
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMatrix metalloproteinase profiling and their roles in disease†
Mayland Chang

Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: mchang@nd.edu
First published on 21st February 2023
Abstract
Matrix metalloproteinases (MMPs) play roles in remodelling of the extracellular matrix that occurs during morphogenesis, repair, and angiogenesis. Dysregulation of extracellular matrix remodelling can lead to cell proliferation, invasion, and tissue fibrosis. Identification of a specific MMP(s) in a disease has been challenging due to the presence of 24 closely-related human MMPs, each existing in three forms, of which only one is active and capable of catalysis. This review focuses on methods for MMP profiling, with particular emphasis on the batimastat affinity resin that binds only to the active forms of MMPs and related ADAMs (a disintegrin and metalloproteinases), which are then identified by mass spectrometry. Use of the batimastat affinity resin has identified targets for intervention in several human diseases.
 Mayland Chang | Mayland Chang is Research Professor at the University of Notre Dame. She received her PhD degree from the University of Chicago, with David Lynn as her mentor. Subsequently, she was an NIH Postdoctoral Fellow at Columbia University with Koji Nakanishi. She worked in the industry as Senior Chemist at Dow Chemical Company, Senior Scientist IV in Drug Metabolism and Pharmacokinetics at Pharmacia Corp., and was Chief Operation Officer of University Research Network Inc., where she managed clinical trials for Wayne State University. Her research interests are in translational medicinal chemistry, understanding of human disease and design of therapeutic interventions. |
1. Introduction
Matrix metalloproteinases (MMPs) are a family of enzymes whose function is to degrade components of the extracellular matrix (collagen, elastin, fibronectin, laminin, proteoglycans).1 The extracellular matrix undergoes remodelling during processes such as morphogenesis, bone remodelling, wound repair, and angiogenesis. Conversely, dysregulated extracellular matrix remodelling can lead to cell proliferation and invasion in cancer, as well as tissue fibrosis. There are 24 human MMPs, which are summarized in Table 1. MMP-4, MMP-5, and MMP-6 not included in Table 1, as they were shown to be identical to other MMPs, and MMP-22 is a chicken MMP. MMPs are classified as collagenases (MMP-1, MMP-8, MMP-13, and MMP-18), gelatinases (MMP-2 and MMP-9), stromelysins (MMP-3, MMP-10, MMP-11, and MMP-19), matrilysins (MMP-7 and MMP-26), membrane-type (MT)-MMPs (MMP-14, MMP-15, MMP-16, MMP-17, MMP-24, and MMP-25), and others.| MMP | Name | Production | Substrate(s) | Role in disease | Other | References |
|---|---|---|---|---|---|---|
| MMP-1 | Interstitial collagenase, collagenase 1 | Secreted: fibroblasts, keratinocytes, endothelial cells, macrophages, hepatocytes | Collagen I, II, III, VII, VIII, X, gelatin | Arthritis, idiopathic pulmonary fibrosis, cancer | Activates proMMP-2 and proMMP-9 | 7 |
| MMP-2 | Gelatinase-A | Secreted: fibroblasts, keratinocytes, endothelial cells, chondrocytes, osteoblasts, leukocytes, platelets, monocytes | Gelatin, collagen I, II, III, IV, VII, X | Cancer | Activates proMMP-9 and proMMP-13 | 8 and 9 |
| MMP-3 | Stromelysin 1 | Secreted: fibroblasts, platelets | Collagen II, IV, V, IX, X, XI, gelatin | Alzheimer's disease, Parkinson's disease | Activates proMMP1 and proMMP-13 | 10 and 11 |
| MMP-7 | Matrilysin | Secreted: epithelial cells, liver, pancreas, prostate, skin | Fibronectin, laminin, collagen IV, gelatin | Idiopathic pulmonary fibrosis | Activates osteopontin | 12 |
| MMP-8 | Neutrophil collagenase, collagenase 2 | Secreted: chondrocytes, endothelial cells, macrophages, smooth muscle cells | Collagen I, II, III, VII, VIII, X, aggrecan, gelatin | Beneficial: wound healing | 13 and 14 | |
| MMP-9 | Gelatinase-B | Secreted: neutrophils, macrophages, osteoblasts, epithelial cells, fibroblasts, dendritic cells, keratinocytes | Gelatin, collagen IV, V | Detrimental: diabetic foot ulcers, cerebral ischemia, traumatic brain injury | 13–17 | |
| MMP-10 | Stromelysin-2 | Secreted: keratinocytes, macrophages, epithelium | Collagen IV, laminin, fibronectin, elastin | Head and neck cancer | Activates proMMP-1 | 18 and 19 |
| MMP-11 | Stromelysin-3 | Secreted: fibroblasts, uterus, placenta, mammary glands | Does not cleave ECM proteins | Cancer | Activated by furin | 20 and 21 |
| MMP-12 | Macrophage elastase | Secreted: chondrocytes, macrophages, osteoblasts, placenta | Elastin, fibronectin, collagen IV | COPD, pulmonary emphysema | Activates protease-activated receptor-1 | 22–24 |
| MMP-13 | Collagenase 3 | Secreted: epithelial cells, neuronal cells, connective tissue | Collagen I, II, III, IV, IX, X, XIV, gelatin | Beneficial: wound healing. Detrimental: osteoarthritis | Activates proMMP-2 and proMMP-9 | 5 and 25 |
| MMP-14 | MT1-MMP | Membrane-associated: fibroblast, platelets, osteoblasts | Gelatin, fibronectin, laminin, collagen I II, III | Detrimental: melanoma | Activates proMMP-2 proMMP-8, proMMP-9, and proMMP-13 | 26 and 27 |
| MMP-15 | MT2-MMP | Membrane-associated: placenta, heart, brain | Gelatin, fibronectin, laminin | Ovarian cancer | Can activate proMMP-2 and proMMP-13 | 28 |
| MMP-16 | MT3-MMP | Membrane-associated: cardiomyocites, lungs, placenta, kidney, ovaries, intestine, prostate, spleen heart, skeletal muscle | Gelatin, fibronectin, laminin | Proliferation, invasion, and migration of cancer cells | Can activate proMMP-2 and proMMP-9 | 29 |
| MMP-17 | MT4-MMP | Glycosylphosphatidylinositor-anchored membrane-associated: leukocytes, brain, colon, ovaries, testicles | Fibrinogen, fibrin | Cancer | Activates ADAMTS-4 | 30 and 31 |
| MMP-18 | Collagenase 4 | Mammary gland, placenta, lung, pancreas, ovary, small intestine, spleen, thymus, prostate, colon, heart | Collagen, gelatin | Macrophage migration | 32 | |
| MMP-19 | Stromelysin- 4 | Leukocytes, colon, intestine, ovary, testis, prostate, thymus, spleen, pancreas, kidney, skeletal muscle, liver, lung, placenta, brain, heart | Aggrecan | Tissue homeostasis, down-regulated during malignant transformation of colon | Can activate proMMP-9 | 33 |
| MMP-20 | Enamelysin | Secreted: dental tissue | Aggrecan | Tooth development | 34 | |
| MMP-21 | Secreted: leukocytes, macrophages, fibroblasts, basal and squamous cells, ovary, kidney, lung, placenta, intestine, neuroectoderm, skin, brain | Unknown | Embryogenesis and tumor progression | 35 and 36 | ||
| MMP-22 (chicken) | Secreted | Casein, gelatin, collagen I | 37 | |||
| MMP-23 | CA-MMP | Membrane-associated: ovary, testicles, prostate | Gelatin | Melanoma | 38 and 39 | |
| MMP-24 | MT5-MMP | Membrane-associated: brain, kidney, pancreas, lung | Gelatin, fibronectin, laminin | Tumor progression and angiogenesis | Can activate proMMP-2 | 40 |
| MMP-25 | MT6-MMP | Membrane-associated: leucocytes, testicles, kidney, skeletal muscle | Gelatin | Cancer | Activates proMMP-2 | 41 |
| MMP-26 | Matrilysin-2 | Collagen IV, fibronectin, fibrinogen, gelatin, vitronectin | Cancer | Activates proMMP-9 | 42 | |
| MMP-27 | B-lymphocytes, macrophages, endometrium, testicles, intestine, lung, skin | Gelatin | Endometrium lesions | 43 | ||
| MMP-28 | Epilysin | Secreted: keratinocytes, epidermis, testis | Casein | Tissue homeostasis and repair | 44 |
MMPs have similar structures, consisting of a propeptide domain, a catalytic domain containing a zinc ion, and a hemopexin domain (Fig. 1). The hemopexin domain is absent in the matrilysins and MMP-23.
