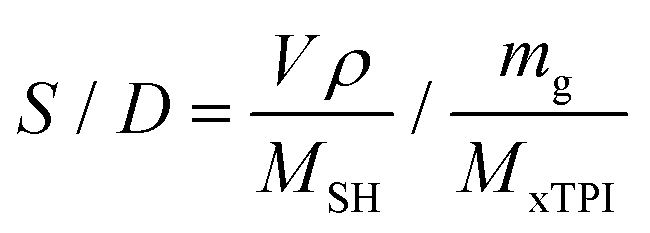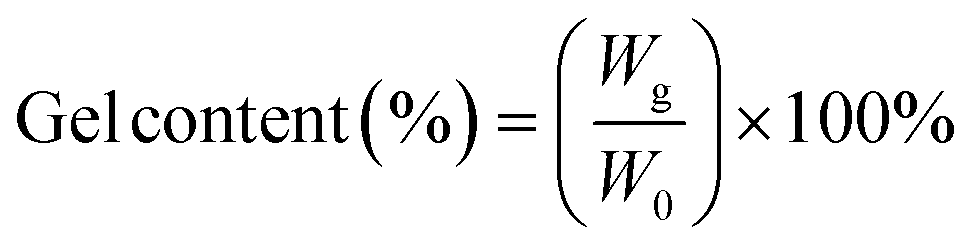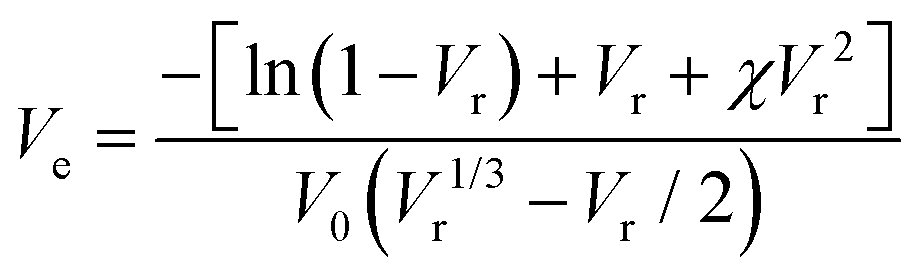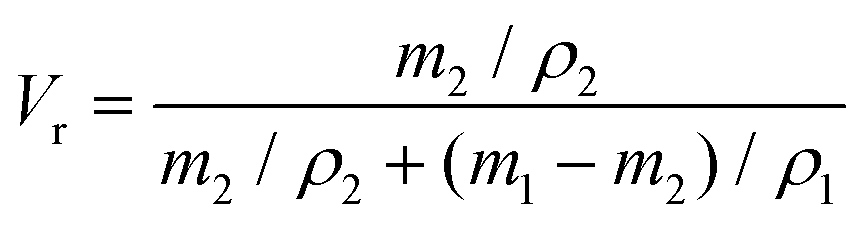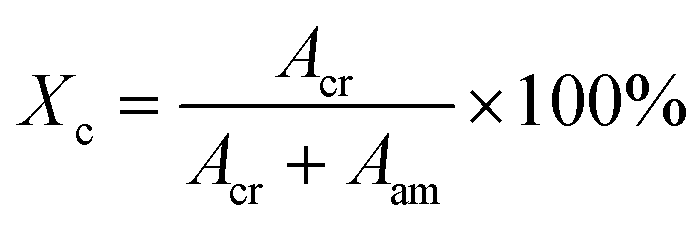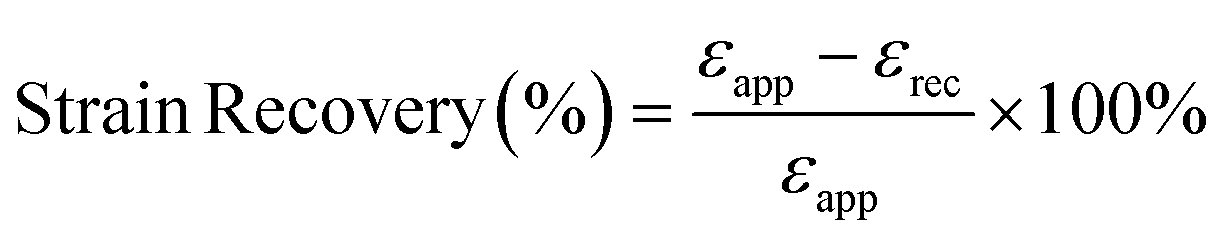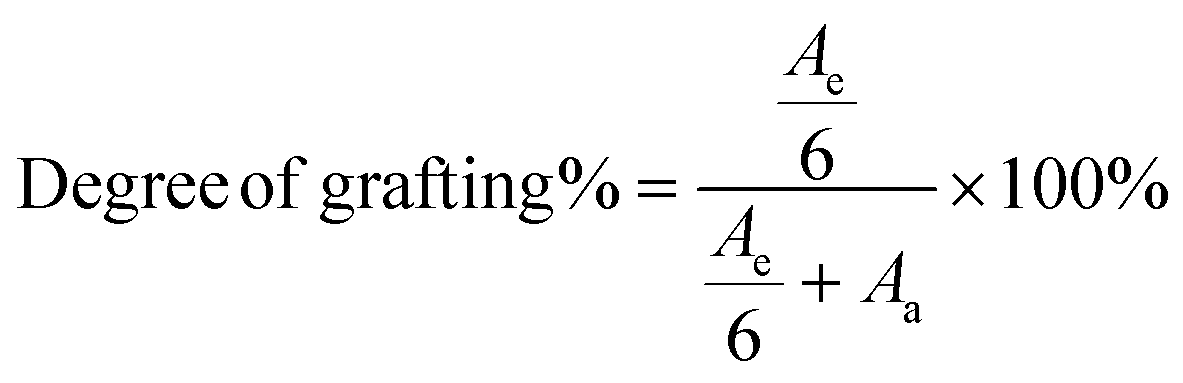DOI:
10.1039/D2PY01218A
(Paper)
Polym. Chem., 2023,
14, 81-91
Achieving high elasticity of trans-1,4-polyisoprene with a combination of radiation crosslinking and thiol–ene grafting†
Received
23rd September 2022
, Accepted 21st November 2022
First published on 22nd November 2022
Abstract
The mechanical properties of semicrystalline polymers are closely related to crystallization. For trans-1,4-polyisoprene (TPI), to explore the possibility of applications as elastomers, crystallization must be suppressed. Previous studies have shown that, although TPI can be crosslinked by sulfur vulcanization or high-energy radiation, the crystallization of TPI cannot be fully eliminated. In this work, we developed a two-step method to modify TPI. In the first step, TPI is irradiated with γ-ray to obtain crosslinked TPI (xTPI), which has decreased crystallinity. Then, the crystallinity of xTPI is further suppressed by grafting n-dodecanethiol to the xTPI chains by thiol–ene click chemistry. The grafted xTPI (g-xTPI) is characterized by nuclear magnetic resonance (NMR), differential scanning calorimetry (DSC), and wide-angle X-ray scattering (WAXS). The effects of the n-dodecanethiol concentration and reaction time on the grafting degree, crystallization behavior, and mechanical properties are systematically investigated. The results show that the crystallinity of g-xTPI decreases with the grafting ratio. The critical grafting ratio for the “semicrystalline-to-amorphous” transition of xTPI is ∼3%. With the increase of the grafting ratio, the strain recovery of g-xTPI increases from 27.5% to 90%, indicating a transformation from a crystalline polymer to an elastomer.
