
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-Alzheimer activity of new coumarin-based derivatives targeting acetylcholinesterase inhibition†
Nahla N. Kamela,
Hanan F. Aly*a,
Ghadha I. Fouad a,
Somaia S. Abd El-Karima,
Manal M. Anwara,
Yasmin M. Syama,
Samia A. Elseginyb,
Kawkab A. Ahmedc,
Hoda F. Boolesd,
Mohamed B. Shalabye,
Wagdy K. B. Khalild,
Rajat Sandhirf,
Sonam Deshwalf and
Maha Z. Rizka
a,
Somaia S. Abd El-Karima,
Manal M. Anwara,
Yasmin M. Syama,
Samia A. Elseginyb,
Kawkab A. Ahmedc,
Hoda F. Boolesd,
Mohamed B. Shalabye,
Wagdy K. B. Khalild,
Rajat Sandhirf,
Sonam Deshwalf and
Maha Z. Rizka
aDepartment of Therapeutic Chemistry, National Research Centre, 12262 El-Bohouth St, Cairo, Egypt. E-mail: Hanan_abduallah@yahoo.com
bGreen Chemistry Department, Chemical Industries Research Division, National Research Centre, P. O. Box 12622, Egypt
cPathology Departments, Faculty of Veterinary Medicine, Cairo University, Giza, 12211, Egypt
dDepartment of Cell Biology, National Research Centre, 12262 El-Bohouth St, Cairo, Egypt
eToxicology Research Department, Research Institute of Medical Entomology (RIME), General Organization of Teaching Hospitals and Institutes (GOTHI), Ministry of Health and Population (MoHP), Dokki, P. O. Box 12311, Cairo, Egypt
fDepartment of Biochemistry, Panjab University, Chandigarh, India
First published on 20th June 2023
Abstract
New 2-oxo-chromene-7-oxymethylene acetohydrazide derivatives 4a–d were designed and synthesized with a variety of bioactive chemical fragments. The newly synthesized compounds were evaluated as acetylcholinesterase (AChE) inhibitors and antioxidant agents in comparison to donepezil and ascorbic acid, respectively. Compound 4c exhibited a promising inhibitory impact with an IC50 value of 0.802 μM and DPPH scavenging activity of 57.14 ± 2.77%. Furthermore, biochemical and haematological studies revealed that compound 4c had no effect on the blood profile, hepatic enzyme levels (AST, ALT, and ALP), or total urea in 4c-treated rats compared to the controls. Moreover, the histopathological studies of 4c-treated rats revealed the normal architecture of the hepatic lobules and renal parenchyma, as well as no histopathological damage in the examined hepatic, kidney, heart, and brain tissues. In addition, an in vivo study investigated the amelioration in the cognitive function of AD-rats treated with 4c through the T-maze and beam balance behavioural tests. Also, 4c detectably ameliorated MDA and GSH, reaching 90.64 and 27.17%, respectively, in comparison to the standard drug (90.64% and 35.03% for MDA and GSH, respectively). The molecular docking study exhibited a good fitting of compound 4c in the active site of the AChE enzyme and a promising safety profile. Compound 4c exhibited a promising anti-Alzheimer's disease efficiency compared to the standard drug donepezil.
1. Introduction
Alzheimer's disease (AD) is one of the most common forms of dementia occurring in the elderly population worldwide, and is the most common fatal age-related neurodegenerative disease, characterized by a decline of cholinergic function, progressive loss of brain function, and memory decline. Approximately 13% of people over the age of 65 and 45% of people over the age of 85 are estimated to have AD.1 According to mounting evidence from genetic, pathological, and functional studies, there is an imbalance between the production and clearance of amyloid β (Aβ) peptides in the brain, resulting in their accumulation and aggregation. The toxic Aβ aggregates in the form of soluble Aβ oligomers, intraneuronal Aβ, and amyloid plaques injure synapses and ultimately cause neurodegeneration and dementia.2 The toxicity of Aβ seems to depend on the presence of the microtubule-associated protein tau, the hyperphosphorylated forms of which aggregate and deposit in AD brains as neurofibrillary tangles and prevent the passage of essential molecules and nutrients.3 This causes axonal transport dysfunction and neuronal loss.4 Moreover, theories such as the neurotoxicity of the excitatory neurotransmitters, altered insulin signaling, oxidative stress, and inflammation can also explain the causes of dementia and provide a new theoretical basis and therapeutic target for the development of novel drugs for AD.5 Acetylcholine esterase (AChE) is the most vital enzyme that maintains the balance of acetylcholine levels. Many studies have suggested a relationship between learning and memory functions and AChE activity in experimental animals.6 Thus, the use of acetylcholinesterase (AChE) inhibitors like rivastigmine or dopenzil that increase the availability of acetylcholine at cholinergic synapses represents a major therapeutic approach to the disease.7 Literature survey showed that the coumarin nucleus, also known as 2H-1-benzopyran-2-one constitutes the main structural component of many natural products and synthetic compounds with a wide range of biological activity. Coumarins are intriguing compounds for drug discovery in the arena of AChEIs because of the possibility of chemical substitutions at numerous places in this core structure.8 The coumarin moiety's potential to inhibit cholinesterase enzymes has been demonstrated in recent in vitro assays. Furthermore, the coumarin nucleus has a number of biological effects on various aspects of Alzheimer's disease, in addition to AChE suppression, including inhibition of the enzyme secretase-1 (BACE-1), cyclooxygenase (COX)/lipoxygenase (LOX) antagonists, antagonists of the cannabinoid receptor 2 (CB2), agonists of the gamma amino butyric acid (GABA) receptor, antagonists of the NMDA receptor, and monoamineoxidase (MAO) inhibitors.9–12 Many coumarin natural products are have anti-Alzheimer activity, such as esculetin I, decursinol II, scopoletin III, and mesuagenin IV.13Docking studies using 2H-chromen-2-one demonstrate this nucleus's ability to attach to the peripheral anionic sites (PAS) of both isoforms of cholinesterase, simulating the enzyme's natural substrate. Because of this value, medicinal chemists working on medicines for AD have viewed coumarin as a preferred scaffold for combining with other pharmacophores that can interact with different targets.14 It has been investigated that the use of benzyloxy group at position 7 of the coumarin scaffold has a significant impact on AChE inhibitory activity, as seen in compounds V, VI,15 and VII.16 In addition, AChE inhibitory activity may have increased as a result of the 4-fluorophenyl ring forming π–π stacking with Trp86, as revealed by a closer investigation of compound VII's binding mechanism.16 Additionally, studies showed that the coumarin's substitution at position 4 emphasizes its AChE inhibitory activity, which is found in compounds VIII,17 and IX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 (Fig. 1).
18 (Fig. 1).
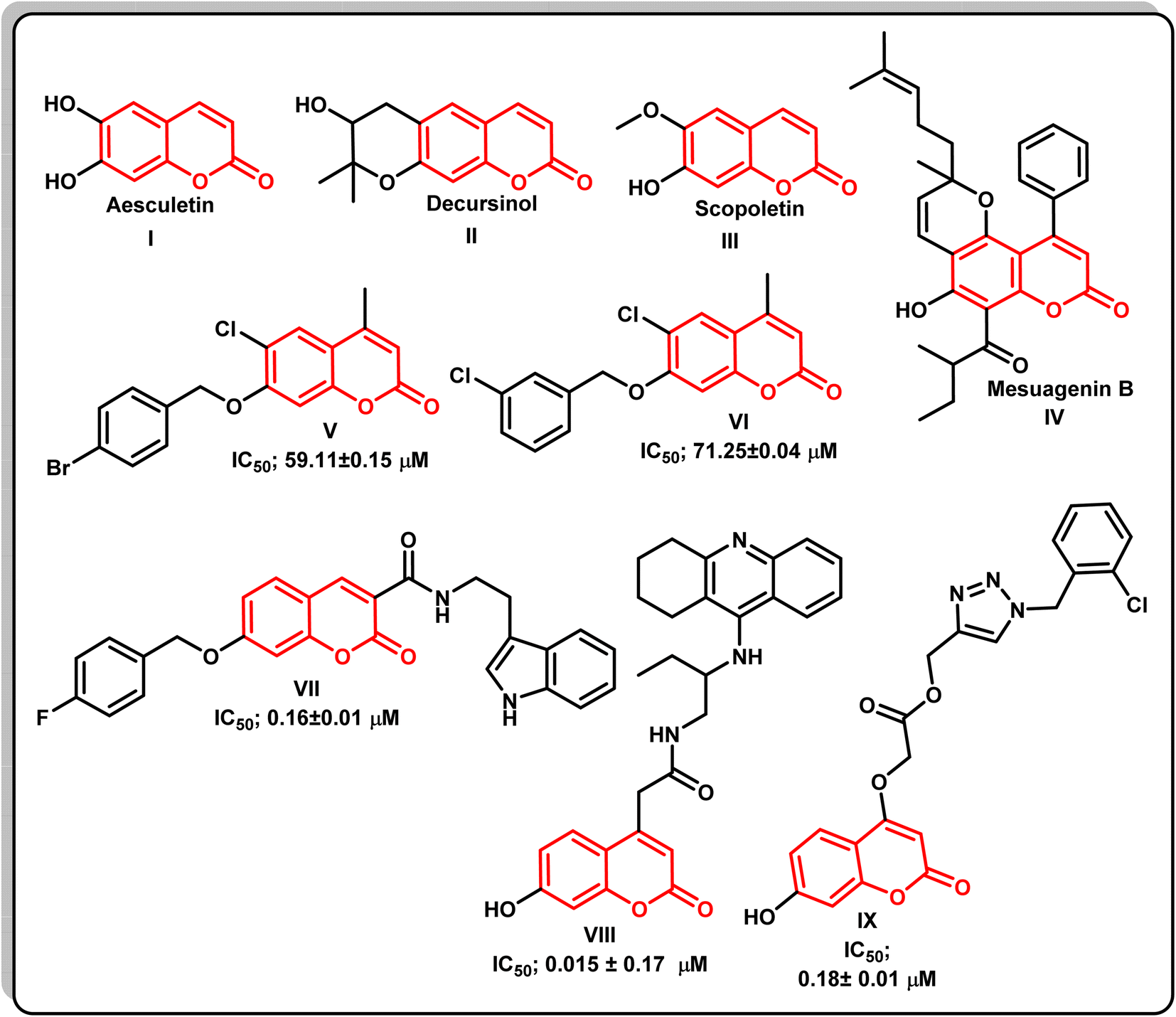 | ||
| Fig. 1 The chemical structures of various naturally occurring and synthetic coumarin compounds with AChE inhibitory activity. | ||
Moreover, different studies exhibited that numerous AChEIs have various aromatic or heterocylic rings conjugated with coumarin cores via nitrogenous linkers, which were intended to stabilize the molecules within the gorge of the AChE enzyme.19 Thus, it was thought valuable to present new compounds as promising potent AChEIs, having the coumarin scaffold conjugated with different distinct aromatic and/or heterocycles at position 7 via an appropriate oxyacetohydrazide linker, in order to gain new compounds with potent AChE inhibitory activity and a safe profile for AD treatment (Fig. 2).
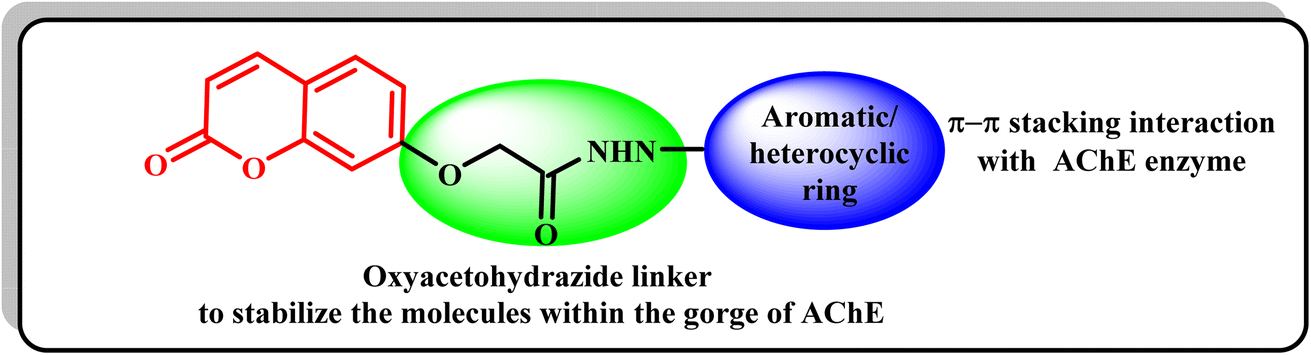 | ||
| Fig. 2 The strategy of the development of new coumarin compounds as AChE inhibitors. | ||
Based on molecular docking study as acetylcholinesterase inhibitors, the target compounds were selected and synthesized. The biosafety of the new compounds was also determined. Furthermore, the inhibitory activity of the most active compound as well as its impact on several biomarkers related to the disease were investigated. In addition, the impact of the most promising compound on the amelioration of the disorders associated with these biomarkers in AD was elucidated. Molecular docking study was carried out to find out the binding mode of the most active compound to interact with the active site of the target enzyme.
