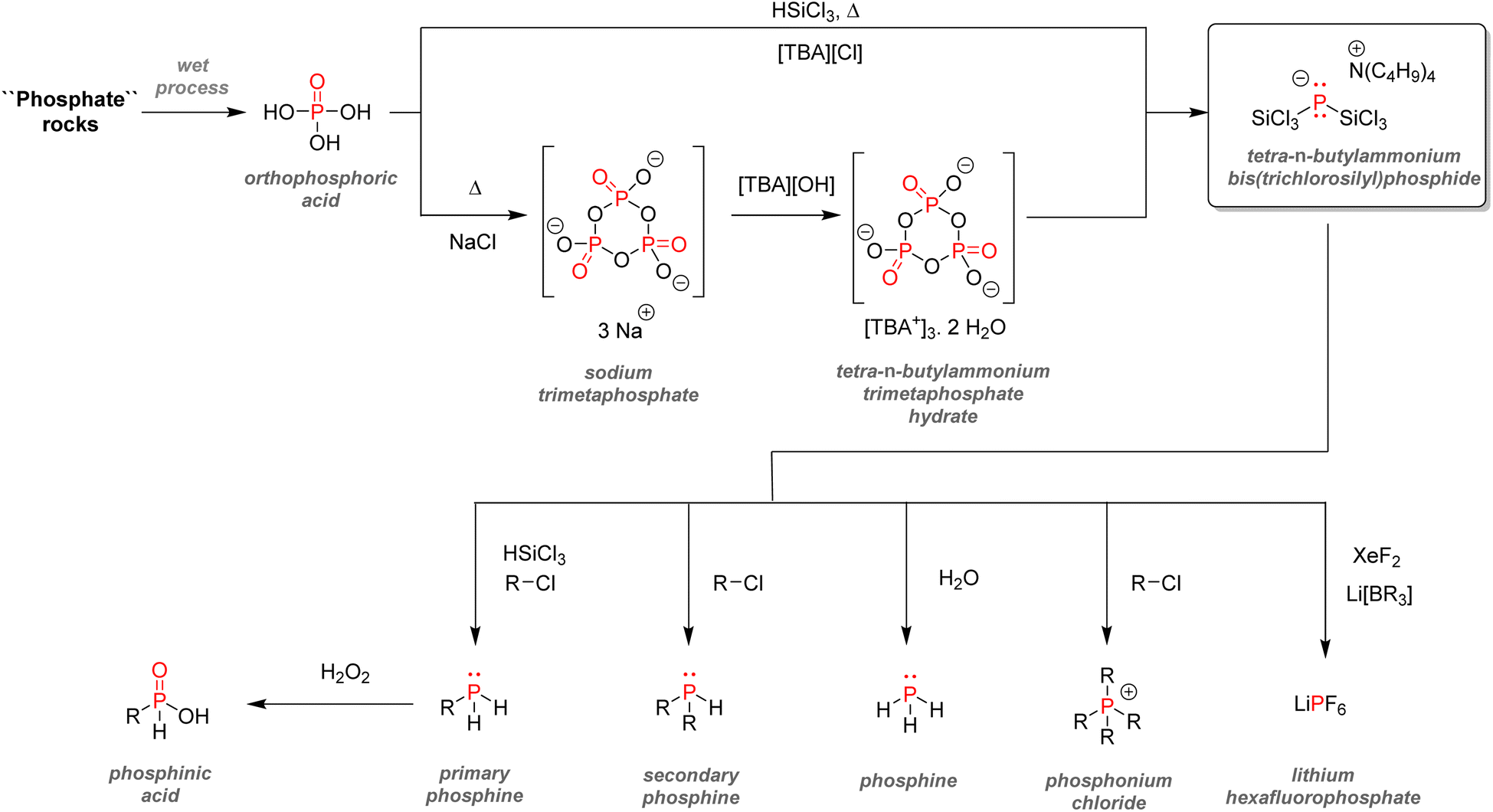 | ||
| Fig. 37 Bis(trichlorosilyl)phosphide salts as intermediates for organophosphorus synthesis. | ||
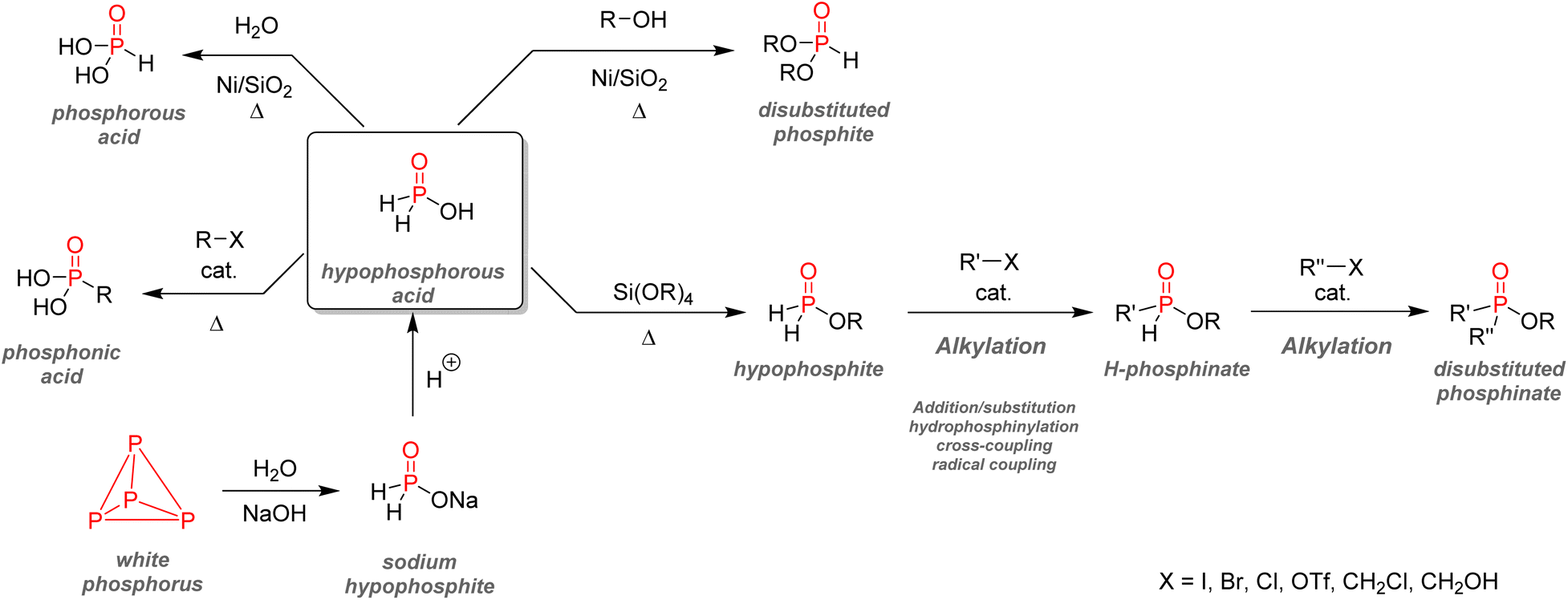 | ||
| Fig. 38 Hypophosphites as intermediates for organophosphorus synthesis. | ||
4. Conclusion
With such a rich diversity in their chemical properties and their wide range of applications in many significant processes, organophosphorus compounds are undoubtedly a valuable class of chemicals worthy of further research and development. Indeed, agriculture, flame retardants, therapeutics, or optoelectronic materials are all fields related in one way or another to the phosphorus industry. However, the latter presently relies on the extraction and transformation of fossil resources unequally distributed around the globe. Furthermore, the future scarcity of phosphate rocks during a potential peak phosphorus event will mechanically damage human activities and raise international tensions by making phosphorus a strategic commodity. As such, efforts to develop a circular economy for phosphorus should be promoted to make this resource less precarious, more sustainable and reduce excess phosphate runoff towards the ocean, which could endanger marine ecosystems and fishing activities. Another issue is linked to the specific chemical processes employed to functionalize organophosphorus: most synthetic routes depend on hazardous P–Cl containing building blocks. They also rely heavily on Grignard or organolithium alkylating agents and energetically costly transformations while creating potentially hazardous halogenated by-products. With the ongoing environmental concern and drive to develop more sustainable processes, a more holistic approach toward the organophosphorus industry is needed: develop safer building blocks and milder alternative procedures featuring a better atom economy and redox efficiency. The phosphide salt route, the hypophosphite route, and the future developments of more efficient P(V) to P(III) reduction or the direct functionalization of phosphoric acid are among the promising new pathways that navigate organophosphorus chemistry with a better redox economy and sustainability.Author contributions