1. Introduction
trans-1,4-Polyisoprene (TPI), known as Gutta Percha or Eucommia ulmoides gum (EUG), is one of the first discovered natural polymers. Unlike its isomer, cis-1,4-polyisoprene (natural rubber), TPI is semicrystalline and has been applied as dental filling materials, medical splints, and damping materials.1–4 In recent years, attention has been focused on the modification of TPI, seeking the possibility of preparing elastomers.5 To this end, the crystallization of TPI has to be suppressed. Modification of TPI includes chlorination,6–8 hydrogenation,9 epoxidation,10–12 grafting,13,14 and Alder–ene reaction.15,16 These methods modify the chemical structure of a certain number of monomers in the TPI chains, which act as defects for crystallization. Therefore, as a general observation, with the increase of the modification degree, the crystallinity and melting temperature decrease. Traditional sulfur vulcanization introduces crosslinks that destroy the regularity of TPI, also resulting in a decrease in crystallinity and melting temperature.17–19 However, there are shortcomings such as insufficient crystallization inhibition19,20 and poor uniformity,21–23 thus limiting the application of TPI as a rubber material.
TPI can be effectively cross-linked by high energy radiation, including γ-ray24,25 or electron beam.26 Upon increasing the absorbed dose, the crystallinity of TPI decreased from 34% to 10%.24 With an absorbed dose of 700 kGy, TPI still exhibits a certain degree of crystallinity (∼18%), indicating that the segments between adjacent crosslinks are still long enough for crystallization. Further increase in the absorbed dose leads to unfavorable degradation and a severe decrease in mechanical properties. In summary, the crosslinking reaction alone is insufficient to reduce the crystallinity of TPI and further modification of the cross-linked TPI is required.
Click chemistry is a term that was proposed by Sharpless27 in 2001 to describe chemical reactions that are high-yielding, highly selective, environmentally-friendly, and simple to perform. Among different click reactions, the thiol–ene click reaction, i.e., the hydrothiolation of a –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bond, is often used in the preparation of functionalized polymers, topological polymers, and surface modification of some polymers.28–38 The thiol–ene reaction generally follows the Michael addition mechanism or a free radical addition mechanism. The former refers to the conjugate addition reaction between the electrophilic conjugate system (electron acceptor) and the nucleophilic negative carbon ion (electron donor), which is generally reacted under the catalysis of an alkali,39–41 while the latter is often photochemically induced. Under such conditions, a thiol is treated with a photoinitiator under UV light, resulting in a thiyl radical (RS˙). Afterward, the RS˙ attacks the –CC– double bond, resulting in an alkyl radical. Then, the alkyl radical captures a hydrogen atom from –S–H to form the thiol–ene addition product and another thiyl radical. Thereby, the reaction occurs continuously.32,33,42,43 Specifically, the thiol–ene reaction has been applied in the modification of semi-crystalline polymers. For example, Gao et al. prepared an unsaturated copolyester with butanediol succinate and butanediol itaconic acid and modified these copolymers with trimethylolpropane-3. It was found that with the increase of itaconic acid content, the crystallinity of the copolymer decreased significantly.44
C– bond, is often used in the preparation of functionalized polymers, topological polymers, and surface modification of some polymers.28–38 The thiol–ene reaction generally follows the Michael addition mechanism or a free radical addition mechanism. The former refers to the conjugate addition reaction between the electrophilic conjugate system (electron acceptor) and the nucleophilic negative carbon ion (electron donor), which is generally reacted under the catalysis of an alkali,39–41 while the latter is often photochemically induced. Under such conditions, a thiol is treated with a photoinitiator under UV light, resulting in a thiyl radical (RS˙). Afterward, the RS˙ attacks the –CC– double bond, resulting in an alkyl radical. Then, the alkyl radical captures a hydrogen atom from –S–H to form the thiol–ene addition product and another thiyl radical. Thereby, the reaction occurs continuously.32,33,42,43 Specifically, the thiol–ene reaction has been applied in the modification of semi-crystalline polymers. For example, Gao et al. prepared an unsaturated copolyester with butanediol succinate and butanediol itaconic acid and modified these copolymers with trimethylolpropane-3. It was found that with the increase of itaconic acid content, the crystallinity of the copolymer decreased significantly.44
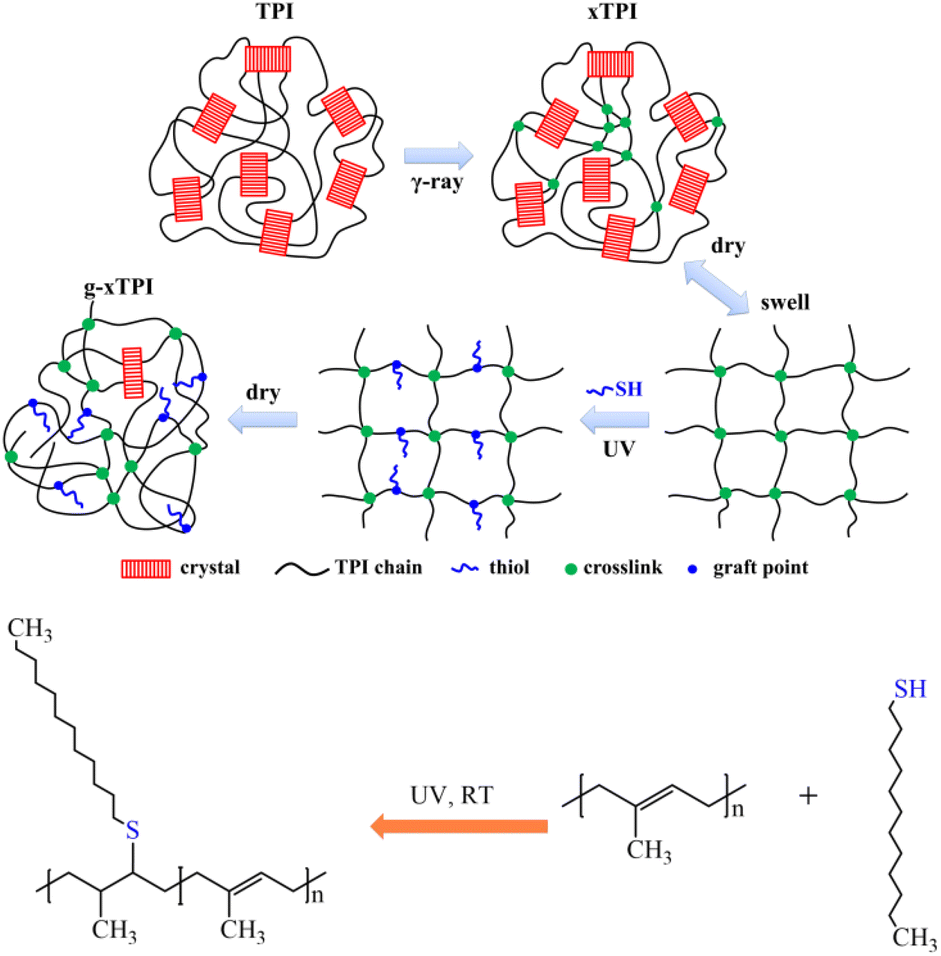
In this work, we designed a two-step methodology to tune the crystallization and physical properties of TPI. The advantages of this new method are (i) separated control of crosslinking and grafting degrees, (ii) tunable physical properties in a broad range, and (iii) high efficiency with no side products. As depicted in Fig. 1, TPI was irradiated with γ-ray to generate crosslinks (referred to as xTPI). Then, xTPI was swollen in its good solvent (toluene). Subsequently, n-dodecylmercaptan was grafted onto the segments between crosslinking points in xTPI by the thiol–ene reaction to obtain grafted xTPI (referred to as g-xTPI). The effects of the reaction time, n-dodecanethiol concentration, and thermal treatment on the crystallization and mechanical properties of the modified TPI were systematically studied. The results show that the two-step grafting method tunes the crystallization and mechanical properties of xTPI in a wide range. In particular, the strain recovery of the material was greatly improved, making the modified TPI suitable for applications as elastomers.