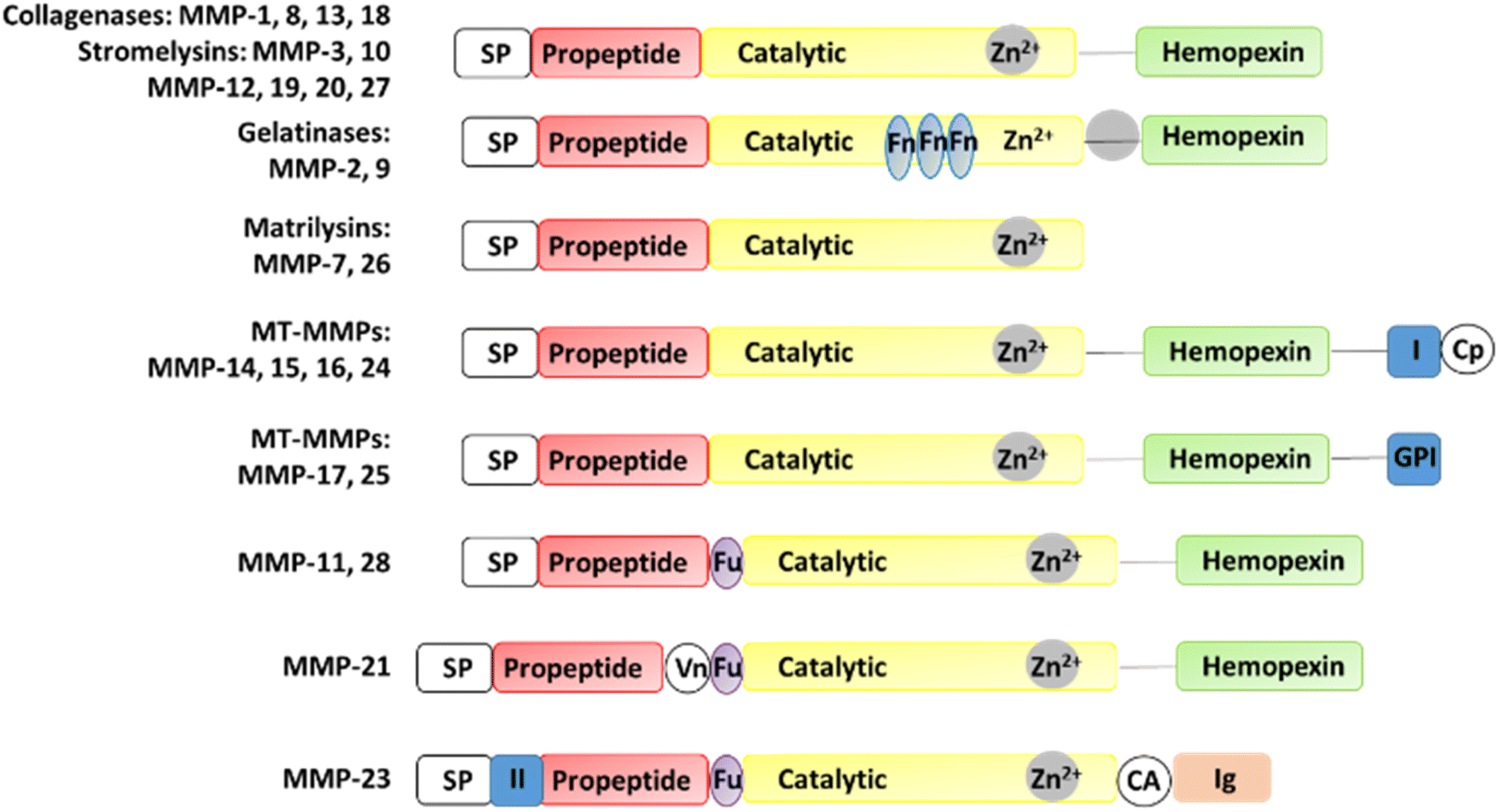 | ||
| Fig. 1 Structures of MMPs. CA, cysteine array region; Cp, cytoplasmic domain; Fn, fibronectin; Fu, furin cleavage site; GPI, GPI anchor; (I) type I transmembrane domain; (II) type II transmembrane domain, Ig, IgG-like domain; SP, signal peptide; Vn, vitronectin. | ||
MMPs are made as inactive zymogens or proMMPs (Fig. 2). Removal of the propeptide domain activates the proMMPs to their active forms. Regulation of MMPs is controlled mainly by tissue inhibitors of metalloproteinases (TIMPs). The TIMP-complexed MMPs are inactive forms of MMPs. Only the active forms of MMPs, and not the proMMPs or TIMP-MMP complexes are capable of catalysis. Thus, only the active MMPs can play roles in physiology or disease pathology. The challenge is identifying which active MMP(s) is involved in a specific disease. Most methods do not distinguish among the three forms of MMPs. One of the most widely used methods is gene expression profiling by measuring mRNA levels of MMPs. However, changes in mRNA levels do not necessarily correlate with protein abundance.2 This article focuses on methods for MMP profiling at the protein level.
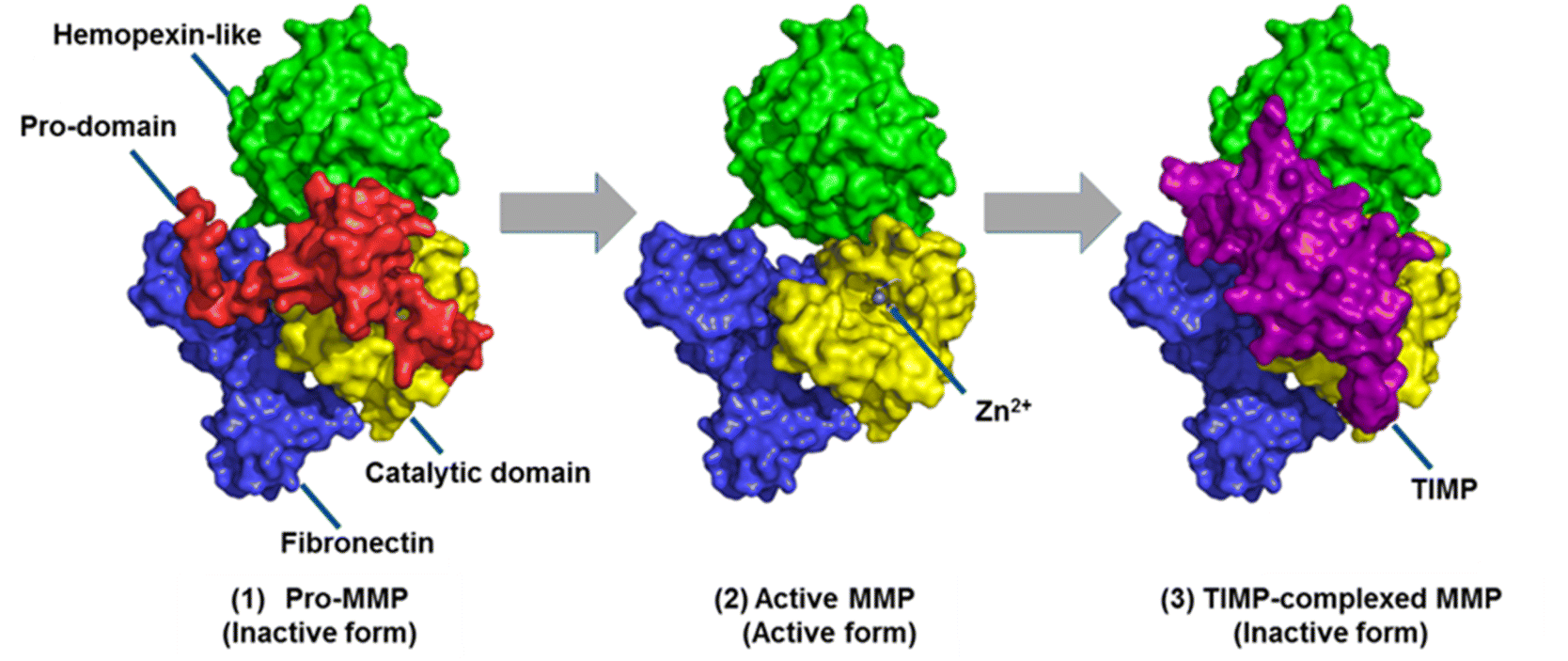 | ||
| Fig. 2 The three forms of MMPs as exemplified by MMP-2. MMPs are made as inactive zymogens or proMMPs. They are activated by removal of the propeptide domain. MMPs are regulated primarily by complexation with TIMPs. This figure has been reproduced from Nguyen et al.6 with permission from IntechOpen, copyright 2016. | ||
2. Methods for MMP profiling
2.1. Western blot
In western blot, proteins are separated by gel electrophoresis, which are then incubated with MMP antibodies. Antibodies bound to the MMP of interest are visible, with the thickness of the protein band correlating to the amount of protein.3 Antibodies are available for 60% of the 24 MMPs: MMP-1, -2, -3, -7, -8, -9, -10, -11, -12, -13, -14, -21, -25, -26, and -28.2.2. Gelatin zymography
In zymography, MMP activity is visualized by substrate conversion and detection of the reaction product or disappearance of the substrate.4 In this method, a substrate is polymerized with acrylamide in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), separating proteins by mass. The most widely used method is gelatin zymography, which detects the gelatinases MMP-2 and MMP-9. This method discriminates proMMPs, however it does distinguish between active unregulated MMPs and TIMP-complexed MMPs because under the denaturing conditions, the TIMP-MMP non-covalent complexes dissociate and appear as active MMP bands. For example, analyses of non-diabetic mouse wounds by gelatin zymography showed the presence of proMMP-9, proMMP-2, and active MMP-2 on days 7 and 10 after wound infliction (Fig. 3A).5 However, active MMP-2 was not observed in these wounds by the batismastat affinity resin/proteomics analysis, suggesting that the observed active MMP-2 band may be in complex with TIMP. To confirm the results, western blot analysis with TIMP-1 and TIMP-2 antibodies showed the presence of TIMP-1 and TIMP-2 in the samples (Fig. 3B and C), suggesting that the active MMP-2 band in Fig. 3A may be MMP-2-TIMP. Immunoprecipitation with TIMP-2 antibody, followed by western blot found that MMP-2 was present together with TIMP-2, indicating that indeed MMP-2 was in complex with TIMP (Fig. 3D and E). Since the TIMP-MMP-2 complex is catalytically incompetent, MMP-2 does not play a role in wound pathology or repair. | ||