Sosthène Ung wrote the review, Prof. C.-J. Li proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Prof. Damha, Prof. Tsantrizos, Prof. Moitessier from the department of Chemistry of McGill University and Prof. Virieux from École Nationale Supérieure de Chimie de Montpellier for their advice and guidance.References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- E. Gajewski, D. K. Steckler and R. N. Goldberg, J. Biol. Chem., 1986, 261, 12733–12737 CrossRef CAS PubMed.
- P. Cohen, Nat. Cell Biol., 2002, 4, E127–E130 CrossRef CAS PubMed.
- M. E. Weeks, J. Chem. Educ., 1933, 10, 302 CrossRef CAS.
- P. Gengembre, Mém. Mathématique Phys., 1783, 10, 651–658 Search PubMed.
- J. L. Gay-Lussac and L. J. Thénard, Gaz. Natl. Ou Monit. Univers., 1808, 40, 581–582 Search PubMed.
- M. F. Crass, J. Chem. Educ., 1941, 18, 277 CrossRef.
- J. P. W. Hughes, R. Baron, D. H. Buckland, M. A. Cooke, J. D. Craig, D. P. Duffield, A. W. Grosart, P. W. J. Parkes and A. Porter, Occup. Environ. Med., 1962, 19, 83–99 CAS.
- D. M. Ivell, Procedia Eng., 2012, 46, 166–171 CrossRef.
- J. B. Readman, US Pat., US417943A, p. 1889 Search PubMed.
- H. Rose, Ann. Chim. Phys., 1832, 51, 1–62 Search PubMed.
- A. W. Hofmann, Justus Liebigs Ann. Chem., 1857, 103, 357–358 CrossRef.
- A. Cahours and H. Gal, Justus Liebigs Ann. Chem., 1870, 155, 223–230 CrossRef.
- A. Cahours and H. Gal, Justus Liebigs Ann. Chem., 1870, 155, 355–359 CrossRef.
- G. A. Petroianu, Pharmazie, 2010, 4, 306–311 Search PubMed.
- B. A. Arbusow, Pure Appl. Chem., 1964, 9, 307–336 CrossRef.
- A. Michaelis and R. Kaehne, Ber. Dtsch. Chem. Ges., 1898, 31, 1048–1055 CrossRef.
- D. E. C. Corbridge, in Phosphorus: Chemistry, Biochemistry and Technology, Taylor & Francis, Boca Raton, 6th edn, 2013, p. 5 Search PubMed.