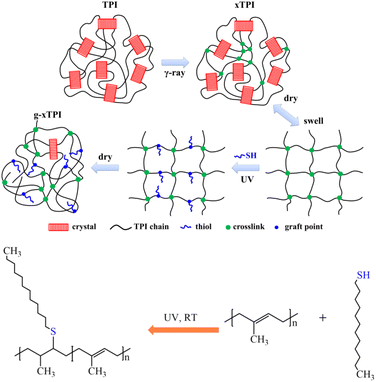 |
| | Fig. 1 Experimental design of the present study and the chemical reaction of TPI grafting. | |
2. Experimental section
2.1 Materials and sample preparation
The TPI used in this study was kindly provided by Qingdao Junxiang Technology Co., Ltd and had a Mooney viscosity of 59. The TPI was crosslinked by γ-ray radiation using a Co59 source at room temperature under the protection of nitrogen. The absorbed dose was 100–1000 kGy and the dose rate was 100 Gy min−1. The irradiated TPI was designated as xTPI.
N-Dodecanethiol was purchased from Beijing Inokay Technology Co., Ltd, with a purity of 98%. Photoinitiator 819 (phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide) with a purity of 99% was purchased from Jiangsu Juming Huagong Technology Co., Ltd. Toluene, chromatographic grade, was purchased from Concord Technology Co., Ltd. The chemicals were used as received without further purification.
About 500 mg of xTPI was added to a glass test tube containing 50 mL of toluene and 10 mg of photoinitiator 819. The thiol was added to the test tube and kept for 24 h under dark condition. The residual air in the test tube was evacuated and the tube was filled with nitrogen. Then, the sample was irradiated with UV at a wavelength of 365 nm, and the distance from the sample to the light source was 15 cm.
The mole ratio of the double bond (–CC–) and thiol bond (–S–H) (referred to as the S/D ratio) can be calculated by eqn (1):
| | 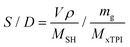 | (1) |
where
MxTPI is the molecular weight of the TPI monomer;
mg is the mass of the xTPI sample; and
MSH,
V, and
ρ are the molecular weight, the volume, and the density of
n-dodecanethiol, respectively. Two series of experiments were carried out to investigate the influence of the S/D ratio or reaction time: (i) the S/D ratio was set as 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
:
3, 1
:
2, 1
:
1, 2
:
1, 3
:
1, and 4
:
1 and the reaction time was fixed at 2 h; and (ii) the reaction time was set to 0.5 h, 1 h, 2 h, 3 h, and 4 h, while the S/D ratio was fixed at 2
:
1.
After the reaction, the g-xTPI samples were taken out and washed in toluene for 5 minutes. Afterward, the samples were washed in ethanol for 5 minutes and this was repeated 3 times. Then, the samples were placed in a fume hood for 24 h to evaporate the solvent and finally placed in a vacuum oven at 40 °C for 24 hours to fully dry the samples.
The crosslinking density was calculated from equilibrium swelling experiments as described in detail in our previous work24 (ESI, section 1†).
2.3 Characterization methods
2.3.1
1H-NMR.
The 1H-NMR spectra of the samples were recorded using a Bruker NMR spectrometer (AVANCE III 400, 350 MHz). The g-xTPI and the initial xTPI were swollen in deuterium chloroform. The data collection number was 64.
2.3.2 Gel content and the crosslinking density (Ve).
The irradiated xTPI samples were extracted using a Soxhlet extractor at 85 °C with chloroform for 24 h. The undissolved fraction was first placed in a fume cupboard to evaporate the majority of the solvent, and then vacuum dried at 40 °C for 24 h to remove the residual solvent. The gel content was calculated by the following formula:| | 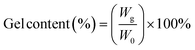 | (2) |
where W0 is the initial weight and Wg is the weight of the dried sample. The values of gel content were the average of three parallel tests.
The xTPI was swollen in toluene for 72 h at room temperature to reach equilibrium. According to the Flory–Rehner45 equation, Ve can be determined by the equilibrium swelling ratio:
| | 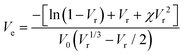 | (3) |
where
χ is the Flory–Huggins polymer–solvent interaction parameter (0.36 for TPI/toluene),
46,47V0 is the molar volume of toluene (106.2 cm
3 mol
−1), and
Vr is the volume fraction of xTPI, which can be calculated according to Bala
et al.48| | 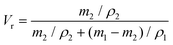 | (4) |
where
m1 and
ρ1 are the weight of the swollen sample and the density of solvent (0.866 g cm
−3), respectively, and
m2 and
ρ2 are the weight of the dried sample and the density of TPI (0.945 g cm
−3), respectively.
2.3.3 Differential scanning calorimetry (DSC).
DSC tests were carried out on a TA Q2000 calorimeter (TA Instruments). The instrument was calibrated with indium before measurements. The samples were protected under N2 atmosphere to avoid oxidation and degradation. The heating/cooling rate was 10 °C min−1.
2.3.4 Wide-angle X-ray scattering (WAXS).
The WAXS measurements were performed on a Xeusss 2.0 SAXS/WAXS system (Xenocs SA, France), equipped with a microfocus CuKα source and a Pilatus 300K semiconductor detector. The wavelength of the X-ray was 1.54 Å. The exposure time was 600 s. The background from air scattering was subtracted from the samples using standard procedures. The two-dimensional images were converted into one-dimensional intensity profiles using the Foxtrot software. All the WAXS tests were carried out at room temperature. The treated sample was first heated to 120 °C at 10 °C min−1 and held for 5 min to erase the thermal history. Then the sample was cooled to −40 °C at 10 °C min−1, and held for 5 min before heating to room temperature.
The degree of crystallinity was calculated by:49
| | 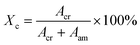 | (5) |
where
Acr and
Aam are the integrated area of the crystalline and amorphous signals, respectively. The fitting of the WAXS profile is illustrated in Fig. S1 (ESI
†).
2.3.5 Mechanical tests.
The samples were cut into mini-tensile bars with a pneumatic cutter. The size of the tensile bars was 25 mm in length and 8 mm in width at both ends. The middle parallel region was 4 mm in length and 3 mm in width. The samples were stretched on a universal material testing machine (Instron 3365) at 22 ± 3 °C. The opening speed of the clamps was 100 mm min−1, corresponding to an initial strain rate of 25 min−1. The results were the average of three parallel experiments.
To measure the strain recovery performance, the specimens were stretched to a strain of 300% at a crosshead speed of 4 mm min−1 (1 min−1 based on the initial gauge length). Then the crosshead moved backward at the same speed till the stress decreased to 0. The strain recovery was calculated according to the formula:50
| | 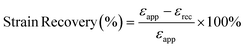 | (6) |
where
εapp is the applied strain (
i.e., 300% in this case) and
εrec is the unrecovered strain.
