
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInitiating abilities of diphosphine- and diamine-ligated Pd complexes/NaBPh4 systems for C1 polymerization of diazoacetates†
Hiroaki
Shimomoto
 *,
Yuto
Miyano
,
Kaito
Kinoshita
,
Tomomichi
Itoh
and
Eiji
Ihara
*
*,
Yuto
Miyano
,
Kaito
Kinoshita
,
Tomomichi
Itoh
and
Eiji
Ihara
*
Department of Materials Science and Biotechnology, Graduate School of Science and Engineering, Ehime University, 3 Bunkyo-cho, Matsuyama 790-8577, Japan. E-mail: shimomoto.hiroaki.mx@ehime-u.ac.jp; ihara@ehime-u.ac.jp; Fax: +81-89-927-9949; Fax: +81-89-927-8547; Tel: +81-89-927-9949 Tel: +81-89-927-8547
First published on 30th January 2023
Abstract
A series of well-defined Pd complexes bearing diphosphines or diamines and a dichloroquinone-derived unit as ligands were newly prepared and their initiating abilities in conjunction with NaBPh4 for the C1 polymerization of diazoacetates were investigated. Among the (diphosphine)Pd(II)Cl(Cl-quinonyl) complexes examined here, square planar cis (diphosphine: diphenylphosphinoferrocene) and distorted square planar trans (diphosphine: xantphos) complexes with NaBPh4 yielded highly syndiotactic polymers from ethyl diazoacetate despite low polymer yields. A series of Pd(0) complexes bearing a bidentate diamine such as N,N,N′,N′-tetramethyl-1,3-propanediamine and 2,3-dichloronaphthoquinone in conjunction with NaBPh4 polymerized diazoacetates to afford moderately syndiotactic polymers in moderate yields, indicating that the (diamine)Pd(0)(dichloroquinone) framework could be a promising general platform for an initiating system with high activity and stereoselectivity for the C1 polymerization of diazoacetates.
Introduction
Polymerization of diazoacetates has been gaining recognition as an effective method for C–C main chain polymer synthesis with the development of effective initiating systems for the polymerization in the last two decades (Scheme 1).1–7 The polymerization affording poly(alkoxycarbonylmethylene)s (PACMs) is classified as a member of “C1 polymerizations”, where the C–C main chain of the products is constructed from “one carbon unit”. In comparison with vinyl polymers obtained from “C2 polymerization”, PACMs are expected to exhibit unique properties or functions derived from the structural feature of having an alkoxycarbonyl group (ester) on each of the main chain carbon atoms; indeed, enhancement in properties such as hydrophilicity, photophysical properties, and so on of PACMs has been observed,8–25 because of the densely-packed ester substituents around the C–C main chain.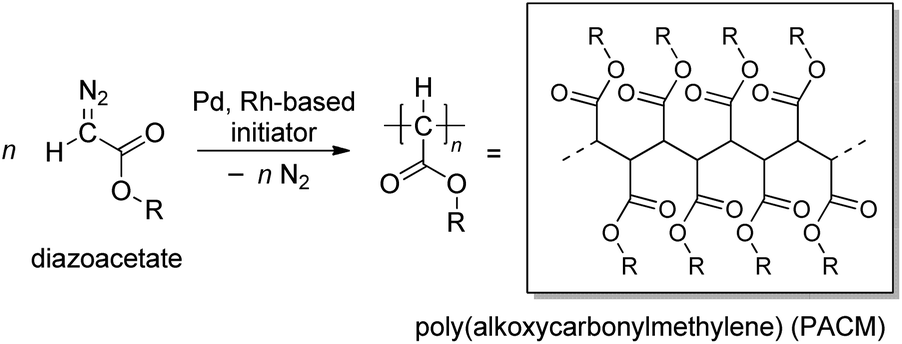 | ||
| Scheme 1 C1 polymerization of diazoacetates. | ||
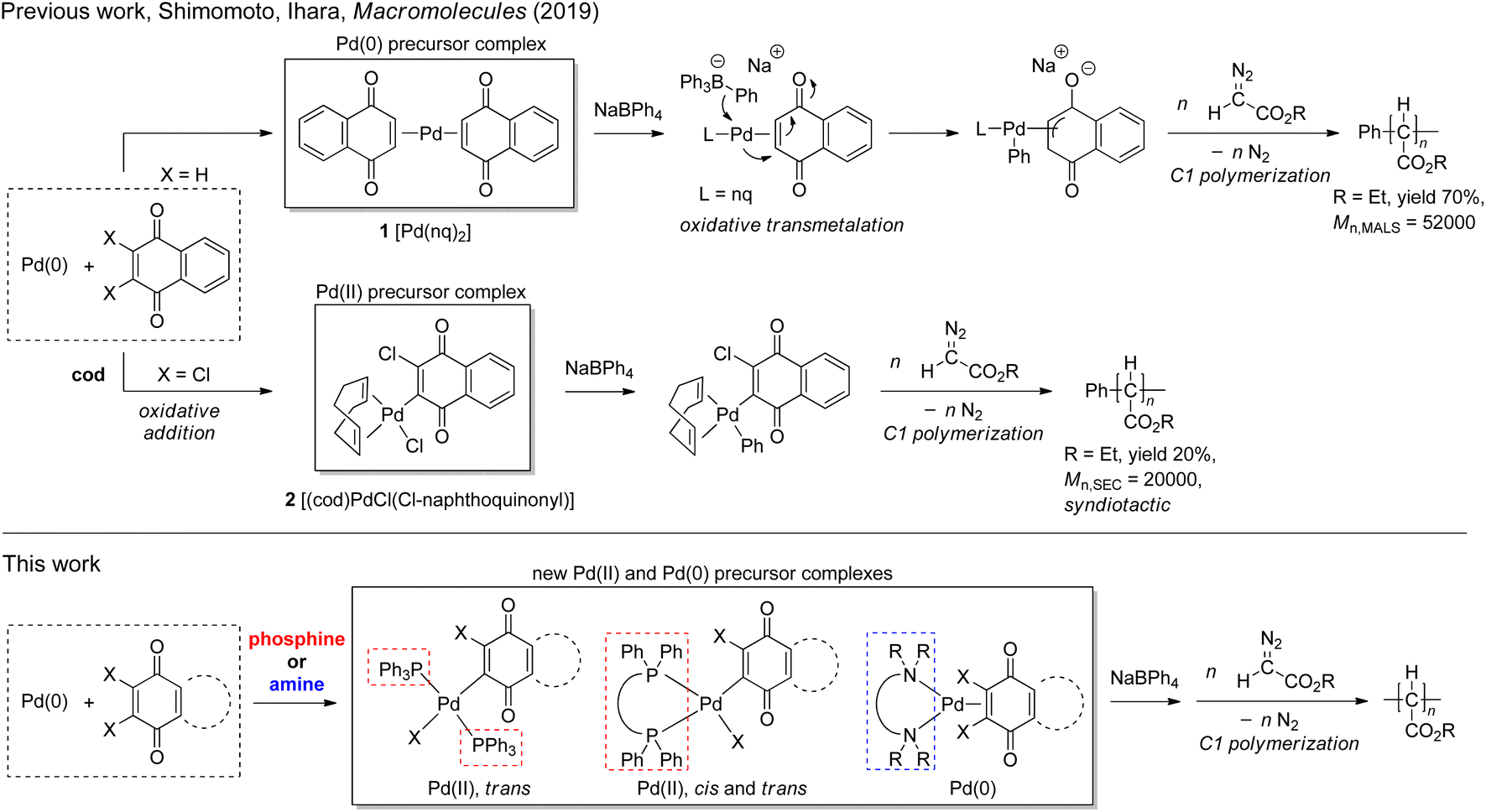
For the preparation of high Mn (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) PACMs, initiators based on some Rh and Pd complexes have been demonstrated to be most effective: for example, de Bruin and coworkers have reported that Rh(diene) complexes can yield high Mn (>200000) polymers with high syndiotacticities,26,27 and we and other groups have demonstrated that some Pd complexes with an η3-anionic ligand are effective for yielding atactic polymers with a variety of ester substituents.28–32 In addition, our recent finding revealed that two types of Pd complexes with naphthoquinone (nq)-based ligands can be utilized as effective initiators for diazoacetate polymerization in conjunction with NaBPh4.33 The first one is zero valent Pd(0)(nq)21, which can afford high Mn polymers in high yields; as shown in Scheme 2, according to a previous literature report on the reactivity of a Pd(0) complex in an analogous system,34 the initiating Pd–Ph group is proposed to be generated via oxidative transmetalation, where the transmetalation of the Ph group occurs from the borate to the Pd center and oxidation of Pd(0) to Pd(II) occurs at the same time; indeed, the high activity of the initiating species thus generated indicates the importance of the presence of an η3-type anionic ligand for the initiator to be highly active.35,36 The second one is divalent (cod)Pd(II)Cl(Cl-naphthoquinonyl) 2 (cod: 1,5-cyclooctadiene) generated via the oxidative addition of one of the two Cl–C bonds of 2,3-dichloronaphthoquinone (dichlone) to the Pd(0) center, which generates an initiating Pd–Ph species via transmetalation with subsequent activation with NaBPh4. Although the polymer yield with this initiating system was rather low (ca. 20%), it is noteworthy that the polymers obtained with this system are highly syndiotactic.
