
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards superior mRNA caps accessible by click chemistry: synthesis and translational properties of triazole-bearing oligonucleotide cap analogs†
Mateusz Kozarskiab,
Karolina Drazkowska b,
Marcelina Bednarczykab,
Marcin Warminskia,
Jacek Jemielity*b and
Joanna Kowalska*a
b,
Marcelina Bednarczykab,
Marcin Warminskia,
Jacek Jemielity*b and
Joanna Kowalska*a
aDivision of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland. E-mail: jkowalska@fuw.edu.pl
bCentre of New Technologies, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland. E-mail: j.jemielity@cent.uw.edu.pl
First published on 25th April 2023
Abstract
Messenger RNA (mRNA)-based gene delivery is a powerful strategy for the development of vaccines and therapeutics. Consequently, approaches that enable efficient synthesis of mRNAs with high purity and biological activity are in demand. Chemically modified 7-methylguanosine (m7G) 5′ caps can augment the translational properties of mRNA; however, efficient synthesis of structurally complex caps, especially on a large scale, is challenging. Previously, we proposed a new strategy to assemble dinucleotide mRNA caps by replacing the traditional pyrophosphate bond formation by copper-catalyzed azide–alkyne cycloaddition (CuAAC). Here, we used CuAAC to synthesize 12 novel triazole-containing tri- and tetranucleotide cap analogs with the aim of exploring the chemical space around the first transcribed nucleotide in mRNA and overcoming some of the limitations previously reported for the triazole-containing dinucleotide analogs. We evaluated the efficiency of incorporation into RNA for these analogs and their influence on the translational properties of in vitro transcribed (IVT) mRNAs in rabbit reticulocyte lysate and JAWS II cultured cells. The incorporation of the triazole moiety within the 5′,5′-oligophosphate of trinucleotide cap produced compounds that were well incorporated into RNA by T7 polymerase while replacing the 5′,3′-phosphodiester bond with triazole impaired incorporation and translation efficiency, despite a neutral effect on the interaction with the translation initiation factor eIF4E. One of the compounds (m7Gppp-tr-C2H4pAmpG), had translational activity and other biochemical properties comparable to natural cap 1 structure, thus being a promising mRNA capping reagent for potential in cellulo and in vivo applications in the field of mRNA-based therapeutics.
Introduction
The 5′ end of eukaryotic messenger RNA (mRNA) terminates with a unique structure called the 5′ cap. The 5′ cap consists of a positively charged 7-methylguanosine (m7G) linked to the first transcribed nucleotide via a 5′,5′-triphosphate bridge.1 The 5′ cap plays crucial roles in many processes in eukaryotic cells, including mRNA maturation, transport, and turnover.2 Moreover, the 5′ cap protects mRNA against premature degradation by 5′-exonucleases3 and facilitates the initiation of protein biosynthesis by enabling recognition of the 5′ end of mRNA by the eukaryotic translation initiation complex 4F (eIF4F), wherein eIF4E is the cap-binding protein.4 The 5′ cap also serves as a tag preventing the recognition of RNA by some elements of the innate immune system, thereby enabling the innate immune system to distinguish between cellular and non-self RNA.2m7G cap analogs have been used as reagents for modifying the 5′ end of mRNA with the aim of designing more potent mRNA-based therapeutics or molecular tools for biological studies.5–8 Chemical modification of the 5′,5′-triphosphate bridge in the m7G cap may increase mRNA half-life and its affinity for eIF4E, resulting in higher protein output.9 To date, several different mRNA cap analogs have been developed with the aim of improving mRNA product quality and translational properties.10,11 However, recent advances in mRNA-based vaccines12–14 have highlighted the demand for synthetic m7G cap analogs that, besides possessing superior biological properties, can be synthesized in bulk. Chemical synthesis of m7G cap analogs is challenging owing to the instability of the positively charged m7G under both acidic and alkaline conditions.15 Additionally, the usual ZnCl2-mediated coupling reaction to form a pyrophosphate bond based on P-imidazolide of 7-methylguanosine diphosphate as the key reagent may be inefficient, time-consuming, and difficult to upscale.16 Therefore, new, faster, and more robust approaches for the synthesis of m7G cap analogs need to be explored.
For addressing this problem, we previously synthesized derivatives of the m7GpppG dinucleotide cap analog, bearing a triazole modification within the 5′,5′-triphosphate bridge, which can be efficiently assembled from two mononucleotide subunits using copper-catalyzed azide–alkyne cycloaddition (CuAAC).17 CuAAC, being the prime example of a ‘click reaction’,18 provides shorter reaction times and minimizes the amount of reagents and organic solvents used and by-products generated during mRNA cap synthesis. After screening multiple caps carrying different triazole-containing polyphosphates, we identified compounds that had a high affinity for eIF4E and improved the efficiency of protein biosynthesis in vitro and in cultured cells.17,19 Unfortunately, we also found several compounds that did not confer favorable translational properties, despite their fairly stable interaction with eIF4E. Notably, the presence of the triazole moiety between the 5′,5′-triphosphate and the 5′ carbon of the first transcribed nucleotide decreased the efficiency of incorporation of the cap analog during transcription initiation, and consequently, decreased capping efficiency of the mRNA product.17 Therefore, such compounds were determined to be unsuitable for mRNA-related application. For instance, m7Gppp-tr-G (Fig. 1) showed higher affinity for eIF4E (KAS = 17.1 ± 1.1 μM−1) than that showed by unmodified m7GpppG (KAS = 12.5 ± 0.3 μM−1). Moreover, capping efficiency of m7Gppp-tr-G was high (79%); however, because of the location of the triazole ring, the compound incorporated into mRNA in the reverse orientation, resulting in a non-functional product. The anti-reverse analog (ARCA) carrying the same modification, m27,2′-OGppp-tr-G, was free of this limitation, but provided unacceptably low RNA capping efficiency (17%).17 Later, an improved version of the triazole-modified dinucleotide cap carrying a triazole-modified tetraphosphate chain, namely m27,2′-OGppp-tr-C2H4pG, was reported (Fig. 1).19 m27,2′-OGppp-tr-C2H4pG showed high affinity for eIF4E (KAS = 49.9 ± 1.3 μM−1) and good chemical stability. The efficiency of translation of mRNA capped with m27,2′-OGppp-tr-C2H4pG was higher than that of mRNA capped with ARCA-capped RNA. However, capping efficiency of m27,2′-OGppp-tr-C2H4pG was notably lower than that observed with reference caps.19
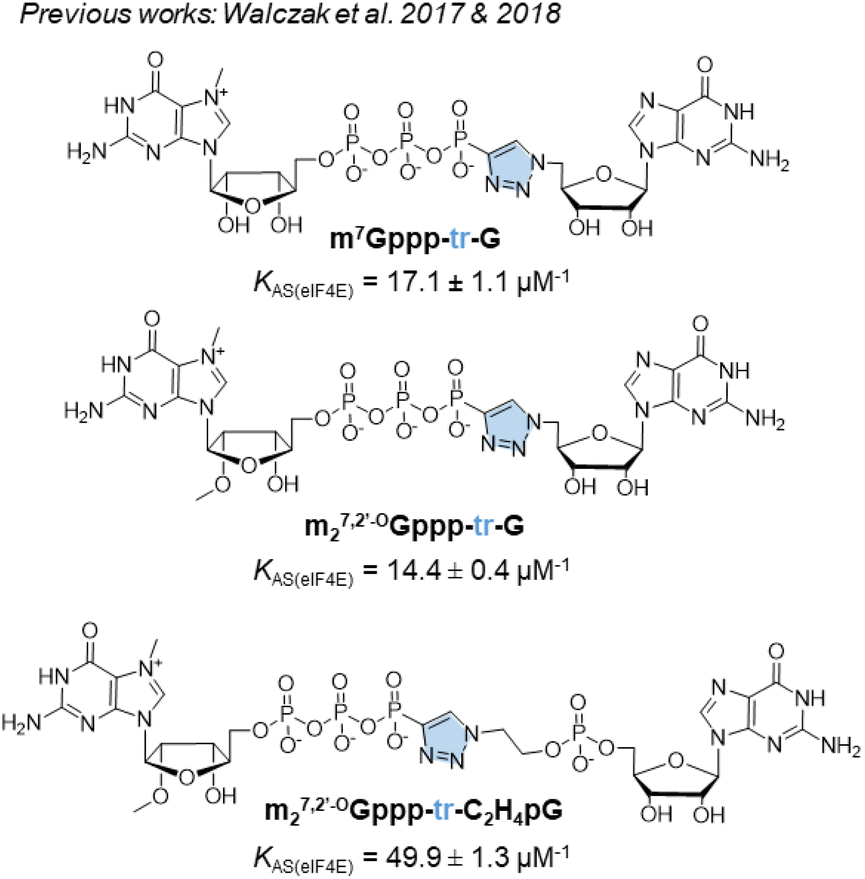 | ||
| Fig. 1 Structures of previously synthesized phosphotriazole dinucleotide cap analogs which have been used as a template in this work.17,19 | ||
In this study, to further explore the potential of triazole-modified capping reagents and address some of the previously identified issues, we developed a novel class of triazole-containing tri- and tetranucleotide cap analogs (Fig. 2). Previous studies have shown that trinucleotide cap analogs based on the general structure of m7GpppNpG initiate transcription from templates containing class III T7 promoter, similar to dinucleotide derivatives of m7GpppG.7,11,20 In the case of dinucleotide cap analogs, RNA polymerase initiates transcription by pairing G in the cap and C in the DNA template. In the case of m7GppApG-like trinucleotides, the transcription is initiated by pairing between G and C at +1 position and additional pairing between A and T at the −1 position in the DNA template. This additional pairing promotes transcription initiation with the cap analog instead of GTP, increasing capping efficiency and reducing the frequency of incorrect (reverse) cap incorporation into RNA, compared to dinucleotides.7 Additionally, the trinucleotide design enables incorporation of epigenetic methylations at the 2′-O position and within the nucleobase of the first transcribed nucleotide, which is impossible with dinucleotide cap analogs. As such, in this study, we explored whether a similar approach can be employed to augment the incorporation of triazole-modified cap analogs into the 5′ end of mRNA. To this end, we developed efficient synthetic pathways for azido- and alkyne-functionalized mono- and dinucleotide subunits, which can be combined in the CuAAC to efficiently form tri- or tetranucleotide caps carrying a triazole moiety within the 5′,5′-oligophosphate bridge or instead of the 5′,3′-phosphodiester bond. The obtained novel cap analogs were incorporated into mRNA encoding Gaussia luciferase by in vitro transcription, and translational properties of capped transcripts were examined in rabbit reticulocyte lysate (RRL) and in cultured cells (JAWS II) to identify structures that were efficient transcription initiators and conferred superior biological properties to mRNA.