| Fig. 3 (A) Gelatin zymography of non-diabetic mouse wounds shows the presence of proMMP-9, proMMP-2, and active MMP-2. (B) Analyses (n = 3 mice per time point) by western blot show the presence of TIMP-1 and TIMP-2. Actin was used as a loading control. (C) Quantification (n = 3 mice per time point) of TIMP-1 and TIMP-2 in (B). (D) Immunoprecipitation of wound homogenates (n = 3 mice per time point) with TIMP-2 antibody, followed by western-blot analysis indicates that MMP-2 is in complex with TIMP-2. (E) Quantification (n = 3 mice per time point) of TIMP-2 and MMP-2 in (D). This figure has been reproduced from Nguyen et al.5 with permission from the American Chemical Society, copyright 2020. | ||
2.3. In situ zymography
In situ zymography detects and localizes MMP activity in tissue sections. In this method, tissue is embedded and cryosectioned, and the sections are placed on glass slides. The slides are immersed in dye quenched (DQ)-collagen for collagenase activity (MMP-1, MMP-8, MMP-13) or DQ-gelatin for gelatinase activity (MMP-2 and MMP-9), and analysed by fluorescence microscopy, where activity appears as green fluorescence. Fig. 4 shows the use of in situ zymography in diabetic wounds.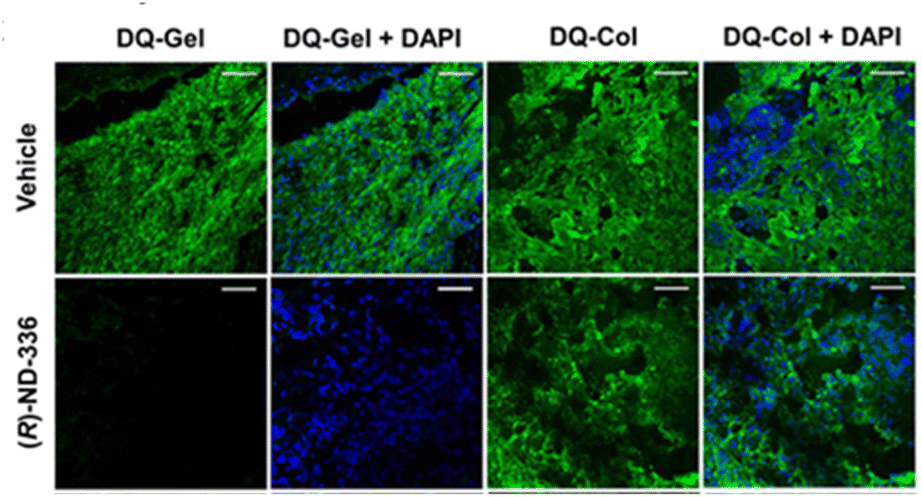 | ||
| Fig. 4 Analyses of diabetic mouse wounds by in situ zymography with DQ-gelatin and DQ-collagen show that the MMP-9 inhibitor (R)-ND-336 inhibits MMP-9 activity (fluorescent green) and does not inhibit MMP-8 activity (fluorescent green). Merged images with nuclear DNA staining by DAPI (blue) as control for the number of nuclei. Images were taken with a 40× lens (scale bars, 50 μm). This figure has been reproduced from Nguyen et al.45 with permission from the American Chemical Society, copyright 2018. | ||
2.4. Activity-based profiling
Activity-based protein profiling (Fig. 5) uses protein engineering to introduce a cysteine nucleophile in the active site proximity that is irreversibly labelled by the affinity-based probe containing a reactive hydroxamate.46 This method images and inhibits the specific MMP. This approach has been demonstrated for MMP-12 and MMP-14.47 While this strategy targets only the active forms of MMPs, it requires protein engineering of MMPs and synthesis of activity-based probes for the specific MMP.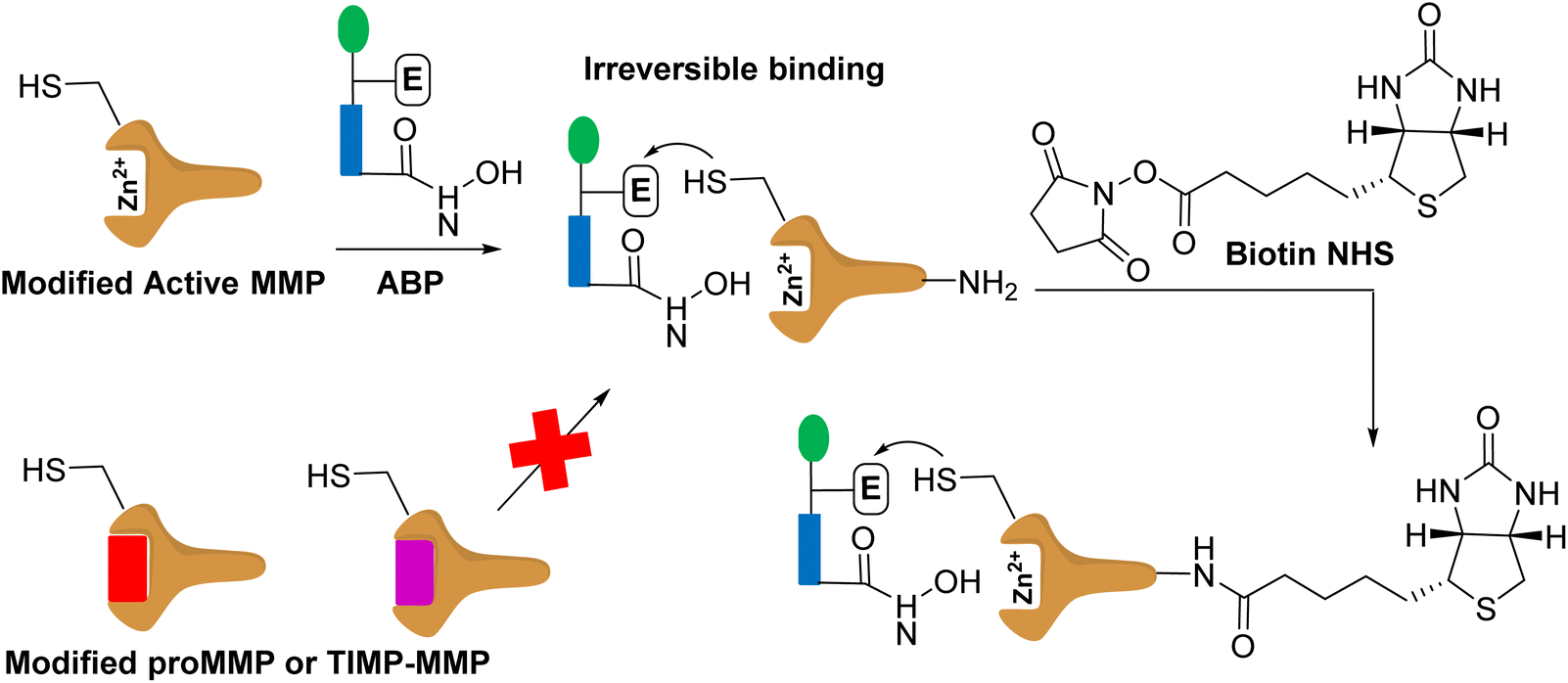 | ||
| Fig. 5 Modified MMP binds to affinity based probe (APB). The modified MMP contains a cysteine. The modified active MMP binds to the ABP irreversibly. The proMMP or TIMP-MMP cannot bind to the ABP as the active site is not accessible as the prodomain and TIMP block it. The labelled active MMP can be treated with biotin for protein purification, and subsequent identification. | ||
In a related approach, the zinc-chelating hydroxamate is coupled to a photocrosslinker that binds covalently only to the active forms of MMPs. The affinity-based probe contains a biotin group that allows for purification of the bound MMPs, which are identified by mass spectrometry.48 Cells were incubated with a cocktail library of affinity-based probes that included six MMPs.49 This approach requires a library of selective MMP-directed probes.