000) PACMs, initiators based on some Rh and Pd complexes have been demonstrated to be most effective: for example, de Bruin and coworkers have reported that Rh(diene) complexes can yield high Mn (>200000) polymers with high syndiotacticities,26,27 and we and other groups have demonstrated that some Pd complexes with an η3-anionic ligand are effective for yielding atactic polymers with a variety of ester substituents.28–32 In addition, our recent finding revealed that two types of Pd complexes with naphthoquinone (nq)-based ligands can be utilized as effective initiators for diazoacetate polymerization in conjunction with NaBPh4.33 The first one is zero valent Pd(0)(nq)21, which can afford high Mn polymers in high yields; as shown in Scheme 2, according to a previous literature report on the reactivity of a Pd(0) complex in an analogous system,34 the initiating Pd–Ph group is proposed to be generated via oxidative transmetalation, where the transmetalation of the Ph group occurs from the borate to the Pd center and oxidation of Pd(0) to Pd(II) occurs at the same time; indeed, the high activity of the initiating species thus generated indicates the importance of the presence of an η3-type anionic ligand for the initiator to be highly active.35,36 The second one is divalent (cod)Pd(II)Cl(Cl-naphthoquinonyl) 2 (cod: 1,5-cyclooctadiene) generated via the oxidative addition of one of the two Cl–C bonds of 2,3-dichloronaphthoquinone (dichlone) to the Pd(0) center, which generates an initiating Pd–Ph species via transmetalation with subsequent activation with NaBPh4. Although the polymer yield with this initiating system was rather low (ca. 20%), it is noteworthy that the polymers obtained with this system are highly syndiotactic.
 | ||
| Scheme 2 Initiating systems with Pd complexes bearing naphthoquinone-based ligands. | ||
With these results in mind, our next strategy examined here for further improving the initiating ability with respect to the polymer yield and Mn and tacticity control is the replacement of the neutral ligands, nq and cod in 1 and 2, respectively, with other neutral ligands in order to affect the reactivity and stereoselectivity of the Pd-containing initiating and propagating species. Thus, as candidates for such neutral ligands, we chose a series of phosphines and amines and attempted to prepare new well-defined Pd precursor complexes with these ligands and investigate the initiating ability of the resulting systems activated with NaBPh4 for diazoacetate polymerization. In the course of the investigation, we have succeeded in finding (N,N,N′,N′-tetramethyl-1,3-propanediamine)Pd(0)(dichlone)/NaBPh4 as a new effective initiating system, which possesses advantages of both systems based on 1 and 2 and can afford polymers with moderate syndioselectivity in moderate yields. The details of the investigation will be described in this paper.
Results and discussion
Preparation of Pd(II) complexes with diphosphine and quinonyl anions as ligands
First of all, we attempted to investigate the possibility of using phosphine as a ligand in place of cyclooctadiene (cod) in the (cod)Pd(II)Cl(Cl-naphthoquinonyl) (2)/NaBPh4 system. For this purpose, we employed three phosphines,37 triphenylphosphine (PPh3), 1,1′-bis(diphenylphosphino)ferrocene (dppf), and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos), and two dichloroquinones, 2,3-dichloronaphthoquinone (dichlone) and 14,15-dichloro-9,10-[1,2]benzenoanthracene-13,16(9H,10H)-dione [Cl2-qu(anth)],38 which was prepared by 2 + 4 addition of anthracene and 1,4-benzoquinone followed by chlorination, and some combinations of these reagents were employed for the syntheses of new Pd(II) complexes following the standard procedure for the preparation of 2 (Scheme 3). Thus, a starting material, Pd(0)2(dba)3·CHCl3, was reacted with an excess amount of phosphine and dichloroquinone in acetone (PPh3 and dppf) or THF (xantphos) at room temperature for 1 h, and as a result of the oxidative addition of a Cl–C bond of dichloroquinone to Pd(0), the expected Pd(II) complexes 3–7 were obtained as air-stable solids, which were identified by NMR and elemental analyses (see the ESI†).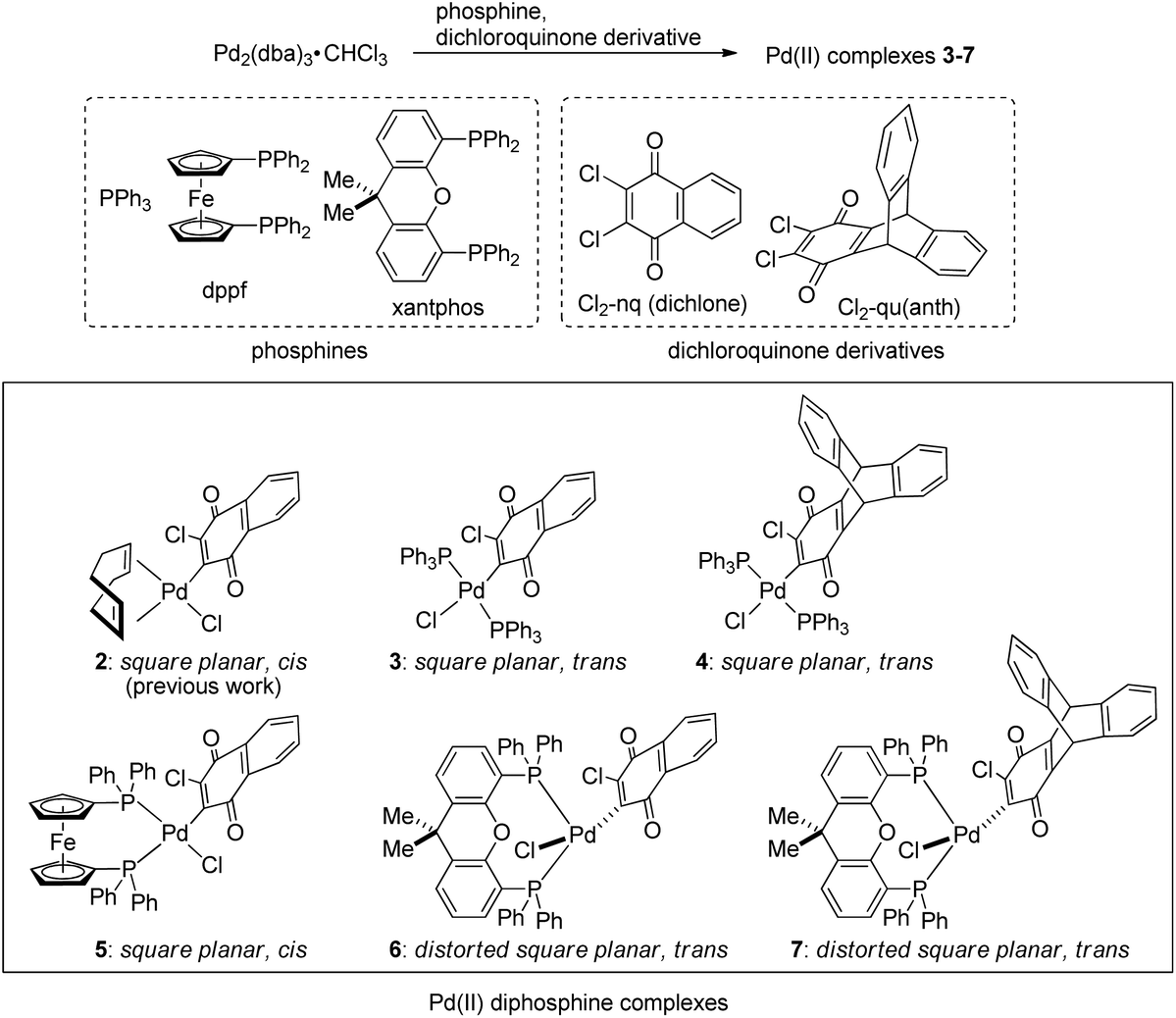 | ||
| Scheme 3 Synthesis of (diphosphine)Pd(II)Cl(Cl-quinonyl) complexes. | ||
Except for 4, solid-state structures of these complexes were identified by X-ray crystal analyses, among which the structures of 3, 5, and 6 are shown in Fig. 1 (for detailed data including the structure of 7, see the ESI†). The Pd center of bis(triphenylphosphine) complex 3 adopts a square planar configuration with two phosphines coordinated in a trans form. In accordance with the solid-state structure, the 31P NMR spectrum of 3 exhibits only one P signal at 22.8 ppm. Although single crystals suitable for X-ray analysis were not obtained for 4, the 31P NMR spectrum of the complex shows only one signal at 22.8 ppm, indicating that 4 also adopts a similar square planar trans configuration to that of 3. Meanwhile, the Pd center in the dppf complex 5 adopts a square planar configuration with the two P atoms coordinated in a cis form as revealed by X-ray analysis. Accordingly, the 31P NMR spectrum of 5 contains two signals at 31.0 and 13.7 ppm, reflecting the presence of two P atoms in different environments. On the other hand, xantphos complexes 6 and 7 adopt a distorted square planar configuration, two P atoms being located in trans positions as confirmed by X-ray analysis. Whereas the C–Pd–Cl angles (174.7° for 6 and 175.7° for 7) are close to the ideal value (180°) for the square planar configuration, the P–Pd–P bite angles of 150.8° and 152.6° for 6 and 7, respectively, are much smaller than the ideal value. In accordance with the solid-state structures, the 31P spectra of 6 and 7 exhibit only one signal at 6.3 ppm. With these Pd(II) complexes ligated with phosphine and oxidatively added dichloroquinone derivatives in hand, we can investigate the relationship of the structures of the Pd(II) complexes and the initiating ability for diazoacetate polymerization with the activation with NaBPh4, hopefully leading to the development of highly active initiating systems for the polymerization.
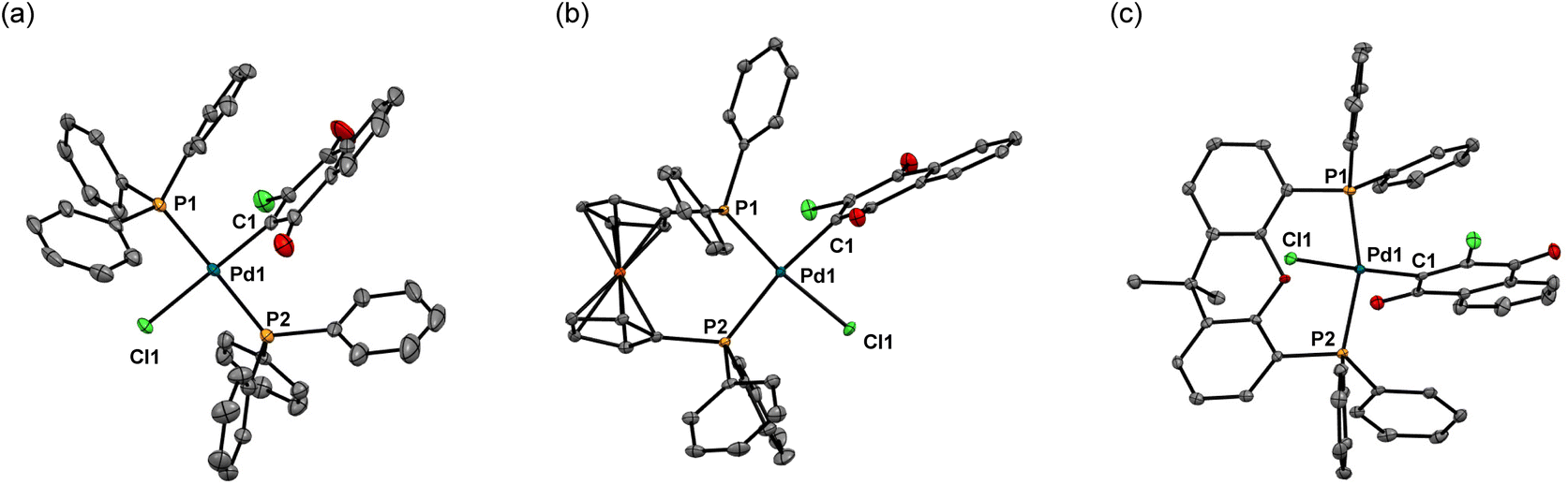 | ||
| Fig. 1 X-ray structures of (a) 3, (b) 5, and (c) 6 with 50% thermal ellipsoids. Hydrogen atoms and solvents have been omitted for clarity. Selected bond lengths (Å) and angles (°): (a) 3: Pd1–P1 = 2.323(1), Pd1–P2 = 2.309(1), Pd1–C1 = 1.991(4), Pd1–Cl1 = 2.370(2), P1–Pd1–P2 = 176.68(4), C1–Pd1–Cl1 = 179.9(1), P1–Pd1–Cl1 = 90.63(4); (b) 5: Pd1–P1 = 2.2448(9), Pd1–P2 = 2.3650(9), Pd1–C1 = 2.029(4), Pd1–Cl1 = 2.356(1), P1–Pd1–P2 = 98.94(4), C1–Pd1–Cl1 = 82.5(1), P1–Pd1–Cl1 = 170.49(4); (c) 6: Pd1–P1 = 2.3261(7), Pd1–P2 = 2.3230(8), Pd1–C1 = 2.013(2), Pd1–Cl1 = 2.4070(7), P1–Pd1–P2 = 150.80(3), C1–Pd1–Cl1 = 174.65(7), P1–Pd1–Cl1 = 87.90(2). | ||
Polymerization of ethyl diazoacetate with (diphosphine)Pd(II)Cl(Cl-quinonyl)/NaBPh4 systems
Following our previous publication on diazoacetate polymerization with 2/NaBPh4,33 polymerization of ethyl diazoacetate (EDA) with 3–7/NaBPh4 was conducted using THF or 1,4-dioxane as the solvent (Table 1). For the effective initiation, one of the Ph groups on the borate should be transferred to Pd, leading to a Pd–Ph initiating species [activation of the Pd(II) precursor], into which carbenes generated from diazoacetates will be inserted efficiently after N2 elimination, resulting in the formation of polymers with a Ph group at the α-chain end. The transmetalation was carried out at −78 °C or 0 °C in THF or 1,4-dioxane respectively. Then, an EDA solution in CH2Cl2 was added to the mixture at a low temperature, followed by polymerization at each temperature mentioned in Table 1.| Run | Pd(II) | Solvent | Temperature | Yieldb (%) |
M
nc |
Đ | Syndiotacticityd |
|---|---|---|---|---|---|---|---|
| a Pd = 0.01 mmol, THF or 1,4-dioxane = 3 mL, polymerization time = 13 h, [EDA]/[Pd] = 100, [NaBPh4]/[Pd] = 1.1; EDA was used as a CH2Cl2 solution with a concentration of 1.5–2.3 M. b After purification with preparative SEC to remove dimers and oligomers. c Determined by SEC using PMMA standards. d Estimated by NMR measurements (Fig. 2).39 e Quoted from our previous work.33 | |||||||
| 1e | 2 | THF | 50 °C | 20 | 20500 |
1.46 | High |
| 2 | 3 | THF | RT | 1.2 | 11300 |
1.46 | Moderate |
| 3 | 3 | THF | 50 °C | 17 | 7600 | 1.58 | Moderate |
| 4 | 3 | 1,4-Dioxane | 50 °C | 24 | 8100 | 2.08 | Moderate |
| 5 | 3 | 1,4-Dioxane | 70 °C | 28 | 10800 |
2.39 | Moderate |
| 6 | 3 | 1,4-Dioxane | 90 °C | 38 | 8100 | 1.83 | Moderate |
| 7 | 4 | THF | 50 °C | 1.6 | 12100 |
1.63 | Low |
| 8 | 5 | THF | 50 °C | 5.7 | 12000 |
2.13 | High |
| 9 | 5 | 1,4-Dioxane | 70 °C | 18 | 10600 |
2.39 | High |
| 10 | 6 | THF | RT | 3.2 | 40600 |
1.62 | Very high |
| 11 | 6 | THF | 50 °C | 21 | 9700 | 2.14 | High |
| 12 | 6 | 1,4-Dioxane | 70 °C | 9.1 | 13500 |
2.23 | High |
| 13 | 7 | THF | 50 °C | 4.1 | 8700 | 1.62 | Low |
The square planar trans-bis(triphenylphosphine) Pd(II) complex 3 with NaBPh4 gave polyEDA′ in a very low yield (1.2%) at room temperature (run 2). The polymer yield increased to the same level (17%) as that with 2/NaBPh4 in the polymerization at 50 °C (run 3). A further increase in the polymer yield was observed in the polymerization at a higher temperature with 1,4-dioxane as the solvent (24–38%) (runs 4–6). Polymer tacticity was evaluated from the peak positions of the main chain methine signals in 1H NMR spectra of polyEDA′s (Fig. 2).39 Although the polymer prepared with the 3/NaBPh4 system (run 5) exhibits a dominant peak at the same position (3.2 ppm) as those for highly syndiotactic polymers obtained with 2/NaBPh4, the presence of relatively large peaks on both sides of the dominant peak suggests that the stereoregularity is not so high (moderate). Another square planar trans-bis(triphenylphosphine) Pd(II) complex 4 with a bulky chloroquinonyl moiety with NaBPh4 was much less active for the polymerization, affording a polymer in a very low yield (1.6%) in the polymerization at 50 °C (run 7). A broad methine proton signal was observed for the product obtained with 4/NaBPh4, suggesting the stereoselectivity of the product is low. These results indicated that the use of two PPh3 groups in a square planar configuration with a trans coordination instead of cis coordinated cod significantly diminishes the syndioselectivity for the polymerization.
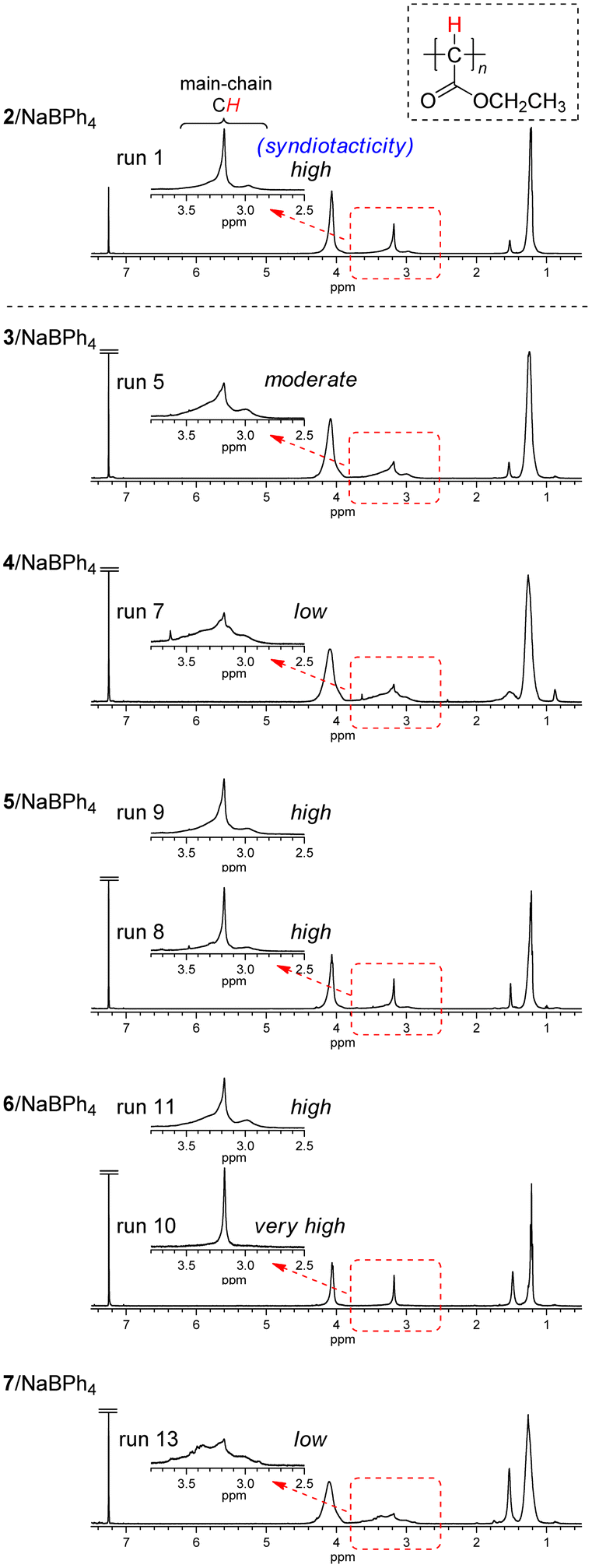 | ||
| Fig. 2 1H NMR spectra of polyEDA′s obtained with 2/NaBPh4, 3/NaBPh4 (run 5 in Table 1), 4/NaBPh4 (run 7 in Table 1), 5/NaBPh4 (runs 8 and 9 in Table 1), 6/NaBPh4 (runs 10 and 11 in Table 1), and 7/NaBPh4 (run 13 in Table 1), recorded in CDCl3 at 50 °C. | ||
Then, the square planar Pd(II) complex with a cis coordinated bidentate phosphine (dppf) 5 with NaBPh4 was employed for EDA polymerization. Polymerization in THF at 50 °C afforded polyEDA′ in a much lower yield (5.7%) compared to that observed with 2/NaBPh4 under the same conditions, while the same level of syndiotacticity as 2/NaBPh4 was observed for the product (run 8, Fig. 2). In addition, the higher syndiotacticity of the product compared with those for the polymers obtained with 3/NaBPh4 and 4/NaBPh4 was maintained for the polymerization in 1,4-dioxane at 70 °C with an increasing polymer yield of 18% (run 9). These results suggest that the cis configuration of a bidentate ligand plays an important role in the syndioselective propagation of EDA.
On the other hand, the distorted square planar trans Pd(II) complex with a xantphos ligand 6 with NaBPh4 afforded polyEDA′ in THF at 50 °C in a similar yield (21%) to that observed with 2/NaBPh4, despite slightly lower syndioselectivity (run 11 in Table 1, Fig. 2); polymerization in 1,4-dioxane at 70 °C resulted in a lower yield of polyEDA′ (9.1%) with the same level of syndioselectivity as that obtained with THF (run 12). Interestingly, the EDA polymerization with 6/NaBPh4 at room temperature afforded polyEDA′ with a slightly higher syndioselectivity than that observed with 2/NaBPh4, although the polymer yield diminished significantly (3.2%) (run 10 in Table 1, Fig. 2). A xantphos ligated Pd(II) complex 7 with a bulky chloroquinone with NaBPh4 afforded polyEDA′ with a low stereoselectivity in a lower yield (4.1%) in THF at 50 °C (run 13 in Table 1, Fig. 2), probably because the bulky chloroquinonyl moiety would sterically prevent the polymerization and exert a negative effect for the stereoselectivity. These results indicated that the distorted square planar trans configuration brought about by the xantphos ligand was not effective for increasing the polymer yield, although under specific conditions a very small amount of a highly syndioselective initiator can be generated.
On the basis of the results described above, although the use of phosphine as a ligand in place of cod in 2/NaBPh4 for diazoacetate polymerization is not so effective for increasing the polymer yield, the same level of activity and syndioselectivity as those of 2/NaBPh4 was observed in some cases. In addition, we can confirm that the steric environment around the Pd center can strongly affect the initiating ability with respect to the polymer yield and stereoselectivity in these systems.
Preparation of Pd(0) complexes with bidentate diamine ligands
Next, we attempted to employ bidentate diamines as a ligand in place of cod and phosphine in the above-described Pd complexes. N,N,N′,N′-tetramethylethylenediamine (tmeda), N,N,N′,N′-tetraethylethylenediamine (teeda), and N,N,N′,N′-tetramethyl-1,3-propanediamine (tmpda) were chosen as bidentate diamines in conjunction with dichlone and its dibromo analogue. For the preparation of Pd complexes, Pd2(dba)3CHCl3 was reacted with excess diamine and dihalonaphthoquinone in acetone (tmeda) or THF (teeda and tmpda) at room temperature for 1 h using a similar procedure employed for the synthesis of phosphine complexes described above (Scheme 4). To our surprise, the products obtained here were not Pd(II), but Pd(0) complexes with a dihalonaphthoquinone moiety coordinated to a Pd center in an η2-mode without the oxidative addition of an X–C bond (X = Cl or Br), as unambiguously identified by X-ray analyses as shown in Fig. 3 for complexes 8 and 10 (for the structure of 11, see the ESI†). The solid-state structures were also supported by 1H NMR, where only two aromatic signals were observed with an equal intensity for two sets of equivalent 2Hs (see the ESI†), indicating that the dihalonaphthoquinones were coordinated in a symmetrical manner in these Pd(0) complexes including 9, whose suitable crystals for X-ray analysis were not obtained. These results indicate that, in contrast to diphosphine ligands, the bidentate diamine ligands render the oxidative addition of an X–C bond in the dihalonaphthoquinones unfavorable. This phenomenon can be reasonably explained by a possible lower electron density on the Pd center in 8–11 than that in the aforementioned diphosphine complexes, owing to less effective electron donation from the two nitrogen atoms in diamines than the phosphorus atoms in diphosphines, because the higher electron density on Pd should be more favorable for the oxidative addition of the X–C bond to proceed.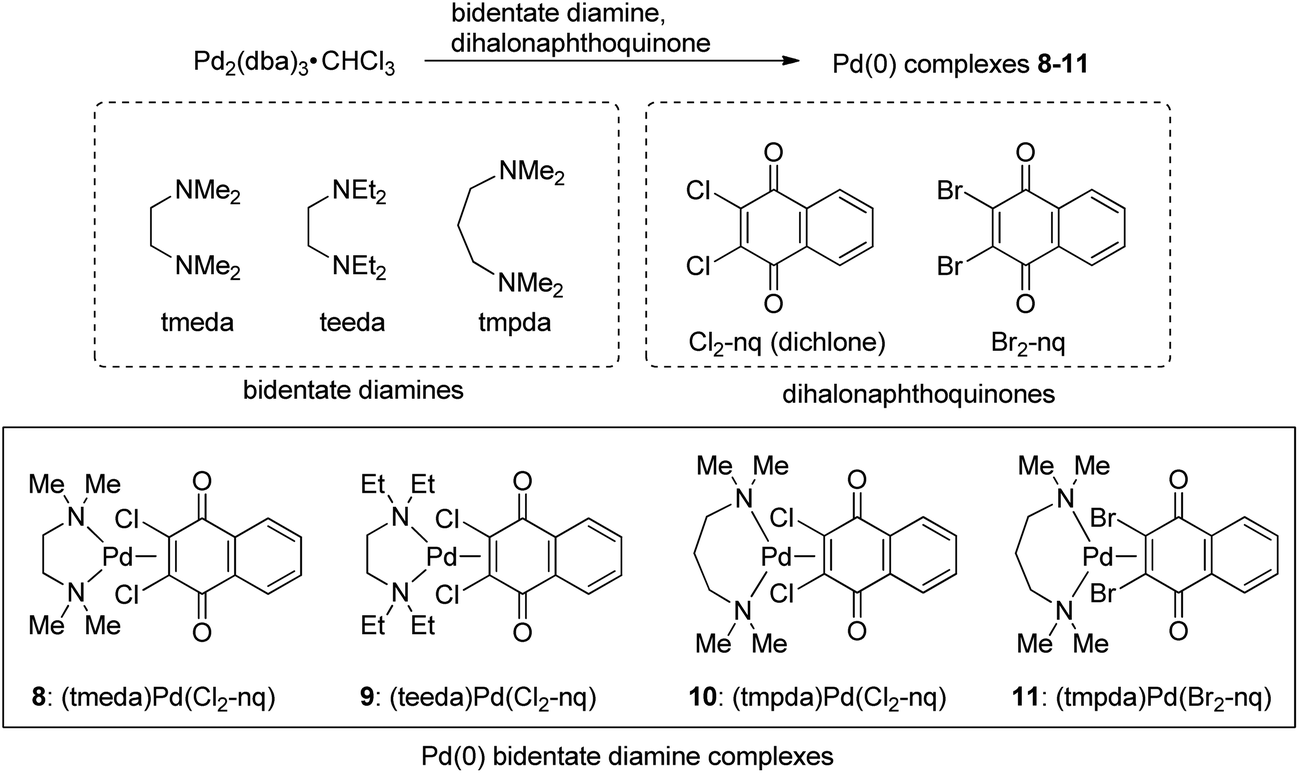 | ||
| Scheme 4 Synthesis of (diamine)Pd(0)(dihalonaphthoquinone) complexes. | ||
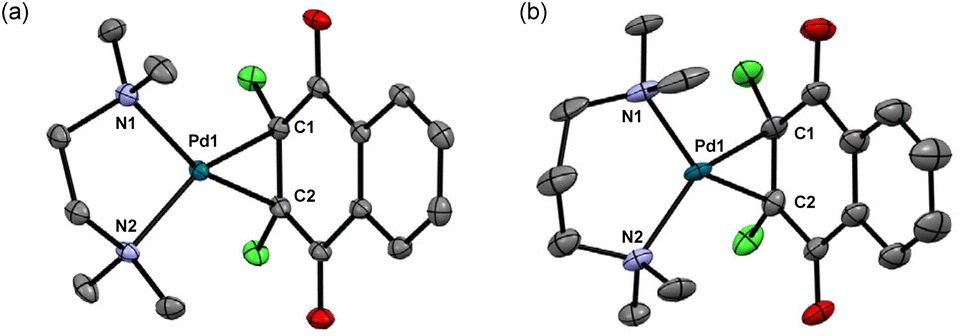 | ||
| Fig. 3 X-ray structures of (a) 8 and (b) 10 with 50% thermal ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): (a) 8: Pd1–N1 = 2.166(2), Pd1–N2 = 2.165(2), Pd1–C1 = 2.074(2), Pd1–C2 = 2.074(2), N1–Pd1–N2 = 83.08(8). (b) 10: Pd1–N1 = 2.177(4), Pd1–N2 = 2.182(3), Pd1–C1 = 2.063(3), Pd1–C2 = 2.078(4), N1–Pd1–N2 = 95.7(1). | ||
According to the mechanism for the formation of the initiating Pd–Ph species described in the Introduction, the unexpectedly obtained Pd(0) complexes with a bidentate diamine would be transformed into an active initiator for the diazoacetate polymerization via a pathway involving the oxidative transmetalation of one of the Ph groups in NaBPh4 to the Pd center, where at the same time an η3-type anionic ligand derived from the dihalonaphthoquinone moiety should be attached as an effective ligand to Pd. In that case, we can expect a high polymer yield as that obtained in the polymerization with the 1/NaBPh4 system, and hopefully, stereoselectivity would be imparted to the polymerization because of the steric effect of the bidentate diamine ligand on the Pd center and additional two Cl atoms on the anionic naphthoquinonyl ligand.
Polymerization of diazoacetates with (diamine)Pd(0)(dihalonaphthoquinone)/NaBPh4 systems
The results of polymerization of EDA with 8–11 in conjunction with NaBPh4 are presented in Table 2. While the polymerization with the Pd(0) complex with tmeda 8 with NaBPh4 at room temperature gave polyEDA′ in a very low yield (2.4%), raising the polymerization temperature to 50 °C resulted in a higher polymer yield of 58% (runs 4 and 5); SEC-estimated Mns of the polyEDA′s were about 10000. The Pd(0) complex with a teeda ligand 9 with NaBPh4 exhibited a similar initiating ability under similar conditions (runs 7 and 8). Polymerization in 1,4-dioxane yielded polyEDA′ with the same level of Mn and yield as in THF at the same temperature (run 9). A further increase in the polymer yield was not observed in the polymerization at a higher temperature of 70 °C (run 10). On the other hand, the tmpda-ligated Pd(0) complex 10 with NaBPh4 afforded polyEDA′ in a higher yield of 34% at room temperature compared to that afforded by 8 and 9 with NaBPh4 (run 11), while at 50 °C the 10/NaBPh4 system showed a similar activity to that exhibited by 8 and 9 with NaBPh4 (run 12). To further confirm the higher activity of 10/NaBPh4, the activities of 8/NaBPh4 and 10/NaBPh4 were compared with the polymerization conducted for 1 h at 50 °C (runs 6 and 13); as a result, while the polymerization with 10/NaBPh4 conducted for 1 h afforded polyEDA′ in a similar yield (49%) to that for 13 h, the yield with 8/NaBPh4 (27%) was much lower in the 1 h reaction period.
| Run | Pd(0) | [EDA]/[Pd] | Solvent | Temperature | Period (h) | Yieldb (%) |
M
nc |
Đ | Syndiotacticityd |
|---|---|---|---|---|---|---|---|---|---|
| a Pd = 0.01 mmol, THF or 1,4-dioxane = 3 mL, [NaBPh4]/[Pd] = 1.1; EDA was used as a CH2Cl2 solution with a concentration of 1.4–2.5 M. b After purification with preparative SEC to remove dimers and oligomers. c Determined by SEC using PMMA standards (values in parentheses were determined by SEC-MALS). d Estimated by NMR measurements (Fig. 4).39 e Quoted from our previous work.33 | |||||||||
| 1e | 2 | 100 | THF | 50 °C | 13 | 20 | 20500 |
1.46 | High |
| 2e | 1 | 100 | THF | RT | 13 | 63 | 17200 |
1.27 | (Atactic) |
| 3e | 1 | 100 | THF | 50 °C | 13 | 72 | 17200 |
1.42 | (Atactic) |
| 4 | 8 | 100 | THF | RT | 13 | 2.4 | 11000 |
1.42 | Moderate |
| 5 | 8 | 100 | THF | 50 °C | 13 | 58 | 9000 | 1.90 | Moderate |
| 6 | 8 | 100 | THF | 50 °C | 1 | 27 | 10700 |
2.20 | Moderate |
| 7 | 9 | 100 | THF | RT | 13 | 3.3 | 12000 |
1.93 | Moderate |
| 8 | 9 | 100 | THF | 50 °C | 13 | 45 | 12100 |
2.09 | Moderate |
| 9 | 9 | 100 | 1,4-Dioxane | 50 °C | 13 | 47 | 13600 |
2.67 | Moderate |
| 10 | 9 | 100 | 1,4-Dioxane | 70 °C | 13 | 42 | 11800 |
2.92 | Moderate |
| 11 | 10 | 100 | THF | RT | 13 | 34 | 17100 |
2.06 | Moderate |
| 12 | 10 | 100 | THF | 50 °C | 13 | 48 | 10800 |
2.16 | Moderate |
| 13 | 10 | 100 | THF | 50 °C | 1 | 49 | 13900 |
2.25 | Moderate |
| 14 | 10 | 200 | THF | 50 °C | 13 | 51 | 13500 |
2.08 | Moderate |
| 15 | 10 | 300 | THF | 50 °C | 13 | 44 | 21300 |
2.26 | Moderate |
| 16 | 10 | 400 | THF | 50 °C | 13 | 36 | 28100 (37100) |
2.13 (1.54) | Moderate |
| 17 | 11 | 100 | THF | 50 °C | 13 | 6.2 | 9400 | 1.53 | High |
The above-described difference in the activity between 8 and 9 bearing a CH2CH2 spacer with NaBPh4 and 10 bearing a CH2CH2CH2 spacer with NaBPh4 should be ascribed to the structural difference in the Pd(II) active species during the initiation and propagation. On comparing the crystal structures of the precursors 8 and 10, the major difference is found to be in the bite angle of N–Pd–N, which is 83.1° and 95.7° for 8 and 10, respectively; other structural parameters being almost the same.
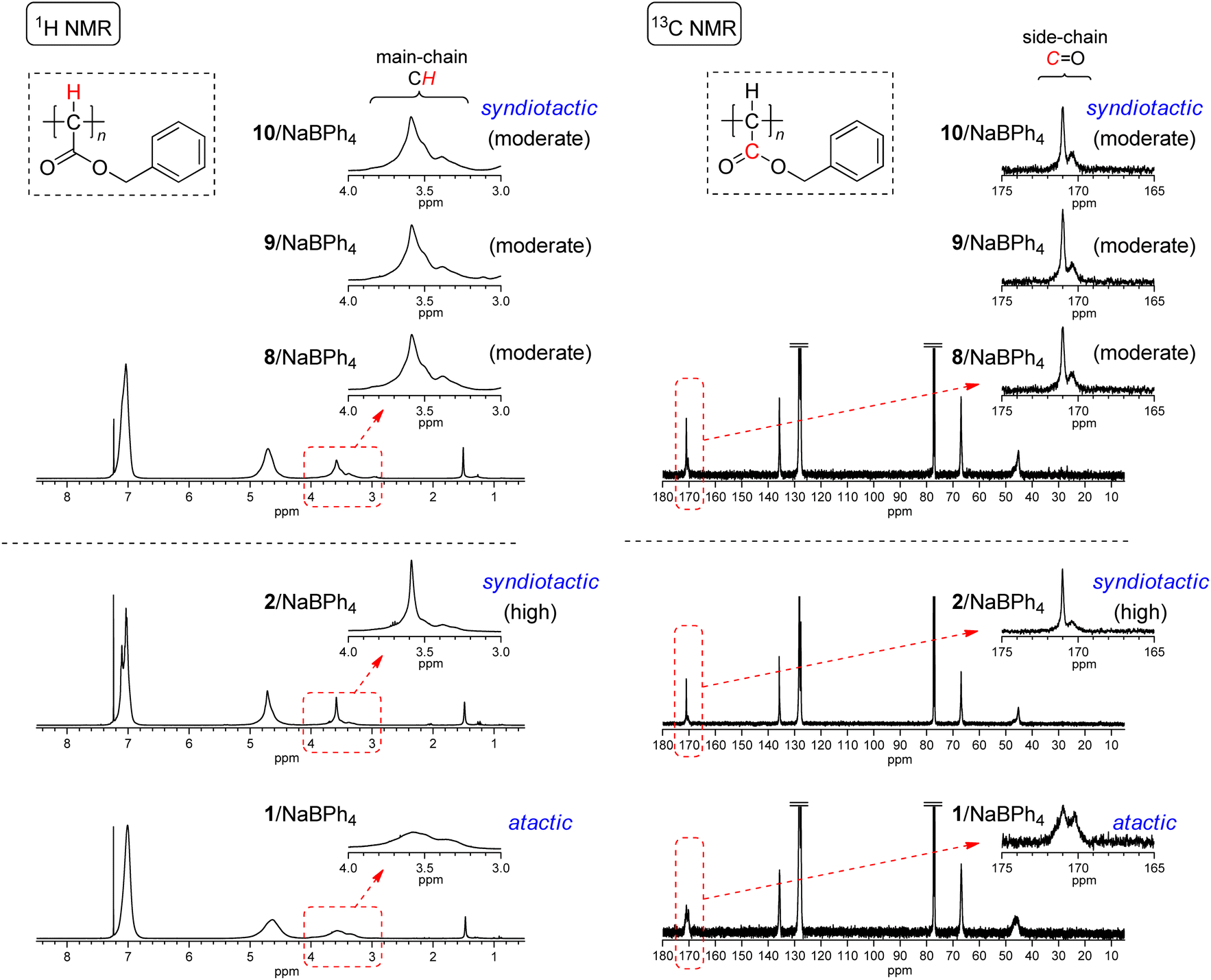
Initiating mechanisms with these 8–11/NaBPh4 systems should be discussed on the basis of the comparison of the polymer yield and tacticity with the polymerization results with 2 and 1 with NaBPh4 as presented in runs 1–3 in Table 2. As described in the Introduction, initiation mechanisms for the two naphthoquinone-based Pd complexes 2 and 1 with NaBPh4 are entirely different.33 In the case of 2, a Ph anion derived from NaBPh4 nucleophilically replaces Cl at the Pd center, resulting in the formation of the Pd–Ph initiating species where the chloronaphthoquinonyl group is attached to Pd with a σ-bond as an anionic ligand. On the other hand, in the case of 1, oxidative transmetalation from NaBPh4 to Pd yields the Pd–Ph initiating species where an η3-type anionic ligand derived from naphthoquinone is attached to Pd. The former system has been revealed to afford highly syndiotactic polymers in low yields, while the latter to afford atactic polymers in high yields as clearly demonstrated in runs 1–3 in Table 2. Even though the polymer yields obtained with 8–10/NaBPh4 systems were lower than that obtained with 1/NaBPh4, moderate yields of polyEDA′s and the isolation of Pd(0) complexes without the oxidative addition of a Cl–C bond in 8–10 indicate that the polymerization with these systems should be initiated via oxidative transmetalation in a manner similar to that of 1/NaBPh4. On the other hand, the appearance of signals of main chain CHs and carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Os in 1H and 13C NMR spectra, respectively, shown in Fig. 4 indicates that the syndiotacticities of the polyEDA′s obtained with 8–10/NaBPh4 systems are apparently higher than those of the atactic polymers obtained with 1/NaBPh4 even in a small extent, suggesting that the coordination of a bidentate diamine ligand and two Cl atoms attached to the naphthoquinone-derived framework of the anionic ligand exert a certain steric effect favorable for the syndioselective propagation (Scheme 5).41
Os in 1H and 13C NMR spectra, respectively, shown in Fig. 4 indicates that the syndiotacticities of the polyEDA′s obtained with 8–10/NaBPh4 systems are apparently higher than those of the atactic polymers obtained with 1/NaBPh4 even in a small extent, suggesting that the coordination of a bidentate diamine ligand and two Cl atoms attached to the naphthoquinone-derived framework of the anionic ligand exert a certain steric effect favorable for the syndioselective propagation (Scheme 5).41
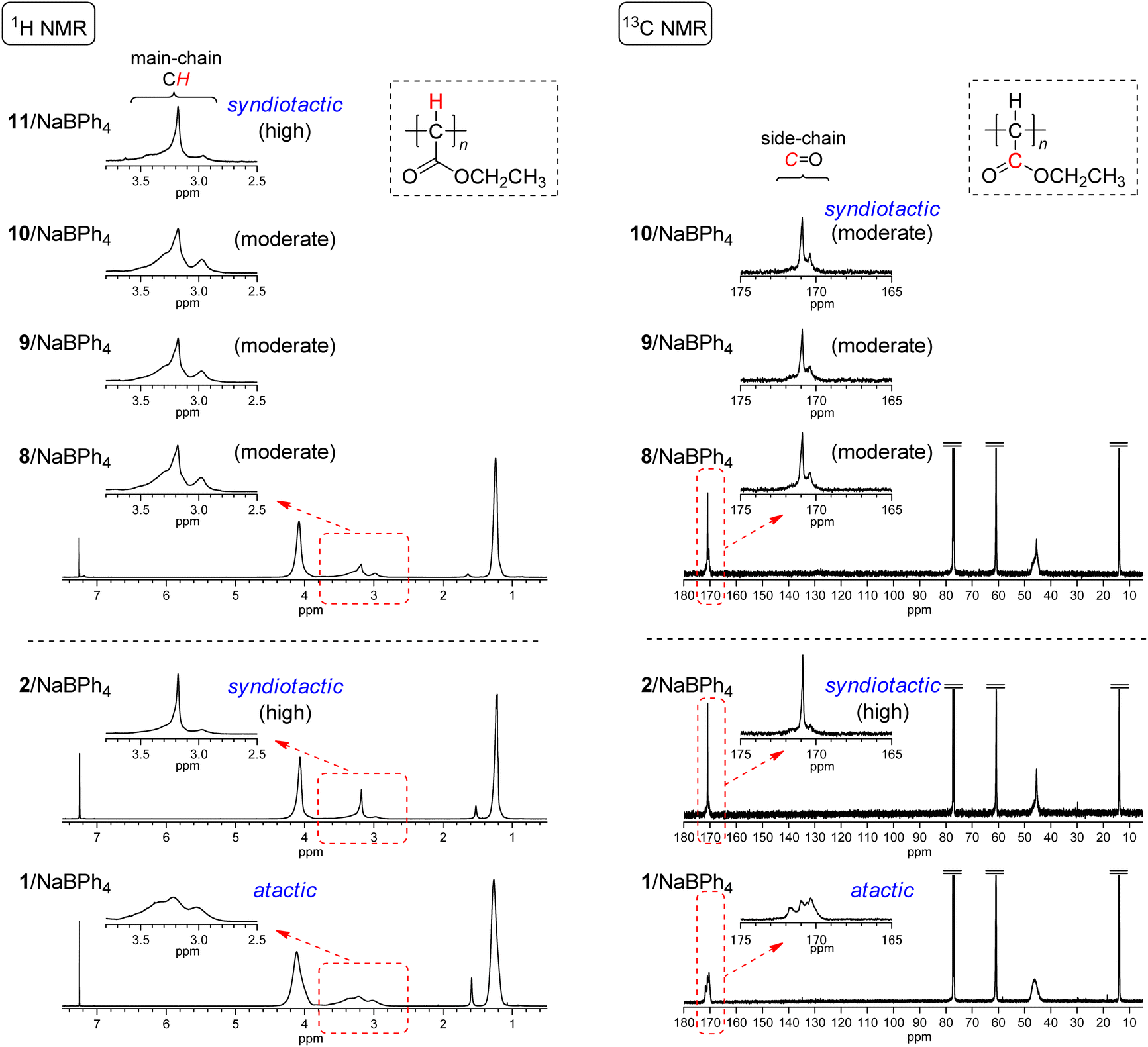 | ||
| Fig. 4 1H and 13C NMR spectra of polyEDA′s obtained with 8/NaBPh4 (run 5 in Table 2), 9/NaBPh4 (run 8 in Table 2), 10/NaBPh4 (run 12 in Table 2), and 11/NaBPh4 (run 17 in Table 2), and with 2/NaBPh4, and 1/NaBPh4, recorded in CDCl3 at 50 °C (1H NMR) or room temperature (13C NMR). | ||
 | ||
| Scheme 5 The proposed mechanism for C1 polymerization of diazoacetates by the (diamine)Pd(0)(dichlone)/NaBPh4 system. | ||
The results obtained with the dibromonaphthoquinone-ligated Pd(0) complex 11 with NaBPh4 are noteworthy, where EDA polymerization furnished a polymer with much lower yield (6.2%) and higher syndiotacticity than those obtained with 8–10/NaBPh4 systems (run 17 in Table 2 and Fig. 4). These results suggest that the polymerization with 11/NaBPh4 proceeded via the same mechanism as that with 2/NaBPh4; thus, we can suppose that the weaker Br–C bond in the dibromonaphthoquinone in 11 resulted in the oxidative addition in the reaction mixture before the polymerization was initiated, even though in isolated 11 in the solid state, the oxidative addition of the Br–C bond did not yet occur; the lower polymer yield than that obtained with 2/NaBPh4 could be ascribed to the steric effect of the larger size of the Br atom in the bromonaphthoquinonyl ligand. However, an alternative possibility that cannot be ruled out here is that oxidative transmetalation occurs for the initiation with 11/NaBPh4 as that with 8–10/NaBPh4 systems, and the larger Br atoms on the η3-type anionic ligand derived from the dibromonaphthoquinone moiety causes the lower polymer yield and higher syndioselectivity.
In any case, the newly developed (diamine)Pd(0)(dichlone)/NaBPh4 system is a unique initiating system for EDA polymerization, which possesses both advantages of 2/NaBPh4 and 1/NaBPh4, affording moderately syndiotactic polymers in moderate yields. Further modification of the structure of the Pd(0) complex will improve both the polymer yield and tacticity.
Runs 14–16 along with run 12 show the relationship between [EDA]/[Pd] feed ratios and Mn of the products in the EDA polymerization with 10/NaBPh4. While the increase of Mn with the increase in the feed ratio was observed to a certain extent, the increase in Mn was not proportional to the feed ratio, and the polymer yield decreased gradually. The highest Mn with the [EDA]/[Pd] feed ratio of 400 was 28100 achieved with SEC and 37100 achieved with SEC-MALS analysis.
The relatively poor controlled behavior of the polymerization with respect to Mn suggested that some undesirable side reactions occurred during the polymerization of EDA with the system, which was confirmed by MALDI-TOF-MS analysis as shown in Fig. 5 for a polyBDA′ sample (BDA: benzyl diazoacetate, Mn = 5200, Đ = 2.24) obtained by BDA polymerization with the 10/NaBPh4 system using a low [BDA]/[Pd] feed ratio of 10 (for BDA polymerization results, see below). The MALDI-TOF-MS spectrum clearly indicates that the chain end structures of the predominant polymer chain are those with Ph and H at α- and ω-chain ends, respectively, which should be generated by the expected initiation with the Pd–Ph species and termination of the propagating chain end with an acidic quencher. Meanwhile, we identified the second largest signal of a polymer structure with chain ends bearing Ph and cyclic ketone frameworks derived by backbiting, at α- and ω-chain ends, respectively. In addition, there appear some minor signals, whose origin cannot be identified. Thus, the MALDI-TOF-MS results agree with the relatively poor controllability of the polymerization because of some undesirable side reactions occurring during the polymerization.
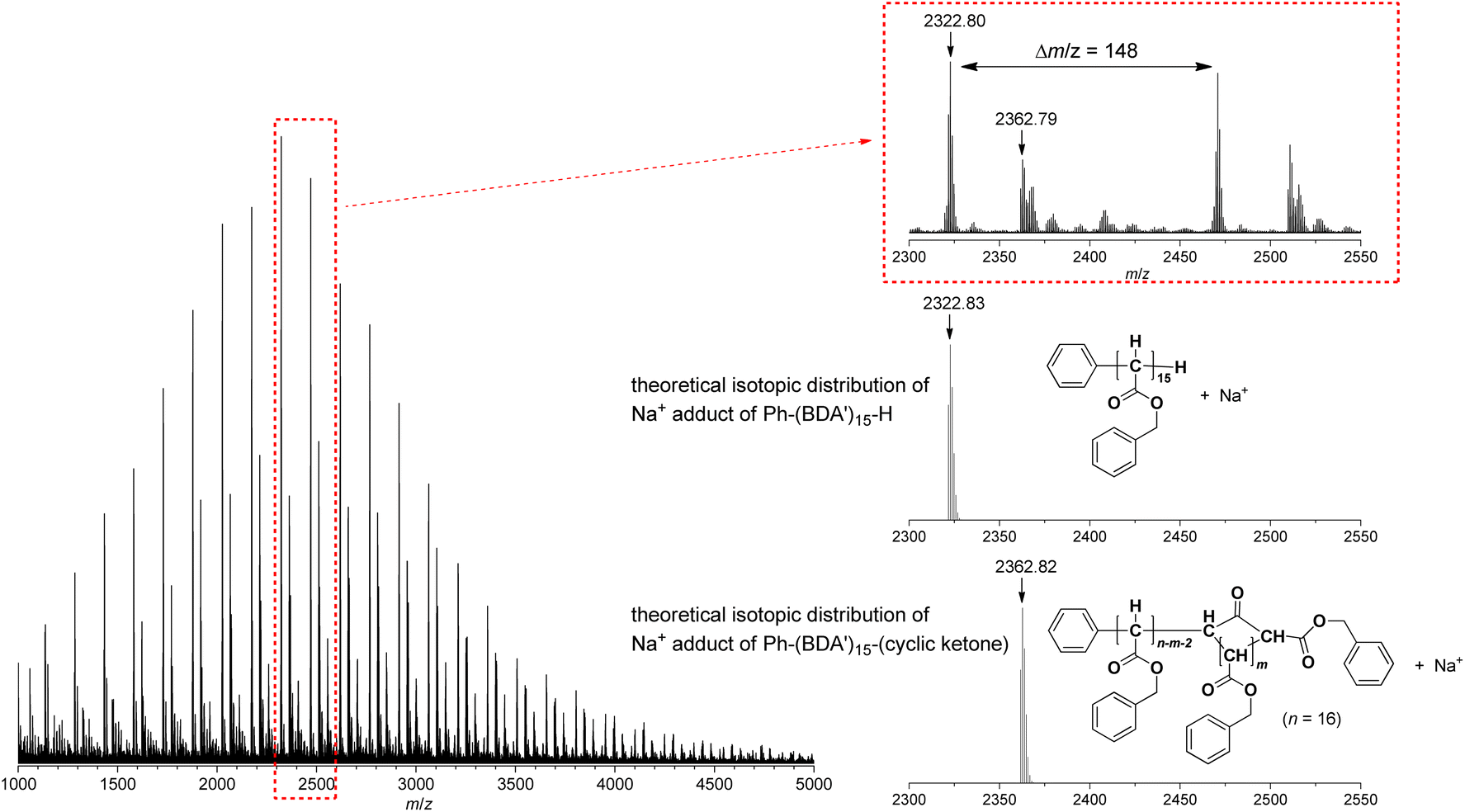 | ||
| Fig. 5 MALDI-TOF-MS spectrum of polyBDA′ obtained with the 10/NaBPh4 initiating system (Mn = 5200, Đ = 2.24). | ||
Polymerization of diazoacetates other than EDA, namely benzyl (BDA), cyclohexyl (c-HDA), and 1-naphthylmethyl (NpCH2DA) diazoacetates was conducted with the (diamine)Pd(0)(dichlone)/NaBPh4 system, and the results were presented in Table 3. As shown in runs 3, 6, and 7, Pd(0) complexes 8–10 in conjunction with NaBPh4 initiated BDA polymerization in a similar efficiency to that with 1/NaBPh4 with respect to Mn and yields of the products. A higher [BDA]/[Pd] ratio of 200 with 10/NaBPh4 as an initiator yielded polyBDA′ with higher Mn (Mn,SEC = 35000, Mn,MALS = 66500) despite a lower yield (32%, run 8), indicating that the side reactions, as mentioned above, with the MALDI-TOF-MS analysis prevented the high yield synthesis of a polymer with high Mn. In addition, as 1H and 13C NMR spectra shown in Fig. 6 indicates, the syndiotacticity of the resulting polyBDA′ is apparently higher than that obtained with 1/NaBPh4, even though it is lower than that obtained with 2/NaBPh4. Thus, the (diamine)Pd(0)(dichlone)/NaBPh4 system is again considered to possess the advantages of both 2/NaBPh4 (syndioselectivity) and 1/NaBPh4 (high polymer yield).
 | ||
| Fig. 6 1H and 13C NMR spectra of polyBDA′s obtained with 2/NaBPh4, 1/NaBPh4, 8/NaBPh4 (run 3 in Table 3), 9/NaBPh4 (run 6 in Table 3), and 10/NaBPh4 (run 7 in Table 3), recorded in CDCl3 at 50 °C (1H NMR) or room temperature (13C NMR). | ||
| Run | Pd(0) | Monomer | [monomer]/[Pd] | Yieldb (%) |
M
nc |
Đ | Syndiotacticityd |
|---|---|---|---|---|---|---|---|
| a At 50 °C in THF (3 mL) for 13 h, Pd = 0.01 mmol, [NaBPh4]/[Pd] = 1.1. b After purification with preparative SEC to remove dimers and oligomers. c Determined by SEC using PMMA standards (values in parentheses were determined by SEC-MALS). d Estimated by NMR measurements (Fig. 6).39 e Quoted from our previous work.33 | |||||||
| 1e | 2 | BDA | 100 | 26 | 11900 |
2.31 | High |
| 2e | 1 | BDA | 100 | 73 | 20900 |
1.57 | (Atactic) |
| 3 | 8 | BDA | 100 | 72 | 11600 |
2.49 | Moderate |
| 4 | 8 | c-HDA | 100 | 2.2 | 5600 | 1.67 | n.d. |
| 5 | 8 | NpCH2DA | 100 | 64 | 8900 | 2.40 | n.d. |
| 6 | 9 | BDA | 100 | 59 | 17600 |
2.31 | Moderate |
| 7 | 10 | BDA | 100 | 74 | 15100 |
2.45 | Moderate |
| 8 | 10 | BDA | 200 | 32 | 35000 (66500) |
1.65 (1.34) | Moderate |
| 9 | 10 | NpCH2DA | 100 | 59 | 12500 |
2.30 | n.d. |
Although 8/NaBPh4 was not effective for c-HDA polymerization probably because the secondary cyclohexyl ester is too bulky for this initiator; NpCH2DA with a larger primary ester than BDA can be successfully polymerized with 8/NaBPh4 (run 5) and 10/NaBPh4 (run 9) to give polymers in moderate yields.
Conclusions
We have demonstrated that diphosphine- and diamine-ligated Pd complexes with a quinone-derived additional ligand can be used in conjunction with NaBPh4 as initiating systems for the C1 polymerization of diazoacetates. As for the (diphosphine)Pd(II)Cl(Cl-quinonyl)-based system, it is significant to find that the steric environment around the Pd center strongly affects the polymerization behavior of diazoacetate; initiating systems with dppf- and xantphos-ligated Pd complexes can yield highly syndiotactic polymers despite low polymer yields. With much improved polymer yields, the (diphosphine)Pd-based system will develop into a highly active initiating system realizing stereospecific polymerization of diazoacetates. On the other hand, (diamine)Pd(0)(dichlone)/NaBPh4 systems have been revealed to be effective initiators for diazoacetate polymerization, yielding moderately syndiotactic polymers in moderate yields; the results support our proposition that the η3-type anionic naphthoquinonyl ligand is essential for achieving high activity. In addition, the moderate syndioselectivity of the resulting polymers suggests that the modification of the diamine structure will further improve the stereoselectivity of the polymerization. We believe that these fundamental investigations and findings hereby reported in this paper are quite important for the development of highly effective initiators for this relatively new and general polymerization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant Numbers JP16K17916, JP18H02021, JP19K05586, JP19K22219, JP21H01988, and JP22K05219) and the Iketani Science and Technology Foundation (Grant Number 0331004-A). The authors thank the Advanced Research Support Center (ADRES) in Ehime University for its assistance in NMR measurements and elemental analyses, and the Ehime Institute of Industrial Technology for its assistance in MALDI-TOF-MS measurements.References
- E. Ihara, Adv. Polym. Sci., 2010, 231, 191–231 CrossRef CAS.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706–1723 RSC.
- N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. de Bruin, Catal. Sci. Technol., 2011, 1, 153–165 RSC.
- C. R. Cahoon and C. W. Bielawski, Coord. Chem. Rev., 2018, 374, 261–278 CrossRef CAS.
- E. Ihara and H. Shimomoto, Polymer, 2019, 174, 234–258 CrossRef CAS.
- H. Shimomoto, Polym. J., 2020, 52, 269–277 CrossRef CAS.
- F. Li, L. Xiao, B. Li, X. Hu and L. Liu, Coord. Chem. Rev., 2022, 473, 214806 CrossRef CAS.
- E. Ihara, R. Okada, T. Sogai, T. Asano, M. Kida, K. Inoue, T. Itoh, H. Shimomoto, Y. Ishibashi and T. Asahi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1020–1023 CrossRef CAS.
- M. Tokita, K. Shikinaka, T. Hoshino, K. Fujii, J. Mikami, N. Koshimizu, K. Sakajiri, S. Kang, J. Watanabe and K. Shigehara, Polymer, 2013, 54, 995–998 CrossRef CAS.
- N. M. G. Franssen, B. Ensing, M. Hegde, T. J. Dingemans, B. Norder, S. J. Picken, G. O. R. Alberda, E. R. van Ekenstein, H. van Eck, J. A. A. W. Elemans, M. Vis, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 11577–11589 CrossRef CAS PubMed.
- H. Shimomoto, E. Itoh, T. Itoh, E. Ihara, N. Hoshikawa and N. Hasegawa, Macromolecules, 2014, 47, 4169–4177 CrossRef CAS.
- H. Shimomoto, H. Asano, T. Itoh and E. Ihara, Polym. Chem., 2015, 6, 4709–4714 RSC.
- H. Shimomoto, K. Shimizu, C. Takeda, M. Kikuchi, T. Kudo, H. Mukai, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Polym. Chem., 2015, 6, 8124–8131 RSC.
- N. Koshimizu, Y. Aizawa, K. Sakajiri, K. Shikinaka, K. Shigehara, S. Kang and M. Tokita, Macromolecules, 2015, 48, 3653–3661 CrossRef CAS.
- H. Shimomoto, A. Oda, M. Kanayama, T. Sako, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1742–1751 CrossRef CAS.
- H. Shimomoto, T. Uegaito, S. Yabuki, S. Teratani, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Solid State Ionics, 2016, 292, 1–7 CrossRef CAS.
- H. Shimomoto, M. Kikuchi, J. Aoyama, D. Sakayoshi, T. Itoh and E. Ihara, Macromolecules, 2016, 49, 8459–8465 CrossRef CAS.
- H. Shimomoto, T. Kudo, S. Tsunematsu, T. Itoh and E. Ihara, Macromolecules, 2018, 51, 328–335 CrossRef CAS.
- K. Shikinaka, K. Suzuki, H. Masunaga, E. Ihara and K. Shigehara, Polym. Int., 2018, 67, 495–499 CrossRef CAS.
- T. Takaya, T. Oda, Y. Shibazaki, Y. Hayashi, H. Shimomoto, E. Ihara, Y. Ishibashi, T. Asahi and K. Iwata, Macromolecules, 2018, 51, 5430–5439 CrossRef CAS.
- D. S. Tromp, M. Lankelma, H. de Valk, E. de Josselin de Jong and B. de Bruin, Macromolecules, 2018, 51, 7248–7256 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Chen, Z. Wu, P. Cheng, Q. Li, J. Fang and D. Chen, Polym. Chem., 2019, 10, 1575–1584 RSC.
- X. Li, B. Mu, C. Chen, J. Chen, J. Liu, F. Liu and D. Chen, Macromolecules, 2019, 52, 6913–6926 CrossRef CAS.
- H. Shimomoto, T. Yamada, T. Itoh and E. Ihara, Polym. J., 2020, 52, 51–56 CrossRef CAS.
- H. Shimomoto, R. Hohsaki, D. Hiramatsu, T. Itoh and E. Ihara, Macromolecules, 2020, 53, 6369–6379 CrossRef CAS.
- D. G. H. Hetterscheid, C. Hendriksen, W. I. Dzik, J. M. M. Smits, E. R. H. van Eck, A. E. Rowan, V. Busico, M. Vacatello, V. V. A. Castelli, A. Segre, E. Jellema, T. G. Bloemberg and B. de Bruin, J. Am. Chem. Soc., 2006, 128, 9746–9752 CrossRef CAS PubMed.
- E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631–11641 CrossRef CAS PubMed.
- E. Ihara, M. Akazawa, T. Itoh, M. Fujii, K. Yamashita, K. Inoue, T. Itoh and H. Shimomoto, Macromolecules, 2012, 45, 6869–6877 CrossRef CAS.
- H. Shimomoto, M. Nakajima, A. Watanabe, H. Murakami, T. Itoh and E. Ihara, Polym. Chem., 2020, 11, 1774–1784 RSC.
- J.-H. Chu, X.-H. Xu, S.-M. Kang, N. Liu and Z.-Q. Wu, J. Am. Chem. Soc., 2018, 140, 17773–17781 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, I. J. Kobylianskii, A. A. Thomas, A. M. Evans, C. P. Delaney, N. C. Flanders, S. E. Denmark, W. R. Dichtel and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 6473–6478 CrossRef CAS PubMed.
- X.-Q. Yao, Y.-S. Wang and J. Wang, Macromolecules, 2021, 54, 10914–10922 CrossRef CAS.
- H. Shimomoto, S. Ichihara, H. Hayashi, T. Itoh and E. Ihara, Macromolecules, 2019, 52, 6976–6987 CrossRef CAS.
- Y. Yamamoto, Adv. Synth. Catal., 2010, 352, 478–492 CrossRef CAS.
- A. J. C. Walters, O. Troeppner, I. Ivanović-Burmazović, C. Tejel, M. Pilar del Río, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2012, 51, 5157–5161 CrossRef CAS PubMed.
- A. J. C. Walters, J. N. H. Reek and B. de Bruin, ACS Catal., 2014, 4, 1376–1389 CrossRef CAS.
- When 1,2-bis(diphenylphosphino)ethane (dppe) was employed as a bidentate phosphine, well-defined Pd complexes could not be isolated in our attempts.
- I. Pochorovski, C. Boudon, J.-P. Gisselbrecht, M.-O. Ebert, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 262–266 CrossRef CAS PubMed.
- Quantitative evaluation of the tacticity for PACMs is not possible at present, because the assignment of NMR signals to the stereoregularity has not yet been established. However, on the basis of de Bruin's reports (ref. 26, 27 and 40) on syndiospecific polymerization, signals at 3.2 ppm in 1H NMR and 171 ppm in 13C NMR have been assigned to the syndiotactic structure of polyEDA′; likewise, signals at 3.6 ppm (1H) and 171 ppm (13C) have been assigned to syndiotactic polyBDA′. Accordingly, the degree of syndiotacticity in this study is qualitatively evaluated to be either very high, high, moderate, or low, depending on the appearance of methine-H signals in 1H NMR and carbonyl-C signals in 13C NMR. With DSC analyses of samples of moderately syndiotactic polyEDA′ and polyBDA′ (Fig. S13 in the ESI†), Tgs of these polymers were observed in the temperature range between Tgs of highly syndiotactic and atactic polymers, demonstrating that the qualitative assessment of the syndiotacticity employed in this study is indeed relevant.
- E. Jellema, A. L. Jongerius, G. A. van Ekenstein, S. D. Mookhoek, T. J. Dingemans, E. M. Reingruber, A. Chojnacka, P. J. Schoenmakers, R. Sprenkels, E. R. H. van Eck, J. N. H. Reek and B. de Bruin, Macromolecules, 2010, 43, 8892–8903 CrossRef CAS.
- As shown in Scheme 5, although the Pd-carbene species generated during the propagation has formally 20 electrons in the Pd center, we think that weak electron-donation from the diamine ligand or temporary detachment of one of the nitrogen atoms from the Pd center would enable the transient species to exist.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, X-ray crystallographic data, DSC measurement results, and NMR spectra. CCDC 2191524, 2191472, 2191475, 2207432, 2191409, 2191416 and 2191419. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2py01548j |
| This journal is © The Royal Society of Chemistry 2023 |