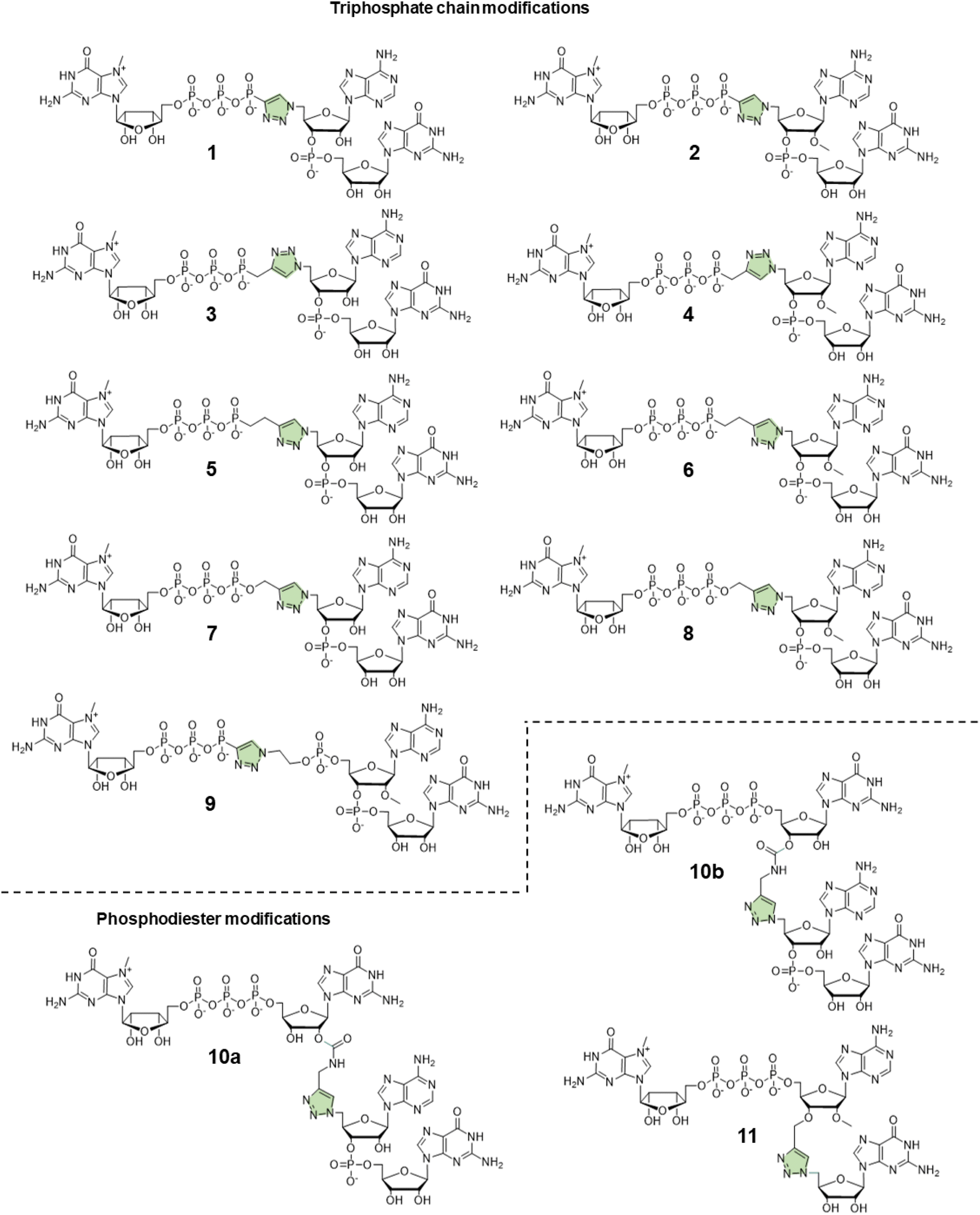 | ||
| Fig. 2 Structures of phosphotriazole trinucleotide cap analogs synthesized in this work. | ||
Results and discussion
Design and synthesis of novel trinucleotide cap analogs bearing triazole moiety
Dinucleotides bearing an azide moiety at the 5′ position of ribose, namely compounds 12a–c, were synthesized through a solid-phase synthesis approach. We synthesized three types of clickable 5′-azidodinucleotides, providing access to cap 0 and cap 1 type RNA ends (Fig. 3).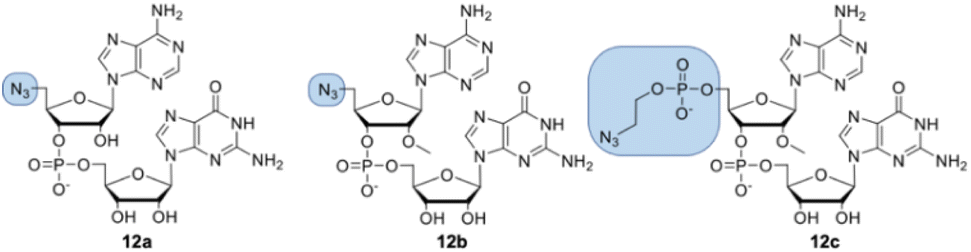 | ||
| Fig. 3 Structures of 5′-azido modified dinucleotides (compounds 12a, 12b, and 12c). | ||
The dinucleotides were synthesized using phosphoramidite approach based on adenosine 2′-O-PivOM-phosphoramidites (Scheme 1).21 In the case of compounds 12a and 12b, the azido group was incorporated on a solid support by treating the 5′-OH deprotected compound with methyltriphenoxyphosphonium iodide, followed by incubation in a saturated solution of sodium azide in DMF.22 After the azidation step, the final dinucleotides 12a and 12b were cleaved from the support, and protecting groups were removed by incubating the compounds in ammonium/methylamine solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) (Scheme 1A). To synthesize dinucleotide 12c, an additional coupling step with 2-cyanoethyl 2-bromoethyl N,N-diisopropylamino-phosphite was performed after 5′-OH deprotection. The obtained dinucleotide carrying 2-bromoethyl phosphoester was converted into 2-azidoethyl phosphoester by treatment with NaN3 in DMF and cleaved from the resin (Scheme 1B). Subsequently, all dinucleotides were isolated by ion-exchange chromatography as triethylammonium salts and used in CuAAC reactions to obtain triazole-bearing trinucleotide cap analogs. Trinucleotide cap analogs 1–9 were synthesized via CuAAC between dinucleotides containing 5′-azido group and m7G 5′-triphosphate analogs containing different terminal alkynes at the γ-phosphate.17,23 Typically, an aqueous solution of 5′-azidodinucleotide (1 equivalent) was mixed with a 5′-triphosphate 7-methylguanosine analog containing alkyne moiety (1–2 equivalents) dissolved in TEA/CH3COOH buffer (pH 7.5), followed by the addition of an aqueous solution of THPTA/CuSO4 (0.5 equivalents) and excess sodium ascorbate. The reaction mixture was mixed for 15–60 minutes and quenched with an aqueous solution of Na2EDTA (Scheme 2A).
:1 v/v) (Scheme 1A). To synthesize dinucleotide 12c, an additional coupling step with 2-cyanoethyl 2-bromoethyl N,N-diisopropylamino-phosphite was performed after 5′-OH deprotection. The obtained dinucleotide carrying 2-bromoethyl phosphoester was converted into 2-azidoethyl phosphoester by treatment with NaN3 in DMF and cleaved from the resin (Scheme 1B). Subsequently, all dinucleotides were isolated by ion-exchange chromatography as triethylammonium salts and used in CuAAC reactions to obtain triazole-bearing trinucleotide cap analogs. Trinucleotide cap analogs 1–9 were synthesized via CuAAC between dinucleotides containing 5′-azido group and m7G 5′-triphosphate analogs containing different terminal alkynes at the γ-phosphate.17,23 Typically, an aqueous solution of 5′-azidodinucleotide (1 equivalent) was mixed with a 5′-triphosphate 7-methylguanosine analog containing alkyne moiety (1–2 equivalents) dissolved in TEA/CH3COOH buffer (pH 7.5), followed by the addition of an aqueous solution of THPTA/CuSO4 (0.5 equivalents) and excess sodium ascorbate. The reaction mixture was mixed for 15–60 minutes and quenched with an aqueous solution of Na2EDTA (Scheme 2A).