- F. Matsumura, in Toxicology of Insecticides, ed. F. Matsumura, Springer US, Boston, MA, 1985, pp. 45–109 Search PubMed.
- J. B. Tucker, War of Nerves: Chemical Warfare from World War I to Al-Qaeda, Anchor Books, First Anchor Books ed., New York, NY, 2007 Search PubMed.
- Chemical Weapons Convention of 1993, https://sites.fas.harvard.edu/%7Ehsp/cwc/cwcbyart.html, accessed 15 July 2020 Search PubMed.
- D. Darling, Terrestrial abundance of elements, https://www.daviddarling.info/encyclopedia/E/elterr.html, accessed 12 July 2020 Search PubMed.
- J. R. Rumble, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics: a Ready-Reference Book of Chemical and Physical Data, CRC Press, Boca Raton, 98th edn, 2017 Search PubMed.
- K. Barbalace, Periodic Table of Elements: Phosphorus – P, https://environmentalchemistry.com/yogi/periodic/P.html, accessed 12 July 2020 Search PubMed.
- I. Hanukoglu, List of Elements of the Periodic Table – Sorted by Abundance in Earth's crust, https://www.science.co.il/elements/?s=Earth, accessed 12 July 2020 Search PubMed.
- Z. Yuan, S. Jiang, H. Sheng, X. Liu, H. Hua, X. Liu and Y. Zhang, Environ. Sci. Technol., 2018, 52, 2438–2450 CrossRef CAS PubMed.
- M. Chen and T. E. Graedel, Glob. Environ. Change, 2016, 36, 139–152 CrossRef.
- D. Cordell, J.-O. Drangert and S. White, Glob. Environ. Change, 2009, 19, 292–305 CrossRef.
- D. Bernhardt and J. F. Reilly II, Mineral Commodity Summaries 2020, U.S. Geological Survey, Reston, VA, 2020 Search PubMed.
- K. Schrödter, G. Bettermann, T. Staffel, T. Klein and T. Hofmann, Phosphoric Acid and Phosphates, in Ullmann's Encyclopedia of Industrial Chemistry, American Chemical Society, 2000 Search PubMed.
- G. Villalba, Y. Liu, H. Schroder and R. U. Ayres, J. Ind. Ecol., 2008, 12, 557–569 CrossRef.
- Y. I. Asa, Fertilizer Industry Handbook 2018, 2018, p. 97 Search PubMed.
- H. Diskowski and T. Hofmann, Phosphorus. in Ullmann's Encyclopedia of Industrial Chemistry, American Chemical Society, 2000 Search PubMed.
- D. E. C. Corbridge, in Phosphorus: Chemistry, Biochemistry and Technology, Taylor & Francis, Boca Raton, 6th edn, 2013, pp. 93–94 Search PubMed.
- G. Bettermann, K. Werner, G. Riess and T. Hofmann, Phosphorus Compounds, Inorganic, in Ullmann's Encyclopedia of Industrial Chemistry, American Chemical Society, 2000 Search PubMed.
- N. N. Greenwood and A. Earnshaw, in Chemistry of the Elements, Butterworth-Heinemann, Oxford; Boston, 2nd edn, 1997, p. 495 Search PubMed.
- E. Wiberg, N. Wiberg and A. F. Holleman, Inorganic Chemistry, Academic Press; De Gruyter, San Diego: Berlin; New York, 1st edn, 2001 Search PubMed.
- J. Svara, N. Weferling and T. Hofmann, Phosphorus Compounds, Organic, in Ullmann's Encyclopedia of Industrial Chemistry, American Chemical Society, 2006 Search PubMed.
- L. M. E. Percival, D. P. G. Bond, M. Rakociński, L. Marynowski, A. v. S. Hood, T. Adatte, J. E. Spangenberg and K. B. Föllmi, Glob. Planet. Change, 2020, 184, 103070 CrossRef.
- D. J. Conley, S. Björck, E. Bonsdorff, J. Carstensen, G. Destouni, B. G. Gustafsson, S. Hietanen, M. Kortekaas, H. Kuosa, H. E. Markus Meier, B. Müller-Karulis, K. Nordberg, A. Norkko, G. Nürnberg, H. Pitkänen, N. N. Rabalais, R. Rosenberg, O. P. Savchuk, C. P. Slomp, M. Voss, F. Wulff and L. Zillén, Environ. Sci. Technol., 2009, 43, 3412–3420 CrossRef CAS PubMed.
- A. J. Watson, T. M. Lenton and B. J. W. Mills, Philos. Trans. R. Soc., A, 2017, 375, 2102 CrossRef PubMed.
- A. M. Michalak, E. J. Anderson, D. Beletsky, S. Boland, N. S. Bosch, T. B. Bridgeman, J. D. Chaffin, K. Cho, R. Confesor, I. Daloğlu, J. V. DePinto, M. A. Evans, G. L. Fahnenstiel, L. He, J. C. Ho, L. Jenkins, T. H. Johengen, K. C. Kuo, E. LaPorte, X. Liu, M. R. McWilliams, M. R. Moore, D. J. Posselt, R. P. Richards, D. Scavia, A. L. Steiner, E. Verhamme, D. M. Wright and M. A. Zagorski, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6448–6452 CrossRef CAS PubMed.
- J. Rockström, W. Steffen, K. Noone, Å. Persson, F. S. Chapin, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. J. Schellnhuber, B. Nykvist, C. A. de Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sörlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. A. Foley, Nature, 2009, 461, 472–475 CrossRef PubMed.