2. Experimental
2.1. Chemical synthesis
The instruments used to determine melting points, spectral data (IR, 1H NMR, 13C NMR, and mass), as well as chemical analyses were included in a detailed description of the in ESI file.†2.1.3.1. N′-(3,4-dihydronaphthalen-1(2H)-ylidene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (4a). White solid, yield 78.0%; IR (KBr, cm−1): 3354 (NH), 1720, 1660 (2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3056 (aromatic C–H), and 2914.5 (alkane C–H); 1H NMR (500 MHz, DMSO-d6): δ 1.82, 2.63, 2.73 (3m, 6H, 3CH2-tetralone ring), 2.39 (s, 3H, CH3), 4.90 (s, 2H, OCH2 minor conformer), 5.35 (s, 2H, OCH2 major conformer), 6.21 (s, 1H, coumarin-H3), 6.94–7.03 (m, 2H, Ar–H), 7.19–7.29 (m, 3H, Ar–H), 7.67–7.71 (m, 1H, Ar–H), 8.08 (dd, 1H, Ar–H, J = 7.5 Hz), 10.62 (br, 1H, NH, minor conformer, D2O exchangeable), 10.90 (br, 1H, NH, major conformer, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6): δ 18.53 (CH3), 21.71, 25.85, 29.29 (3CH2-tetralone ring), 66.08 (–OCH2), 101.98, 111.62, 112.78, 113.74, 124.99, 126.71, 126.77, 128.93, 129.51, 132.51, 140.27, 148.86, 153.75, 155.00, 160.53, 161.88 (CO), 169.63 (CO); anal. calcd for: C22H20N2O4: C, 70.20; H, 5.36; N, 7.44, found: C, 70.52; H, 5.06; N, 7.15.
O), 3056 (aromatic C–H), and 2914.5 (alkane C–H); 1H NMR (500 MHz, DMSO-d6): δ 1.82, 2.63, 2.73 (3m, 6H, 3CH2-tetralone ring), 2.39 (s, 3H, CH3), 4.90 (s, 2H, OCH2 minor conformer), 5.35 (s, 2H, OCH2 major conformer), 6.21 (s, 1H, coumarin-H3), 6.94–7.03 (m, 2H, Ar–H), 7.19–7.29 (m, 3H, Ar–H), 7.67–7.71 (m, 1H, Ar–H), 8.08 (dd, 1H, Ar–H, J = 7.5 Hz), 10.62 (br, 1H, NH, minor conformer, D2O exchangeable), 10.90 (br, 1H, NH, major conformer, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6): δ 18.53 (CH3), 21.71, 25.85, 29.29 (3CH2-tetralone ring), 66.08 (–OCH2), 101.98, 111.62, 112.78, 113.74, 124.99, 126.71, 126.77, 128.93, 129.51, 132.51, 140.27, 148.86, 153.75, 155.00, 160.53, 161.88 (CO), 169.63 (CO); anal. calcd for: C22H20N2O4: C, 70.20; H, 5.36; N, 7.44, found: C, 70.52; H, 5.06; N, 7.15.
2.1.3.2. 2-((4-Methyl-2-oxo-2H-chromen-7-yl)oxy)-N′-(2-oxoindolin-3-ylidene) acetohydrazide (4b). White solid, yield 67.0%; IR (KBr, cm−1); 3355 (NH), 1720.5, 1695, 1660 (3C
O), 3062 (aromatic C–H), and 2920 (alkane C–H); 1H NMR (500 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 5.01 (s, 2H, OCH2, major conformer), 5.45 (s, 2H, OCH2, minor conformer), 6.25 (s, 1H, coumarin-H3), 6.96 (d, 1H, Ar–H, J = 7.5 Hz), 7.09–7.10 (m, 3H, Ar–H), 7.39 (t, 1H, Ar–H, J = 7.5 Hz), 7.56 (br, 1H, Ar–H), 7.75 (br, 1H, Ar–H), 11.29 (s, 1H, NH, D2O exchangeable), 12.58 (br, 1H, NH, minor conformer, D2O exchangeable), 13.70 (s, 1H, NH, major conformer, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6): δ 18.53 (CH3), 66.54 (–OCH2), 102.78, 111.63, 112.16, 112.84, 120.04, 121.45, 123.08, 127.05, 132.35, 143.08, 153.66, 154.99, 160.40, 162.97 (CO), 167.00 (CO); anal. calcd for: C20H15N3O5: C, 63.66; H, 4.01; N, 11.14; found: C, 63.46; H, 4.31; N, 11.54.
2.1.3.3. N′-(2,3-Dihydro-1H-inden-1-ylidene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (4c). White solid, yield 77.0%; IR (KBr, cm−1); 3340 (NH), 1715, 1660 (2C
O), 3057 (aromatic C–H), and 2920 (alkane C–H); 1H NMR (500 MHz, DMSO-d6): δ 2.39 (s, 3H, CH3), 2.80–2.87 (m, 2H, CH2 indane ring), 3.07–3.09 (m, 2H, CH2, indane ring), 4.88 (s, 2H, OCH2, minor conformer), 5.30 (s, 2H, OCH2, major conformer), 6.22 (s, 1H, coumarin-H3), 6.94–7.04 (m, 2H, Ar–H), 7.30–7.41 (m, 3H, Ar–H), 7.65–7.73 (m, 2H, Ar–H), 10.49 (s, 1H, NH, minor conformer, D2O exchangeable), 10.82 (s, 1H, NH, major conformer, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6): δ 18.55 (CH3), 27.48, 28.55 (2CH2, indane ring), 65.87 (–OCH2), 101.97, 111.64, 112.76, 112.84, 121.75, 126.13, 126.82, 127.37, 127.50, 130.97, 137.98, 153.79, 155.02, 158.61, 160.55, 161.91 (CO), 169.21 (CO); anal. calcd for: C21H18N2O4: C, 69.60; H, 5.01; N, 7.73; found: C, 69.21; H, 5.41; N, 7.52.
2.1.3.4. 4-((2-(2-((4-Methyl-2-oxo-2H-chromen-7-yl)oxy)acetyl) hydrazineylidene)methyl) benzoic acid (4d). White solid, yield 75.0%; IR (KBr, cm−1); 3445 (OH), 3360 (NH), 1710, 1695, 1665 (3C
O), 3054 (aromatic C–H), and 2920 (alkane C–H); 1H NMR (500 MHz, DMSO-d6): δ 2.39 (s, 3H, CH3), 4.84 (s, 2H, OCH2, minor conformer), 5.33 (s, 2H, OCH2, major conformer), 6.21 (s, 1H, coumarin-H3, major conformer), 6.24 (s, 1H, coumarin-H3, minor conformer), 6.99–7.06 (m, 2H, Ar–H), 7.69 (d, 1H, Ar–H, J = 8.5 Hz, major conformer), 7.73 (d, 1H, Ar–H, J = 8.5 Hz, minor conformer), 7.82 (d, 2H, Ar–H, J = 8.5 Hz, minor conformer), 7.85 (d, 2H, Ar–H, J = 8.5 Hz, major conformer), 7.97–8.00 (m, 2H, Ar–H), 8.07 (s, 1H, –NCH, major conformer), 8.38 (s, 1H, –NCH, minor conformer), 11.82 (s, 1H, NH, D2O exchangeable, minor conformer), 11.83 (s, 1H, NH, D2O exchangeable, major conformer); 12.63 (br, H, COOH, D2O exchangeable); 13C NMR (125 MHz, DMSO-d6): δ 18.54 (CH3), 65.70 (–OCH2), 102.05, 111.68, 112.83, 113.82, 126.79, 127.00, 127.45, 127.62, 130.12, 132.11, 138.39, 143.38, 153.76, 155.03, 160.54, 161.79 (CO), 167.32 (CO), 169.10 (CO); anal. calcd for: C20H16N2O6: C, 63.16; H, 4.24; N, 7.37.
2.2. Biological evaluation
2.2.1.1. Acetylcholinesterase inhibition screening protocol. Acetylcholinesterase (AChE) inhibitory activity was measured using quantitative colorimetric assay according to the method developed by Ellman et al.21 More details are in the ESI file.†
2.2.1.2. Antioxidant assay by DPPH. In vitro estimation of antioxidant activity according to Gulcin et al.22
2.2.1.2.1. Calculation. Inhibition (%) = read of control − read of test/read of control × 100.
2.2.2.1. Animal care. Wistar albino rats weighing between 180–200 g were housed in the animal house of National Research Centre. More details are in the ESI file.†
2.2.2.2. Biosafety assay. Different concentrations of the most promising synthetic compounds were used to determine LD50 in experimental animals for 24 hours. LD50 is the dose that produces mortality in 50% of animals. For each concentration six animals were used.
2.2.2.3. Toxicity study. Selected 8 Wistar rats of uniform weight were taken and divided into two groups of 4 rats each for each concentration. The synthesized compound 4c was given orally to rats in doses 50 and 100 mg per kg b.wt as described by Desoukey et al.23 More details are in the ESI file.†
2.2.2.3.1. Acute toxicity. No mortality was observed at the dose of 50 mg per kg b.wt. While, one rat of 4 in the group administered 100 mg per kg b.wt died post 24 h (25%). No further death or any behaviour changes were observed for one month. Thus, the applied dose for the compound 4c was considered to be (10 mg per kg b.wt), which is the dose applied for the reference drug donepezil.
2.2.2.3.2. Chronic toxicity. In chronic toxicity, we used 1/10 of the LD50 (10 mg per kg b.wt) supplemented orally to male Wistar rats for one month and didn't see any indication of harmfulness (no mortality, no going bald, no loose bowels, and so on). Moreover, irregularities in conduct, food and water admissions, and well-being status among the treated creatures weren't noticed. Toxicity tests on liver and kidney functions, as well as a complete blood picture, were also performed.
2.2.2.4. Histopathological investigation. Specimens of liver, kidneys, heart, and brain were harvested from all mice/group, immediately fixed in 10% neutral buffered formalin, and routinely processed according to Suvarna et al.24 Sections of 4–5 μm thickness were prepared and stained with Hematoxylin and Eosin (H&E), for histopathological examination by a light microscope (Olympus BX50, Tokyo, Japan).
2.2.2.5. Biochemical analyses.
2.2.2.5.1. In vivo studies. After one month, rats were anaesthetized with diethyl ether, blood was drawn via sublingual vein rupture, and rats were decapitated. Blood samples were centrifuged at 3000 rpm, and the clear serum that was separated was frozen at −80 °C for biochemical analysis.25–29 More details are in the ESI file.†
2.2.2.5.2. Induction of AlCl3 and grouping of animals. Induction of AlCl3-induced Alzheimer's disease. AlCl3 solutions were made freshly at the beginning of each experiment. AlCl3 was dissolved in drinking water and administered orally to rats daily for two months at 0.5 mL per 100 g b.wt at a dose of 100 mg kg−1.30
2.2.2.5.3. Behaviour studies. The neurocognitive function of rats was estimated by T-maze test (constructed in the NRC, Egypt) according to Deacon and Rawlins.31 Before performing this experiment, the animals were left without food for 24 h, with only water to drink.32,33 More details are in the ESI file.†
2.2.2.6. In vivo determination of oxidative stress.
2.2.2.6.1. Blood sample preparation. The rats were fasted overnight. Then, they were anaesthetized by inhaling 1.9% diethyl ether on a small piece of cotton (0.08 mL per litre of container volume). Blood samples were collected after anaesthetizing the animals using a cardiac puncture in a clean and dry test tube. Blood was left to clot for 10 minutes and centrifuged at 3000 rpm (1.1750g) to obtain serum. The separated serum was stored at −80 °C for biochemical analyses.