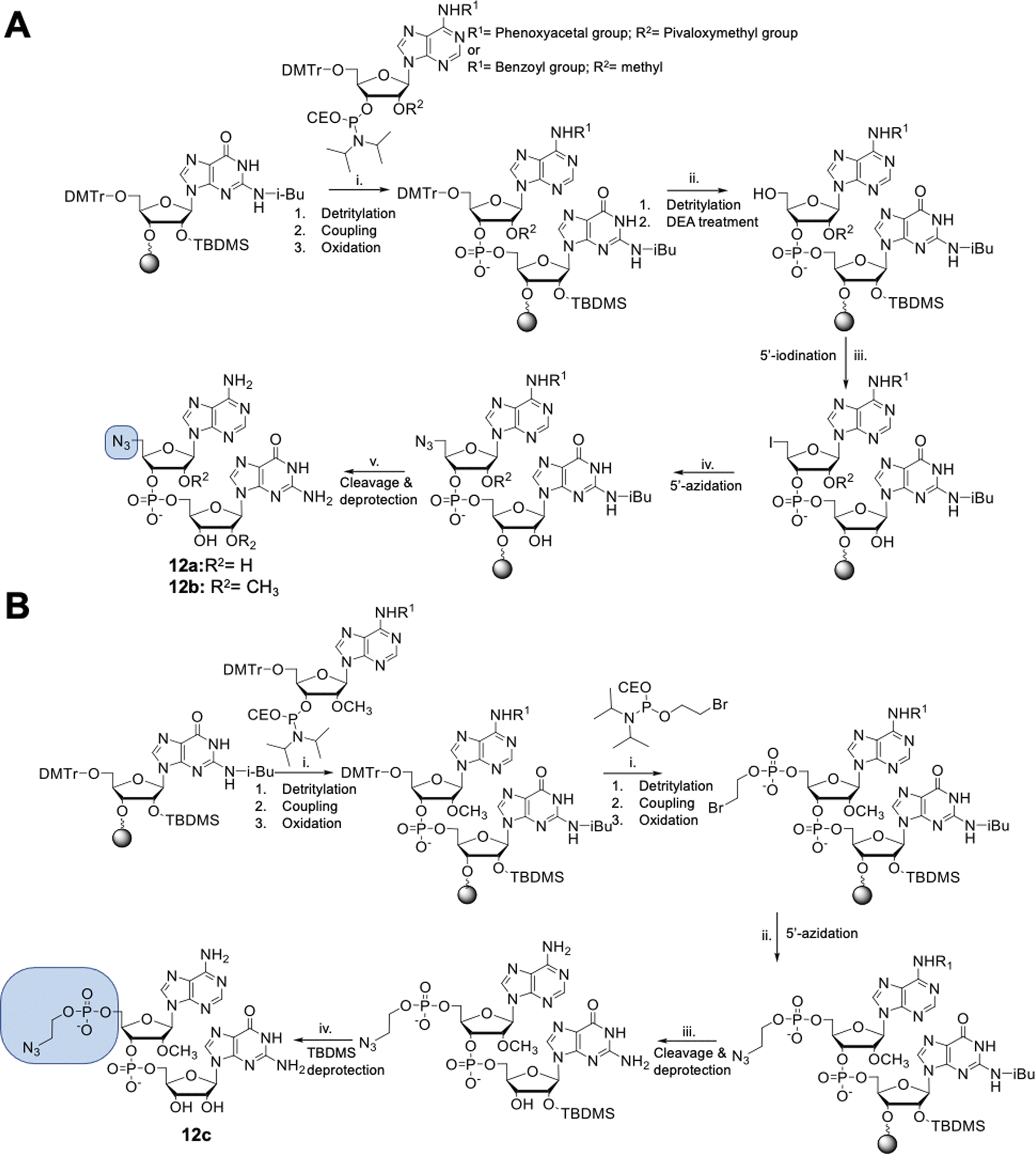 | ||
| Scheme 1 Solid phase synthesis of 5′-azido modified dinucleotides. (A) Solid phase synthesis of compounds 12a and 12b; (i) (1) 3% trichloroacetic acid in dichloromethane, (2) 2 equivalents of phosphoramidites in acetonitrile 0.2 M and BTT Activator 0.3 M, (3) 50 mM iodine solution in pyridine/water (9:1, v/v); (ii) (1) 3% trichloroacetic acid in dichloromethane, (2) 20% (v/v) diethylamine in acetonitrile; (iii) 0.6 M (PhO)3PCH3I in anhydrous DMF, 15 minutes, rt; (iv) saturated solution of NaN3 in DMF, 1 hour, 60 °C; (v) 33% ammonium hydroxide and 40% methylamine in water (1/1, v/v), 1 hour, 50 °C; (B) synthesis of compound 12c; (i) (1) 3% trichloroacetic acid in dichloromethane, (2) 2 equivalents of phosphoramidites in acetonitrile 0.2 M and BTT Activator 0.3 M, (3) 50 mM iodine solution in pyridine/water (9:1, v/v); (ii) saturated solution of NaN3 in DMF, 1 hour, 60 °C; (iii) 33% ammonium hydroxide and 40% methylamine in water (1/1, v/v), 1 hour, 50 °C; (iv) TEA, TEA*3HF, DMSO, 3 h, 65 °C. | ||
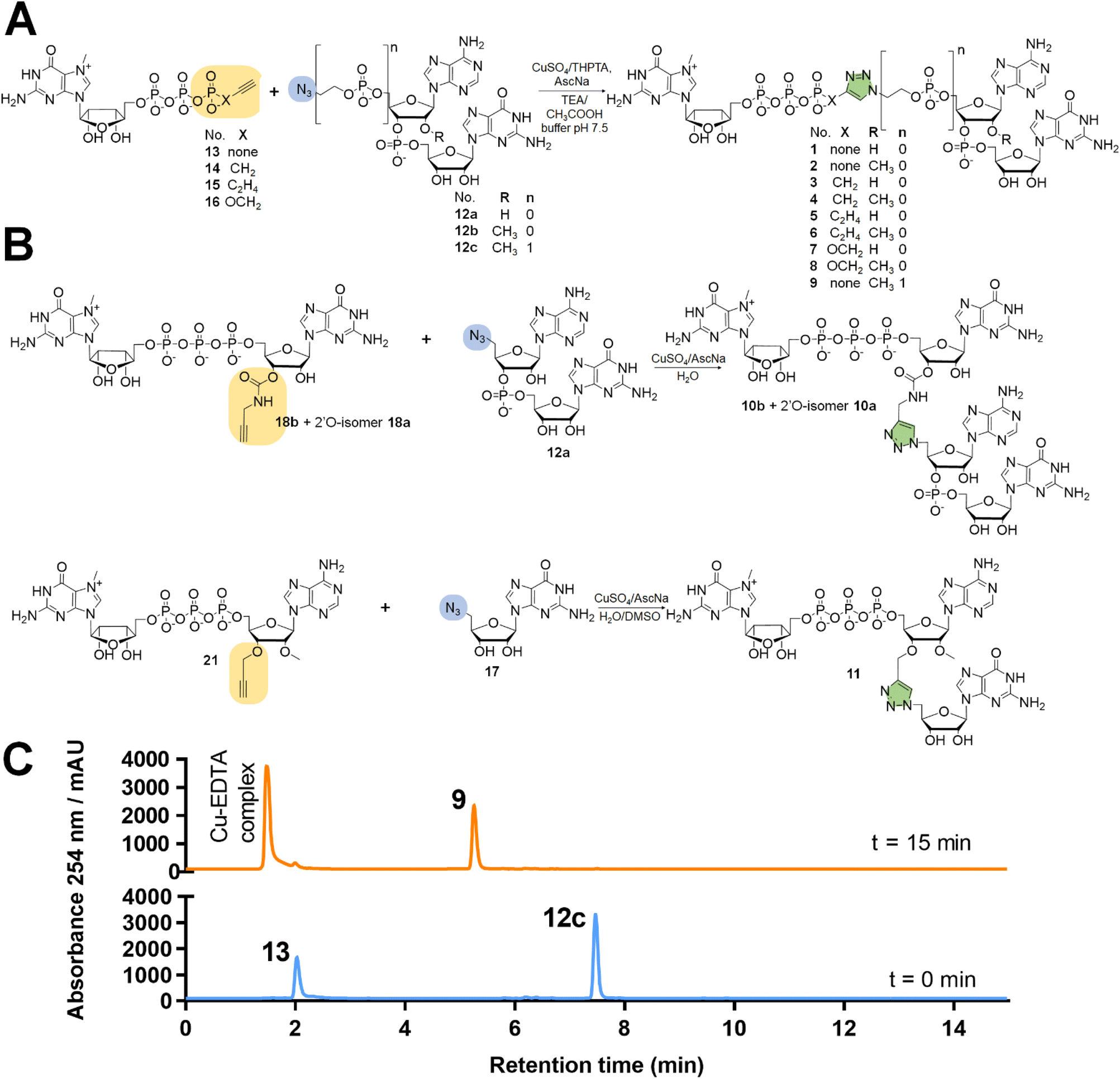 | ||
| Scheme 2 Synthesis of tri- and tetranucleotide cap analogs by CuAAC. (A) Synthesis of trinucleotide cap analogs with phosphate bridge modification. (B) Synthesis of tri- and tetranucleotide cap analogs with phosphodiester modification. (C) Representative RP-HPLC chromatogram obtained for the synthesis of compound 9. | ||
Tri- and tetranucleotide cap analogs in which the phosphodiester bond was replaced with a triazole moiety, namely compounds 10a, 10b, and 11, were synthesized via different routes (Scheme 2B). For this purpose, we synthesized 2′- and 3′-alkyne functionalized cap analogs and linked them to 5′-azidonucleoside 17 or dinucleotide 12a via CuAAC to obtain functional triazole-bearing tri- or tetranucleotide cap analogs.
To obtain tetranucleotide cap analogs, we used alkyne-functionalized dinucleotides, m7GpppG derivative functionalized with carbamoyl moiety at either the 2′ or 3′ position of guanosine, 18a and 18b.24 The mixture of 18a and 18b (1 equivalent) was reacted with 5′-azidodinucleotide 12a (1 equivalent) in an aqueous solution to obtain a mixture of compounds 10a and 10b. After CuAAC, compounds 10a and 10b were separated using RP-HPLC (Scheme 2B). Finally, we synthesized a trinucleotide cap analog, compound 11, carrying a 3′-OCH2-triazole moiety and methyl group at the 2′ position of adenosine. The starting material was 2′-O-methyl-3′-O-propargyl-adenosine (compound 19), which was obtained in one step by treating commercially available 2′-O-methyl-adenosine with propargyl bromide in the presence of NaH and TBAI.25 Compound 19 was converted into 2′-O-methyl-3′-O-propargyl-adenosine 5′-monophosphate (20) using phosphoryl chloride in trimethyl phosphate.26,27 The product was isolated by ion-exchange chromatography and linked with m7GDP imidazole-derivative using MgCl2-mediated coupling reaction to obtain 3′-alkyne-functionalized m7GpppAm (21). Finally, compounds 21 and 17 (5′-azido-5′-deoxyguanosine) were used to synthesize compound 11 via CuAAC. Compounds 21 and 17 were mixed in dimethyl sulfoxide (DMSO)/water, followed by the addition of copper sulfate (0.3 equivalent) and sodium ascorbate (10 equivalents). The reaction mixture was then stirred for 1 h at 25 °C (Scheme 2B). The reaction progress was monitored using RP-HPLC with an absorption detector (Scheme 2C). The 5′-azido dinucleotides were converted into desired products (compounds 1–9, 10a–b, and 11) in high yields (76–100%), based on RP-HPLC analyses. Triazole-bearing tri- and tetranucleotide cap analogs were isolated from the reaction mixtures by ion-exchange chromatography and by preparative RP-HPLC. The structures and purities of all the compounds were confirmed by HRMS and NMR.