- A. R. Jupp, S. Beijer, G. C. Narain, W. Schipper and J. C. Slootweg, Chem. Soc. Rev., 2021, 50, 87–101 RSC.
- S. Marque and P. Tordo, Reactivity of Phosphorus Centered Radicals, Springer, Berlin Heidelberg, 2005 Search PubMed.
- A. C. Gaumont and J. M. Denis, Chem. Rev., 1994, 94, 1413–1439 CrossRef CAS.
- E. H. Krenske, K. N. Houk, B. A. Arndtsen and D. J. S. Cyr, J. Am. Chem. Soc., 2008, 130, 10052–10053 CrossRef CAS PubMed.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed.
- T. E. Gier, J. Am. Chem. Soc., 1961, 83, 1769–1770 CrossRef CAS.
- F. Mathey, in Modern Heterocyclic Chemistry, John Wiley & Sons, Ltd, 2011, pp. 2071–2116 Search PubMed.
- R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 3090–3093 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, Hoboken, New Jersey, 6th edn, 2014 Search PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC.
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Sato and A. Suzuki, J. Am. Chem. Soc., 1989, 111, 314–321 CrossRef CAS.
- S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544–4568 CrossRef CAS PubMed.
- M. R. Netherton, C. Dai, K. Neuschütz and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 10099–10100 CrossRef CAS PubMed.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS.
- T. Ohta, H. Takaya and R. Noyori, Inorg. Chem., 1988, 27, 566–569 CrossRef CAS.
- R. Noyori, M. Tokunaga and M. Kitamura, Bull. Chem. Soc. Jpn., 1995, 68, 36–55 CrossRef CAS.
- W. S. Knowles, Acc. Chem. Res., 1983, 16, 106–112 CrossRef CAS.
- S. Akutagawa, Appl. Catal., A, 1995, 128, 171–207 CrossRef CAS.
- D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, Amsterdam, Netherlands, 2007 Search PubMed.
- D. Morales-Morales, Pincer Compounds: Chemistry and Applications, Elsevier, Amsterdam, Netherlands, 2018 Search PubMed.
- R. A. Gossage and G. van Koten, The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, Springer International Publishing, Cham, 1st edn, 2016 Search PubMed.
- G. van Koten and D. Milstein, Organometallic Pincer Chemistry, Springer Berlin, Heidelberg, 2013 Search PubMed.
- K. J. Szabó and O. F. Wendt, Pincer and Pincer-type Complexes: Applications in Organic Synthesis and Catalysis, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef.
- M. Feller, C. Gunanathan, A. Kumar, R. Langer, M. Montag, T. Schaub, M. Vogt and T. Zell, David Milstein: Shaping Organometallic Catalysis Over Five Decades, ChemViews, 2022 Search PubMed.
- C. Li, J. Kan, Z. Qiu, J. Li, L. Lv and C. Li, Angew. Chem., Int. Ed., 2020, 59, 4544–4549 CrossRef CAS PubMed.
- M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC.
- H. Valdés, J. M. Germán-Acacio, G. van Koten and D. Morales-Morales, Dalton Trans., 2022, 51, 1724–1744 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- V. V. Grushin, Chem. Rev., 2004, 104, 1629–1662 CrossRef CAS PubMed.
- T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771–803 CrossRef CAS.
- L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley, New York, 2000 Search PubMed.
- D. B. Chesnut, J. Phys. Chem. A, 2003, 107, 4307–4313 CrossRef CAS.
- D. B. Chesnut and A. Savin, J. Am. Chem. Soc., 1999, 121, 2335–2336 CrossRef CAS.
- A. E. Reed and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 1434–1445 CrossRef CAS.
- E. N. Tsvetkov and A. A. Korkin, Theor. Exp. Chem., 1985, 21, 36–42 CrossRef.
- K. M. Diemoz and A. K. Franz, J. Org. Chem., 2019, 84, 1126–1138 CrossRef CAS PubMed.
- B. Klabuhn, H. Goetz, P. Steirl and D. Alscher, Tetrahedron, 1976, 32, 603–607 CrossRef CAS.
- A. W. Johnson and H. L. Jones, J. Am. Chem. Soc., 1968, 90, 5232–5236 CrossRef CAS.
- K. Gluza and P. Kafarski, in Drug Discovery, ed. H. El-Shemy, InTech, 2013 Search PubMed.
- P. Finkbeiner, J. P. Hehn and C. Gnamm, J. Med. Chem., 2020, 63, 7081–7107 CrossRef CAS PubMed.
- A. Emadi, R. J. Jones and R. A. Brodsky, Nat. Rev. Clin. Oncol., 2009, 6, 638–647 CrossRef CAS PubMed.
- W. H. Organization, Model List of Essential Medicines: 21st List, 2019, https://www.who.int/publications/i/item/WHOMVPEMPIAU2019.06, accessed 2020-07-31 Search PubMed.
- A. C. Dijkmans, N. V. O. Zacarías, J. Burggraaf, J. W. Mouton, E. Wilms, C. van Nieuwkoop, D. J. Touw, J. Stevens and I. M. C. Kamerling, Antibiotics, 2017, 6, 24 CrossRef PubMed.
- E. Helgstrand, B. Eriksson, N. G. Johansson, B. Lannero, A. Larsson, A. Misiorny, J. O. Noren, B. Sjoberg, K. Stenberg, G. Stening, S. Stridh and B. Oberg, Science, 1978, 201, 819–821 CrossRef CAS PubMed.