2.2.2.6.2. Brain tissue sampling preparation. At the end of the experiment, the rats were fasted overnight, subjected to anaesthesia, and scarified. The whole brain of each rat was rapidly dissected, washed with isotonic saline, and dried on filter paper.34,35 More details are in the ESI file.†
2.3. Molecular docking studies
The crystal structure of acetylcholinesterase (PDB: 1EVE) was retrieved from Protein Data Bank (http://www.rcsb.org). Protein is prepared with Molecular Operating Environment (MOE) software; the water was removed, and the hydrogen atoms were added using MOE. More details are in the ESI file.†3. Results and discussion
3.1. Chemistry
A new series of 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)-N′-substituted (methylene)acetohydrazide derivatives 4a–d were synthesized according to the synthetic route adopted in Scheme 1. The key starting compound 7-hydroxy-4-methyl-2H-chromen-2-one (1) was refluxed with ethyl bromoacetate in the presence of anhydrous potassium carbonate in acetone to give the corresponding ester 2. Hydrazinolysis of compound 2 with hydrazine hydrate produced the corresponding acetohydrazide 3 in a good yield.20 Furthermore, condensation of compound 3 with different aromatic ketone and aldehydes such as; α-tetralone, isatin, 1-indanone, and 4-carboxybenzaldehyde in refluxing ethanol led to the formation of the corresponding target Schiff's bases 4a–d. The molecular structures of the new compounds were confirmed via micro and spectral analyses. The FT-IR spectra of compounds 4a–d showed absorption bands at 3355–3340 cm−1 related to the NH group, 1720–1710 cm−1 (lactonic –CO carbonyl stretching), and 1671–1650 cm−1 (amide –CO carbonyl stretching). The 1H-NMR and 13C-NMR spectral data of compounds 4a–d confirmed the presence of these derivatives as trans and cis conformers in ratios ranging between 2:1 and 3:1. The 1H-NMR and 13C-NMR spectral data of compounds 4a–d showed that they are present as trans and cis isomers. The 1H-NMR spectra of compounds 4a–d exhibited three singlets at δ 2.39–2.40, 4.84–5.45, and 6.21–6.25 ppm related to CH3, O–CH2, and coumarin-H3, respectively. In addition, the methine proton of the NCH group of compound 4d appeared as a singlet signal at δ 8.07 and 8.38 ppm, while the NH proton appeared as an exchangeable signal at δ 10.62–13.70 ppm. The aromatic protons appeared as multiplet signals in the range δ 6.94–8.08 ppm. 13C-NMR spectra of compounds 4a–d showed signals at δ 18.53–18.55 and 65.70–66.54 ppm due to CH3 and OCH2 carbons alongside different signals at the range δ 101.97–160.40 ppm representing coumarin-C3, NCH, and the aromatic carbons. The carbonyl groups appeared as two singlets at δ 161.79 and 169.63 ppm. The mass spectra of the new compound 4a–d represented their molecular ion peaks which were in agreement with their molecular formulae (Scheme 1).
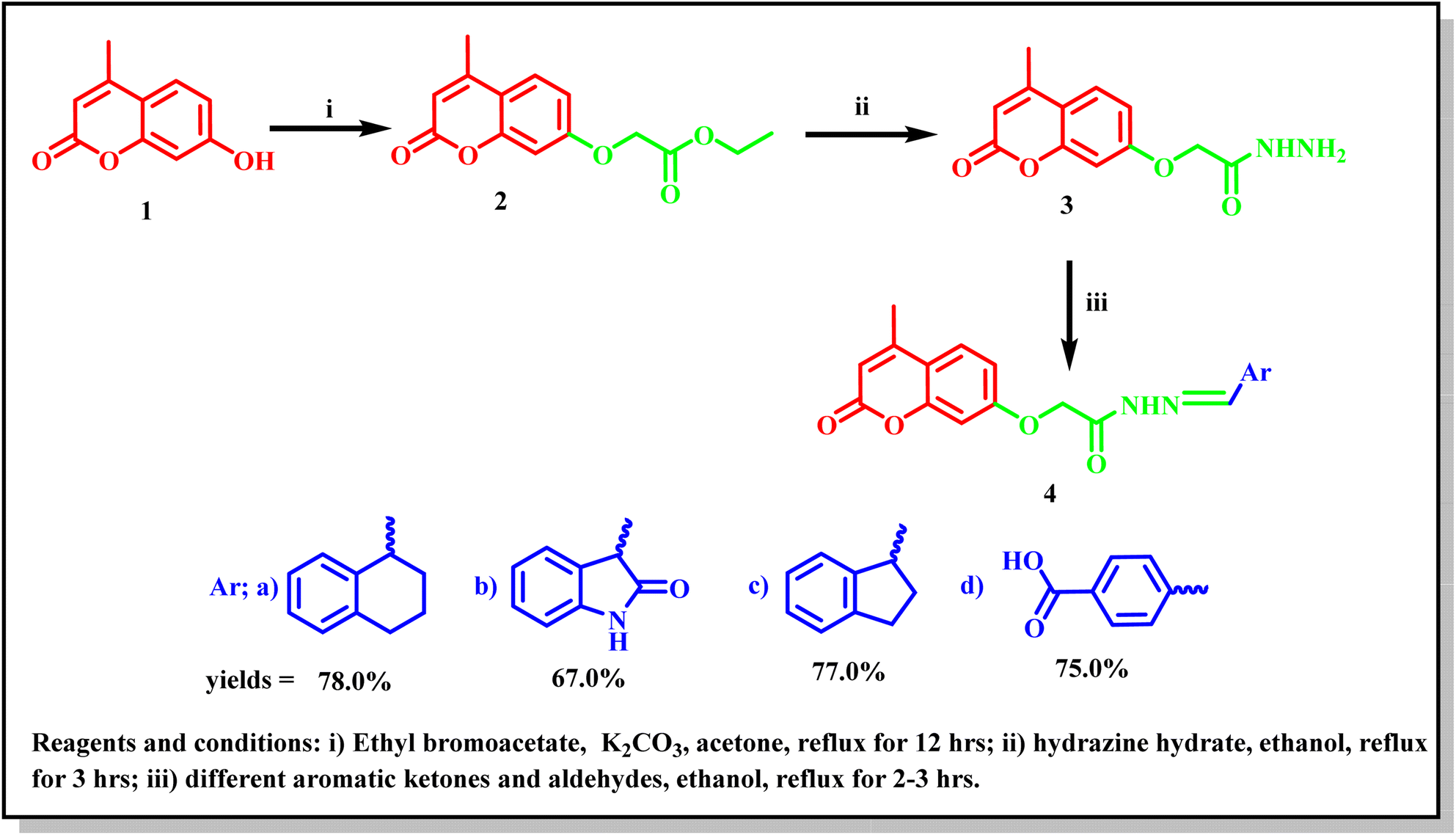 | ||
| Scheme 1 Synthetic route for the synthesis of the target 4-methyl-2-oxo-2H-chromene Schiff's bases 4a–d. | ||
3.2. In vitro studies
| Compound no. | AChE IC50 (μg mL−1) | SD (±) |
|---|---|---|
| a Data are mean ± of three replicate in each group. | ||
| 4a | 4.941 | 0.25 |
| 4b | 7.869 | 0.4 |
| 4c | 0.802 | 0.04 |
| 4d | 1.20 | 0.06 |
| Donepezil | 0.155 | 0.01 |
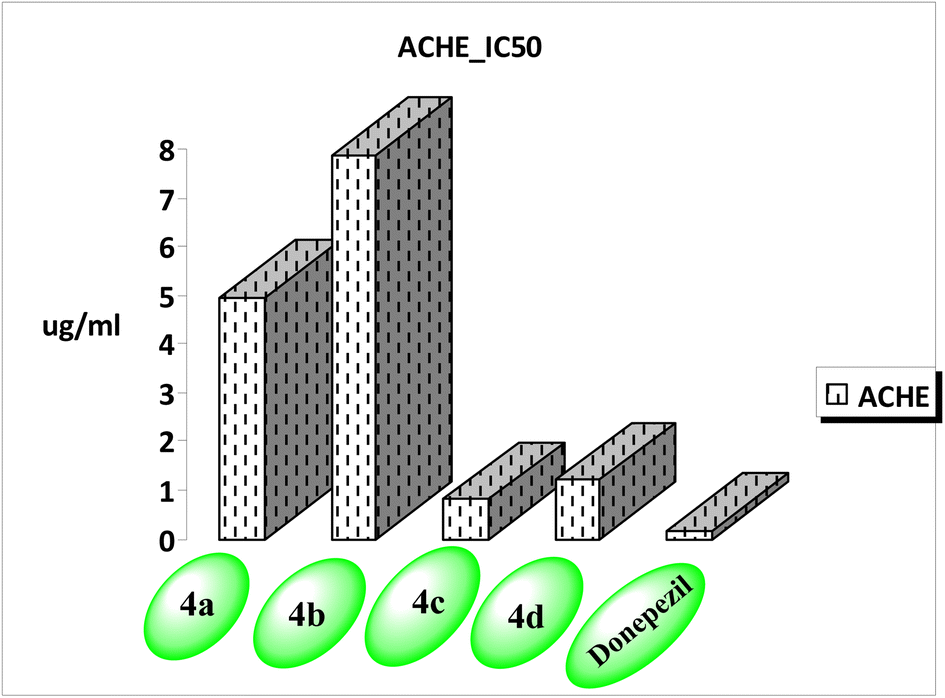 | ||
| Fig. 3 AChE inhibiting activity of the new compounds. | ||
In addition, the new compounds 4a–d were subjected to DPPH scavenging assay to evaluate their antioxidant activity in comparison with vitamin C as a reference drug. Table 2 shows the percentage of free radical scavenging activity at two different concentrations of 0.01 mg, and 0.05 mg. The results showed that the activity of the tested analogues increased in a dose dependent manner. The most promising antioxidant activity was obtained by compound 4c in comparison with vitamin C, with a DPPH percentage of inhibition 57.14 ± 2.77%, and 89.00 ± 6.99%, respectively, at 0.05 mg. Low inhibition activities were obtained by compounds 4a and 4b with percentages of 40.9 ± 3.1, and 35.00 ± 1.00%, respectively. Compound 4d appeared to be inactive as an antioxidant agent, showing a percentage of inhibition 10.3 ± 0.33%.