Evaluation of RNA capping efficiency
The efficiency of incorporation of the cap analogs into RNA (capping efficiency) during in vitro transcription was assessed. Short RNA molecules were generated by in vitro transcription in the presence of cap analogs at concentrations 6-fold higher than GTP. The transcripts were purified by HPLC, treated with DNAzyme to reduce 3′-end heterogeneity, and analyzed in a high-resolution polyacrylamide gel (Fig. 4B). Previously reported di- and trinucleotide cap analogs, ARCA, and m7GpppAmpG (cap 1), respectively, were used as reference (Fig. 4B). The expected length of uncapped RNA obtained using this protocol was 25 nt. The length of capped RNA varied from 26 to 28 nt, depending on the pairing scheme with the promoter sequence (Fig. 4A).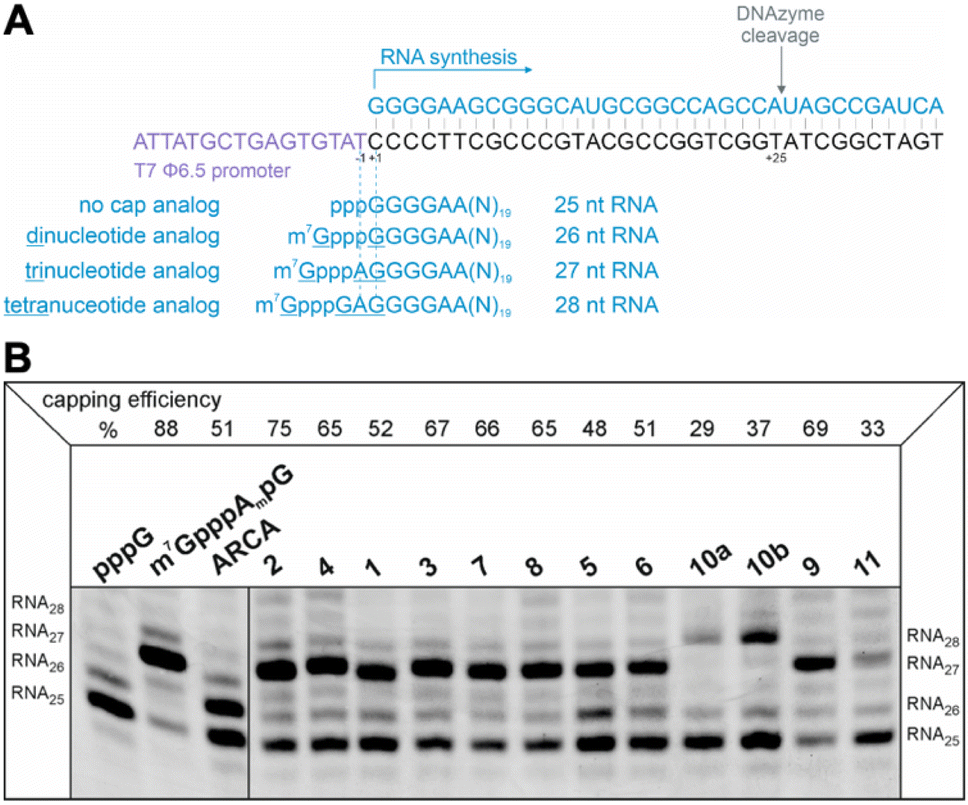 | ||
| Fig. 4 (A) Cap analog pairing with the template sequence which initiates transcription. The particular type of analog incorporated into short RNA, i.e., di-, tri-, or tetranucleotide, defines the length of the final transcript after DNAzyme trimming as 26, 27, or 28 nucleotides, respectively. Control uncapped RNA is 25 nucleotides long. (B) Analysis of capping efficiency. Short RNAs obtained by in vitro transcription from a template containing φ6.5 promoter, capped with different analogs during the reaction (with ATP, CTP, UTP, 2 mM each; 0.5 mM GTP; 3 mM cap analog), were analyzed in 15% PAA after trimming by DNAzyme. Capping efficiency values based on densitometric quantification of bands corresponding to capped and uncapped RNAs in each sample are given above the gel. | ||
Capping efficiency for control trinucleotide analog m7GpppAmpG and dinucleotide analog ARCA was observed to be 88% and 51%, respectively. As expected, control trinucleotide analogs were incorporated into the transcripts more efficiently than control dinucleotide analogs under the same conditions. The capping efficiency of the novel analogs ranged between 29% and 75%. Significantly lower capping efficiencies (compared to compounds 1–9), i.e., 29%, 37%, and 33% were observed for compounds 10a, 10b, and 11, respectively, which carry a triazole moiety in place of the first phosphodiester bond. The lowest capping efficiency was observed for compound 10a, a tetranucleotide, in which triazole moiety was attached to 2′ position of the first transcribed nucleotide. Analog 2 showed the highest capping efficiency (75%) among all the triazole-bearing cap analogs, significantly higher than that of its dinucleotide counterpart (17%).17 Relatively high capping efficiency was also observed for compounds 9 (69%), 3 (67%), 7 (66%), 4 (65%), and 8 (65%); consequently, these analogs seemed to be promising molecules for further testing. Since compounds 10a and 11 showed low capping efficiencies, they were excluded from further analysis.
Exploring the translational properties of mRNA capped with triazole-containing cap analogs
After assessing the capping efficiencies of the novel triazole-bearing oligonucleotide cap analogs, we explored the translational properties of mRNA carrying them. The typical in vitro translation experiment was performed in RRL programmed with mRNA at one of four concentrations (see Experimental section for details). The determined relative translation efficiencies are shown in Fig. 5 and numerical data are shown in Table 1.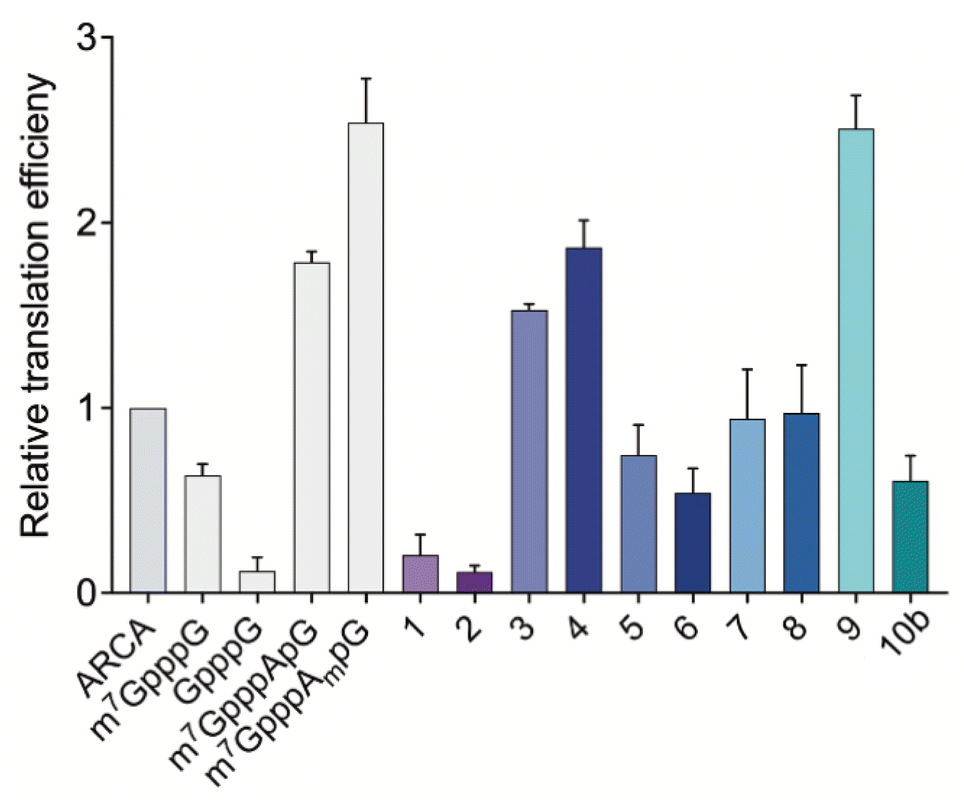 | ||
| Fig. 5 Translation efficiencies for mRNAs capped with novel triazole-bearing trinucleotide cap analogs from IVT reaction with ATP, CTP, UTP, 2 mM each; 0.5 mM GTP; 3 mM cap analog, measured in vitro in rabbit reticulocyte lysate system. Gaussia luciferase luminescence signal was measured as a function of mRNA concentration and the linear regression coefficients were obtained from those dependencies. The coefficients obtained for each capped-mRNA were normalized to the coefficient obtained for ARCA-mRNA to determine relative translational efficiencies. Four different concentrations of mRNA in translation reactions were tested (see Experimental section for details). mRNAs capped with ARCA and other unmodified m7G caps were used as positive controls and GpppG was used as a negative control. Data represent mean value ± SD calculated for three replications. | ||
| Translational properties of capped mRNA | ||
|---|---|---|
| Capped-RNA | RRL | JAWS II |
| Translation efficiency ± SD | Normalized protein output ± SEM | |
| ARCA (m27,2′-OGpppG) | 1 | 1 |
| m7GpppG | 0.64 ± 0.06 | n.d. |
| pppG | n.d. | 0.10 ± 0.09 |
| GpppG | 0.12 ± 0.07 | n.d. |
| m7GpppApG | 1.79 ± 0.06 | n.d. |
| m7GpppAmpG | 2.54 ± 0.24 | 5.93 ± 0.42 |
| 1 | 0.20 ± 0.11 | 0.21 ± 0.09 |
| 2 | 0.12 ± 0.03 | 0.43 ± 0.11 |
| 3 | 1.53 ± 0.03 | 0.22 ± 0.08 |
| 4 | 1.87 ± 0.14 | 0.42 ± 0.19 |
| 5 | 0.75 ± 0.16 | 0.25 ± 0.09 |
| 6 | 0.54 ± 0.13 | 0.21 ± 0.05 |
| 7 | 0.94 ± 0.27 | 0.15 ± 0.05 |
| 8 | 0.97 ± 0.26 | 0.37 ± 0.14 |
| 9 | 2.51 ± 0.18 | 4.79 ± 0.10 |
| 10b | 0.61 ± 0.14 | 0.15 ± 0.07 |
In the RRL experiment, mRNAs capped with previously reported di- and trinucleotide cap analogs, ARCA, m7GpppG, GpppG, and m7GpppApG (cap0) m7GpppAmpG (cap1), were used as references. In contrast to ARCA, m7GpppG can be incorporated into RNA in both the reverse and forward orientations, resulting in non-functional and functional mRNA, respectively.28 Therefore, translation efficiency of m7GpppG-RNA was observed to be 1.3-fold lower than that of ARCA-capped RNA. Relative translation efficiency for GpppG-RNA (negative control) was observed to be 0.11. In contrast to mRNA capped with dinucleotide cap analogs, mRNA capped with trinucleotide cap analogs (m7GpppApG and m7GpppAmpG) were more active in RRL. The translation efficiencies of m7GpppApG-capped RNA and m7GpppAmpG-capped RNA (88% capping efficiency) were 2-fold and 2.5-fold higher, respectively than that of ARCA-capped RNA (51% capping efficiency). Among all the novel triazole-containing cap analogs, the highest translation efficiency (2.51 ± 0.18) was observed for mRNA capped with compound 9, in which the triazole moiety was located within the 5′,5′-tetraphosphate chain between α and β phosphates. The translation efficiency of mRNA bearing this analog was 2.5-fold higher than that of ARCA-capped RNA and was comparable with m7GpppAmpG-capped mRNA, despite lower capping efficiency. mRNA capped with compounds 1–8, in which the triazole moiety was located at the 5′ position of the ribose, showed lower translation efficiency than that observed for mRNA capped with m7GpppAmpG. The lowest translation efficiency was observed for mRNAs capped with compounds 1 and 2 (0.20 ± 0.11 and 0.12 ± 0.03) (Table 1), in which the triazole ring was directly connected to the phosphate group. This finding contrasted with the previous finding that the triazole moiety at this position in the dinucleotide does not disturb the interaction of mRNA with eIF4E.17 This may be attributed to the conformational rigidity of the modified 5′,5′-triphosphate chain that reveals itself after incorporation into RNA, but not for ‘free’ caps. In agreement with this hypothesis, the insertion of a methylene linker between the triazole and phosphate groups in compounds 3 and 4 significantly increased the translation efficiency of mRNA (1.53 ± 0.03 and 1.87 ± 0.14, respectively). The translation efficiency of mRNAs capped with compounds 3 and 4 was nearly 2-fold higher than that of ARCA-capped RNA and comparable with that of m7GpppApG-capped mRNA (1.79 ± 0.06). However, elongating the methylene linker by inserting a second methylene group (compounds 5 and 6) decreased the translation efficiency (0.75 ± 0.16 and 0.54 ± 0.13, respectively), which may be attributed to steric effects combined with slightly lower capping efficiencies of these analogs. In addition, the introduction of a phosphoester group instead of phosphonate (compounds 7 and 8) did not improve the translational properties of capped mRNAs. The translation of transcripts capped with these compounds was comparable with that of ARCA-capped mRNA.