- A. Canestri, J. Ghosn, M. Wirden, F. Marguet, N. Ktorza, I. Boubezari, S. Dominguez, P. Bossi, E. Caumes, V. Calvez and C. Katlama, Antiviral Ther., 2006, 11, 561–566 CrossRef CAS.
- E. M. Lewiecki, Ther. Adv. Chronic Dis., 2010, 1, 115–128 CrossRef CAS PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef CAS PubMed.
- K. S. E. Tanaka, E. Dietrich, S. Ciblat, C. Métayer, F. F. Arhin, I. Sarmiento, G. Moeck, T. R. Parr and A. R. Far, Bioorg. Med. Chem. Lett., 2010, 20, 1355–1359 CrossRef CAS PubMed.
- B. Badet, K. Inagaki, K. Soda and C. T. Walsh, Time-dependent inhibition of Bacillus stearothermophilus alanine racemase by (1-aminoethyl)phosphonate isomers by isomerization to noncovalent slowly dissociating enzyme-(1-aminoethyl)phosphonate complexes, https://pubs.acs.org/doi/pdf/10.1021/bi00359a029, accessed 31 July 2020 Search PubMed.
- J. T. Whitteck, W. Ni, B. M. Griffin, A. C. Eliot, P. M. Thomas, N. L. Kelleher, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2007, 46, 9089–9092 CrossRef CAS PubMed.
- J. Krapcho, C. Turk, D. W. Cushman, J. R. Powell, J. M. DeForrest, E. R. Spitzmiller, D. S. Karanewsky, M. Duggan and G. Rovnyak, J. Med. Chem., 1988, 31, 1148–1160 CrossRef CAS PubMed.
- E. Skordalakes, G. G. Dodson, D. S. C. Green, C. A. Goodwin, M. F. Scully, H. R. Hudson, V. V. Kakkar and J. J. Deadman, J. Mol. Biol., 2001, 311, 549–555 CrossRef CAS PubMed.
- R. Bone, N. S. Sampson, P. A. Bartlett and D. A. Agard, Biochemistry, 1991, 30, 2263–2272 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- T. Usui, Y. Oiso, A. Tomita, E. Ogata, T. Uchida, K. Ikeda, T. Watanabe and S. Higuchi, Int. J. Clin. Pharmacol. Ther., 1997, 35, 239–244 CAS.
- C. M. Shipman, P. I. Croucher, R. G. Russell, M. H. Helfrich and M. J. Rogers, Cancer Res., 1998, 58, 5294–5297 CAS.
- P. Anderson, Expert Opin. Pharmacother., 2006, 7, 1475–1486 CrossRef CAS PubMed.
- M. Fellner, R. P. Baum, V. Kubíček, P. Hermann, I. Lukeš, V. Prasad and F. Rösch, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 834 CrossRef PubMed.
- E. D. Clercq, Nat. Rev. Microbiol., 2004, 2, 704–720 CrossRef PubMed.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648–1661 CrossRef CAS PubMed.
- R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Cent. Sci., 2020, 6, 672–683 CrossRef CAS PubMed.
- A. Fung, Z. Jin, N. Dyatkina, G. Wang, L. Beigelman and J. Deval, Antimicrob. Agents Chemother., 2014, 58, 3636–3645 CrossRef PubMed.
- S. Safrin, J. Cherrington and H. S. Jaffe, Rev. Med. Virol., 1997, 7, 145–156 CrossRef CAS PubMed.
- C. P. Holstege, M. Kirk and F. R. Sidell, Crit. Care Clin., 1997, 13, 923–942 CrossRef CAS PubMed.
- F. Matsumura, Toxicology of Insecticides, Springer US, Boston, MA, 1985 Search PubMed.
- R. L. Metcalf (deceased) and A. R. Horowitz, Insect Control, 2. Individual Insecticides, in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2014, pp. 1–94 Search PubMed.
- D. Atwood and C. Paisley-Jones, Pesticides Industry Sales and Usage 2008-2012 Market Estimates, U.S. environmental Protection Agency, 2017, https://www.epa.gov/pesticides/pesticides-industry-sales-and-usage-2008-2012-market-estimates, accessed 2020-07-31 Search PubMed.
- R. L. Metcalf, Insect Control, in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2000 Search PubMed.
- M. Eddleston, N. A. Buckley, P. Eyer and A. H. Dawson, Lancet, 2008, 371, 597–607 CrossRef CAS.
- S. Berg and E. A. Bittner, The MGH Review of Critical Care Medicine, Lippincott Williams & Wilkins, 2013 Search PubMed.
- M. A. Dar, G. Kaushik and J. F. Villareal Chiu, in Abatement of Environmental Pollutants, ed. P. Singh, A. Kumar and A. Borthakur, Elsevier, 2020, pp. 25–66 Search PubMed.
- F. Müller and A. P. Applebyki, Weed Control, 2. Individual Herbicides. in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2011 Search PubMed.
- S. O. Duke and S. B. Powles, Pest Manag. Sci., 2008, 64, 319–325 CrossRef CAS PubMed.
- C. Tomlin, The Pesticide Manual: a World Compendium, British Crop Protection Council, Alton, Hampshire, 15th edn, 2009 Search PubMed.
- B. K. Singh and A. Walker, FEMS Microbiol. Rev., 2006, 30, 428–471 CrossRef CAS PubMed.
- S. M. Bradberry, A. T. Proudfoot and J. A. Vale, Toxicol. Rev., 2004, 23, 159–167 CrossRef CAS PubMed.
- D. Cressey, Widely used herbicide linked to cancer, Nat. News, 2015 DOI:10.1038/nature.2015.17181.