2.5. Affinity resins
Affinity resins consisting of attachment of the broad-spectrum inhibitors batimastat (Fig. 6A) and TAPI-2 (Fig. 6B) inhibitors of MMPs have been covalently attached to sepharose resin.50,51 Only the active forms of MMPs bind to the affinity resins.52 In the case of the TAPI-2 resin, the bound MMPs are eluted with buffer containing EDTA, and following trypsin digestion the peptides are identified by mass spectrometry. TAPI-2 inhibits the related proteinases called ADAMs (a disintegrin and metalloproteinase): ADAM8 (Ki 10 ± 1 μM), ADAM10 (Ki 3 ± 2 μM), ADAM12 (Ki >100 μM), ADAM17 (Ki 120 ± 30 nM).53 TAPI-2 also inhibits MMP-12 with a Ki value of 12 μM.50 Given the Ki values, it appears that TAPI-2 is more selective towards ADAM17.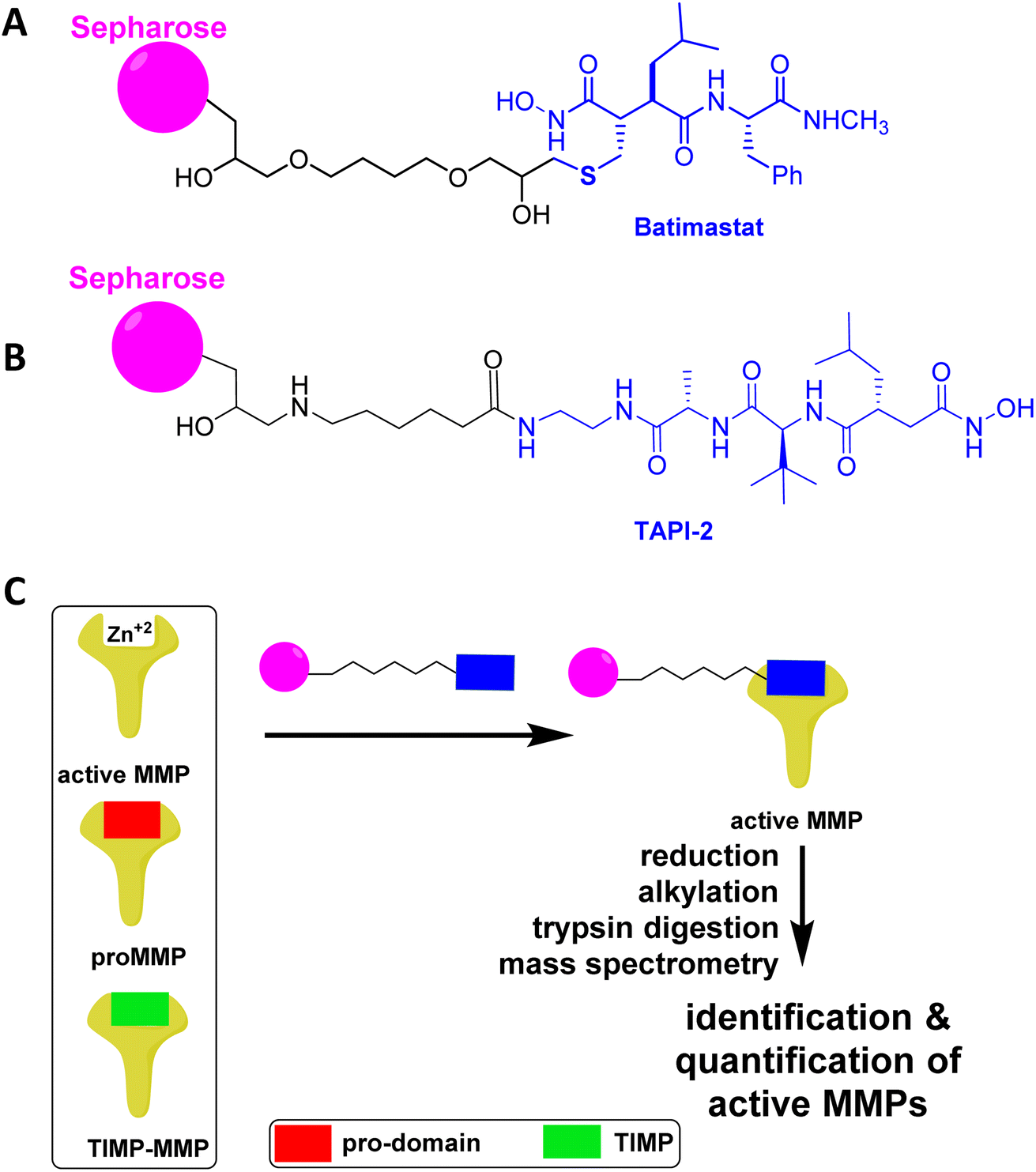 | ||
| Fig. 6 Affinity resin for binding of active MMPs based on the broad-spectrum MMP inhibitor (A) batimastat or (B) TAPI-2. (C) Only the active MMPs bind to the affinity resin, as proMMPs and TIMP-MMPs do not have accessible the active site for complexation of the hydroxamate with zinc ion. (C) Analysis of the active MMPs by reduction of cysteines, alkylation of the resulting thiols, trypsin digestion, and mass spectrometry identification and quantification. Panel C is reproduced from Peng et al.54 with permission from the American Chemical Society, copyright 2022. | ||
For the batimastat resin, cysteines in MMPs are reduced and alkylated, followed by trypsin digestion, and analysis of the resulting peptides by tandem mass spectrometry.14,54 Batimastat is a potent broad-spectrum MMP/ADAM inhibitor with IC50 values of 3 nM for MMP-1,55 4 nM for MMP-2,55 20 nM for MMP-3,55 6 nM for MMP-7,56 10 nM for MMP-8,55 10 nM for MMP-9,55, 1 nM for MMP-13,56 2.8 nM for MMP-14,56 29 nM for MMP-16,57 50 nM for ADAM8,58 50 nM for ADAM9,59 and 230 nM for ADAM17.60 The batimastat affinity resin has a limit of quantification of 6 femtomoles and recoveries for MMPs and ADAMs are similar, ranging from 75% to 100%.61 Based on the inhibition data, the batimastat affinity resin should be more sensitive than the TAPI-2 affinity resin. Both the TAPI-2 and batimastat affinity resins are not commercially available.
3. MMP profiling of human diseases and their roles
MMPs have been implicated in numerous diseases, including cancer, neurological diseases (Alzheimer's, Parkinson's, stroke, traumatic brain injury, spinal cord injury), pulmonary diseases such as COPD, pulmonary emphysema, idiopathic pulmonary fibrosis, and arthritis among others. Table 1 summarizes the roles of MMPs in disease.Only the affinity-based protein profiling, the TAPI-2 affinity resin, and the batimastat affinity resin can “fish” out active MMPs, which can then be identified by mass spectrometry. An advantage of these methods is the ability to identify active MMPs in diseased tissue, rather than analyzing for specific MMPs by zymography and ELISA. The batimastat affinity resin has been used recently to identify the related proteinases ADAMs.62,63 Bioanalytical methods have been developed to quantify the levels of active MMPs and ADAMs identified by the batimastat affinity resin.14,54,62,63
Once the active MMPs and ADAMs are identified in human diseased tissues and quantified relative to control tissues, the roles of the identified MMPs/ADAMs can be ascertained by use of selective inhibitors for that particular MMP/ADAM in in vitro or in vivo models of the disease. A challenge is that the 24 human MMPs and 25 human ADAMs are closely related, so selective inhibitors may not be available. Adding to the complexity is that MMPs/ADAMs can play beneficial (such as repair) and detrimental (such as pathology) roles in a disease. Confirmation of the MMP role in disease can be ascertained with MMP gene knockouts. However, gene ablation of a particular MMP may be accompanied by other effects. For example, MMP-2 knockout mice and gene ablation of MMP-8 have a compensatory increase in MMP-9.64,65
3.1. Diabetic foot ulcers (DFUs)
Most studies in human DFUs have used wound fluid analyzed for specific MMPs. Wound fluid from healed (n = 7) and non-healed (n = 9) DFUs were analyzed for MMP-2 and MMP-9 by zymography, and for MMP-1, MMP-8, and TIMP-1 by ELISA.66 Active MMP-9 was not observed in the healed DFUs at inclusion, increasing to 20.85 pg μg−1 protein in non-healed ulcers, indicating that elevated levels of MMP-9 were detrimental to wound healing. Likewise, analysis of wound fluid from 23 healed and 39 non-healed DFUs by zymography found higher levels of active MMP-9 in non-healers (2.90 ± 1.64, p < 0.05) compared to non-healers (1.18 ± 1.21 μg mL−1),67 indicating that higher levels of MMP-9 can predict poor wound healing.Wound tissue from patients with DFUs (n = 25) and non-diabetic controls (n = 23) were analyzed with the batimastat affinity resin. Only two MMPs in their active forms were identified: MMP-8 and MMP-9.45 The analysis was stratified by wound severity and infection, with Wagner grade (WG) 1 as a superficial ulcer, 2 as a deeper full-thickness ulcer, 3 as deep abscess or osteomyelitis, and 4 as partial gangrene of the foot. MMP-8 was present at 10-fold higher levels in DFUs compared to controls; similar levels were observed regardless of wound severity and infection (Fig. 7A). In contrast, active MMP-9 concentrations increased with DFU severity and infection: 7-fold higher in WG1-2, and 34-fold higher in WG3-4 (Fig. 7B). The role of MMP-9 was found to be detrimental to wound healing using selective MMP-9 inhibitors and MMP-9 knockout mice made diabetic.13,14 On the other hand, MMP-8 was shown to be beneficial in wound repair by the use of a selective MMP-8 inhibitor and application of exogenous recombinant MMP-8.13,14