Protein output from capped mRNAs in JAWS II cells
Protein-production efficiency (output) from mRNAs capped with the triazole-bearing analogs was investigated in JAWS II cells. mRNAs encoding Gaussia luciferase were prepared during the IVT reaction utilizing a 6-fold higher cap analog concentration than GTP. Gaussia luciferase is secreted outside the cells, which enables convenient luminescence analysis in the collected cell culture medium at different time points without cell lysis.29 Because the presence of RNA impurities may significantly affect the outcome of cell-culture experiments with in vitro transcribed mRNA,7 uncapped impurities were enzymatically removed from the samples (apart from pppG control), followed by HPLC purification to remove dsRNA. mRNAs before and after HPLC purification were analyzed using a TBE agarose gel (Fig. S1A†). To confirm the removal of dsRNAs, we performed dot-blot analysis using a dsRNA-recognizing antibody (Fig. S1B†). The purified mRNAs were used for transfection of JAWS II cells (lipofection), followed by luciferase activity measurement at different time points (16, 40, 64, and 88 h).mRNAs capped with triazole-bearing cap analogs 1–9 and 10b were evaluated. The time-dependent expression is shown in Fig. 6A (representative results from a single replicate), whereas the cumulative luminescence calculated as the sum of all four time points (overall protein output) is shown in Fig. 6B (results from four biological replicates). Surprisingly, the results of the cell culture experiment did not correlate quantitatively with results from the studies in RRL. A significant difference was particularly visible for mRNA capped with compound 4. The translation efficiency of mRNA capped with compound 4 in RRL was 2-fold higher than that of ARCA-capped RNA, whereas, in living cells, mRNA capped with compound 4 was approximately 50% less active than ARCA-capped RNA. Overall, the majority of the triazole-bearing analogs showed notably lower protein production in JAWS II cells than the reference mRNAs. However, mRNA capped with compound 9 was an exception. Protein output from mRNA capped with compound 9 was comparable to that of m7GpppAmpG-capped RNA. Notably, compound 9 had also the highest translation efficiency in the RRL.
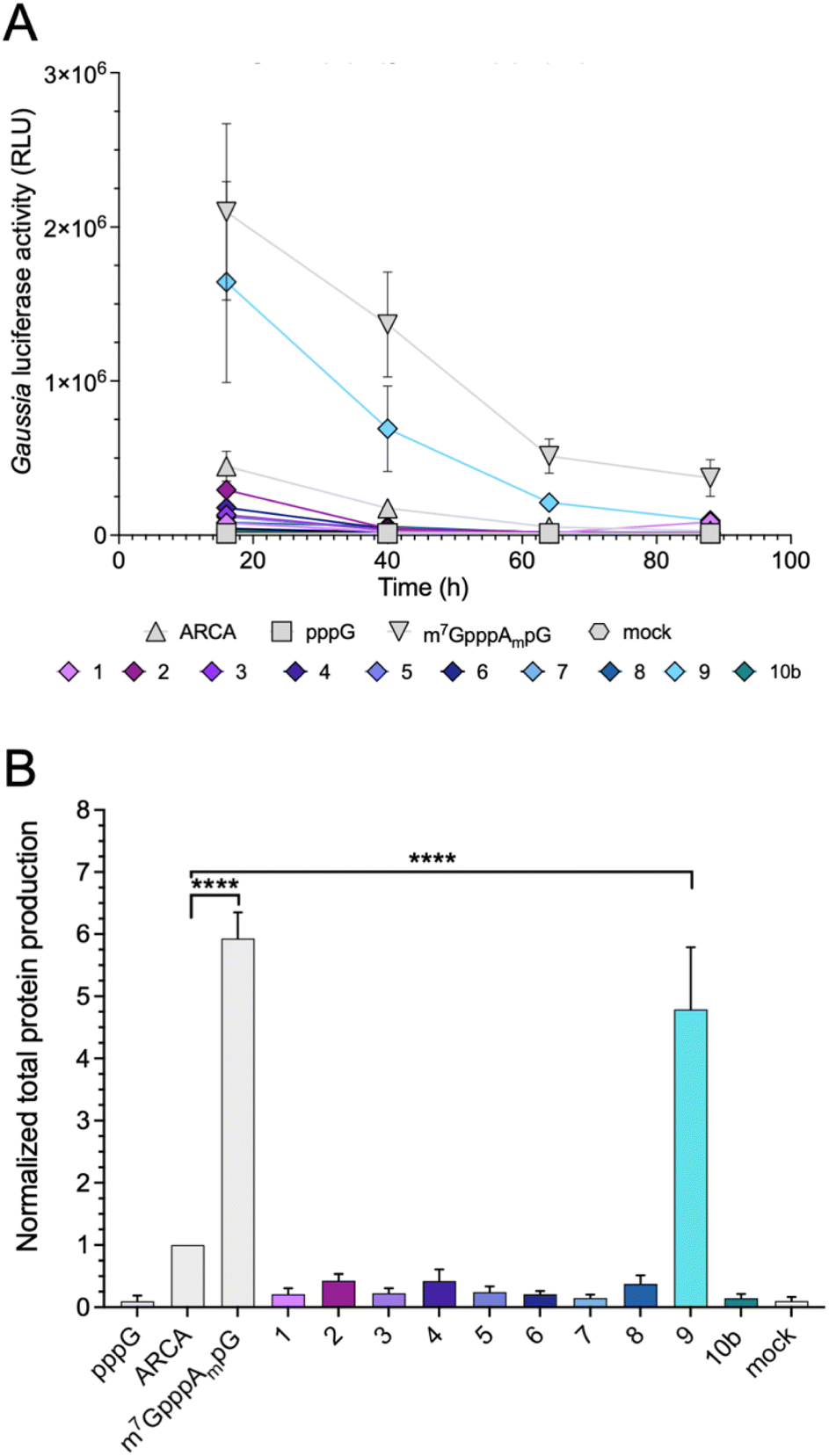 | ||
| Fig. 6 Protein output from HPLC-purified mRNAs capped with various analogs. (A) Gaussia luciferase activity was measured in medium collected from JAWS II cultures at particular-time points: 16, 40, 64, and 88 h after cell transfection with luciferase mRNAs capped with various analogs. Medium was replaced with fresh one; therefore, results from each time point show activity of newly produced luciferase. Measurements for technical triplicates from single biological replicate are presented. (B) Total protein production (cumulative luminescence) over four days in JAWS II cells transfected with mRNA capped with various analogs (results from tetraplicate experiment). Bars represent mean value ± SEM normalized to ARCA-capped RNA. Statistical significance: ****P < 0.0001 (one-way ANOVA with Turkey's multiple comparison test). | ||
The affinity for eIF4E and susceptibility to degradation by the hDCP1/DCP2 complex
Interestingly, as explained previously, we observed a divergence between protein expression in RRL and living JAWS II cells, especially for mRNA capped with compound 4. To determine a plausible explanation for the discrepancies, we tested the biochemical properties of the selected compounds (2, 4, 6, 8, 9 and 10b). First, we determined the affinities for eIF4E of triazole-bearing trinucleotide cap analogs, namely compounds 2, 4, 6, 8, and 9, tetranucleotide cap analog 10b and compared them to the affinity of m7GpppAmpG. To that end, we conducted a fluorescence quenching titration experiment, wherein a decrease in protein fluorescence emission was observed upon binding of the cap analogs to eIF4E.30,31 We found that the KAS values of nearly all the triazole-bearing cap analogs were comparable with or slightly lower that the KAS value determined for m7GpppAmpG, with the exception of compound 9 (KAS = 136.1 ± 13.2 μM−1). KAS value for compound 9 was more than 4-fold higher than that for m7GpppAmpG (KAS = 29.6 ± 2.3 μM−1) (Fig. 7 and Table S2†). This was expected because of the presence of an additional, negatively-charged phosphate group in compound 9.32 Overall, the results suggest that increased affinity of triazole-bearing cap analog for eIF4E, as observed for compound 9, is a necessary factor for achieving high translational activity of capped-mRNA.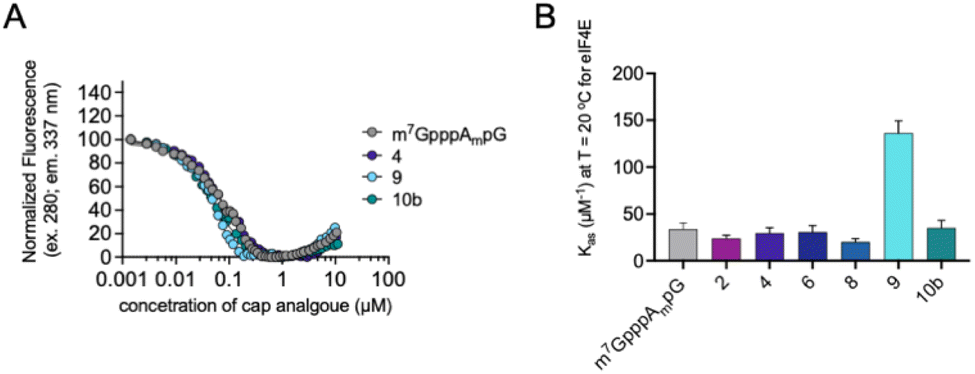 | ||
| Fig. 7 The affinities for eIF4E determined using FQT experiments. (A) Example curves obtained for triazole cap analogs. (B) Cumulative KAS values for 2′-O-methyl triazole cap analogs. | ||
To further characterize compound 9, we also tested the susceptibility of 27-nt capped RNA to degradation by the human DCP1/DCP2 complex (hDCP1/DCP2). To this end, we synthesized short, 27-nt RNAs capped with compound 9, compound 4, which relatively high translational affinity in RRL but not in JAWS II cells, and m7GpppAmpG, which was used as a reference (see Experimental section for details). The synthesized RNAs were incubated with hDCP1/DCP2 complex at 37 °C and reaction progress was analyzed at different time points (0, 15, 30, and 60 min) by polyacrylamide gel-electrophoresis (PAGE; Fig. 8A).