3. Results and discussion
3.1 Crosslinking of TPI
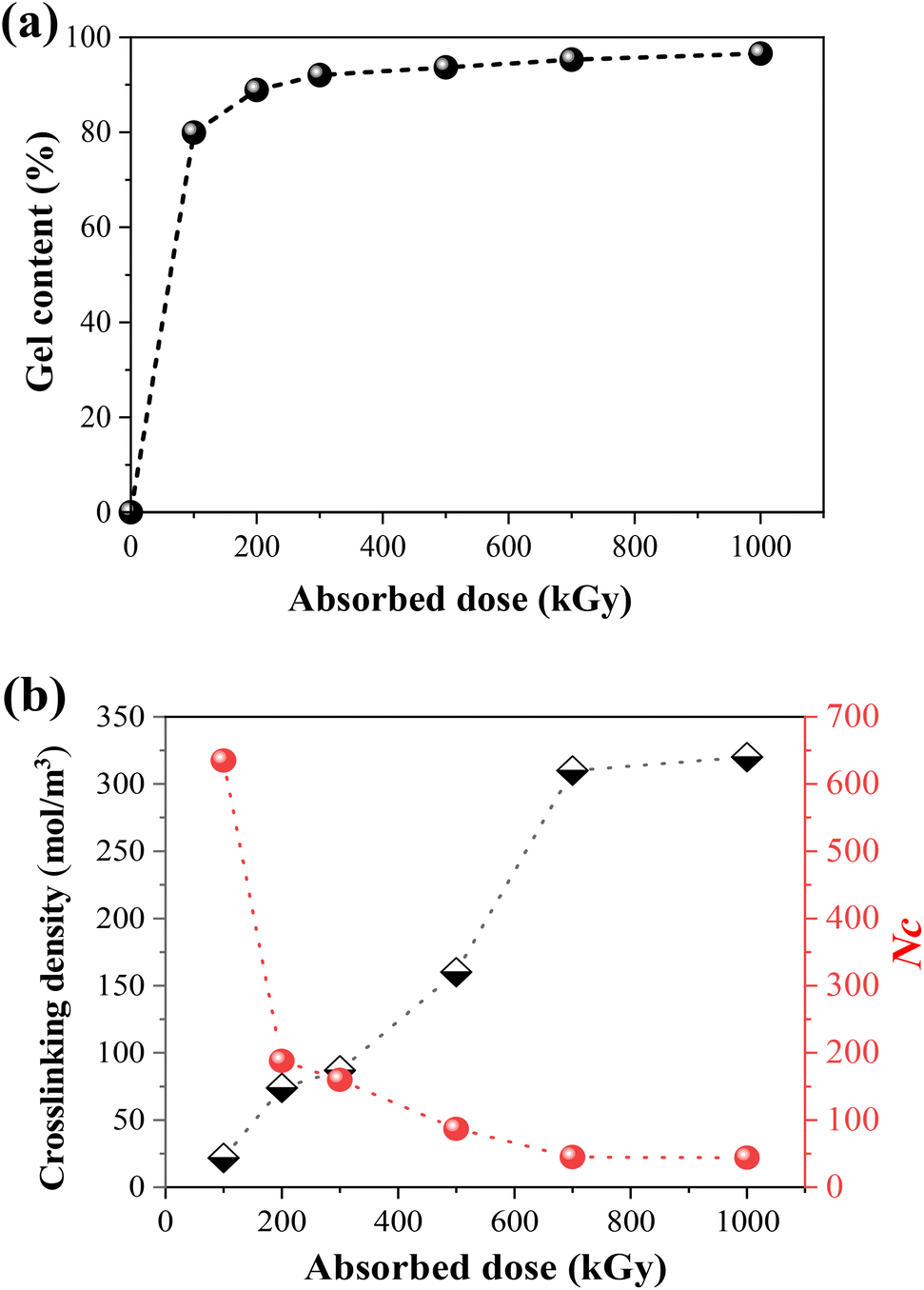
It has been confirmed in our previous study24 that TPI belongs to the irradiation cross-linking type polymer. As shown in Fig. 2a, the gel content and crosslinking density of the samples increase with the absorbed dose. As for semi-crystalline polymers, the crosslinking reaction mainly occurs in the amorphous region. However, there are still chain segments that are sufficiently long for crystallization. Fig. 2b shows the crosslinking degree and number of repeating units between the crosslinks (Nc) under different absorbed doses, the latter of which was calculated by the following formula:51where M0 is the molecular weight of the TPI monomer (68 g mol−1), ρ1 represents the density of TPI (0.945 g cm−3), and Ve represents the crosslinking density of TPI after irradiation.
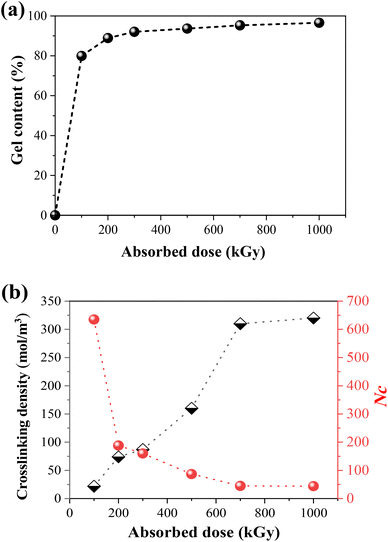 |
| | Fig. 2 (a) The gel content of irradiated TPI under an N2 atmosphere with different absorbed doses; (b) change of crosslinking density and monomer numbers between crosslinking points as a function of absorbed dose. | |
3.2 Grafting of x-TPI: the effect of the crosslinking degree
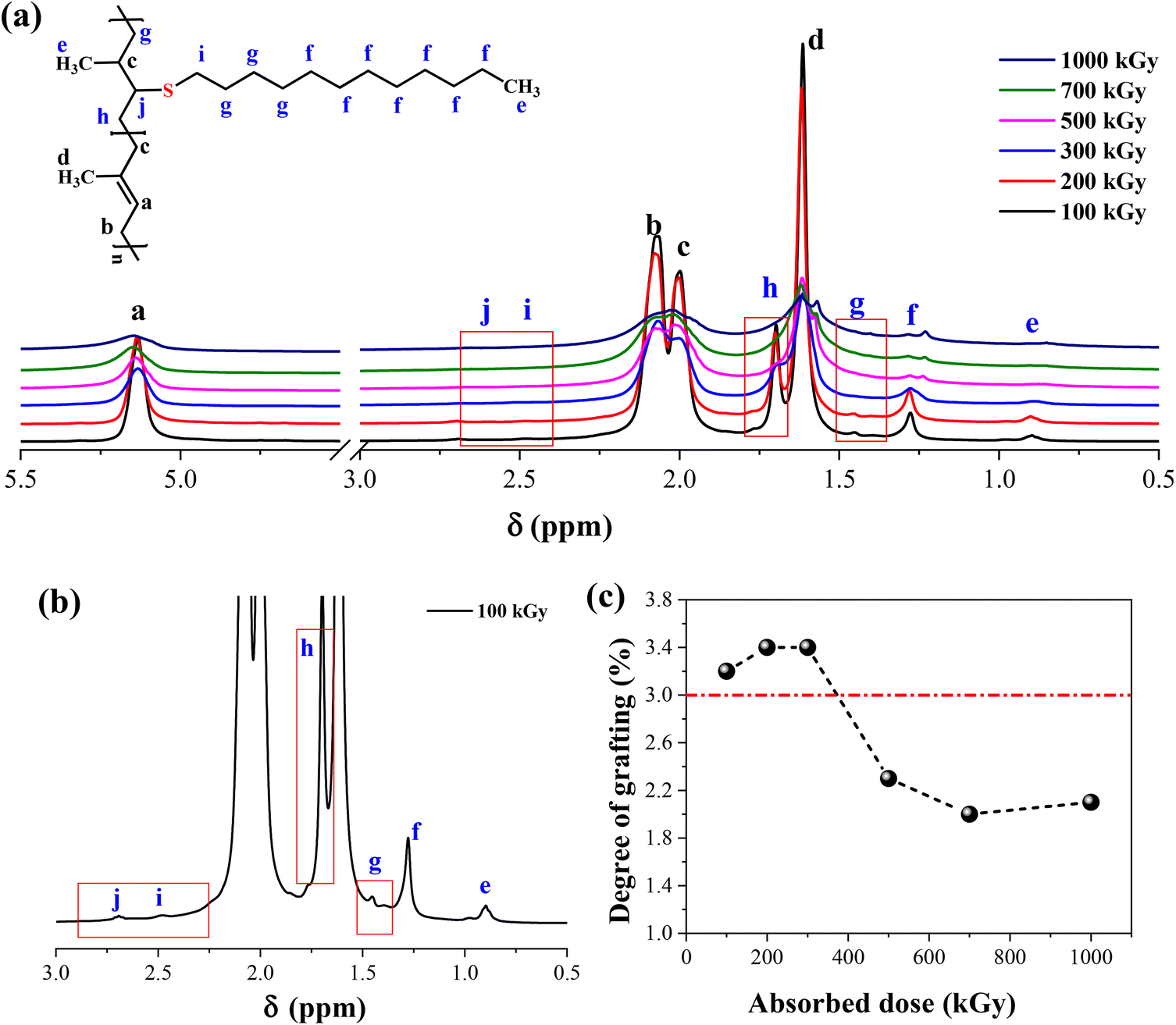
As shown in Fig. 1, the methodology needs a network structure. The first parameter to consider is the crosslinking degree of xTPI. Two factors must be considered. First, the gel content should be high so that during the second step, the weight loss of xTPI is low. Second, the network should not be too dense to hinder the diffusion of thiol. Fig. 3 shows the 1H-NMR spectra of the modified TPI. Three resonance peaks were found in the 1H-NMR spectrum of the initial xTPI. According to the previous studies,15,52–54 the peak at 5.14 ppm corresponds to the proton in C–H, marked as a. The peaks at 2.07 ppm and 1.99 ppm for the proton in –CH2–, marked as b and c, are slightly different in chemical shifts. The peak at 1.61 ppm corresponds to the proton in –CH3, marked as d as shown in Fig. 3a.
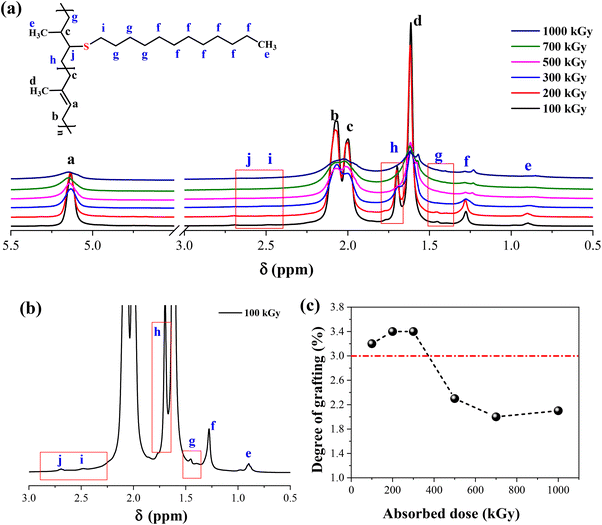 |