- L. N. Vandenberg, B. Blumberg, M. N. Antoniou, C. M. Benbrook, L. Carroll, T. Colborn, L. G. Everett, M. Hansen, P. J. Landrigan, B. P. Lanphear, R. Mesnage, F. S. vom Saal, W. V. Welshons and J. P. Myers, J. Epidemiol. Community Health, 2017, 71, 613–618 CrossRef PubMed.
- S. O. Duke, Weed Sci., 2020, 68, 201–207 CrossRef.
- D. Schwartz, S. Berger, E. Heinzelmann, K. Muschko, K. Welzel and W. Wohlleben, Appl. Environ. Microbiol., 2004, 70, 7093–7102 CrossRef CAS PubMed.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Appl. Phys. Lett., 2009, 94, 013301 CrossRef.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Mater., 2010, 22, 1872–1876 CrossRef CAS PubMed.
- S. Shao, J. Ding, T. Ye, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 3570–3574 CrossRef CAS PubMed.
- C. Liu, Y. Li, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2013, 25, 3320–3327 CrossRef CAS.
- C. Liu, Y. Gu, Q. Fu, N. Sun, C. Zhong, D. Ma, J. Qin and C. Yang, Chem.–Eur. J., 2012, 18, 13828–13835 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 20290–20293 CrossRef CAS PubMed.
- S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233–4243 RSC.
- N. Lin, J. Qiao, L. Duan, H. Li, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 19451–19457 CrossRef CAS.
- N. Lin, J. Qiao, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2014, 118, 7569–7578 CrossRef CAS.
- M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- C.-H. Lin, C.-W. Hsu, J.-L. Liao, Y.-M. Cheng, Y. Chi, T.-Y. Lin, M.-W. Chung, P.-T. Chou, G.-H. Lee, C.-H. Chang, C.-Y. Shih and C.-L. Ho, J. Mater. Chem., 2012, 22, 10684–10694 RSC.
- D. Joly, D. Tondelier, V. Deborde, B. Geffroy, M. Hissler and R. Réau, New J. Chem., 2010, 34, 1603–1611 RSC.
- Y. Matano, Y. Hayashi, H. Nakano and H. Imahori, Heteroat. Chem., 2014, 25, 533–547 CrossRef CAS.
- A. Kira, Y. Shibano, S. Kang, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2010, 39, 448–450 CrossRef CAS.
- C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710–11717 CrossRef CAS PubMed.
- M. Stolar, J. Borau-Garcia, M. Toonen and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 3366–3371 CrossRef CAS PubMed.
- C. Reus and T. Baumgartner, Dalton Trans., 2016, 45, 1850–1855 RSC.
- X.-D. Jiang, J. Zhao, D. Xi, H. Yu, J. Guan, S. Li, C.-L. Sun and L.-J. Xiao, Chem.–Eur. J., 2015, 21, 6079–6082 CrossRef CAS PubMed.
- X. Chai, X. Cui, B. Wang, F. Yang, Y. Cai, Q. Wu and T. Wang, Chem.–Eur. J., 2015, 21, 16754–16758 CrossRef CAS PubMed.
- A. Fukazawa, S. Suda, M. Taki, E. Yamaguchi, M. Grzybowski, Y. Sato, T. Higashiyama and S. Yamaguchi, Chem. Commun., 2016, 52, 1120–1123 RSC.
- H. Ogasawara, M. Grzybowski, R. Hosokawa, Y. Sato, M. Taki and S. Yamaguchi, Chem. Commun., 2018, 54, 299–302 RSC.
- P. Hindenberg, A. López-Andarias, F. Rominger, A. de Cózar and C. Romero-Nieto, Chem.–Eur. J., 2017, 23, 13919–13928 CrossRef CAS PubMed.
- E. Regulska and C. Romero-Nieto, Dalton Trans., 2018, 47, 10344–10359 RSC.
- Market Report Flame Retardants: Industry Analysis, Trends, https://www.ceresana.com/en/market-studies/chemicals/flame-retardants/, accessed 12 August 2020 Search PubMed.
- A. Innes and J. Innes, in Applied Plastics Engineering Handbook, ed. M. Kutz, William Andrew Publishing, Oxford, 2011, pp. 469–485 Search PubMed.
- S. Shaw, Rev. Environ. Health, 2010, 25(10), 261–305 CAS.
- B. Schartel, Materials, 2010, 3, 4710–4745 CrossRef CAS PubMed.
- E. D. Weil and S. V. Levchik, Phosphorus Flame Retardants, in Kirk-Othmer Encyclopedia of Chemical Technology, American Cancer Society, 2017, pp. 1–34 Search PubMed.
- F. Çelebi, O. Polat, L. Aras, G. Gündüz and I. M. Akhmedov, J. Appl. Polym. Sci., 2004, 91, 1314–1321 CrossRef.
- M. Rakotomalala, S. Wagner and M. Döring, Materials, 2010, 3, 4300–4327 CrossRef CAS PubMed.
- J. Zheng, Y. Yu, L. Zhang, X. Zhen and Y. Zhao, Aust. J. Chem., 2014, 67, 1688–1692 CrossRef CAS.
- J. Zheng, X. Li, Y. Yu, X. Feng and Y. Zhao, J. Therm. Anal. Calorim., 2014, 117, 319–324 CrossRef CAS.
- A. Blum, M. Behl, L. S. Birnbaum, M. L. Diamond, A. Phillips, V. Singla, N. S. Sipes, H. M. Stapleton and M. Venier, Environ. Sci. Technol. Lett., 2019, 6, 638–649 CrossRef CAS PubMed.