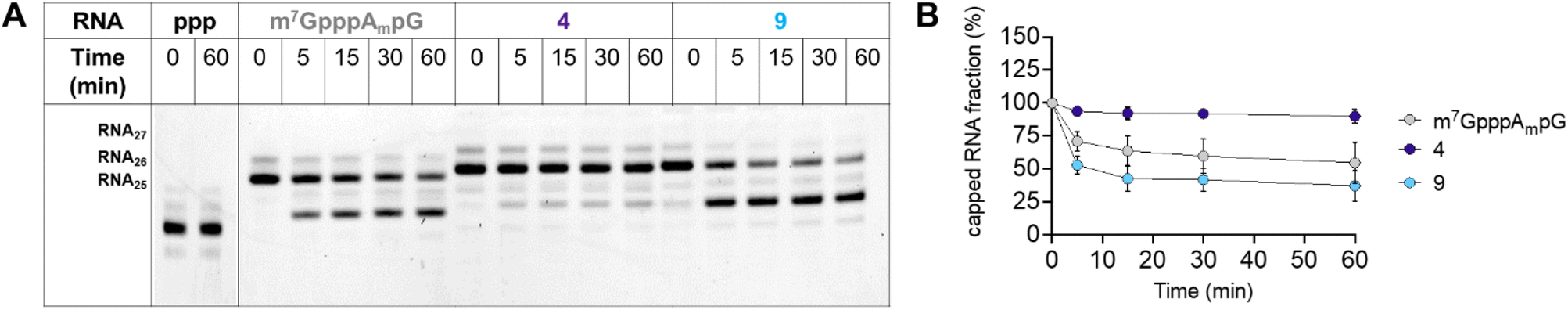 | ||
| Fig. 8 Susceptibility to degradation of capped RNAs by hDCP1/DCP2 complex. (A) Representative PAGE analysis; RNA25: uncapped RNA. RNA27 – RNA capped with m7GpppAmpG, compound 4, or compound 9. RNA26 – RNA27 decapped by hDCP1/2. (B) Comparison of degradation of RNA capped with m7GpppAmpG, 4, and 9 in time. Data represents mean value ± SD calculated for two replicates. | ||
We found that RNA capped with analog 4 was completely resistant to degradation by the hDCP1/DCP2 complex under the experimental conditions. Surprisingly, RNA capped with analog 9, which was efficiently expressed in both the RRL system and JAWS II living cells, was degraded faster than the reference (m7GpppAmpG-capped RNA). These findings juxtaposed with the determined binding affinities for eIF4E, and translational properties of mRNAs capped with 4 and 9, suggest that for the triazole-modified compounds affinity for eIF4E and conformational flexibility may have greater influence on translational properties of mRNA in living cells than susceptibility to hydrolysis by hDCP1/DCP2.
Conclusions
Herein, we combined a ‘click-chemistry’-based approach to nucleotide assembly with tri- and tetranucleotide-based mRNA capping strategies to achieve an alternative approach to assembly of oligonucleotide cap analogs conferring high translational activity to mRNA. We successfully combined trinucleotide mRNA capping technology, enhancing both capping efficiency and protein production in cultured cells, with CuAAC-based assembly, which may facilitate synthesis of mRNA cap analogs in bulk. Overall, we synthesized 12 triazole-bearing mRNA cap analogs (compounds: 1–9, 10a, 10b and 11) in 15–62% yield. Evaluating the capping efficiencies for all the synthesized cap analogs revealed that analogs bearing the triazole moiety within the 5′,5′-phosphate bridge were relatively well incorporated into short mRNA (50–75% capping efficiency), compared to the unmodified reference (88%), whereas modification within the first phosphodiester bond severely impaired incorporation. Further studies identified compound 9 (m7Gppp-tr-C2H4pAmpG) as the cap analog that provided superior properties to mRNA in terms of affinity for eIF4E and translational properties in vitro (in RRL system) and in cultured cells (JAWS II). mRNA capped with compound 9 showed biological properties comparable with mRNA capped with unmodified cap 1 (m7GpppAmpG) commercially available as Clean Cap AG®. Therefore, analog 9, which bears a triazole modification within the oligophosphate bridge, is a promising candidate for further evaluation as an mRNA capping reagent in cultured cells and in vivo models for application in mRNA-based therapeutics. Furthermore, due to synthetic accessibility via click chemistry, this structural design may be useful for the development of strategies for post-transcriptional chemical capping of mRNA.Experimental section
Chemical synthesis
:1, v/v) for 1 h at 55 °C. The obtained suspension was filtered off, and the resin was washed with water. The aqueous fractions were combined and freeze-dried. The final products were isolated by ion-exchange chromatography using a DEAE Sephadex™ column, wherein the final products were eluted with a linear gradient of triethylammonium bicarbonate (TEAB) in deionized water (0–0.6 M). 2′-O-TBDMS deprotection was omitted because all acidic protecting groups were removed during the 5′-iodination step.1H NMR: δH (500.24 MHz; D2O; TSP) 8.19 (1H, s), 8.08 (1H, s), 7.91 (1H, s), 5.92 (1H, d, J = 4.3, H1′), 5.78 (1H, d, J = 4.6, H1′), 4.75–4.73 (1H, m, H2′), 4.68–4.62 (2H, m, H2′, H3′), 4.51–4.47 (1H, m, H3′), 4.37–4.33 (1H, m, H4′), 4.32–4.24 (1H, m, H4′, H5′), 4.20–4.14 (1H, m, H5′′), 3.66 (1H, dd, J = 13.7, 3.2, H5′), 3.60 (1H, dd, J = 13.7, 5.1, H5′′), 3.19 (9H, q, J = 7.4, TEA), and 1.27 (14H, t, 7.4, TEA). 31P NMR: δP (202.49 MHz, D2O, H3PO4) −0.73 (1P, s). HRMS (ESI−) calculated for [C20H23N13O10P−]: 636.1434; m/z found: 636.1437.
1H NMR: δH (500.24 MHz; D2O; TSP) 8.25 (1H, s), 8.14 (1H, s), 7.93 (1H, s), 6.04 (1H, d, J = 4.7, H1′), 5.82 (1H, d, J = 4.9, H1′), 4.76–4.72 (1H, m, H2′), 4.54–4.48 (2H, m, H2′, H3′), 4.32 (1H, dd, J = 8.5, 4.7, H3′), 4.22–4.18 (2H, m, H4′), 3.59 (2H, dd, J = 4.3, 2.9, H5′, H5′′), 3.49 (3H, s, CH3), 3.57–3.51 (1H, m, H5′), and 3.46–3.41 (1H, m, H5′′). 31P NMR: δP (202.49 MHz; D2O; H3PO4) −0.15 (1P, d, J = 8.4). HRMS (ESI−) calculated for [C21H25N13O10P−]: 650.1590; m/z found: 650.1599.
:1, v/v; 10 mL) and shaken for 1 h at 55 °C. Next, the suspension was filtered using a syringe with frit. The resin was then washed with water (20 mL). The collected dinucleotide fractions were evaporated and freeze-dried to remove ammonia. After freeze-drying, 5′-azido dinucleotide was dissolved in DMSO (250 μL) and heated to 65 °C. Then, triethylamine (125 μL) was added, followed by TEA·3HF (65 μL). The final solution was shaken for 4 h at 65 °C. Next, the pH of the reaction mixture was adjusted to 6–7 with an aqueous solution of 0.05 M NaHCO3. 5′-Azido-dinucleotide was isolated by ion-exchange chromatography using a DEAE Sephadex™ column, wherein 5′-azido-dinucleotide was eluted with a linear gradient of TEAB (0–0.6 M).1H NMR: δH (500.24 MHz; D2O; TSP) 8.25 (1H, s), 8.41 (1H, s), 8.23 (1H, s), 7.97 (1H, s), 6.10 (1H, d, J = 5.6, H1′), 5.86 (1H, d, J = 5.5, H1′), 4.96 (1H, ddd, J = 8.2, 4.8, 3.6, H3′), 4.55–4.48 (2H, m, H2′, H3′), 4.48–4.43 (1H, m, H4′), 4.37–4.32 (1H, m, H4′), 4.22 (2H, t, J = 4.0, H5′, H5′′), 4.06 (1H, dt, J = 11.7, 3.6, H5′), 3.98 (1H, ddd, J = 11.7, 4.9, 2.6, H5′′), 3.95–3.86 (m, CH2), 3.53 (2H, q, J = 7.3, EtOH), 3.48 (3H, s, CH3), 3.44–3.34 (2H, m, CH2), 3.20 (5H, q, J = 7.3, TEA), 1.33 (t, J = 7.3, EtOH), and 1.28 (t, J = 7.3, TEA). 31P NMR: δP (202.49 MHz; D2O; H3PO4) −0.14 (1P, s), 0.58 (1P, s). HRMS (ESI−) calculated for [C23H30N13O14P2−]: 774.1516; m/z found 774.1520.
:1) were added. The reaction was initiated by adding an aqueous solution of sodium ascorbate (10 equivalents). The reaction mixture was shaken for 15–60 minutes at 25 °C or 37 °C. The reaction was quenched by adding an aqueous solution of Na2EDTA (1.0–1.5 equivalents). The obtained compounds were purified by ion-exchange chromatography using a DEAE Sephadex™ A-25 column (HCO3− form) and by RP-HPLC with a semi-preparative column. The structures of the obtained cap analogs were confirmed by high-resolution mass spectrometry and NMR (1H, 31P, COSY, HSQC).1H NMR: δH (399.90 MHz; D2O; TSP) 9.13 (1H, s, H8 m7G), 8.21 (1H, s, H8), 8.20 (1H, s, H8), 8.18 (1H, s, H2), 8.01 (1H, s, Htriazol), 5.95 (1H, d, J = 3.5, H1′), 5.93 (1H, d, J = 3.5 Hz, H1′), 5.82 (1H, d, J = 4.9, H1′), 5.00–4.87 (1H, m, H3′), 4.68–4.59 (2H, m, H2′ one proton H2′ is overlapped with water), 4.56 (1H, d, J = 5.1, H3′), 4.49 (2H, dt, J = 15.0, 4.4, H3′, H4′), 4.35 (1H, s, H4′), 4.33–4.12 (7H, m, H5′, H5′′, H4′), and 4.03 (3H, s, CH3). 31P NMR: δP (161.89 MHz; D2O; H3PO4): −0.74 (1P, s, ApG), −7.94 (d, J = 22.2), −11.49 (d, J = 19.1), and −23.32 (1P, t, J = 20.6, Pβ). HRMS (ESI−) calculated for [C33H40N18O23P4−]: 1181.1548; m/z found: 1181.1563.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.12 (1H, s, H8 m7G), 8.21 (1H, s, H8), 8.19 (1H, s, H8), 8.16 (1H, s, H2), 7.94 (1H, s, Htriazol), 6.02 (1H, d, J = 3.7, H1′), 5.94 (1H, d, J = 3.7, H1′), 5.80 (1H, d, = 5.2, H1′), 5.00 (1H, dt, J = 8.4, 5.7, H3′), 4.61 (1H, dd, J = 4.9, 3.7, H2′), 4.56 (1H, dd, J = 10.0, 5.7, H3′), 4.50 (dd, J = 5.0, 4.8, H4′), 4.46 (1H, t, J = 5.0, H3′), 4.39 (1H, dd, J = 5.2, 3.7, H2′), 4.36–4.33 (1H, m, H4′), 4.31–4.29 (1H, m, H4′), 4.27–4.12 (5H, m, H5′, H5′′), 4.02 (3H, s, CH3 m7G), and 3.47 (3H, m, CH3, 2′-OMe). 31P NMR: δP (161.89 MHz; D2O; H3PO4) −1 (1P, s, ApG), −7.96 (d, J = 22.2), −11.49 (d, J = 17.3), and −23.35 (1P, t, J = 22.0, Pβ). HRMS (ESI−) calculated for [C34H43N18O23P4−]: 1195.1704; m/z found: 1195.1712.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.17 (1H, s, H8), 8.21 (1H, s, H8), 8.10 (1H, s, H8), 7.94 (1H, s, H2), 7.77 (1H, d, J = 2.2, Htriazol), 5.96 (1H, d, J = 3.6, H1′), 5.92 (1H, d, J = 3.4, H1′), 5.80 (1H, d, J = 4.7, H1′), 4.89 (1H, dt, J = 7.8, 5.6, H3′), 4.75 (1H, t, J = 5.0, H2′), 4.72–4.66 (3H, m, H2′, H3′, H4′), 4.63 (1H, dd, J = 4.9, 3.6, H2′), 4.61–4.54 (1H, m, H4′), 4.50 (2H, q, J = 5.1, H3′, H4′), 4.38–4.26 (4H, m, H5′,H5′′), 4.25–4.14 (1H, m, H5′′,H5′), 4.03 (3H, s, CH3), 3.59 (dd, J = 13.2, 7.0), and 3.14 (2H, dq, J = 20.5, 15.5, HCH2). 31P NMR: δP (161.89 MHz; D2O; H3PO4) 10.56 (1P, dd, J = 45.3, 24.6), −0.80 (1P, s, ApG), −11.56 (1P, d, J = 19.5), and −23.10 (1P, dd, J = 25.5, 19.3, Pβ). HRMS (ESI−) calculated for [C34H42N19O23P4−]: 1195.1704; m/z found 1195.1722.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.17 (1H, s, H8 m7G), 8.23 (1H, s, H8), 8.12 (1H, s, H8), 7.95 (1H, s, H2), 7.74 (1H, d, J = 2.1, HTriazol), 6.04 (1H, d, J = 3.5, H1′), 5.97 (1H, d, J = 3.6, H1′), 5.82 (1H, d, J = 5.0, H1′), 5.02 (1H, dt, J = 8.1, 5.6, H3′), 4.68 (2H, d, J = 4.8, H2′, H3′), 4.64 (1H, dd, J = 4.9, 3.7, H2′), 4.56–4.47 (5H, m, H2′, H3′, H4′), 4.36–4.30 (3H, m, H5′, H5′′), 4.26–4.15 (3H, m, H5′, H5′′), 4.03 (3H, s, CH3), 3.49 (3H, s, CH3 2′-OMe), and 3.22–3.05 (2H, m, CH2). 31P NMR: δP (161.89 MHz; D2O; H3PO4) 10.39 (1P, q, J = 22.4), −1.04 (1P, s, ApG), −11.56 (1P, d, J = 19.5), and −23.10 (1P, dd, J = 25.1, 19.5, Pβ). HRMS (ESI−) calculated for [C35H45N18O23P4−]: 1209.1861; m/z found 1209.1865.