| | Fig. 3 (a) The 1H-NMR spectra of the modified xTPI vary with absorbed doses (fixed S/D 3:1, reaction time 2 h); (b) magnified spectrum of xTPI with absorbed dose = 100 kGy; (c) the grafting ratio of the modified xTPI as a function of absorbed dose. | |
After the grafting reaction, new resonance peaks appear at 0.89 ppm, 1.28 ppm, 1.45 ppm, 1.69 ppm, 2.52 ppm, and 2.69 ppm, marked as e, f, g, h, i, and j, respectively, which can be assigned to the protons in the grafted monomers (inset of Fig. 3a and b). Fig. 3b shows the magnified spectrum of g-xTPI, highlighting peaks g, i, and j. With the increase of the concentration of thiol, the intensity of the new peaks increased accordingly. The grafting degree of g-xTPI can be calculated by:
| | 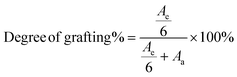 | (8) |
where
Ae is the area of peak e, a summation of the protons in the grafted thiol-terminated methyl group and the methyl group on the saturated single bond (0.88 ppm), and
Aa is the area of peak a, corresponding to the unsaturated double bond (5.14 ppm). The results show that the grafting degree slightly increases in the beginning and then decreases with the absorbed dose. The sample with 300 kGy has the highest grafting degree while maintaining a high gel content. Therefore, in the following sections, xTPI with 300 kGy was selected.
3.3 Grafting of x-TPI: the effect of the concentration of thiol and the reaction time
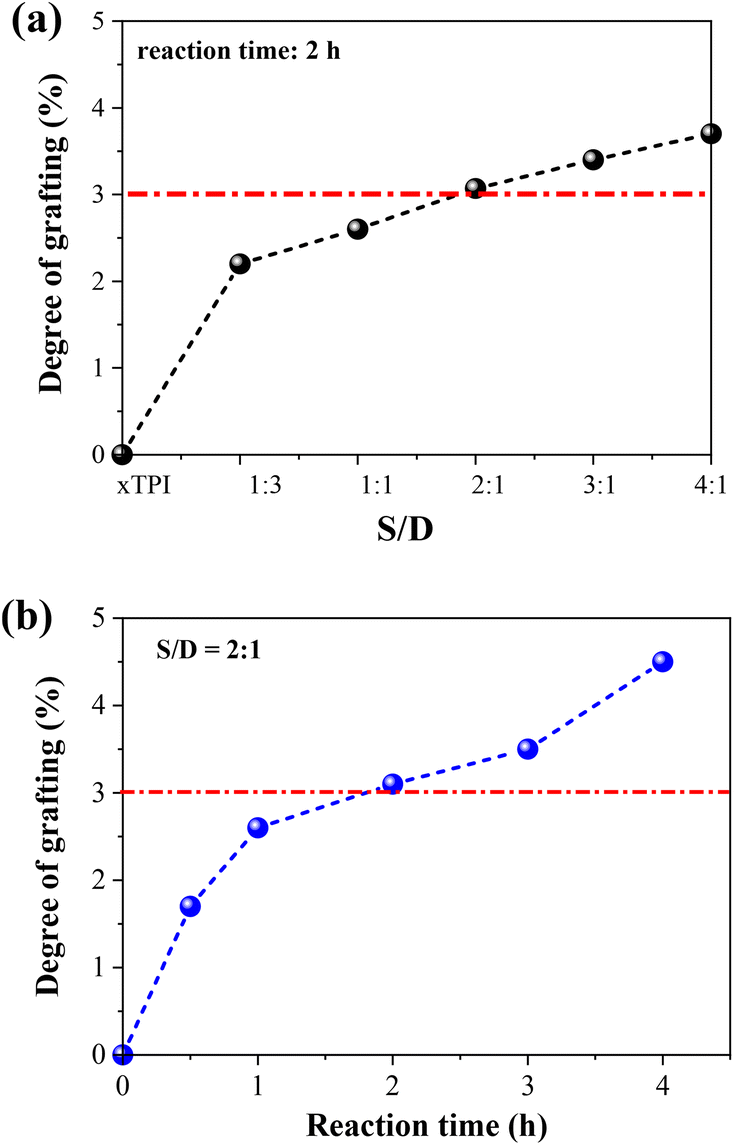
1H-NMR spectra were applied to characterize the influence of the concentration of thiol and the reaction time on the grafting degree (Fig. S2, ESI†). Two sets of experiments were carried out: (i) change of the S/D ratio with a fixed reaction time of 2 h; (ii) change of the reaction with a fixed S/D ratio (2:1). As shown in Fig. 4, the grafting degree of xTPI increases with the increase of the S/D ratio. Meantime, with the increase of the reaction time, the grafting degree also increases. The red line labels the critical value of the grafting degree as will be shown in the following sections. The g-xTPI samples exhibit similar thermal stability, as shown in Fig. S3 (ESI†).