- J. Alongi, F. Cuttica and F. Carosio, ACS Sustainable Chem. Eng., 2016, 4, 3544–3551 CrossRef CAS.
- J. Alongi, A. Di Blasio, J. Milnes, G. Malucelli, S. Bourbigot, B. Kandola and G. Camino, Polym. Degrad. Stab., 2015, 113, 110–118 CrossRef CAS.
- J. Alongi, F. Carosio and G. Malucelli, Polym. Degrad. Stab., 2014, 106, 138–149 CrossRef CAS.
- M. Rahman and C. S. Brazel, Prog. Polym. Sci., 2004, 29, 1223–1248 CrossRef CAS.
- P. Walters, D. F. Cadogan and C. J. Howick, Plasticizers, in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2020, pp. 1–27 Search PubMed.
- M. Ehrich and B. S. Jortner, in Hayes' Handbook of Pesticide Toxicology, ed. R. Krieger, Academic Press, New York, 3rd edn, 2010, pp. 1479–1504 Search PubMed.
- D. N. Brooke, M. J. Crookes, P. Quarterman and J. Burns, J. Environmental Risk Evaluation Report: 2-Ethylhexyl Diphenyl Phosphate (CAS No. 1241-94-7), UK Environment Agency, 2009 Search PubMed.
- M. Rahman and C. S. Brazel, Polym. Degrad. Stab., 2006, 91, 3371–3382 CrossRef CAS.
- A. E. Lemire, A. F. Janzen and K. Marat, Inorg. Chim. Acta, 1985, 110, 237–241 CrossRef CAS.
- C. A. Blake, D. J. Crousse, C. F. Coleman, K. B. Brown and A. D. Kelmers, Progress report: Further studies of the dialkylphosphoric acid extraction (DAPEX) process for uranium, Oak Ridge National Laboratory, 1957 Search PubMed.
- J. R. Kumar, J.-S. Kim, J.-Y. Lee and H.-S. Yoon, Sep. Purif. Rev., 2011, 40, 77–125 CrossRef CAS.
- C. V. S. B. Rao, T. G. Srinivasan and P. R. V. Rao, Solvent Extr. Ion Exch., 2010, 28, 202–224 CrossRef CAS.
- D. Beltrami, G. Cote, H. Mokhtari, B. Courtaud, B. A. Moyer and A. Chagnes, Chem. Rev., 2014, 114, 12002–12023 CrossRef CAS PubMed.
- G. Koch, S. Träger, A. Max, W.-D. Krebs, W. Stoll, W. Heit, E. Warnecke, P. Brennecke and E. Merz, Nuclear Technology, 3. Fuel Cycle, in Ullmann's Encyclopedia of Industrial Chemistry, American Cancer Society, 2011 Search PubMed.
- J. S. Preston, P. M. Cole, W. M. Craig and A. M. Feather, Hydrometallurgy, 1996, 41, 1–19 CrossRef CAS.
- X. Huang, J. Li, Z. Long, Y. Zhang, X. Xue and Z. Zhu, J. Rare Earths, 2008, 26, 410–413 CrossRef.
- K. Yoshizuka, H. Kosaka, T. Shinohara, K. Ohto and K. Inoue, Bull. Chem. Soc. Jpn., 1996, 69, 589–596 CrossRef CAS.
- X. Wang, W. Li, S. Meng and D. Li, J. Chem. Technol. Biotechnol., 2006, 81, 761–766 CrossRef CAS.
- E. Jorjani and M. Shahbazi, Arabian J. Chem., 2016, 9, S1532–S1539 CrossRef CAS.
- O. Klimchuk, L. Atamas, S. Miroshnichenko, V. Kalchenko, I. Smirnov, V. Babain, A. Varnek and G. Wipff, J. Inclusion Phenom. Macrocyclic Chem., 2004, 49, 47–56 CrossRef CAS.
- L. Chen, Y. Wang, X. Yuan, Y. Ren, N. Liu, L. Yuan and W. Feng, Sep. Purif. Technol., 2018, 192, 152–159 CrossRef CAS.
- K. Kiegiel, L. Steczek and G. Zakrzewska-Trznadel, Application of Calixarenes as Macrocyclic Ligands for Uranium(vi), https://www.hindawi.com/journals/jchem/2013/762819/, accessed 14 August 2020 Search PubMed.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- M. B. Geeson and C. C. Cummins, ACS Cent. Sci., 2020, 6, 848–860 CrossRef CAS PubMed.
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed.
- R. Engel, C–P bond formation using nucleophilic trivalent phosphorus reagents, in Synthesis of Carbon–Phosphorus Bonds, CRC Press, Boca Raton, 2003 Search PubMed.
- M. Caporali, M. Serrano-Ruiz and M. Peruzzini, in Chemistry beyond Chlorine, ed. P. Tundo, L.-N. He, E. Lokteva and C. Mota, Springer International Publishing, Cham, 2016, pp. 97–136 Search PubMed.
- J. E. Borger, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Chem.–Eur. J., 2017, 23, 11738–11746 CrossRef CAS PubMed.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- R. Engel, Synthesis of Carbon–Phosphorus Bonds, CRC Press, Boca Raton, 2003 Search PubMed.
- S. Hörold, N. Weferling and H. P. Breuer, Eur. Pat., EP0969008A2, 2000 Search PubMed.
- N. Weferling, O. Stelzer, G. Kolbe, Can. Pat., CA2311950A1, 1999 Search PubMed.
- M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 471–473 CrossRef CAS.
- M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 473–477 CrossRef CAS.