1H NMR: δH (399.90 MHz; D2O; TSP) 8.07 (1H, s), 7.93 (1H, s), 7.77 (1H, s), 7.53 (1H, s), 7.84 (1H, s, Htriazol), 5.91–5.89 (2H, m, H1′), 5.80 (1H, d, J = 4.5, H1′), 4.89 (1H, s), 4.74–4.69 (2H, m, H2′, overlapped with water), 4.64 (1H, dd, J = 5.1, 2.1, H2′), 4.61–4.58 (1H, m), 4.55 (3H, s), 4.49 (2H, dd, J = 10.2, 5.3), 4.36–4.30 (4H, m), 4.23–4.16 (2H, m), 4.02 (3H, s, CH3), 2.81–2.70 (2H, m, CH2), and 2.05–1.91 (2H, m, CH2, overlapped with acetone). 31P NMR: δP (161.89 MHz; D2O; H3PO4) 17.20–16.82 (1P, m, Pα), −0.80 (1P, s, 3′P), −11,52 (1P, d, J = 18.4, Pγ), and −23.02 (1P, dd, J = 25.2, 19.0, Pβ). HRMS (ESI−) calculated for [C35H45N18O23P4−]: 1209.1861; m/z found 1209.1861.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.17 (1H, s, H8 m7G), 8.24 (1H, s, H8), 8.10 (1H, s, H8), 7.98 (1H, s, H2), 7.48 (1H, d, J = 2.1, HTriazol), 6.08 (1H, d, J = 2.1, H1′), 5.98 (1H, d, J = 3.5, H1′), 5.82 (1H, d, J = 4.9, H1′), 5.08 (1H, dt, J = 7.2, 5.0, H3′), 4.69–4.67 (1H, m, H2′), 4.57–4.53 (2H, m, H2′, H3′), 4.50–4.44 (4H, m, H3′, H4′), 4.39–4.34 (3H, m, H5′, H5′′), 4.30–4.24 (2H, m, H5′, H5′′), 4.20–4.12 (1H, m, H5′, H5′′), 4.07 (3H, s, CH3), 3.53 (3H, s, CH3 2′-OMe), 2.74–2.68 (2H, m, CH2), and 1.99–1.91 (2H, m, CH2). 31P NMR: δP (161.89 MHz; D2O; H3PO4) 17.10–16.50 (1P, m, Pγ), −0.95–(−0.93) (1P, m, ApG), −11.45 (1P, d, J = 18.7, Pα), and −22.83–(−23.11) (1P, m, Pβ). HRMS (ESI−) calculated for [C36H47N18O23P4−]: 1223.2017; m/z found 1223.2014.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.14 (1H, s, H8 m7G), 8.16 (1H, s, H8), 8.11 (1H, s, H8), 7.95 (1H, s, C2), 7.84 (1H, s, Htriazol), 5.97 (1H, d, J = 3.7, H1′), 5.94 (1H, d, J = 2.4, H1′), 5.81 (1H, d, J = 4.7, H1′), 5.02–4.97 (1H, m, H3′), 4.89–4.88 (2H, m, CH2), 4.77–4.75 (1H, m, H2′, overlapped with water), 4.73 (1H, dd, J = 5.2, 2.5, H2′), 4.66 (1H, dd, J = 4.9, J2 = 3.8, H2′), 4.63–4.62 (2H, m, H3′), 4.54–4.49 (3H, m, H4′), 4.38–4.16 (5H, m, H5′, H5′′), and 4.06 (3H, s, CH3). 31P NMR: δP (161.89 MHz; D2O; H3PO4) −0.73 (1P, s, ApG), −11.43–(−11.75) (2P, m, Pα, Pγ), and −22.88 (1P, t, J = 18.8, Pβ). HRMS (ESI−) calculated for [C34H43N18O24P4−]: 1211.1654; m/z found 1211.1655.
1H NMR: δH (399.90 MHz; D2O; TSP) 9.14 (1H, s, H8 m7G), 8.18 (1H, s, H8), 8.11 (1H, s, H8), 7.95 (1H, s, C2), 7.83 (1H, s, Htriazol), 6.05 (1H, d, J = 2.5, H1′), 5.97 (1H, d, J = 3.7, H1′), 5.81 (1H, d, J = 5.0, H1′), 5.14–5.07 (1H, m, H3′), 4.88 (2H, d, J = 5.2), 4.66 (1H, dd, J = 4.9, 3.7, H2′), 4.60 (2H, d, J = 3.8, H3′), 4.53–4.46 (4H, m, H2′, H4′), 4.40–4.32 (3H, m, H5′, H5′′), 4.31–4.13 (3H, m, H5′, H5′′), 4.05 (3H, s, CH3 m7G), and 3.51 (3H, s, CH3, 2′-OMe). 31P NMR: δP (161.89 MHz; D2O; H3PO4) −0.11 (1P, m, 3′P), −10.69 (1P, d, J = 18.8, Pα or Pγ), −10.89 (1P, d, J = 18.3, Pα or Pγ), and −22.87 (1P, dd, J = 18.8, 18.3, Pβ). HRMS (ESI−) calculated for [C35H45N18O24P4−]: 1225.1810; m/z found 1225.1810.
1H NMR: δH (500.24 MHz; D2O; TSP) 9.12 (1H, s, H8 m7G), 8.38 (1H, s, H8), 8.26 (1H, s, H8, Htriazol), 8.21 (1H, s, H2), 7.97 (1H, s, H8), 6.05 (1H, d, J = 5.2, H1′), 5.96 (1H, d, J = 3.4, H1′), 4.94 (1H, dt, J = 8.3, 4.4, H3′), 4.61 (1H, dd, J = 4.8, 3.6, H1′), 4.56 (2H, q), 4.53–4.49 (2H, m), 4.47 (1H, t, J = 5.1), 4.44–4.38 (1H, m), and 4.37–4.31 (2H, m). 31P NMR: δP (202.53 MHz; D2O; H3PO4) δ −0.36 (1P, s), −0.93 (1P, s), −7.46 (1P, d, J = 21.6, Pα or Pγ), −11.45 (1P, d, J =17.5, Pα or Pγ), −23.14 (1P, t, J = 20.3, Pβ). HRMS (ESI−) calculated for [C35H45N18O24P4−]: 1225.1810; m/z found 1225.1810.
Compound 10a. 1H NMR: δH (500.24 MHz; D2O; TSP) 9.02 (s, 1H), 8.20 (s, 1H), 7.94 (s, 1H), 7.89 (s, 1H), 7.81 (s, 1H), 7.63 (s, 1H), 5.88–5.86 (m, 3H), 5.83 (1H, d, J = 4.2), 5.72 (1H, d, J = 4.1), 5.44 (1H, t, J = 5.5), 4.76–4.68 (5H, m), 4.65 (1H, t, J = 5.1), 4.62–4.54 (6H, m), 4.52 (1H, d, J = 5.6 ), 4.49 (1H, d, J = 4.9), 4.45 (2H, t, J = 5.0), 4.41–4.21 (15H, m), 4.20–4.11 (3H, m), 4.03 (s, 3H). 31P NMR: δP (202.53 MHz; D2O; H3PO4) 0.16 (1P, d, J = 9.5, Pα), −10.58 (2P, dd, J = 19.4, 12.0, 3′P and Pγ), −22.11 (1P, t, J = 19.3, Pβ).
Compound 10b. 1H NMR: δH (500.24 MHz; D2O; TSP) 9.05 (1H, s), 8.23 (1H, s), 8.01 (1H, s), 7.96 (2H, s), 7.92 (1H, s), 7.72 (1H, s), 5.93 (1H, d, J = 3.0), 5.88 (1H, d, J = 3.5), 5.80 (1H, d, J = 4.7), 5.73 (1H, d, J = 7.2), 5.25 (1H, t, J = 10.8), 4.74 (1H, d, J = 3.0), 4.72–4.65 (4H, m), 4.57 (3H, dd, J = 8.1, 5.0), 4.49 (1H, t, J = 5.3), 4.45 (2H, t, J = 5.2), 4.43–4.15 (12H, m), 4.02 (3H, s). 31P NMR: δP (202.53 MHz; D2O; H3PO4) δ 0.23 (1P, d, J = 10.6, Pα), −10.54 (2H, dd, J = 19.3, 11.3, 3′P and Pγ), −21.99 (1P, t, J = 19.2). HRMS (ESI−) calculated for [C35H45N18O24P42−] 759.1225; m/z found 759.1229 (compound 10a), 759.1225 (compound 10b).
1H NMR: δH (399.90 MHz; DMSO; TSP) 8.39 (1H, s, ade), 8.14 (1H, s, ade), 7.39 (2H, s, Ade NH2), 6.01 (1H, d, J = 6.4, H1′), 5.56 (1H, dd, J = 7.0, 4.7, H4′), 4.56 (1H, dd, J = 6.4, 4.9, H2′), 4.36 (1H, dd, J = 4.9, 2.8, H3′), 4.33 (2H, t, J = 2.2), 4.16 (1H, q, J = 3.3, H4′), 3.70 (1H, ddd, J = 12.1, 4.7, 3.7, H5′), 3.58 (1H, ddd, J = 12.1, 7.0, 3.4, H5′′), 3.51 (1H, t, J = 2.4), and 3.31 (3H, s, 2′-OMe).
1H NMR: δH (500.24 MHz; D2O; TSP) (1H, s, ade), 8.26 (1H, s, ade), 6.20 (1H, d, J = 5.7, H1′), 4.67–4.61 (2H, m, H2′, H3′), 4.56–4.53 (1H, m, H4′), 4.45 (1H, dd, J = 16.0, 2.4, CH2), 4.41 (1H, dd, J = 16.0, 2.4, CH2), 3.51 (1H, q, J = 7.3, solvent), 3.47 (3H, s, 2′-Ome), 3.20 (7H, q, J = 7.3, TEA), 3.07, and 2.96 (1H, t, J = 2.4, C3H3). 31P NMR: δP (161.89 MHz; D2O; H3PO4). HRMS (ESI−) calculated for [C14H17N5O7P−] 398.0871; m/z found: 398.0872.
1H NMR: δH (500.24 MHz; D2O; TSP) 9.05 (1H, s, C8 m7G), 8.46 (1H, s, base), 8.24 (1H, s, base), 6.09 (1H, d, J = 6.2, H1′), 5.90 (1H, d, J = 3.8, H1′), 4.62 (1H, dd, J = 5.3, 3.3), 4.57–4.52 (3H, m, H2′ and H3′), 4.49–4.41 (3H, m), 4.41–4.35 (2H, m), 4.33–4.22 (3H, m), 4.05 (3H, s, CH3, m7G), 3.42 (3H, s, 2′-OMe), and 2.96 (1H, t, J = 2.4, CH). 31P NMR: δP (161.89 MHz; D2O; H3PO4) −10.89 (2P, dd, J = 19.3, 13.3, Pα, Pγ), and −22.44 (1P, t, J = 19.3, Pβ). HRMS (ESI−) calculated for [C25H32N10O17P3−] 837.1165; m/z found: 837.1166.
1H NMR: δH (500.24 MHz; D2O; TSP) 9.11 (1H, s, H8 m7G), 8.42 (1H, s, H8), 8.26 (1H, s, H8), 7.93 (1H, s, H2), 7.55 (1H, s, Htriazol), 6.06 (1H, d, J = 5.2, H1′), 5.89 (1H, d, J = 3.4, H1′), 5.76 (1H, d, J = 2.8, H1′), 4.85 (2H, dd, J = 6.2, 2.8), 4.76, 4.72–4.69 (1H, m, H2′), 4.62–4.57 (2H, m, H2′ and H3′), 4.52 (1H, t, J = 5.3, 5.1, H4′), 4.46–4.43 (4H, m), 4.41 (1H, dd, J = 4.1, 2.6), 4.38–4.33 (2H, m), 4.30–4.23 (3H, m), 4.02 (3H, s, CH3 m7G), and 3.25 (3H, s, CH3 2′-OMe). 31P NMR: δP (161.89 MHz; D2O; H3PO4) −0.74 (1P, s, ApG), −7.94 (d, J = 22.2), −11.49 (d, J = 19.1), and −23.32 (1P, t, J = 20.6, Pβ). HRMS (ESI−) calculated for [C35H45N18O24P4−] 1145.2147; m/z found: 1145.2154.
Biological and biochemical experiments
The translation efficiency of non-HPLC purified mRNA encoding Gaussia luciferase was determined using an RRL system (Promega). A typical in vitro translation reaction (10 μL) contained 4 μL of RRL mixture, 0.05 μL of amino acid mixture excluding leucine, 0.05 μL of amino acid mixture excluding methionine, 1.9 μL of 1 M potassium acetate, 0.4 μL of 25 mM magnesium chloride, 0.2 μL of Ribolock RNase Inhibitor (Thermo Fisher Scientific), 2.4 μL of water, and 1 μL of one of tested mRNA. Four stock solutions of capped mRNAs were prepared. For mRNAs capped with ARCA, m7GpppG, GpppG, m7GpppApG, 1, 2, 5, 6, 7, 8, and 10B, solutions of concentrations 93.75, 70.31, 46.88, and 23.44 pg μL−1 were used. For mRNAs capped with m7GpppAmpG, 3, 4, and 9, 280, 140, 93.75, and 70.31 pg μL−1 stocks were used. The translation reactions were pre-incubated for 1 h at 30 °C. Following incubation, the reaction mixtures were cooled on ice, and 1 μL of tested mRNAs encoding Gaussia luciferase from the stocks was added, followed by incubation for 1 h at 30 °C. The translation reaction was stopped by freezing the sample in liquid nitrogen.
Author contributions
M. K., J. K. and J. J. designed the study, M. K. performed chemical syntheses, RRL experiments and fluorescence quenching titration experiments, K. D. performed experiments in cells, M. B. performed decapping assays, M. W. provided chemical reagents. M. K., K. D., J. K. and wrote the first draft of the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Foundation for Polish Science (TEAM POIR.04.04.00-00-20A2/16 to J. J.) and the National Science Centre (2018/31/B/ST5/03821 to J. K.) is gratefully acknowledged.Notes and references
- C. M. Wei, A. Gershowitz and B. Moss, Cell, 1975, 4, 379–386 CrossRef CAS PubMed.
- A. Galloway and V. H. Cowling, Biochim. Biophys. Acta, Gene Regul. Mech., 2019, 1862, 270–279 CrossRef CAS PubMed.
- F. Borbolis and P. Syntichaki, FEBS J., 2021, 1457–1475 Search PubMed.
- A. Ramanathan, G. B. Robb and S. H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 CrossRef.
- M. Shanmugasundaram, A. Senthilvelan and A. R. Kore, Chem. Rec., 2022, 22(8), 27 CrossRef.
- M. Warminski, P. J. Sikorski, J. Kowalska and J. Jemielity, Topics in Current Chemistry Collections, Springer International, 2017, pp. 211–239 Search PubMed.
- P. J. Sikorski, M. Warminski, D. Kubacka, T. Ratajczak, D. Nowis, J. Kowalska and J. Jemielity, Nucleic Acids Res., 2020, 48, 1607–1626 CrossRef CAS.
- N. Klöcker, F. P. Weissenboeck, M. van Dülmen, P. Špaček, S. Hüwel and A. Rentmeister, Nat. Chem., 2022, 14, 905–913 CrossRef PubMed.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745–1755 CrossRef CAS PubMed.
- R. Wojcik, M. R. Baranowski, L. Markiewicz, D. Kubacka, M. Bednarczyk, N. Baran, A. Wojtczak, P. J. Sikorski, J. Zuberek, J. Kowalska and J. Jemielity, Pharmaceutics, 2021, 13, 1941–1960 CrossRef CAS PubMed.
- A. Senthilvelan, T. Vonderfecht, M. Shanmugasundaram, I. Pal, J. Potter and A. R. Kore, Org. Lett., 2021, 23, 4133–4136 CrossRef CAS.
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS.
- R. Verbeke, I. Lentacker, S. C. De Smedt and H. Dewitte, J. Controlled Release, 2021, 333, 511–520 CrossRef CAS.
- S. Kwon, M. Kwon, S. Im, K. Lee and H. Lee, Arch. Pharmacal Res., 2022, 45, 245–262 CrossRef CAS PubMed.
- R. C. Moschel, W. R. Hudgins and A. Dipple, J. Org. Chem., 1984, 49, 363–372 CrossRef CAS.
- M. Shanmugasundaram, A. Senthilvelan and A. R. Kore, Chem. Rec., 2022, 22, 26 CrossRef PubMed.
- S. Walczak, A. Nowicka, D. Kubacka, K. Fac, P. Wanat, S. Mroczek, J. Kowalska and J. Jemielity, Chem. Sci., 2017, 8, 260–267 RSC.
- L. Y. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- S. Walczak, P. J. Sikorski, R. Kasprzyk, J. Kowalska and J. Jemielity, Org. Biomol. Chem., 2018, 16, 6741–6748 RSC.
- M. Ishikawa, R. Murai, H. Hagiwara, T. Hoshino and K. Suyama, Nucleic Acids Symp. Ser., 2009, 129–130 CrossRef CAS.
- M. Warminski, J. Kowalska and J. Jemielity, Curr. Protoc. Nucleic Acid Chem., 2020, 82, e112 CAS.
- M. Warminski, J. Kowalska and J. Jemielity, Org. Lett., 2017, 19, 3624–3627 CrossRef CAS PubMed.
- P. Wanat, S. Walczak, B. A. Wojtczak, M. Nowakowska, J. Jemielity and J. Kowalska, Org. Lett., 2015, 17, 3062–3065 CrossRef CAS PubMed.
- M. Warminski, Z. Warminska, J. Kowalska and J. Jemielity, Eur. J. Org. Chem., 2015, 2015, 6153–6169 CrossRef CAS.
- A. M. Jawalekar, N. Meeuwenoord, J. G. O. Cremers, H. S. Overkleeft, G. A. van der Marel, F. Rutjes and F. L. van Delft, J. Org. Chem., 2008, 73, 287–290 CrossRef CAS.
- M. Yoshikawa, T. Kato and T. Takenishi, Tetrahedron Lett., 1967, 50, 5065–5068 CrossRef CAS PubMed.
- M. Yoshikawa, T. Kato and T. Takenishi, Bull. Chem. Soc. Jpn., 1969, 42, 3505–3508 CrossRef CAS.
- J. Stepinski, C. Waddell, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2001, 7, 1486–1495 CAS.
- T. Wurdinger, C. Badr, L. Pike, R. de Kleine, R. Weissleder, X. O. Breakefield and B. A. Tannous, Nat. Methods, 2008, 5, 171–173 CrossRef CAS PubMed.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS PubMed.
- A. Niedzwiecka, J. Stepinski, J. M. Antosiewicz, E. Darzynkiewicz and R. Stolarski, in Translation Initiation: Reconstituted Systems and Biophysical Methods, ed. J. Lorsch, Elsevier Academic Press Inc., San Diego, 2007, vol. 430, pp. 209–245 Search PubMed.
- J. Zuberek, J. Jemielity, A. Jablonowska, J. Stepinski, M. Dadlez, R. Stolarski and E. Darzynkiewicz, Biochemistry, 2004, 43, 5370–5379 CrossRef CAS PubMed.
- N. Hebert, A. Beck, R. B. Lennox and G. Just, J. Org. Chem., 1992, 57, 1777–1783 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC methods, NMR and HRMS spectra, HPLC profiles. See DOI: https://doi.org/10.1039/d3ra00026e |
| This journal is © The Royal Society of Chemistry 2023 |