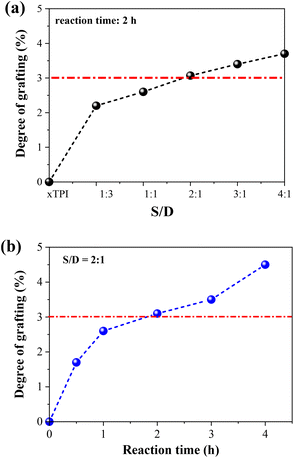 |
| | Fig. 4 The grafting degree of g-xTPI as a function of (a) S/D ratio and (b) reaction time. | |
3.4 Crystallization and melting behavior of g-xTPI
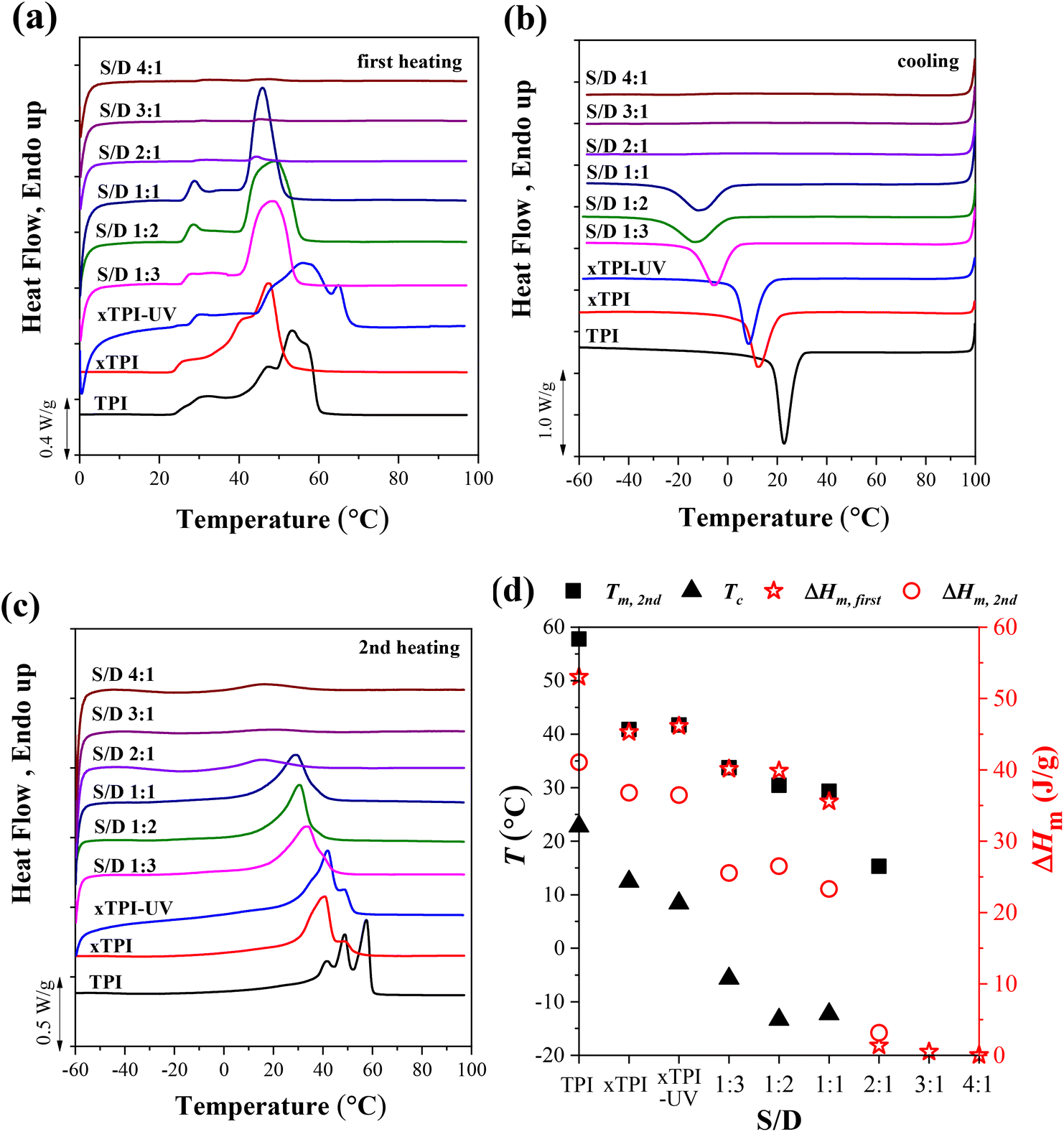
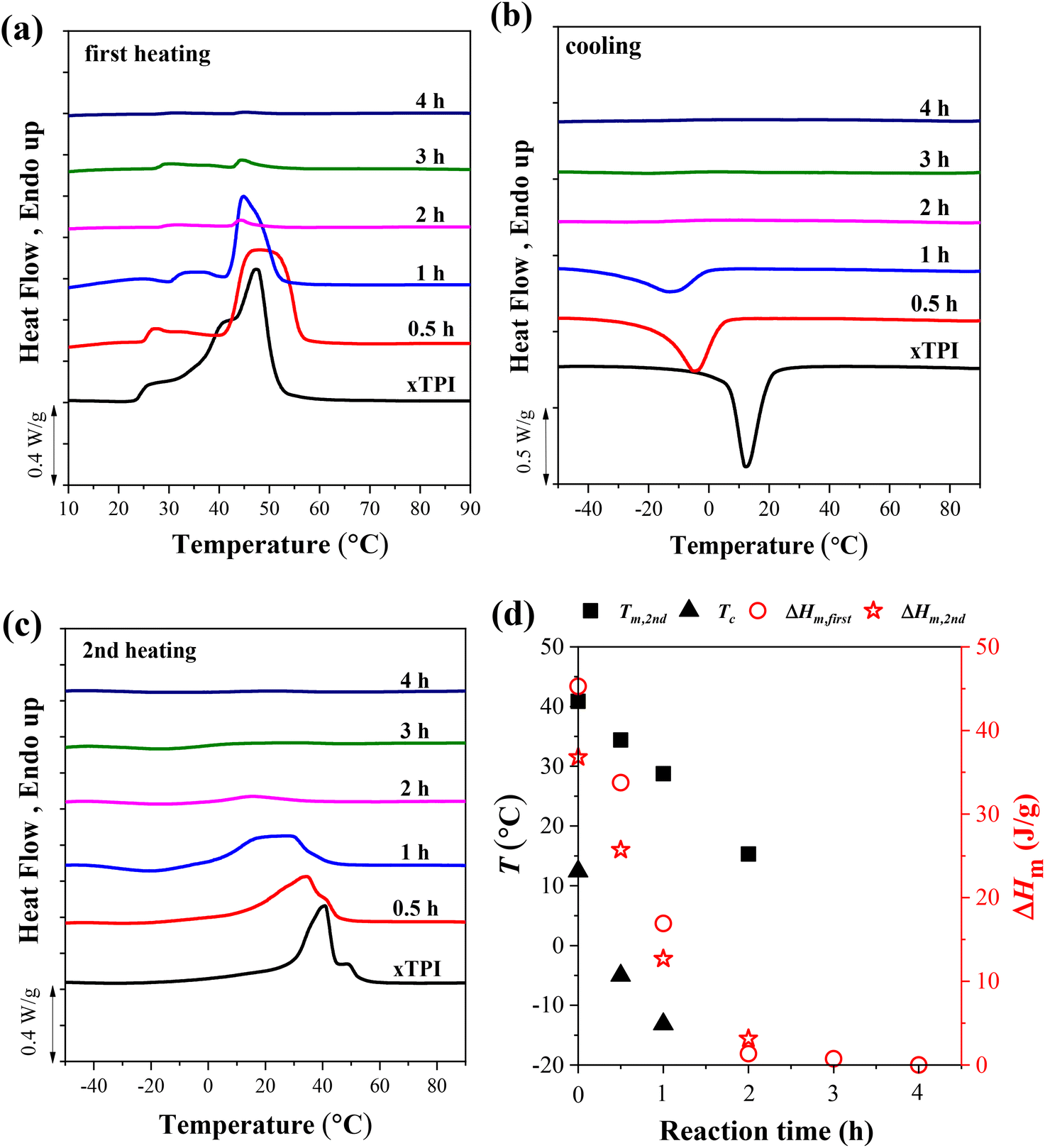
Fig. 5 shows the DSC curves of g-xTPI with different S/D ratios, together with three control samples, i.e., TPI, xTPI, and xTPI-UV. xTPI-UV has nearly the same treatment as g-xTPI but without the addition of thiol. In the first heating curve (Fig. 5a), xTPI-UV shows the highest melting temperature (Tm), even higher than TPI and xTPI. This is because of its solution crystallization history. In the subsequent cooling and second heating runs (Fig. 5b and c), TPI shows the highest crystallization temperature (Tc) and Tm, as expected. xTPI-UV shows lower Tc and comparable Tm as compared to xTPI. The Tc and Tm of g-xTPI are lower than the control samples and decrease with the S/D ratio. As summarized in Fig. 5d, Tc gradually decreases from 22.8 °C for the initial TPI to −10 °C for g-xTPI with S/D = 1:1. With a further increase in the S/D ratio, no crystallization and melting of xTPI can be observed by DSC. The results indicate that the grafting degree for the S/D ratio = 2:1 is a value for the transition from crystalline to amorphous. In Fig. 3c, the critical grafting ratio for the transition is labeled as a dotted line (∼3%).
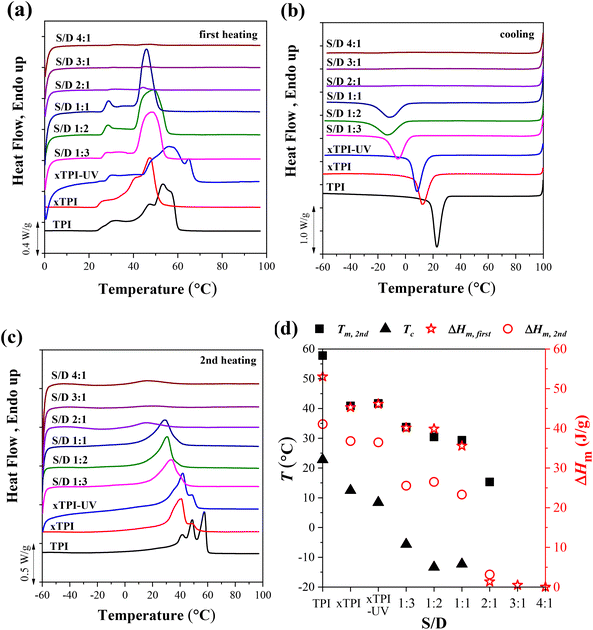 |
| | Fig. 5 The DSC curves of the modified xTPI with fixed reaction time: (a) the first heating, (b) cooling, and (c) the second heating DSC curves. (d) Tm, Tc, and ΔHm as a function of S/D. The heating/cooling rate is 10 °C min−1. The UV irradiation time was fixed at 2 h. | |
To explore the effect of the reaction time on the crystallization behavior of xTPI, DSC measurements were carried out on samples with different reaction times and a fixed S/D ratio (Fig. 6). Similar to the results shown in Fig. 5a, the melting behavior in the first heating curves is strongly influenced by the thermal history, evidenced by the fact that g-xTPI with a reaction time of 0.5 or 1 h shows a higher Tm as compared to xTPI. The cooling and second heating curves reflect the real crystallization and melting behavior of the samples. As shown in Fig. 6b and c, the Tm and Tc of the samples decrease with the reaction time. No crystallization or melting signal was detected when the reaction time was longer than 2 h. The results are summarized in Fig. 6d.