| Compounds | Concentration (0.01 mg) | Concentration (0.05 mg) |
|---|---|---|
| a Data are means ± SD of three replicates in each group. | ||
| 4a | 31.8 ± 0.2 | 40.9 ± 3.1 |
| 4b | 31.8 ± 1.2 | 35.00 ± 1.00 |
| 4c | 28.00 ± 0.4 | 57.14 ± 2.77 |
| 4d | 4.5 ± 0.4 | 10.3 ± 0.33 |
| Vitamin C | 80.00 ± 5.00 | 89.00 ± 6.99 |
| Blood components | Control rats | 4c-treated rats |
|---|---|---|
| a Data are means ± SD of 8 rats in the treated group. Statistical analysis was carried out using co-state and SPSS computer programs (version 8), where the unshared letter is significant at P ≤ 0.05. | ||
| Hemoglobin (Hb) (g L−1) | 14.55 ± 0.14a | 14.58 ± 0.59a |
| Red blood cells (RBCs) (million cells per mm) | 7.88 ± 0.22a | 8.00 ± 0.08a |
| Haematocrit (%) | 45.90 ± 0.11a | 45.00 ± 1.87a |
| Mean corpuscular volume (MCV) (fL) | 55.50 ± 0.20a | 59.00 ± 0.03a |
| Mean corpuscular hemoglobin (MCH) (pg) | 17.00 ± 0.90a | 17.78 ± 0.88a |
| Mean corpuscular hemoglobin concentration (MCHC) (g dL−1) | 34.78 ± 0.29a | 33.98 ± 0.25a |
| RDW-CV (%) | 16.00 ± 0.60a | 15.80 ± 0.19a |
| Platelets (103 mm−1) | 449.00 ± 14.80a | 465.00 ± 12.00a |
| MPV (fL) | 7.19 ± 0.11a | 8.66 ± 0.19a |
| White blood cells WBCs (103 cmm−1) | 10.00 ± 0.10a | 10.89 ± 1.90a |
| Neutrophils (103 cm−1) | 2.59 ± 0.07a | 2.39 ± 0.09a |
| Lymphocyte (103 cm−1) | 7.88 ± 0.22a | 7.62 ± 0.66a |
| Monocyte (103 cm−1) | 1.87 ± 0.08a | 1.93 ± 0.06a |
| Groups | ALT (U/l) | AST (U/l) | ALP (U/l) |
|---|---|---|---|
| a Data are means ± SD of 8 rats in the treated group. Statistical analysis was carried out using Co-state and SPSS computer programs (version 8), where the unshared letter is significant at P ≤ 0.05. | |||
| Control male rats | 68.00 ± 3.00a | 29.00 ± 2.00a | 134.00 ± 8.00a |
| Treated male rats | 79.00 ± 4.40a | 32.00 ± 1.20a | 136.00 ± 9.00a |
a
| Groups | Urea (mg dL−1) | Creatinine (mg dL−1) |
|---|---|---|
| a Data are means ± SD of 15 rats in the treated group. Statistical analysis was carried out using co-state and SPSS computer programs (version 8), where the unshared letter is significant at P ≤ 0.05. | ||
| Control male rats | 18.60 ± 1.22 | 2.40 ± 0.10 |
| Treated male rats | 20.00 ± 1.57 | 2.80 ± 0.07 |
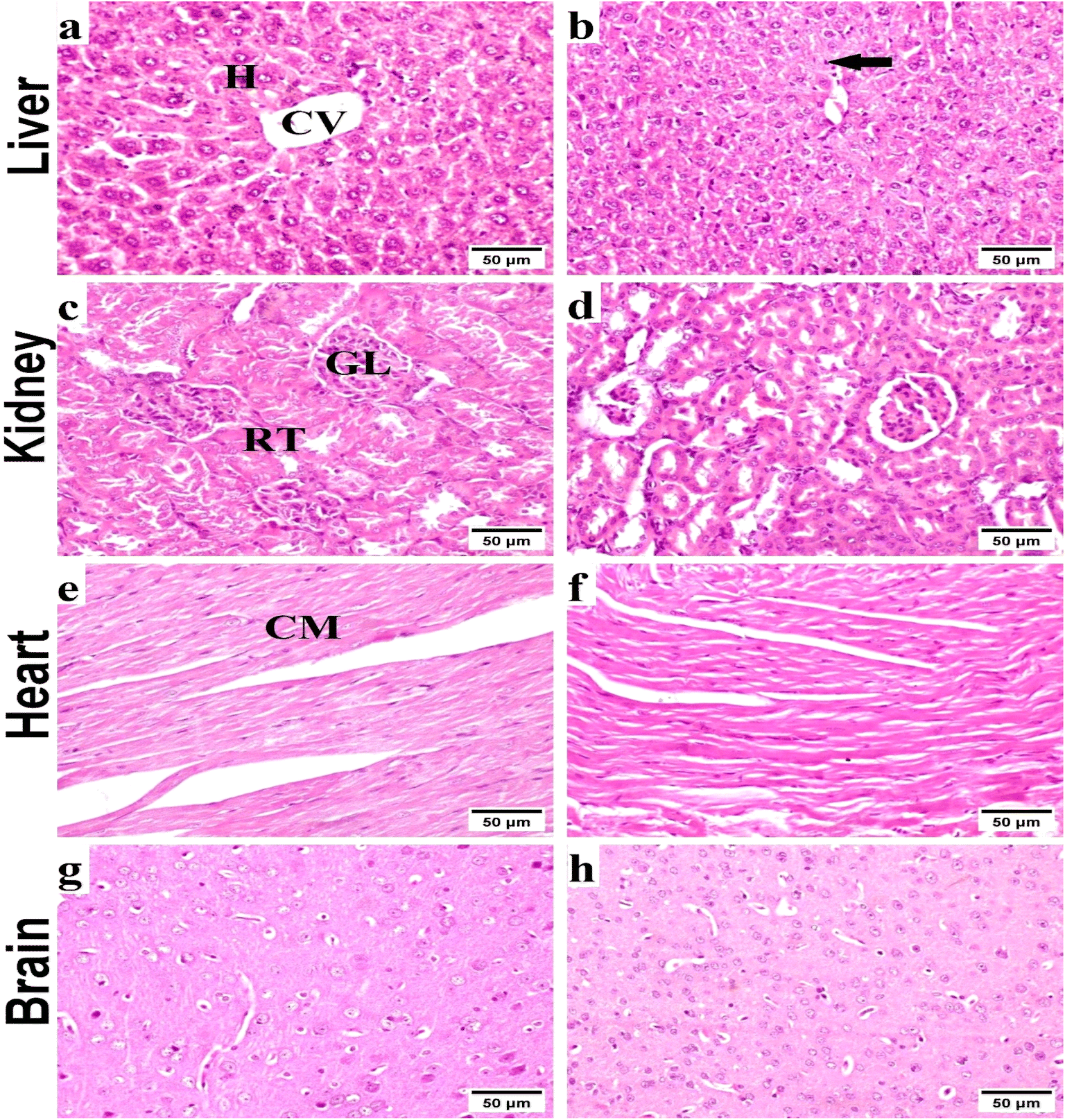 | ||
| Fig. 4 Representative photomicrographs exhibited H & E-stained sections of liver (a and b), kidney (c and d), heart (e and f) and brain (g and h) respectively (scale bar, 50 μm); (a) normal control liver with normal hepatic tissue histoarchitecture: normal central vein (CV) and normal hepatocytes (H). (b) Received compound 4c, showing slight Kupffer cell activation (arrow). (c) Normal control kidneys, showing the histological structure of renal parenchyma; normal glomeruli (GL), and renal tubules (RT). (d) Received compound 4c, showing no histopathological alterations. (e) Normal control heart exhibiting normal cardiac myocytes. (f) Received compound 4c, showing normal cardiac tissue. (g and h) Normal control brain extracts, showing histologically normal brain tissues with no histopathological alterations. | ||
| Tested groups | Baseline (Sc) | Induction two months (Sc) | Treatment 4 weeks (Sc) |
|---|---|---|---|
| a Data are expressed as mean SD (n = 10) in seconds. Groups with similar letters are not significantly different, while those with different letters are significantly different at p ≤ 0.05. | |||
| Control | 13.30 ± 1.00 | 14.80 ± 1.00 | 14.90 ± 1.00 |
| AlCl3-AD | 15.30 ± 1.10 | 50.90 ± 3.00 | — |
| % change | 1.53 | 243.92 | |
| AD-4c | — | — | 28.20 ± 1.80 |
| % improvement | 148.37 | ||
| Donepezil | — | — | 23.10 ± 1.70 |
| % improvement | 181.70 | ||
| Tested groups | Baseline (Sc) | Induction 2 months (Sc) | Treatment 4 weeks (Sc) |
|---|---|---|---|
| Control | 9.60 ± 0.87 | 9.80 ± 0.66 | 10.15 ± 0.22 |
| AlCl3-AD | 9.00 ± 1.10 | 2.90 ± 0.70 | — |
| % change | 6.25 | 70.41 | |
| AD-4c | — | — | 8.99 ± 1.00 |
| % improvement | 60.00 | ||
| Donepezil standard drug | — | — | 9.80 ± 0.88 |
| % improvement | 67.98 |
| Group/parameters | MDA (mg per g tissues) | GSH (mg per g tissues) |
|---|---|---|
| a Data are expressed in seconds as mean ± SD (n = 8). Groups with similar letters are not significantly different, while those with different letters are significantly different at p ≤ 0.05. | ||
| Control | 11.75 ± 0.43 | 29.66 ± 0.55 |
| AD | 33.08 ± 2.10 | 13.60 ± 0.91 |
| % change | 181.54 | 54.15 |
| 4c-AD treated | 22.43 ± 1.66 | 21.66 ± 1.21 |
| % change to disease | 32.19 | 59.26 |
| % improvement | 90.64 | 27.17 |
| Donepezil-AD treated | 22.43 ± 2.00 | 23.99 ± 2.00 |
| % change to disease | 32.19 | 76.40 |
| % improvement | 90.64 | 35.03 |
In the same line, we have found that AD rats exhibited down-regulation of Bcl-2 expression and overexpression of the Bax and Tau genes. However, AD rats treated with compound 4c showed improved expression of Bcl-2 and reduced expression of Bax and Tau genes (Fig. 5–7).