- D. G. Yakhvarov, Y. S. Ganushevich and O. G. Sinyashin, Mendeleev Commun., 2007, 17, 197–198 CrossRef CAS.
- K. M. Armstrong and P. Kilian, Eur. J. Inorg. Chem., 2011, 2011, 2138–2147 CrossRef.
- D. H. R. Barton and J. Zhu, J. Am. Chem. Soc., 1993, 115, 2071–2072 CrossRef CAS.
- S. K. Ghosh, C. C. Cummins and J. A. Gladysz, Org. Chem. Front., 2018, 5, 3421–3429 RSC.
- P. Barbaro, A. Ienco, C. Mealli, M. Peruzzini, O. J. Scherer, G. Schmitt, F. Vizza and G. Wolmershäuser, Chem.–Eur. J., 2003, 9, 5195–5210 CrossRef CAS PubMed.
- D. G. Yakhvarov, E. V. Gorbachuk and O. G. Sinyashin, Eur. J. Inorg. Chem., 2013, 2013, 4709–4726 CrossRef CAS.
- Y. H. Budnikova, D. G. Yakhvarov and O. G. Sinyashin, J. Organomet. Chem., 2005, 690, 2416–2425 CrossRef CAS.
- Y. H. Budnikova, S. A. Krasnov, T. V. Graznova, A. P. Tomilov, V. V. Turigin, I. M. Magdeev and O. G. Sinyashin, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 513–518 CrossRef CAS.
- M. Pecht and Y. Deng, Microelectron. Reliab., 2006, 46, 53–62 CrossRef CAS.
- U. Braun and B. Schartel, Macromol. Chem. Phys., 2004, 205, 2185–2196 CrossRef CAS.
- Y. Zhu, J. Ren, X. Zhang and D. Yang, Nanoscale, 2020, 12, 13297–13310 RSC.
- B. Trofimov, N. Gusarova and L. Brandsma, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 109, 601–604 CrossRef.
- N. K. Gusarova, S. I. Shaikhudinova, A. M. Reutskaya, N. I. Ivanova, A. A. Tatarinova and B. A. Trofimov, Russ. Chem. Bull., 2000, 49, 1320 CrossRef CAS.
- T. M. Wrodnigg, A. E. Stütz and S. G. Withers, Tetrahedron Lett., 1990, 31, 5463–5466 CrossRef.
- M. C. J. M. V. Hooijdonk, G. Gerritsen and L. Brandsma, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 162, 39–49 CrossRef.
- L. Brandsma, S. Arbuzova, R.-J. D. Lang, N. Gusarova and B. Trofimov, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 126, 125–128 CrossRef CAS.
- D. Albouy, G. Etemad-Moghadam and M. Koenig, Eur. J. Org. Chem., 1999, 1999, 861–868 CrossRef.
- N. K. Gusarova, S. N. Arbuzova and B. A. Trofimov, Pure Appl. Chem., 2012, 84, 439–459 CAS.
- B. A. Trofimov, S. N. Arbuzova and N. K. Gusarova, Russ. Chem. Rev., 1999, 68, 215 CrossRef CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Green Chem., 2012, 14, 2699–2702 RSC.
- M. M. Rauhut, H. A. Currier, A. M. Semsel and V. P. Wystrach, J. Org. Chem., 1961, 26, 5138–5145 CrossRef CAS.
- R. H. Hall and H. G. Khorana, J. Am. Chem. Soc., 1954, 76, 5056–5060 CrossRef CAS.
- M. Smith and H. G. Khorana, J. Am. Chem. Soc., 1958, 80, 1141–1145 CrossRef CAS.
- R. F. Long and A. L. Morrison, J. Chem. Soc., 1954, 3854–3856 RSC.
- E. Cherbuliez and J.-P. Leber, Helv. Chim. Acta, 1952, 35, 644–664 CrossRef CAS.
- E. Cherbuliez and J. Rabinowitz, Helv. Chim. Acta, 1958, 41, 1168–1175 CrossRef CAS.
- E. Cherbuliez and J. Rabinowitz, Helv. Chim. Acta, 1956, 39, 1455–1461 CrossRef CAS.
- F. D. Popp and W. E. McEwen, Chem. Rev., 1958, 58, 321–401 CrossRef CAS.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383–1385 CrossRef CAS PubMed.
- D. Pham Minh, J. Ramaroson, A. Nzihou and P. Sharrock, Ind. Eng. Chem. Res., 2012, 51, 3851–3854 CrossRef CAS.
- C. J. Besecker, V. W. Day and W. G. Klemperer, Organometallics, 1985, 4, 564–570 CrossRef CAS.
- M. B. Geeson, P. Ríos, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 6375–6384 CrossRef CAS PubMed.
- R. Gilmour, Phosphorus Compounds, in Kirk-Othmer Encyclopedia of Chemical Technology, American Cancer Society, 2019, pp. 1–57 Search PubMed.
- S. Deprèle and J.-L. Montchamp, J. Organomet. Chem., 2002, 643–644, 154–163 CrossRef.
- E. E. Nifant'ev and L. P. Levitan, J. Gen. Chem. USSR, 1965, 35, 762 Search PubMed.
- J.-L. Montchamp, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 66–75 CrossRef CAS.
- J.-L. Montchamp, Pure Appl. Chem., 2019, 91, 113–120 CrossRef CAS.
- J.-L. Montchamp, J. Organomet. Chem., 2005, 690, 2388–2406 CrossRef CAS.
- H. C. Fisher, L. Prost and J.-L. Montchamp, Eur. J. Org. Chem., 2013, 2013, 7973–7978 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |