
An efficient cyclodepolymerization route for the chemical recycling of poly(ethylene adipate)†
 *b
and
Zhiping
Li
*a
*b
and
Zhiping
Li
*a
Abstract
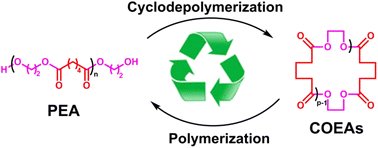
Cyclodepolymerization is a special chemical recycling (upcycling) method for polyesters based on ring–chain equilibria in solution, yet it is usually carried out in halogenated solvents at temperatures around 200 °C. We present here a green cyclodepolymerization route for the efficient chemical recycling of poly(ethylene adipate) (PEA) to its cyclic oligomers, cyclic oligo(ethylene adipate)s (COEAs), using dibutyltin oxide as a catalyst and toluene as a solvent. The effect of solvent on the COEAs yield was studied, and toluene was found to be a better solvent than chlorobenzene since it provides higher yields in addition to the facile purification of COEAs from the reaction mixture due to the solubility difference between PEA and COEAs. The effects of PEA concentration, reaction time and catalyst amount on the yield of COEAs are thoroughly studied. Our results show that the yield of COEAs increases with reduced PEA concentration, prolonged reaction time and increase of catalyst amount. Since COEAs are readily polymerized to pristine PEA, our strategy affords a closed-loop chemical recycling way of PEA.
- This article is part of the themed collection: Plastic Conversion
 Please wait while we load your content...
Please wait while we load your content...