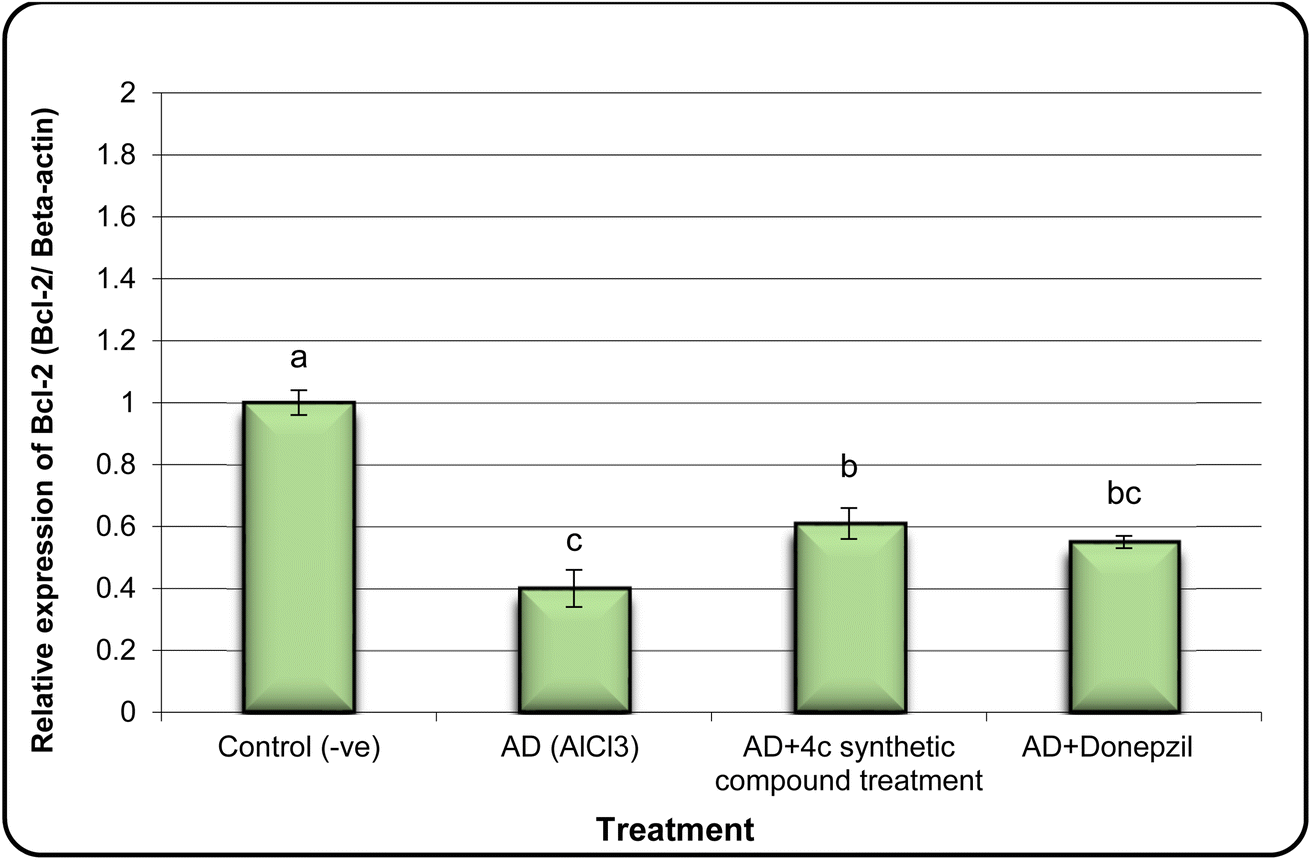 | ||
| Fig. 5 The expression alterations of Bcl-2 gene in brain samples of AD induced rats treated with compound 4c. Means with different superscripts (a, b, c) between groups in the same treatment are significantly different at P < 0.05. Data are presented as mean ± SD. | ||
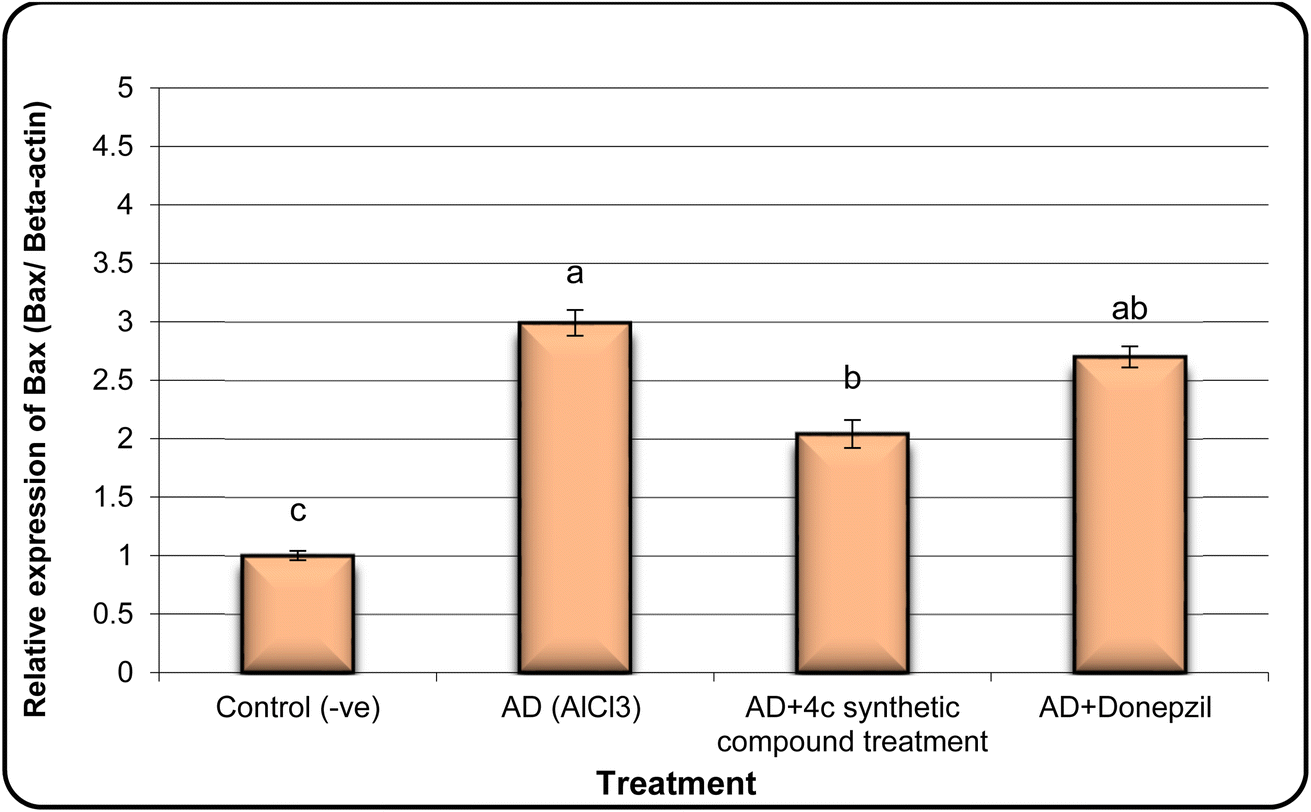 | ||
| Fig. 6 The expression alterations of Bax gene in brain samples of AD induced rats treated with the compound 4c. Means with different superscripts (a, b, c) between groups in the same treatment are significantly different at P < 0.05. Data are presented as mean ± SD. | ||
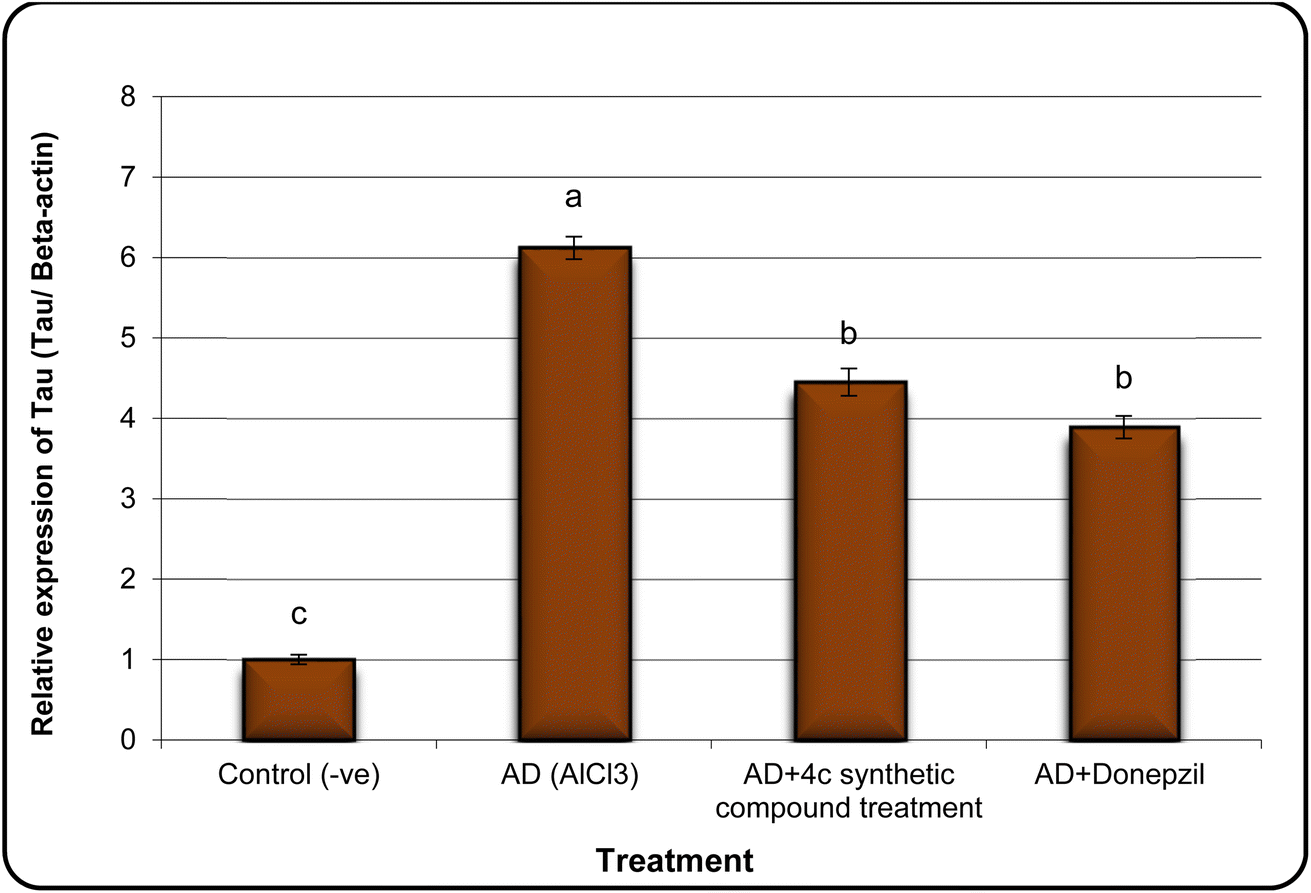 | ||
| Fig. 7 The expression alterations of Tau gene in brain samples of AD induced rats treated with the compound 4c. Means with different superscripts (a, b, c) between groups in the same treatment are significantly different at P < 0.05. Data are presented as mean ± SD. | ||
3.3. Molecular docking studies
In order to validate our rational design, compounds 4a–4d were docked against the acetylcholine esterase enzyme. The crystal structure of the acetylcholinesterase complex with donepezil (PDB: 1EVE) was used as a target for the molecular modelling analysis.41,42 In order to validate the docking protocol, the native ligand (donepezil) was re-docked using MOE. The self-docking result showed that the RMSD value between the native ligand and the re-docked pose ≤2 Å which indicated the reliability of the docking protocol. The docking results of donepezil revealed interactions between the di-methoxy phenyl group and Trp279, while the phenyl group demonstrated an arene–arene interaction with Trp84. The N atom of the piperidine ring showed arene–cation interactions with Trp84 and Phe330. Hydrophobic interactions were found between donepezil and Leu282, Phe288 and Phe290, and Phe331. Tyr121 showed hydrophilic interactions with the carbonyl group (Fig. 8).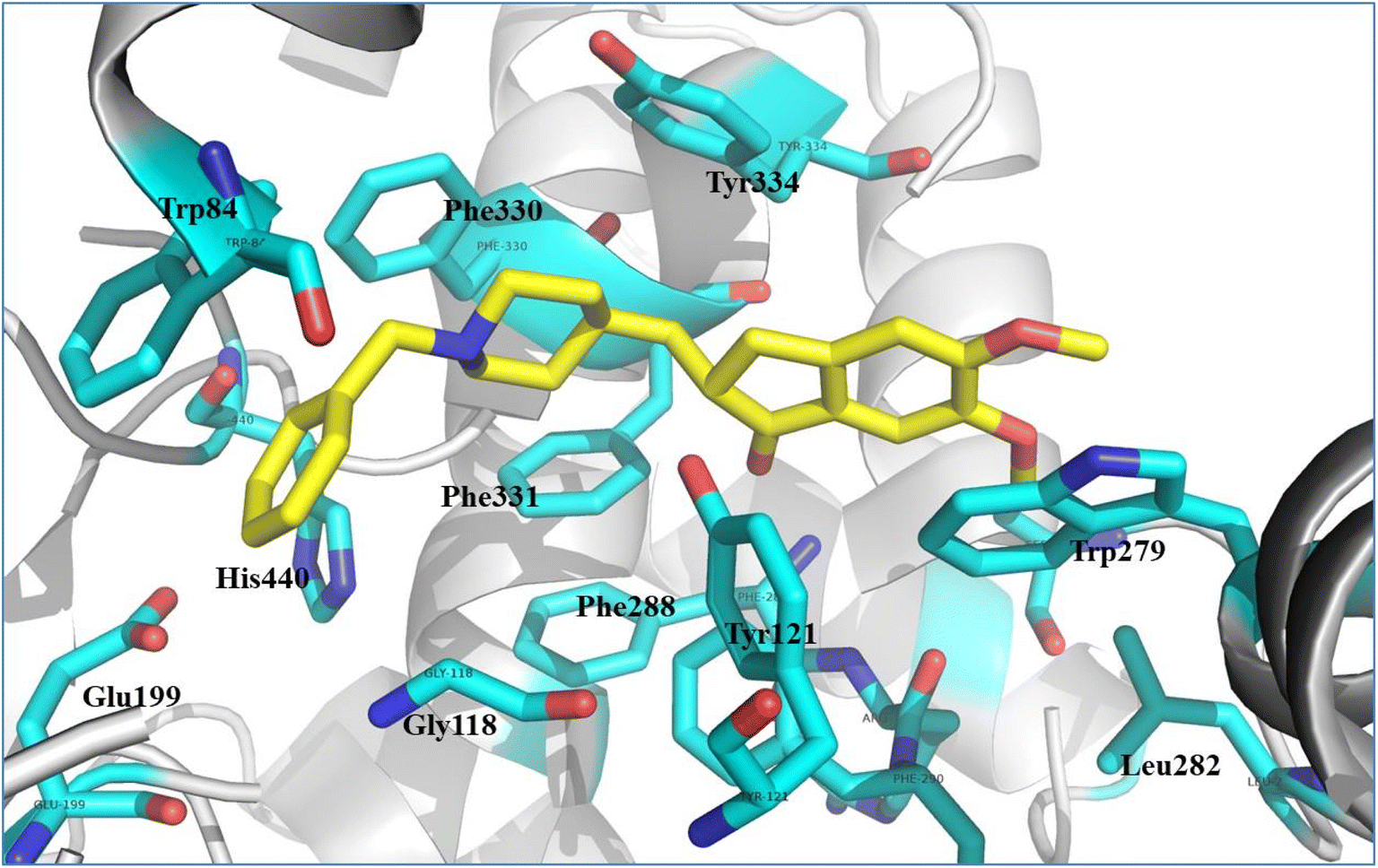 | ||
| Fig. 8 Illustrated interaction of the native ligand (yellow stick) with amino acid residues (cyan stick), the protein is represented as a white cartoon. | ||
We studied the binding mode and the interactions between the compounds 4a–4d and the active site of the enzyme. Compounds 4a and 4b showed good binding modes within the active site in comparison with the native ligand (Fig. 9a and 10a, respectively). The two compounds nearly showed the same binding mode with binding scores of −8.7 and −7.4 kcal mol−1, respectively (Table 9). Compound 4a showed H-bonds with OH-Tyr121 and OH-Tyr130 at a distance (2.5 and 3.2 Å), respectively. The chromen and naphthalene moieties showed arene–arene (π–π) interactions with Trp84 and Trp279, respectively. The naphthalene moiety showed hydrophobic interactions with Phe331 and the hydrophobic part of Tyr334 (Fig. 9b). While compound 4b showed a H-bond with OH-Tyr121 at a distance 3.1 Å, arene–arene interactions with Trp48 and Trp279. The chromen moiety showed hydrophobic interactions with Phe290, Phe331, and Tyr334 (Fig. 10b).
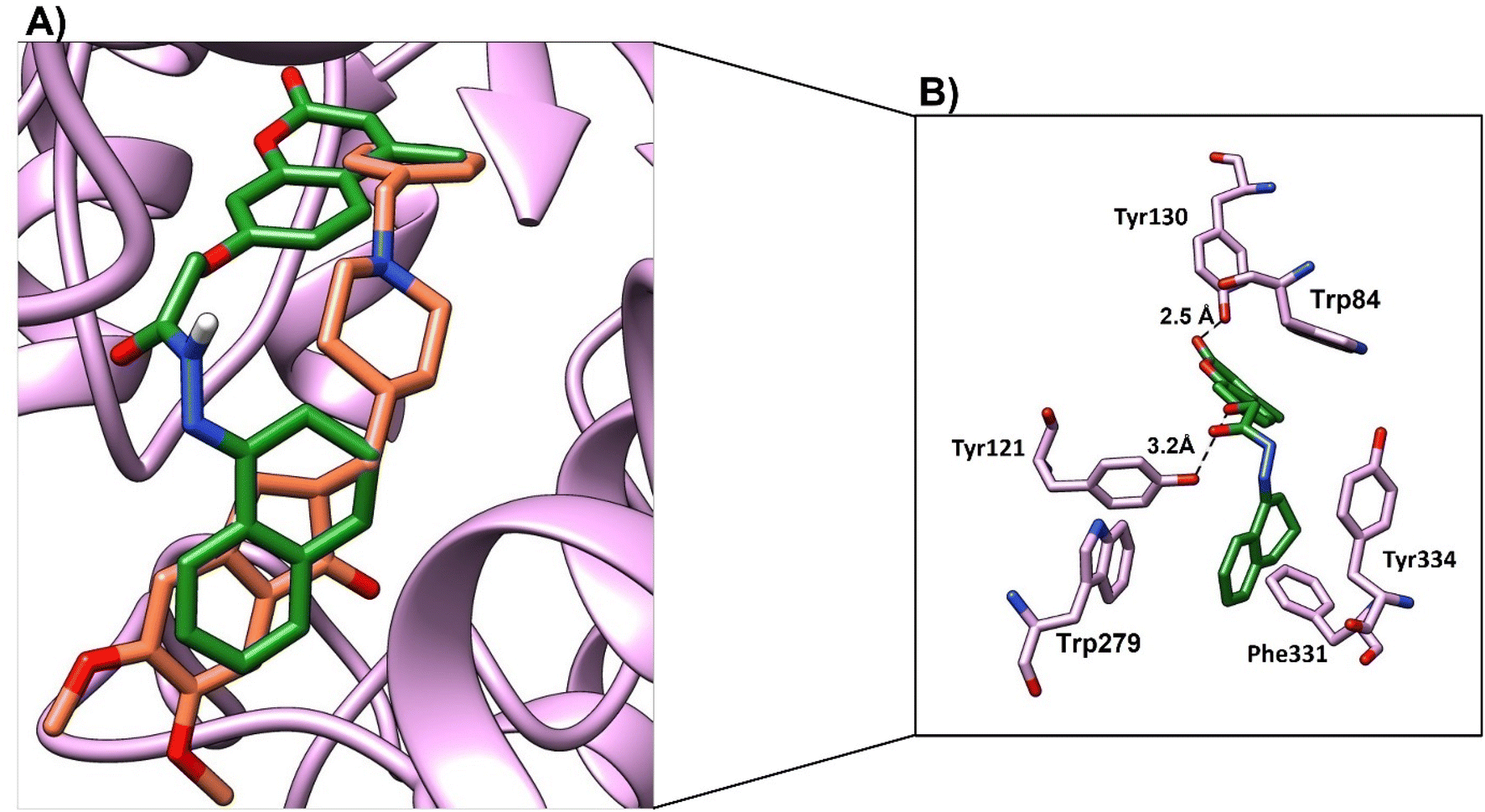 | ||
| Fig. 9 (A) Binding mode of 4a (green, stick) and native ligand (brown, stick) within the active site. (B) Interactions of compound 4a (green) and key residues (purple), H-bond was represented as a black-dotted line. | ||
 | ||
| Fig. 10 (A) Binding mode of 4b (green, stick) and native ligand (brown, stick) within the active site. (B) Interactions of compound 4b (green) and the key residues (purple), H-bond was represented as a black-dotted line. | ||
| Compd no. | Moieties from compounds | Residue-ID | Distance (Å) | Type of interactions | Score (kcal mol−1) |
|---|---|---|---|---|---|
| 4a | (1) Oxygen atom of 4-methyl-2-oxo-2H-chromen-7-yl)oxy | OH-Tyr121 | 3.2 | H-bond | −8.7 |
| (2) CO of 4-methyl-2-oxo-2H-chromen moiety |
OH-Tyr130 | 2.5 | H-bond | ||
| (3) Chromen moiety | Trp84 | π–π | |||
| (4) Naphthalene moiety | Trp279 | π–π | |||
| Phe331, Tyr334 | Hydrophobic | ||||
| 4b | (1) Oxygen atom of 4-methyl-2-oxo-2H-chromen-7-yl)oxy | OH-Tyr121 | 3.1 | H-bond | −7.4 |
| Trp279 | π–π | ||||
| (2) Chromen moiety | Phe290, Phe331, Tyr334 | Hydrophobic | |||
| (3) Oxoindoline moiety | Trp84 | π–π | |||
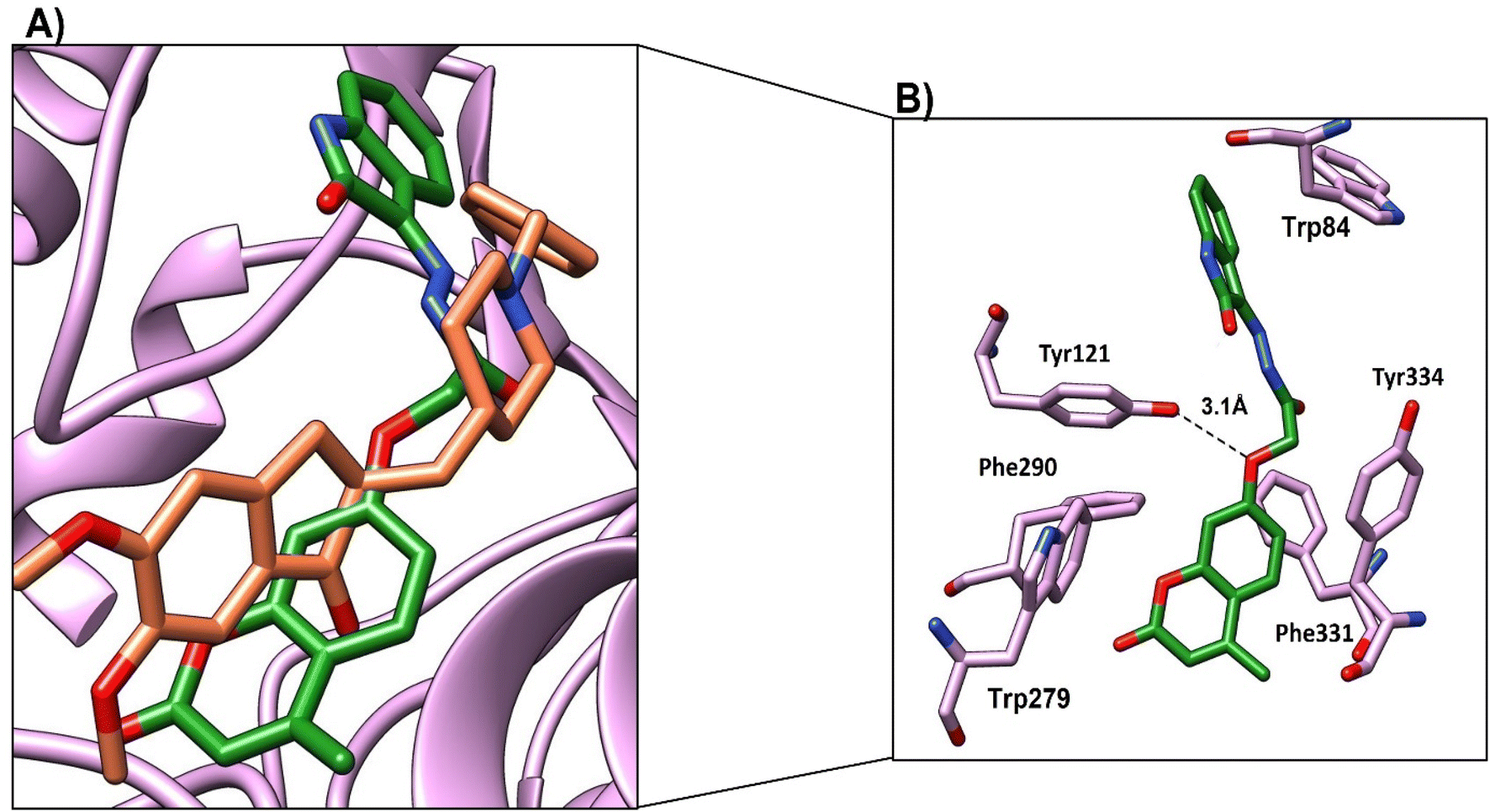
| 4c | (1) Carbonyl of 4-methyl-2-oxo-2H-chromen-7-aryloxy | OH-Tyr130 | 2.5 | H-bond | −9.1 |
| (2) CO of acetohydrazide |
OH-Tyr121 | 2.9 | H-bond | ||
| (3) Chromen moiety | Trp84 | π–π | |||
| (4) Indene moiety | Phe330, Phe331, Tyr334 | Hydrophobic | |||
| Trp279 | π–π | ||||
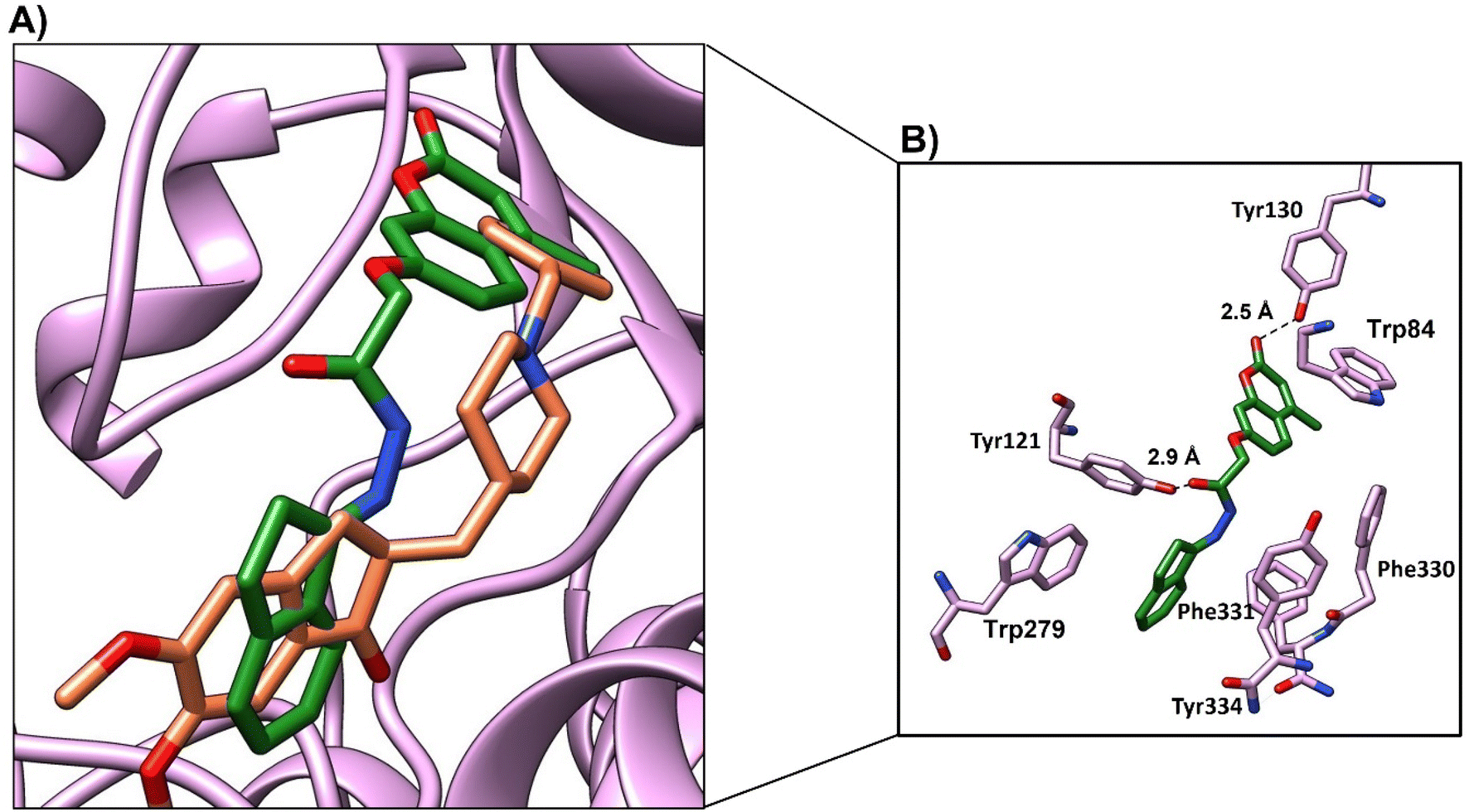
| 4d | (1) Oxygen atom of 4-methyl-2-oxo-2H-chromen-7-yl)oxy | OH-Tyr121 | 3.0 | H-bond | −8.1 |
| (2) Chromen | Trp279 | π–π | |||
| (3) CO group of benzoic acid |
Phe330, Phe331, Tyr334 | Hydrophobic | |||
| (4) Phenyl group of benzoic acid | OH-Tyr130 | 2.2 | H-bond | ||
| Trp84 | π–π |
Compound 4c, the most promising compound, showed the best docking score (−9.1 kcal mol−1) and the best binding mode (Fig. 11a). Compound 4c showed H-bonds between CO of chromen and OH-Tyr130 at a distance of 2.5 Å and CO of acetohydrazide and Tyr121 at a distance 2.9 Å (Fig. 11b). The chromen moiety showed arene–arene interactions with Trp84, while the indene moiety showed arene–arene interactions with Trp279 and showed hydrophobic interactions with Phe330, Phe331 and Tyr 334 (Fig. 11b and Table 9).
 | ||
| Fig. 11 (A) Binding mode of 4c (green, stick) and native ligand (brown, stick) within the active site. (B) Interactions of compound 4c (green) and the key residues (purple), H-bond represented as a black-dotted line. | ||
Compound 4d showed a moderate binding mode (binding score of −8.1 kcal mol−1) as the chromen moiety moved a little away from the binding site (Fig. 12a). However, compound 4d showed good interactions with the active site residue; the oxygen atom of 4-(methyl-2-oxo-2H-chromen-7-yl)oxy formed a H-bond with OH-Tyr121 at distance (3 Å), and CO of benzoic acid formed a H-bond with Tyr130 at distance (2.2 Å) (Fig. 11b and Table 9). The chromen moiety showed arene–arene interactions with Trp279 and hydrophobic interactions with Phe330 and Phe 331.
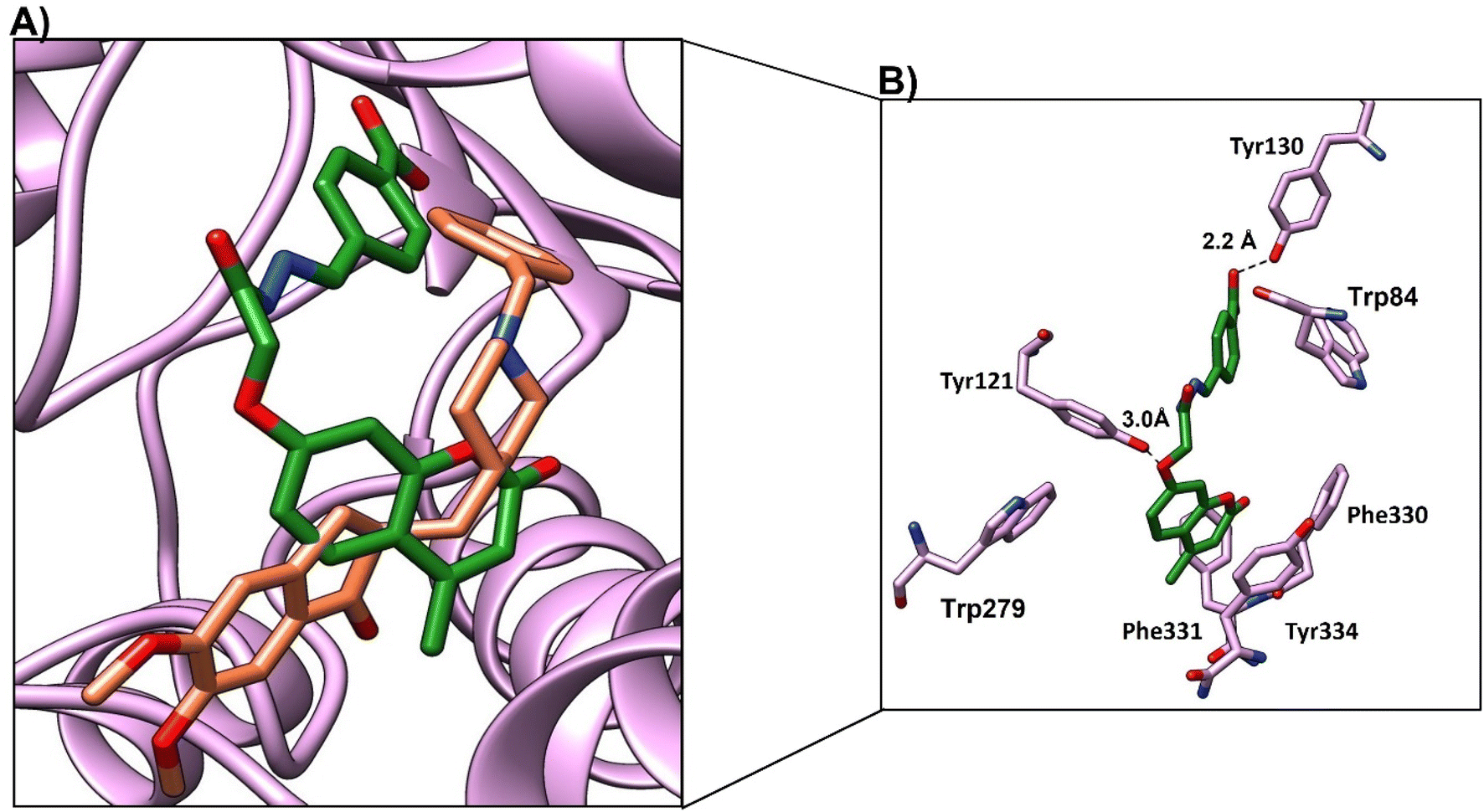 | ||
| Fig. 12 (A) Binding mode of 4d (green, stick) and native ligand (brown, stick) within the active site. (B) Interactions of compound 4d (green) and key residues (purple), H-bond represented as a black-dotted line. | ||
In conclusion, the four compounds showed good binding modes and nearly similar poses within the active site of the cholinesterase enzyme. However, compound 4c showed the best binding mode and the best binding score, which indicates the stability of the compounds within the acetylcholine esterase pocket. This result may explain the promising activity of 4c. All compounds showed good interactions with Tyr121, Tyr130, and hydrophobic residues Phe330 and Phe 331. Besides, the arene–arene interactions with Trp84 and trp279.
The drug-likeness, Lipinski's, and ADM/Tox (absorption, distribution, metabolism, and toxicology) properties of compound 4c were assessed using SwissADME, pkCSM,44 and Data Warrior.
It was observed that compound 4c followed Lipinski's rule (compound 4c showed molecular Clogp < 5, molecular weight ≤500, H-bonds acceptor <10, and H-bonds donor <5) and showed total polar surface area = 80.90 Å2 which was ≤140 Å2. These results indicate 4c has good oral bioavailability and absorbance. Also, compound 4c passed the PAINS filter, and the synthetic accessibility of 4c is 3.54, which means it is an easily synthesized compound (Table 10).
a
| Drug likeness parameters | Compound 8 |
|---|---|
| a MW = molecular weight, logPo/w = lipophilicity, HBD = hydrogen bond donor, HBA = hydrogen bond acceptor, NRB = no. of rotatable bonds, MR = molecular refractivity, SA = synthetic accessibility (from 1 (very easy) to 10 (very difficult)) TPSA = topological polar surface area, good ≤140 Å2. |
|
| Smile | [O+]1c2c(CC1c1nn(cc1C1C(C#N)C(O)N(N)C(N)C1C#N)-c1ccccc1)cccc2 |
| MW | 362.39 |
| logPo/w |
2.95 |
| HBD | 1 |
| HBA | 5 |
| NRB | 4 |
| MR | 102.39 |
| TPSA | 80.90 Å2 |
| Lipinski | Yes (no violation) |
| Bioavailability score | 3.54 |
| PAINS filtrate | 0 alert |
| Synthetic accessibility | 3.54 |
Table 11 summarized the absorption, distribution, metabolism, and excretion of compound 4c. It was observed that compound 4c has good human intestinal absorption (92.5%), moderate Caco-2 permeability (0.82 cm s−1), and low skin permeability, which indicates that compound 4c is not suitable for transdermal preparations.
a
| a HIA = human intestinal absorption [0–20 (poor), 20–70 (moderate), 70–100 (well)], Caco2 = In vitro Caco2 cell permeability, SP = skin permeability, BBB = blood–brain barrier permeability. | |
|---|---|
| Absorption | |
| Model name | Predicted value |
| Water solubility | −3.823 mol L−1 |
| Human intestinal absorption (HIA) | 92.5 |
| Caco2 permeability | 0.82 cm s−1 |
| Skin permeability | −2.69 |
|
|
| Distribution | |
| BBB permeability | −0.21 |
| CNS permeability | −2.18 |
|
|
| Metabolism | |
| CYP1A2 inhibitor | Yes |
| CYP2D6 inhibitor | No |
| CYP3A4 substrate | Yes |
|
|
| Excretion | |
| Total clearance | 0.87 mL min−1 kg−1 |
| Renal OCT2 substrate | No |
Cytochrome P450 is an important detoxification enzyme in humans, many drugs are activated or inhibited by cytochrome P450. It was found that compound 4c is a CYP1A2 and CYP2D6 inhibitor. At the same time, compound 4c is predicted to be deactivated by the CYP3A4 enzyme. The total clearance rate of compound 4c is 0.87 mL min−1 kg−1. Toxicity evaluation of compound 4c showed it is not tumorigenic, or mutagenic, and it is not a skin irritant, which indicated it has a safe profile. Also, it has a high reproductive effect.
In conclusion, compound 4c has good binding within the active site, which is considered the most stable binding mode among other compounds. Also, compound 4c showed the lowest binding energy score. The pharmacokinetic profile showed that it followed Lipinski's rule and had a good total polar surface area value, which revealed its good oral bioavailability. The pharmacokinetic profile of compound 4c showed its high human intestinal absorption. The CYP3A4 enzyme is responsible for the deactivation of compound 4c. Table 12 showed that compound 4c is not mutagenic, not tumorigenic, and not an irritant. Fortunately, it revealed a safe toxicity profile.
| Test | Carcinogenicity (mouse) |
|---|---|
| Mutagen | None |
| Tumorigenic | None |
| Irritant | None |
| Reproductive effect | High |
4. Conclusion
This study represents the design and synthetic pathway for the synthesis of new 2-oxo-coumarin-7-oxymethylene acetohydrazide derivatives 4a–d. The chemical structures of the new analogues were confirmed via IR, 1H-NMR, 13C-NMR, and Mass spectra. The newly synthesized compounds were evaluated as acetylcholinesterase (AChE) inhibitors and antioxidant agents in comparison to donepezil and ascorbic acid, respectively, as reference drugs. Compound 4c represented the most promising AChE inhibitory impact, with an IC50 value of 0.802 μM and DPPH scavenging activity of 57.14 ± 2.77%. Moreover, the biochemical and haematological studies showed that compound 4c produced an insignificant change in the blood profile levels, the hepatic enzyme levels (AST, ALT, and ALP-1), and the total urea between the control and 4c-treated rats. Furthermore, normal architecture of hepatic lobules and normal renal parenchyma, as well as no histopathological damage in the examined hepatic, kidney, heart, and brain tissues, were obtained by the histopathological studies in 4c-treated rats. In addition, the T-maze test investigated that AD-groups treated with 4c exhibited a remarkable reduction in the time taken by the rats to reach food in comparison to donepezil, with percentages of improvement reaching 148.37% compared to the standard drug (181.70%). Also, 4c detectably ameliorated MDA and GSH, reaching 90.64 and 27.17%, respectively, in comparison to the standard drug, which recorded improvement percentages of 90.64% and 35.03% for MDA and GSH, respectively. Also, compound 4c increased the expression of Bcl-2 gene and down-regulated the expression Bax and Tau genes in the brain samples of AD-induced rats treated with compound 4c in a similar manner to that obtained by the reference drug donepezil.In silico studies showed that compound 4c has a promising binding mode within the active site of acetylcholine esterase and is considered the most stable binding mode among other compounds 4a, 4b, and 4d. Also, compound 4c exhibited the lowest binding energy score. In addition, the pharmacokinetic profile showed that compound 4c followed Lipinski's rule, revealing its good oral bioavailability, its high human intestinal absorption, and that the CYP3A4 enzyme is responsible for the deactivation of compound 4c. Compound 4c is not mutagenic, not tumorigenic, and not an irritant. Fortunately, it revealed a safe toxicity profile. Accordingly, the coumarin analogue 4c could be considered a basic nucleus in the field of drug discovery for new multi-targeted directed ligands against Alzheimer.
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of National Research Centre, Dokki, Giza, Egypt (NRC) and Experiments were approved by the Animal Ethics Committee of The Medical Research Ethics Committee (MREC) of NRC no. 19301.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed through the project “Molecular Simulation to Explore Therapeutic Efficacy of Novel Compounds for Prevention and Management of Alzheimer's Disease” funded by the Egyptian Academy of Scientific Research and Technology under an agreement programme between the academy and the Indian Academy of Science.References
- R. L. Frozza, V. Mychael, M. V. Lourenco and F. G. De Felice, Front. Neurosci., 2018, 12, 1–13 CrossRef PubMed.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 4, 306–318 CrossRef PubMed.
- R. J. O'brien and P. C. Wong, Annu. Rev. Neurosci., 2011, 34, 185–204 CrossRef PubMed.
- B. P. Greenwood, V. Tolstikov, P. P. Narain, J. Chaufty, V. R. Akmaev, V. V. ishnudas, S. Gesta, E. J. Nestler, R. Sarangarajan, N. R. Narain and M. A. Kiebish, Handbook of Biomarkers and Precision Medicine, Chapman and Hall/CRC, 1st edn, 2019, 529–537 Search PubMed.
- Z. Breijyeh and R. Karaman, Molecules, 2020, 25(24), 5789 CrossRef CAS PubMed.
- G. Weinstein and S. Seshadri, Alzheimer's Res. Ther., 2014, 6(1), 1–10 CrossRef PubMed.
- M. Pandareesh, T. Anand and F. Khanum, Neurochem. Res., 2016, 41, 985–999 CrossRef CAS PubMed.
- N. George, B. Al Sabahi, M. AbuKhader, K. Al Balushi, M. J. Akhtar and S. A. Khan, J. King Saud Univ., Sci., 2022, 34(4), 101977 CrossRef.
- S.-Y. Lee, Y.-J. Chiu, S.-M. Yang, C.-M. Chen, C.-C. Huang, G.-J. Lee-Chen, W. Lin and K.-H. Chang, CNS Neurosci. Ther., 2018, 24(12), 1286–1298 CrossRef CAS PubMed.
- A. Stefanachi, F. Leonetti, L. Pisani, M. Catto and A. Carotti, Molecules, 2018, 23(2), 250 CrossRef PubMed.
- F. Annunziata, C. Pinna, S. Dallavalle, L. Tamborini and A. Pinto, Int. J. Mol. Sci., 2020, 21(13), 4618 CrossRef CAS PubMed.
- G. Moya-Alvarado, O. Yañez, N. Morales, A. González-González, C. Areche, M. T. Núñez, A. Fierro and O. García-Beltrán, Molecules, 2021, 26(9), 2430 CrossRef CAS PubMed.
- E. Jameel, T. Umar, J. Kumar and N. Hoda, Chem. Biol. Drug Des., 2016, 87(1), 21–38 CrossRef CAS PubMed.
- K. M. Amin, D. E. Rahman, H. A. Allam and H. H. El-Zoheiry, Bioorg. Chem., 2021, 110, 104792 CrossRef CAS PubMed.
- A. Rehman, S. Magsi, M. A. Abbasi, S. Rasool, A. Malik, G. Hussain, M. Ashraf and N. Khalid, Pak. J. Pharm. Sci., 2014, 27, 271–278 Search PubMed.
- S. Ghanei-Nasab, M. Khoobi, F. Hadizadeh, A. Marjani, A. Moradi, H. Nadri, S. Emami, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2016, 121, 40–46 CrossRef CAS PubMed.
- S. Hamulakova, L. Janovec, M. Hrabinova, K. Spilovska, J. Korabecny, P. Kristian, K. Kuca and J. Imrich, J. Med. Chem., 2014, 57(16), 7073–7084 CrossRef CAS PubMed.
- S. M. Bagheri, M. Khoobi, H. Nadri, A. Moradi, S. Emami, L. J. Baleh, F. Jafarpour, F. H. Moghadam, A. Foroumadi and A. Shafiee, Chem. Biol. Drug Des., 2015, 86(5), 1215–1220 CrossRef CAS PubMed.
- A. S. Alpan, S. Parlar, L. Carlino, A. H. Tarikogullari, V. Alptüzün and H. S. Güneş, Synthesis, biological activity and molecular modeling studies on 1H-benzimidazole derivatives as acetylcholinesterase inhibitors, Bioorg. Med. Chem., 2013, 21(17), 4928–4937 CrossRef CAS PubMed.
- Y. M. Syam, S. S. El-Karim, T. Nasr, S. A. Elseginy, M. M. Anwar, M. M. Kamel and H. F Ali, Bioorg. Med. Chem., 2019, 19(3), 250–269 CAS.
- G. L. Ellman, D. K. Courtney, V. Andreas and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- I. Gulcin, H. A. Alici and M. Cesur, Chem. Pharm. Bull., 2005, 53(3), 281–285 CrossRef PubMed.
- S. Y. Desoukey, W. M. El Kady, A. A. Salama, E. G. Hagag, S. M. El-Shenawy and M. El-Shanawany, Int. J. Pharmacogn. Phytochem. Res., 2016, 8(7), 1121–1131 Search PubMed.
- J. D. Bancroft, Bancroft's Theory and Practice of Histological Techniques E-Book, ed. K. S. Suvarna, C. Layton and J. D. Bancroft, 2019 Search PubMed.
- S. Reitman and S. Frankel, Am. J. Clin. Pathol., 1957, 28(1), 56–63 CrossRef CAS PubMed.
- A. Belfield and D. Goldberg, Enzyme, 1971, 12, 561–573 CrossRef CAS PubMed.
- H. Bartels, M. Böhmer and C. Heierli, Clin. Chim. Acta, 1972, 37, 193–197 CrossRef CAS PubMed.
- J. K. Fawcett and J. E. A. Scott, J. Clin. Pathol., 1960, 13, 156–1591 CrossRef CAS PubMed.
- M. A. Thrall, D. C. Baker, and E. D Lassen, Veterinary Hematology and Clinical Chemistry, Lippin-cott Williams and Wilkins, Philadelphia, USA, 2004 Search PubMed.
- M. Kawahara, M. Kato-Negishi and K. Tanaka, Metallomics, 2017, 9, 619–633 CrossRef CAS PubMed.
- R. M. J. Deacon and J. N. P. Rawlins, Nat. Protoc., 2006, 1(1), 7–12 CrossRef PubMed.
- E. Y. Pioli, B. N. Gaskill, G. Gilmour, M. D. Tricklebank, S. L. Dix, D. Bannerman and J. P. Garner, Brain Res., 2014, 261, 249–257 Search PubMed.
- M. Altun, E. Bergman, E. Edström, H. Johnson and B. Ulfhake, Physiol. Behav., 2007, 92(5), 911–923 CrossRef CAS PubMed.
- E. Beutler, O. Duron and B. M. Kelly, Clin. Med., 1963, 61, 882–888 CAS.
- H. Ohkawa, N. Ohishi and K. Yagi, Anal. Biochem., 1979, 95, 351–358 CrossRef CAS PubMed.
- M. Callens, N. Kraskovskaya, K. Derevtsova, W. Annaert, G. Bultynck, I. Bezprozvanny and T. Vervliet, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868(6), 118997 CrossRef CAS PubMed.
- H. Borai, M. K. Ezz, M. Z. Rizk, H. F. Aly, M. El-Sherbiny, A. A. Matloub and G. I. Fouad, Biomed. Pharmacother., 2017, 93, 837–851 CrossRef PubMed.
- A. H. Elmaidomy, U. R. Abdelmohsen, F. Alsenani, H. F. Aly, S. G. E Shams, E. A. Younis, K. A. Ahmed, A. M. Sayed, A. I. Owis, N. Afifi and D. El Amir, RSC Adv., 2022, 12, 11769–11785 RSC.
- H. Aly, N. Elrigal, S. Ali, M. Rizk and N. Ebrahim, J. Mater. Environ. Sci., 2018, 9, 1931–1941 CAS.
- M. Z. Rizk, I. H. Borai, M. K. Ezz, M. El-Sherbiny, H. F. Aly, A. Matloub and G. I. Fouad, J. Mater. Environ. Sci., 2018, 9(7), 2098–2108 CAS.
- S. A. Elseginy and M. M. Anwar, ACS Omega, 2021, 7(1), 150–1164 Search PubMed.
- S. A. Elseginy, J. Biomol. Struct. Dyn., 2022, 40(24), 13658–13674 CrossRef CAS PubMed.
- T. Matsubara, E. Yoshihara, T. Iwata, Y. Tochino and Y. Hachino, Biotransformation of coumarin derivatives (1) 7-alkoxycoumarin O-dealkylase in liver microsomes, Jpn. J. Clin. Pharmacol. Ther., 1982, 32(1), 9–21 CAS.
- D. E. Pires, T. L. Blundell and D. B. Ascher, pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures, J. Med. Chem., 2015, 58(9), 4066–4072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02344c |
| This journal is © The Royal Society of Chemistry 2023 |