 | ||
| Fig. 7 Identification and quantification of (A) active MMP-8 and (B) active MMP-9 in patients with DFUs WG1–4 (n = 25) and in dermal samples from non-diabetic (control) patients (n = 23) using the batimastat affinity resin coupled with proteomics. Higher levels of active MMP-9 are observed in WG3–4 (n = 13) when compared to WG1–2 (n = 12). Mean ± SEM; **p < 0.01 using Mann–Whitney U two-tailed test. This figure has been reproduced from Nguyen et al.45 with permission from the American Chemical Society, copyright 2018. | ||
3.2. Pressure ulcers (PUs)
Analysis of wound fluid from 56 patients with PUs for MMP-2, MMP-9, TIMP-1, and TIMP-2 found that the ratio of MMP-9 to TIMP-1 significantly decreased in healing PUs, supporting that high levels of MM-9 activity and low levels of TIMP-1 are detrimental to wound healing.68Analyses of wound tissue from patients with PUs ulcer stage 3–4 (where stage 3 is full-thickness deep open ulcer and stage 4 is full-thickness with muscle or bone exposure) using the batimastat affinity resin and proteomics, revealed the presence of active MMP-8 and MMP-9.54 MMP-8 was present at 16-fold higher levels in PUs relative to controls, while MMP-9 was 14-fold higher (Fig. 8).54 The amounts of MMP-9 decreased with improvement of PUs, indicating that MMP-9 played a detrimental role in PUs. The higher levels of MMP-8 and MMP-9 in PUs were not surprising, given that PUs share similar pathology with DFUs; in both diseases wound repair is stalled with persistent inflammation, and development of both wounds are due to neuropathy and trauma. The likelihood of developing PUs is twice as high for patients with diabetes.69 In addition, MMP-1 and MMP-14 in their active forms were present in PUs (Fig. 8). The levels of MMP-1 in PUs were similar to those in the controls, while MMP-14 concentrations were 46-fold higher in PUs compared to the controls. Inhibition of MMP-9 and MMP-14 accelerated wound healing and improved ulcer stage in a mouse model of PUs.54
 | ||
| Fig. 8 Identification and quantification of active MMPs in patients with PUs using the batimastat affinity resin with proteomics. Active MMP-1, MMP-8, MMP-9, and MMP-14 were identified in wound tissue of patients at ulcer stage 3–4 PUs (n = 15) and in control (healthy) patients (n = 23). Mean ± SEM, *p < 0.05, ***p < 0.001 by Mann–Whitney U two-tails. This figure has been reproduced from Peng et al.54 with permission from the American Chemical Society, copyright 2022. | ||
3.3. Idiopathic pulmonary fibrosis (IPF)
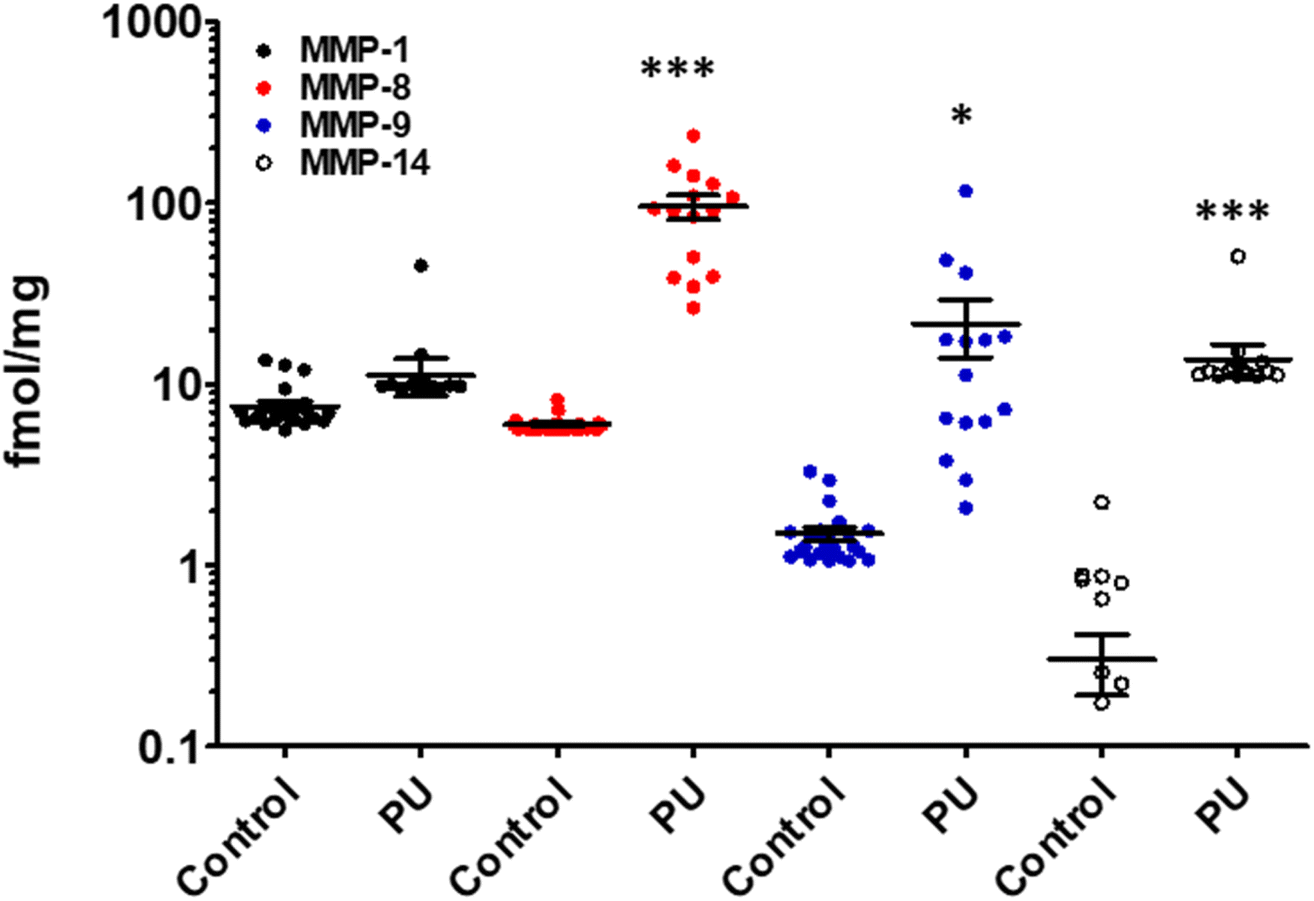
MMP-9 has been found in lungs and bronchoalveolar lavage fluid from patients with IPF.70 MMP-9 expression increased in airway basal cells from patients with IPF relative to those from normal lungs.71 In a mouse model of IPF, mice treated with andecaliximab, a monoclonal antibody that inhibits MMP-9, showed improved outcomes.71,72Lung tissue from patients with IPF were analyzed with the batimastat affinity resin coupled with proteomics. Six proteinases MMP-1, MMP-8, MMP-14, ADAM9, ADAM10, and ADAM17 were identified.63 Levels of MMP-1 were 1.5-fold higher in IPF compared to control (Fig. 9A), while MMP-8 levels were lower in IPF than control (Fig. 9B) and those of MMP-14 were similar in IPF and control (Fig. 9C). ADAM9 (Fig. 9D), ADAM10 (Fig. 9E), and ADAM17 (Fig. 9F) levels increased in IPF compared to controls. Inhibition of MMP-1 and ADAM10 decreased fibrosis in an in vitro assay,63 indicating the potential for therapeutic intervention. Bleomycin induced lung-injured mice administered the ADAM-10 inhibitor GI254023X showed significantly reduced mortality.73
 | ||
| Fig. 9 Identification and quantification of active MMPs and ADAMs in lungs of patients with IPF using the batimastat affinity resin and proteomics. Higher levels of MMP-1, ADAM9, ADAM10, and ADAM17 were observed in IPF lungs. Concentrations of (A) active MMP-1, (B) active MMP-8, (C) active MMP-14, (D) active ADAM9, (E) active ADAM10, and (F) active ADAM17. IPF lung tissue (n = 26 patients) and healthy control lungs (n = 4); mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 relative to control by Student's t-test with two-tail distribution and unequal variance. This figure has been reproduced from Peng et al.63 with permission from the American Chemical Society, copyright 2022. | ||
3.4. Pterygium
MMP-1 and MMP-3 were reported in cultured human pterygium fibroblasts by mRNA, ELISA, and western blot.74 MMP-2 and MMP-9 were observed in pterygium tissues from 15 patients by mRNA and gelatin zymography.75 Elevated levels of MMP-7 were reported in cultured pterygia using monoclonal antibodies.76 MMP-1, MMP-2, and MMP-9 were found in pterygium cells by immunohistochemistry using antibodies.77The batimastat affinity resin and mass spectrometry was used to identify active MMP-14, ADAM9, ADAM10, and ADAM17 in pterygium (benign ocular tumor) tissue (Fig. 10).62 Normal human conjunctiva fibroblasts were used as control due to the unavailability of control human eye tissue for ethical reasons. Active MMP-14 and ADAM10 were about 70 times higher in patients with pterygia than controls. Inhibition of MMP-14 decreased fibroblast migration and collagen contraction in in vitro assays,62 indicating a potential therapeutic approach to treat pterygium. Current treatment of pterygium is recurrent surgery.
 | ||
| Fig. 10 Identification and quantification of active MMPs and ADAMs in pterygium tissue by the batimastat affinity resin and proteomics. Primary pterygium tissue (n = 28 patients); mean ± SEM, ND = not detectable, ***p < 0.001 by Student's t-test. This figure has been reproduced from Masitas et al.62 with permission from the American Chemical Society, copyright 2022. | ||
3.5. Glioblastoma multiforme
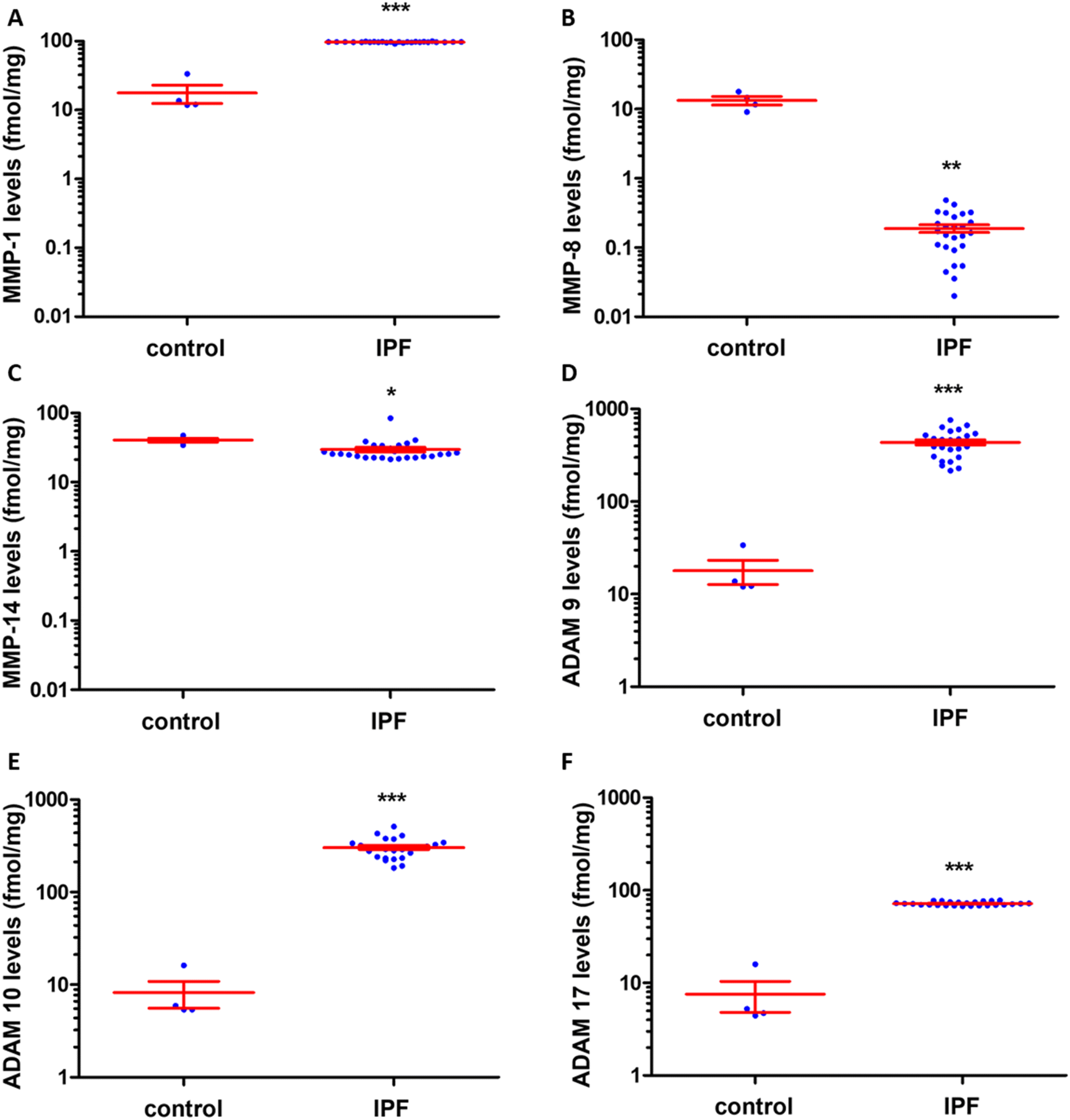
MMP-14 (also known as membrane-type 1-matrix metalloproteinase (MT1-MMP)), has been implicated in the pathology of gliomas.78 MMP-14 expression has been correlated to glioma cell invasiveness, with higher levels of MMP-14 expression in the most invasive U251 glioma cells compared to other cell lines.79 In addition, MMP-14 plays a role in tumor angiogenesis through activation of MMP-2 and MMP-9.80Brain tissue from patients with glioblastoma multiforme were analyzed by the batimastat affinity resin, followed by proteomics. Only MMP-14 in its active form was identified and quantified.81 Levels of active MMP-14 were non-quatifiable in control brains and were 4.0 ± 0.8 fmol mg−1 tissue in brains of patients with glioblastoma multiforme (Fig. 11). Inhibition of MMP-14 with a selective inhibitor prolonged survival in mice with glioblastoma.81
 | ||
| Fig. 11 Identification and quantification of active MMP-14 in brain tissue by the batimastat affinity resin and proteomics. Glioblastoma multiforme patients (n = 15), healthy controls (n = 4); mean ± SEM, **p < 0.001 by Student's t-test. This figure has been graphed from data in Thakur et al.81 | ||
4. MMP inhibitors in development
Recent reviews describe new strategies for targeting MMPs,82–84 including integration of computational design and yeast surface display to engineer a TIMP-mutant that is a potent and specific inhibitor of MMP-14 (ref. 85) and MMP-responsive drug delivery and tumor targeting.86 This section reviews MMP inhibitors in development.The role of MMPs in promoting cancer progression prompted the development of small-molecule MMP inhibitors. At the time it was thought that all MMPs were involved in cancer progression. Thus, the initial inhibitors were broad-spectrum hydroxamate derivatives that inhibited all MMPs by chelating to zinc. Numerous MMP inhibitors were studied in clinical trials for cancer, but all failed. The reasons for failure were numerous,87 including poor clinical trial design, broad-spectrum inhibition of MMPs and ADAMs, lack of knowledge of which MMP(s) should be targeted, undesirable side effects, poor pharmacokinetic properties, poor knowledge of MMP biology, animal models not paralleling the human disease.
To date, Periostat® (doxycycline hyclate, Fig. 12) is the only MMP-inhibitor on the market; it is approved for treatment of periodontitis. Doxycycline is a weak broad-spectrum MMP inhibitor, with IC50s of >400 μM for MMP-1, 56 μM for MMP-2, 32 μM for MMP-3, 28 μM for MMP-7, 26–50 μM for MMP-8, and 2–50 μM for and MMP-13.87
 | ||
| Fig. 12 Structures of MMP inhibitors. | ||
A clinical trial was completed (NTC 02744703) in 10 patients receiving caffeic acid phenethyl ester (Fig. 12) as an MMP inhibitor for bonding of resin material and tooth substrate. Pre-treatment improved the bond strength of resin restoration to dentin. Caffeic acid phenethyl ester inhibits MMP-1 (IC50 187 μM), MMP-2 (IC50 5 μM), MMP-3 (IC50 224 μM), MMP-7 (IC50 335 μM), and MMP-9 (IC50 2 μM).88 Additional clinical trials with caffeic acid phenethyl ester have been started.89 Two clinical trials, a phase 1 study assessing the safety and tolerability of multiple oral doses in 21 subjects with amyotrophic lateral sclerosis (NTC 03049046), and a phase 3 clinical trial in 300 patients with esophageal cancer receiving oral caffeic acid phenethyl ester (NTC 03070262) are listed in https://clinicaltrials.gov; no results are reported.
Andecaliximab (GS-5745) is a monoclonal antibody from Gilead Sciences, Inc. that inhibits MMP-9. In a phase 1b study, andecaliximab was evaluated alone and in combination with mFOLFOX6 (fluoracil). Encouraging results were observed in patients with gastric or gastroesophageal junction adenocarcinoma.90 However, a phase 3 clinical trial in 432 patients with gastric or gastroesophageal junction adenocarcinoma did not improve overall survival.91 In 2019, Gilead terminated clinical development of andecaliximab for oncology.
(R)-ND-336 (Fig. 12) is a thiirane mechanism-based inhibitor selective for MMP-2 (Ki 127 nM), MMP-9 (Ki 19 nM), and MMP-14 (Ki 119 nM); it inhibits other MMPs poorly (KiMMP-8 8590 nM, all other MMPs Ki > 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM).45 (R)-ND-336 accelerates wound healing in diabetic mice,45 including in an infected model,92 as well as in a mouse model of pressure ulcers.54 As MMP-9 is detrimental to diabetic wound healing, an MMP-9 siRNA hydrogel has shown efficacy in diabetic rats. Targeting MMP-14 sensitized glioblastoma to radiation; inhibition of MMP-14 with (R)-ND-336 significantly increased survival in a mouse model of glioblastoma multiforme93 (survival of 77 days after irradiation compared to 148 days with combination of irradiation plus (R)-ND-336, p = 0.02).81 (R)-ND-336 has completed Investigational New Drug-enabling studies.
000 nM).45 (R)-ND-336 accelerates wound healing in diabetic mice,45 including in an infected model,92 as well as in a mouse model of pressure ulcers.54 As MMP-9 is detrimental to diabetic wound healing, an MMP-9 siRNA hydrogel has shown efficacy in diabetic rats. Targeting MMP-14 sensitized glioblastoma to radiation; inhibition of MMP-14 with (R)-ND-336 significantly increased survival in a mouse model of glioblastoma multiforme93 (survival of 77 days after irradiation compared to 148 days with combination of irradiation plus (R)-ND-336, p = 0.02).81 (R)-ND-336 has completed Investigational New Drug-enabling studies.
5. Conclusions
MMP profiling is challenging due to the presence of 24 closely-related human MMPs and 25 human ADAMs, each existing in three forms, of which only one is active and capable of catalysis. This challenge has been overcome with development of affinity resins that incorporate a broad-spectrum MMP/ADAM inhibitor. In particular, the batimastat affinity resin is a potent and broad-spectrum chemical tool that captures MMPs and ADAMs in their active forms, which are subsequently identified by mass spectrometry. The batimastat affinity resin has proven useful in identifying targets for intervention in several human diseases.Author contributions
M. C. wrote the manuscript.Conflicts of interest
The author declares the following competing financial interests: US Patent 9,604,957, US Patent 9,951,035, US Patent 10,253,103, European Patent 3107905, Japan Patent 6,5,21,995, and China Patent ZL201580023723.6 have been issued for (R)-ND-336 (assigned to the University of Notre Dame, with M. C. as an inventor. M. C. reports involvement in an organization with a financial interest in (R)-ND-336.Acknowledgements
M. C. was supported in part by Department of Defense Therapeutic Development Award W81XWH-19-1–0493.Notes and references
- H. Nagase, R. Visse and G. Murphy, Cardiovasc. Res., 2006, 69, 562–573 CrossRef CAS PubMed.
- D. Greenbaum, C. Colangelo, K. Williams and M. Gerstein, Genome Biol., 2003, 4, 117 CrossRef PubMed.
- T. Mahmood and P. C. Yang, Am. J. Med. Sci., 2012, 4, 429–434 Search PubMed.
- J. Vandooren, N. Geurts, E. Martens, P. E. Van den Steen and G. Opdenakker, Nat. Methods, 2013, 10, 211–220 CrossRef CAS PubMed.
- T. T. Nguyen, W. R. Wolter, B. Anderson, V. A. Schroeder, M. Gao, M. Gooyit, M. A. Suckow and M. Chang, ACS Pharmacol. Transl. Sci., 2020, 3, 489–495 CrossRef CAS PubMed.
- T. T. Nguyen, S. Mobashery and M. Chang, Roles of matrix metalloproteinases in cutaneous wound healing, in Wound Healing: New Insights into Ancient Challenges, ed. V. A. Alexandrescu, InTech, 2016, pp. 37–71 Search PubMed.
- A. Pardo and M. Selman, Int. J. Biochem. Cell Biol., 2005, 37, 283–288 CrossRef CAS PubMed.
- G. Klein, E. Vellenga, M. W. Fraaije, W. A. Kamps and E. S. de Bont, Crit. Rev. Oncol./Hematol., 2004, 50, 87–100 CrossRef CAS PubMed.
- R. Fridman, M. Toth, D. Pena and S. Mobashery, Cancer Res., 1995, 55, 2548–2555 CAS.
- Y. C. Chung, Y. S. Kim, E. Bok, T. Y. Yune, S. Maeng and B. K. Jin, Mediators Inflammation, 2013, 2013, 370526 Search PubMed.
- Y. Yoshiyama, M. Asahina and T. Hattori, Acta Neuropathol., 2000, 99, 91–95 CrossRef CAS PubMed.
- A. Pardo, K. Gibson, J. Cisneros, T. J. Richards, Y. Yang, C. Becerril, S. Yousem, I. Herrera, V. Ruiz, M. Selman and N. Kaminski, PLoS Med., 2005, 2, e251 CrossRef PubMed.
- M. Gao, T. T. Nguyen, M. A. Suckow, W. R. Wolter, M. Gooyit, S. Mobashery and M. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15226–15231 CrossRef CAS PubMed.
- M. Gooyit, Z. Peng, W. R. Wolter, H. Pi, D. Ding, D. Hesek, M. Lee, B. Boggess, M. M. Champion, M. A. Suckow, S. Mobashery and M. Chang, ACS Chem. Biol., 2014, 9, 105–110 CrossRef CAS PubMed.
- Z. Gu, J. Cui, S. Brown, R. Fridman, S. Mobashery, A. Y. Strongin and S. A. Lipton, J. Neurosci., 2005, 25, 6401–6408 CrossRef CAS PubMed.
- O. Hadass, B. N. Tomlinson, M. Gooyit, S. Chen, J. J. Purdy, C. R. I. Robinson, D. S. Shin, V. A. Schroeder, M. A. Suckow, A. Simonyi, G. Y. Sun, S. Mobashery, J. Cui, M. Chang and Z. Gu, PLoS One, 2013, 8, e76904 CrossRef CAS PubMed.
- P. E. Van den Steen, B. Dubois, I. Nelissen, P. M. Rudd, R. A. Dwek and G. Opdenakker, Crit. Rev. Biochem. Mol. Biol., 2002, 37, 375–536 CrossRef CAS PubMed.
- W. B. Saunders, K. J. Bayless and G. E. Davis, J. Cell Sci., 2005, 118, 2325–2340 CrossRef CAS PubMed.
- E. M. Deraz, Y. Kudo, M. Yoshida, M. Obayashi, T. Tsunematsu, H. Tani, S. B. Siriwardena, M. R. Keikhaee, G. Qi, S. Iizuka, I. Ogawa, G. Campisi, L. Lo Muzio, Y. Abiko, A. Kikuchi and T. Takata, PLoS One, 2011, 6, e25438 CrossRef CAS PubMed.
- X. Zhang, S. Huang, J. Guo, L. Zhou, L. You, T. Zhang and Y. Zhao, Int. J. Oncol., 2016, 48, 1783–1793 CrossRef CAS PubMed.
- W. Pan, M. Arnone, M. Kendall, R. H. Grafstrom, S. P. Seitz, Z. R. Wasserman and C. F. Albright, J. Biol. Chem., 2003, 278, 27820–27827 CrossRef CAS PubMed.
- I. K. Demedts, A. Morel-Montero, S. Lebecque, Y. Pacheco, D. Cataldo, G. F. Joos, R. A. Pauwels and G. G. Brusselle, Thorax, 2006, 61, 196–201 CrossRef CAS PubMed.
- S. Molet, C. Belleguic, H. Lena, N. Germain, C. P. Bertrand, S. D. Shapiro, J. M. Planquois, P. Delaval and V. Lagente, Inflammation Res., 2005, 54, 31–36 CrossRef CAS PubMed.
- H. H. Hou, H. C. Wang, S. L. Cheng, Y. F. Chen, K. Z. Lu and C. J. Yu, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2018, 315, L432–L442 CrossRef CAS PubMed.
- G. Tardif, P. Reboul, J. P. Pelletier and J. Martel-Pelletier, Mod. Rheumatol., 2004, 14, 197–204 CrossRef CAS PubMed.
- M. Toth, I. Chvyrkova, M. M. Bernardo, S. Hernandez-Barrantes and R. Fridman, Biochem. Biophys. Res. Commun., 2003, 308, 386–395 CrossRef CAS PubMed.
- C. Marusak, I. Bayles, J. Ma, M. Gooyit, M. Gao, M. Chang and B. Bedogni, Pharmacol. Res., 2016, 113, 515–520 CrossRef CAS PubMed.
- A. Lin, H. H. Xu, D. P. Xu, X. Zhang, Q. Wang and W. H. Yan, Hum. Immunol., 2013, 74, 439–446 CrossRef CAS PubMed.
- H. Wang, C. Qi and D. Wan, Ann. Transl. Med., 2021, 9, 124 CrossRef CAS PubMed.
- Y. Itoh, M. Kajita, H. Kinoh, H. Mori, A. Okada and M. Seiki, J. Biol. Chem., 1999, 274, 34260–34266 CrossRef CAS PubMed.
- C. Yip, P. Foidart, A. Noel and N. E. Sounni, Int. J. Mol. Sci., 2019, 20, 354 CrossRef PubMed.
- J. Cossins, T. J. Dudgeon, G. Catlin, A. J. Gearing and J. M. Clements, Biochem. Biophys. Res. Commun., 1996, 228, 494–498 CrossRef CAS PubMed.
- V. O. Bister, M. T. Salmela, M. L. Karjalainen-Lindsberg, J. Uria, J. Lohi, P. Puolakkainen, C. Lopez-Otin and U. Saarialho-Kere, Dig. Dis. Sci., 2004, 49, 653–661 CrossRef CAS PubMed.
- Y. Lu, P. Papagerakis, Y. Yamakoshi, J. C. Hu, J. D. Bartlett and J. P. Simmer, Biol. Chem., 2008, 389, 695–700 CrossRef CAS PubMed.
- Y. Pu, L. Wang, H. Wu, Z. Feng, Y. Wang and C. Guo, Oncol. Rep., 2014, 31, 2644–2650 CrossRef CAS PubMed.
- G. N. Marchenko, N. D. Marchenko and A. Y. Strongin, Biochem. J., 2003, 372, 503–515 CrossRef CAS PubMed.
- M. Yang and M. Kurkinen, Chicken matrix metalloproteinase 22, Handbook of Proteolytic Enzymes, 2004 Search PubMed.
- D. Pei, T. Kang and H. Qi, J. Biol. Chem., 2000, 275, 33988–33997 CrossRef CAS PubMed.
- D. Moogk, I. P. da Silva, M. W. Ma, E. B. Friedman, E. V. de Miera, F. Darvishian, P. Scanlon, A. Perez-Garcia, A. C. Pavlick, N. Bhardwaj, P. J. Christos, I. Osman and M. Krogsgaard, J. Transl. Med., 2014, 12, 342 CrossRef PubMed.
- E. Llano, A. M. Pendas, J. P. Freije, A. Nakano, V. Knauper, G. Murphy and C. Lopez-Otin, Cancer Res., 1999, 59, 2570–2576 CAS.
- S. Kojima, Y. Itoh, S. Matsumoto, Y. Masuho and M. Seiki, FEBS Lett., 2000, 480, 142–146 CrossRef CAS PubMed.
- H. Yamamoto, A. Vinitketkumnuen, Y. Adachi, H. Taniguchi, T. Hirata, N. Miyamoto, K. Nosho, A. Imsumran, M. Fujita, M. Hosokawa, Y. Hinoda and K. Imai, Carcinogenesis, 2004, 25, 2353–2360 CrossRef CAS PubMed.
- A. Cominelli, H. P. Gaide Chevronnay, P. Lemoine, P. J. Courtoy, E. Marbaix and P. Henriet, Mol. Hum. Reprod., 2014, 20, 767–775 CrossRef CAS PubMed.
- J. Lohi, C. L. Wilson, J. D. Roby and W. C. Parks, J. Biol. Chem., 2001, 276, 10134–10144 CrossRef CAS PubMed.
- T. T. Nguyen, D. Ding, W. R. Wolter, R. L. Perez, M. M. Champion, K. V. Mahasenan, D. Hesek, M. Lee, V. A. Schroeder, J. I. Jones, E. Lastochkin, M. K. Rose, C. E. Peterson, M. A. Suckow, S. Mobashery and M. Chang, J. Med. Chem., 2018, 61, 8825–8837 CrossRef CAS PubMed.
- N. Amara, M. Tholen and M. Bogyo, ACS Chem. Biol., 2018, 13, 2645–2654 CrossRef CAS PubMed.
- M. Morell, T. Nguyen Duc, A. L. Willis, S. Syed, J. Lee, E. Deu, Y. Deng, J. Xiao, B. E. Turk, J. R. Jessen, S. J. Weiss and M. Bogyo, J. Am. Chem. Soc., 2013, 135, 9139–9148 CrossRef CAS PubMed.
- A. Saghatelian, N. Jessani, A. Joseph, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10000–10005 CrossRef CAS PubMed.
- S. A. Sieber, S. Niessen, H. S. Hoover and B. F. Cravatt, Nat. Chem. Biol., 2006, 2, 274–281 CrossRef CAS PubMed.
- J. R. Freije and R. Bischoff, J. Chromatogr. A, 2003, 1009, 155–169 CrossRef CAS PubMed.
- D. Hesek, M. Toth, V. Krchnak, R. Fridman and S. Mobashery, J. Org. Chem., 2006, 71, 5848–5854 CrossRef CAS PubMed.
- D. Hesek, M. Toth, S. O. Meroueh, S. Brown, H. Zhao, W. Sakr, R. Fridman and S. Mobashery, Chem. Biol., 2006, 13, 379–386 CrossRef CAS PubMed.
- M. L. Moss and F. H. Rasmussen, Anal. Biochem., 2007, 366, 144–148 CrossRef CAS PubMed.
- Z. Peng, T. T. Nguyen, M. Wang, B. Anderson, M. M. Konai, V. A. Schroeder, W. R. Wolter, T. Page-Mayberry, C. E. Peterson, S. Mobashery and M. Chang, ACS Chem. Biol., 2022, 17, 1357–1363 CrossRef CAS PubMed.
- I. Botos, L. Scapozza, D. Zhang, L. A. Liotta and E. F. Meyer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2749–2754 CrossRef CAS PubMed.
- H. Laronha, I. Carpinteiro, J. Portugal, A. Azul, M. Polido, K. T. Petrova, M. Salema-Oom and J. Caldeira, Biomolecules, 2020, 10, 717 CrossRef CAS PubMed.
- F. Grams, H. Brandstetter, S. D'Alo, D. Geppert, H. W. Krell, H. Leinert, V. Livi, E. Menta, A. Oliva, G. Zimmermann, F. Gram and V. E. Livi, Biol. Chem., 2001, 382, 1277–1285 CAS.
- U. Schlomann, D. Wildeboer, A. Webster, O. Antropova, D. Zeuschner, C. G. Knight, A. J. Docherty, M. Lambert, L. Skelton, H. Jockusch and J. W. Bartsch, J. Biol. Chem., 2002, 277, 48210–48219 CrossRef CAS PubMed.
- T. Maretzky, S. Swendeman, E. Mogollon, G. Weskamp, U. Sahin, K. Reiss and C. P. Blobel, Biochem. J., 2017, 474, 1467–1479 CrossRef CAS PubMed.
- H. P. Hansen, B. Matthey, S. Barth, T. Kisseleva, T. Mokros, S. J. Davies, R. P. Beckett, R. Foelster-Holst, H. H. Lange, A. Engert and H. Lemke, Int. J. Cancer, 2002, 98, 210–215 CrossRef CAS PubMed.
- J. I. Jones, T. T. Nguyen, Z. Peng and M. Chang, Pharmaceuticals, 2019, 12, 79 CrossRef CAS PubMed.
- C. Masitas, Z. Peng, M. Wang, M. M. Konai, L. F. Avila-Cobian, L. Lemieux, J. Hovanesian, J. E. Grady, S. Mobashery and M. Chang, ACS Pharmacol. Transl. Sci., 2022, 5, 555–561 CrossRef CAS PubMed.
- Z. Peng, M. M. Konai, L. F. Avila-Cobian, M. Wang, S. Mobashery and M. Chang, ACS Pharmacol. Transl. Sci., 2022, 5, 555–561 CrossRef PubMed.
- J. Y. Hsu, R. McKeon, S. Goussev, Z. Werb, J. U. Lee, A. Trivedi and L. J. Noble-Haeusslein, J. Neurosci., 2006, 26, 9841–9850 CrossRef CAS PubMed.
- A. Gutierrez-Fernandez, M. Inada, M. Balbin, A. Fueyo, A. S. Pitiot, A. Astudillo, K. Hirose, M. Hirata, S. D. Shapiro, A. Noel, Z. Werb, S. M. Krane, C. Lopez-Otin and X. S. Puente, FASEB J., 2007, 21, 2580–2591 CrossRef CAS PubMed.
- M. Muller, C. Trocme, B. Lardy, F. Morel, S. Halimi and P. Y. Benhamou, Diabetic Med., 2008, 25, 419–426 CrossRef CAS PubMed.
- Y. Liu, D. Min, T. Bolton, V. Nube, S. M. Twigg, D. K. Yue and S. V. McLennan, Diabetes Care, 2009, 32, 117–119 CrossRef CAS PubMed.
- G. P. Ladwig, M. C. Robson, R. Liu, M. A. Kuhn, D. F. Muir and G. S. Schultz, Wound Repair Regen., 2002, 10, 26–37 CrossRef PubMed.
- P. Liu, W. He and H. L. Chen, J. Wound Ostomy Continence Nurse, 2012, 39, 495–499 CrossRef PubMed.
- A. Pardo and M. Selman, Proc. Am. Thorac. Soc., 2006, 3, 383–388 CrossRef CAS PubMed.
- M. S. Espindola, D. M. Habiel, A. L. Coelho, B. Stripp, W. C. Parks, J. Oldham, F. J. Martinez, I. Noth, D. Lopez, A. Mikels-Vigdal, V. Smith and C. M. Hogaboam, Am. J. Respir. Crit. Care Med., 2021, 203, 458–470 CrossRef CAS PubMed.
- E. Ahrman, O. Hallgren, L. Malmstrom, U. Hedstrom, A. Malmstrom, L. Bjermer, X. H. Zhou, G. Westergren-Thorsson and J. Malmstrom, J. Proteomics, 2018, 189, 23–33 CrossRef PubMed.
- D. Lagares, P. Ghassemi-Kakroodi, C. Tremblay, A. Santos, C. K. Probst, A. Franklin, D. M. Santos, P. Grasberger, N. Ahluwalia, S. B. Montesi, B. S. Shea, K. E. Black, R. Knipe, M. Blati, M. Baron, B. Wu, H. Fahmi, R. Gandhi, A. Pardo, M. Selman, J. Wu, J. P. Pelletier, J. Martel-Pelletier, A. M. Tager and M. Kapoor, Nat. Med., 2017, 23, 1405–1415 CrossRef CAS PubMed.
- D. Q. Li, S. B. Lee, Z. Gunja-Smith, Y. Liu, A. Solomon, D. Meller and S. C. Tseng, Arch. Ophthalmol., 2001, 119, 71–80 CAS.
- S. F. Yang, C. Y. Lin, P. Y. Yang, S. C. Chao, Y. Z. Ye and D. N. Hu, Invest. Ophthalmol. Visual Sci., 2009, 50, 4588–4596 CrossRef PubMed.
- N. Di Girolamo, M. T. Coroneo and D. Wakefield, Invest. Ophthalmol. Visual Sci., 2001, 42, 1963–1968 CAS.
- N. Dushku, M. K. John, G. S. Schultz and T. W. Reid, Arch. Ophthalmol., 2001, 119, 695–706 CrossRef CAS PubMed.
- I. Ulasov, R. Yi, D. Guo, P. Sarvaiya and C. Cobbs, Biochim. Biophys. Acta, 2014, 1846, 113–120 CAS.
- T. Abe, T. Mori, K. Kohno, M. Seiki, T. Hayakawa, H. G. Welgus, S. Hori and M. Kuwano, Clin. Exp. Metastasis, 1994, 12, 296–304 CrossRef CAS PubMed.
- T. Itoh, M. Tanioka, H. Yoshida, T. Yoshioka, H. Nishimoto and S. Itohara, Cancer Res., 1998, 58, 1048–1051 CAS.
- V. Thakur, V. S. Thakur, B. Aguila, T. I. Slepak, M. Wang, W. Song, M. Konai, S. Mobashery, M. Chang, A. B. Rana, D. Wang, J. T. de Freitas, S. Humayun Gultekin, S. M. Welford, M. E. Ivan and B. Bedogni, Neurooncol Adv., 2022, 4, vdac147 Search PubMed.
- G. B. Fields, Cells, 2019, 8, 984 CrossRef CAS PubMed.
- S. Mondal, N. Adhikari, S. Banerjee, S. A. Amin and T. Jha, Eur. J. Med. Chem., 2020, 194, 112260 CrossRef CAS PubMed.
- A. Winer, S. Adams and P. Mignatti, Mol. Cancer Ther., 2018, 17, 1147–1155 CrossRef CAS PubMed.
- V. Arkadash, G. Yosef, J. Shirian, I. Cohen, Y. Horev, M. Grossman, I. Sagi, E. S. Radisky, J. M. Shifman and N. Papo, J. Biol. Chem., 2017, 292, 3481–3495 CrossRef CAS PubMed.
- Q. Yao, L. Kou, Y. Tu and L. Zhu, Trends Pharmacol. Sci., 2018, 39, 766–781 CrossRef CAS PubMed.
- R. E. Vandenbroucke and C. Libert, Nat. Rev. Drug Discovery, 2014, 13, 904–927 CrossRef CAS PubMed.
- T. W. Chung, S. K. Moon, Y. C. Chang, J. H. Ko, Y. C. Lee, G. Cho, S. H. Kim, J. G. Kim and C. H. Kim, FASEB J., 2004, 18, 1670–1681 CrossRef CAS PubMed.
- Y. Yordanov, Pharmacia, 2019, 66, 107–114 CrossRef CAS.
- M. A. Shah, A. Starodub, S. Sharma, J. Berlin, M. Patel, Z. A. Wainberg, J. Chaves, M. Gordon, K. Windsor, C. B. Brachmann, X. Huang, G. Vosganian, J. D. Maltzman, V. Smith, J. A. Silverman, H. J. Lenz and J. C. Bendell, Clin. Cancer Res., 2018, 24, 3829–3837 CrossRef CAS PubMed.
- M. A. Shah, G. Bodoky, A. Starodub, D. Cunningham, D. Yip, Z. A. Wainberg, J. Bendell, D. Thai, J. He, P. Bhargava and J. A. Ajani, J. Clin. Oncol., 2021, 39, 990–1000 CrossRef CAS PubMed.
- Z. Peng, T. T. Nguyen, W. Song, B. Anderson, W. R. Wolter, V. A. Schroeder, D. Hesek, M. Lee, S. Mobashery and M. Chang, ACS Pharmacol. Transl. Sci., 2021, 4, 107–117 CrossRef CAS PubMed.
- B. Lan, L. Zhang, L. Yang, J. Wu, N. Li, C. Pan, X. Wang, L. Zeng, L. Yan, C. Yang and M. Ren, J. Nanobiotechnol., 2021, 19, 130 CrossRef CAS PubMed.
Footnote |
| † This paper is dedicated to Professor David Lynn on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2023 |