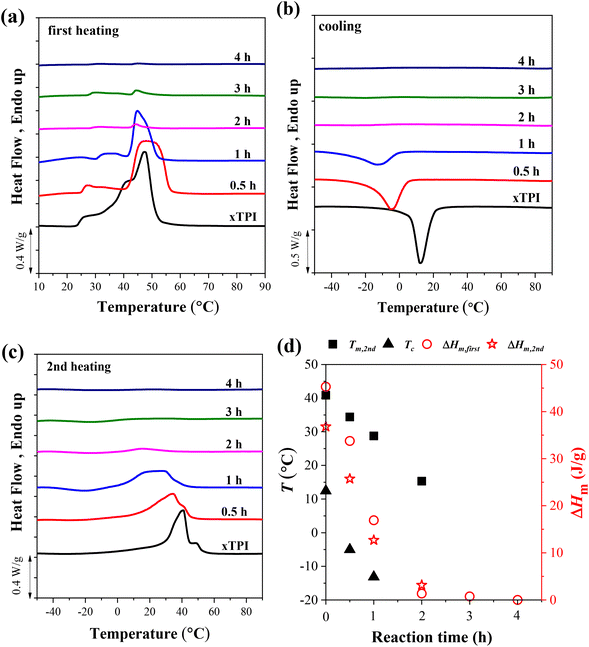 |
| | Fig. 6 The DSC curves of g-xTPI with different reaction times and a fixed S/D ratio (2:1): (a) the first heating, (b) cooling, and (c) the second heating curves. (d) Tm, Tc and ΔHm as a function of reaction time. The heating/cooling rate is 10 °C min−1. | |
3.5 Crystallization structure and crystallinity of g-xTPI
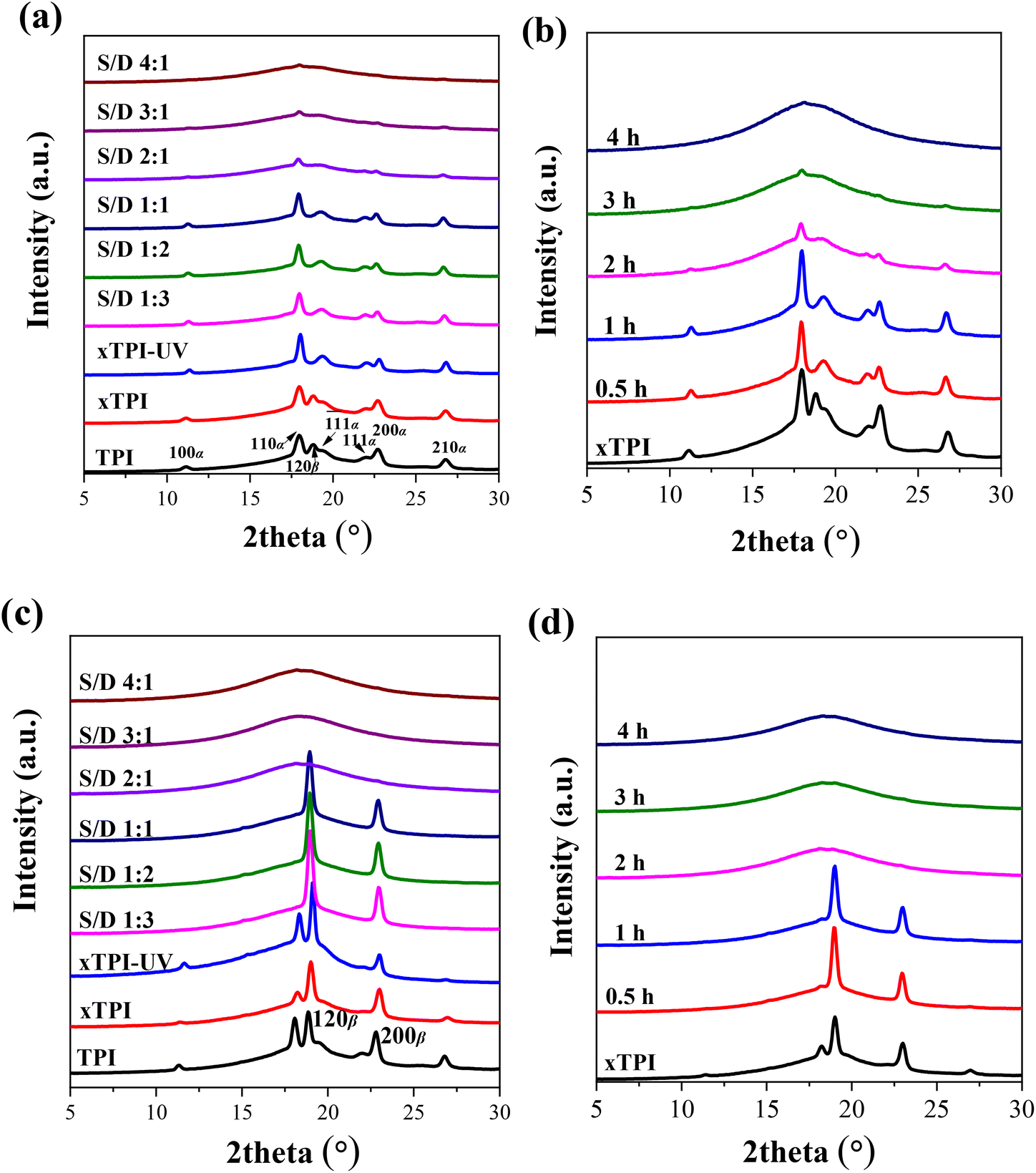
TPI mainly exhibits two crystal forms, namely, the stable α form and the metastable β form.55–58Fig. 7a and b show the WAXS curves of the as-prepared modified xTPI with different S/D ratios (fixed reaction time) and different reaction times (fixed S/D ratio). The initial xTPI sample is mainly composed of the α form because of the appearance of characteristic reflection peaks of (100), (110), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1), (111), (200), and (210). Along with the DSC results, because of the thermal history of the as-prepared samples, the WAXS curves of xTPI with S/D ratios of 1:3 to 1:1 (Fig. 7a) and xTPI with a reaction time of 0.5 or 1 h (Fig. 7b) were similar to those of the initial xTPI. Significant drops in diffraction intensities were observed with S/D ratio higher than 1:1 or with a reaction time longer than 1 h. With a further increase in the S/D ratio or reaction time, the intensity of the diffraction peaks of the modified xTPI decreases to ∼0. In the previous study, we found that xTPI crystallizes into the β form after melting and recrystallization.24Fig. 7c and d show the WAXS curves of recrystallized samples. The characteristic peaks of β form were observed: (120) and (200). Under this condition, g-xTPI with a S/D ratio higher than 2:1 or with the reaction time longer than 2 h are completely amorphous.
1), (111), (200), and (210). Along with the DSC results, because of the thermal history of the as-prepared samples, the WAXS curves of xTPI with S/D ratios of 1:3 to 1:1 (Fig. 7a) and xTPI with a reaction time of 0.5 or 1 h (Fig. 7b) were similar to those of the initial xTPI. Significant drops in diffraction intensities were observed with S/D ratio higher than 1:1 or with a reaction time longer than 1 h. With a further increase in the S/D ratio or reaction time, the intensity of the diffraction peaks of the modified xTPI decreases to ∼0. In the previous study, we found that xTPI crystallizes into the β form after melting and recrystallization.24Fig. 7c and d show the WAXS curves of recrystallized samples. The characteristic peaks of β form were observed: (120) and (200). Under this condition, g-xTPI with a S/D ratio higher than 2:1 or with the reaction time longer than 2 h are completely amorphous.
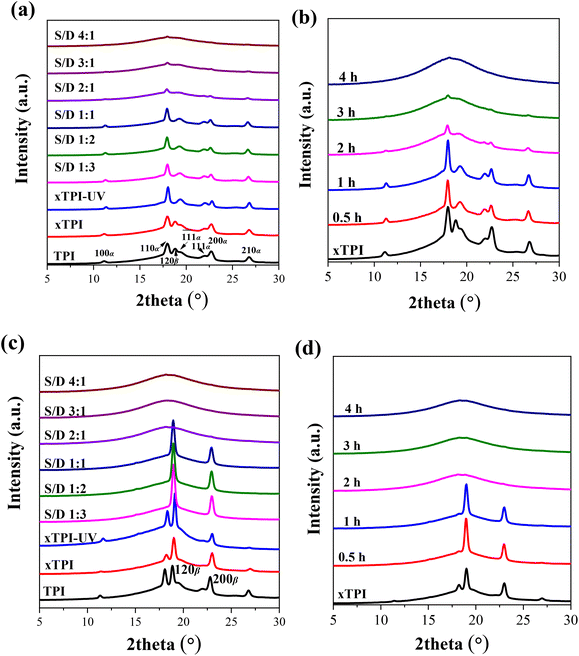 |
| | Fig. 7 WAXS of the modified xTPI: (a) and (c) samples with the varied S/D ratio and fixed reaction time (2 h); (b) and (d) samples with different reaction times and fixed S/D ratio (2:1). The thermal history of the samples is different: (a) and (b) are as prepared samples; (c) and (d) are samples that melted at 120 °C and cooled to −40 °C at 10 °C min−1 (for details, see the Experimental section). All the WAXS curves were taken at room temperature. | |
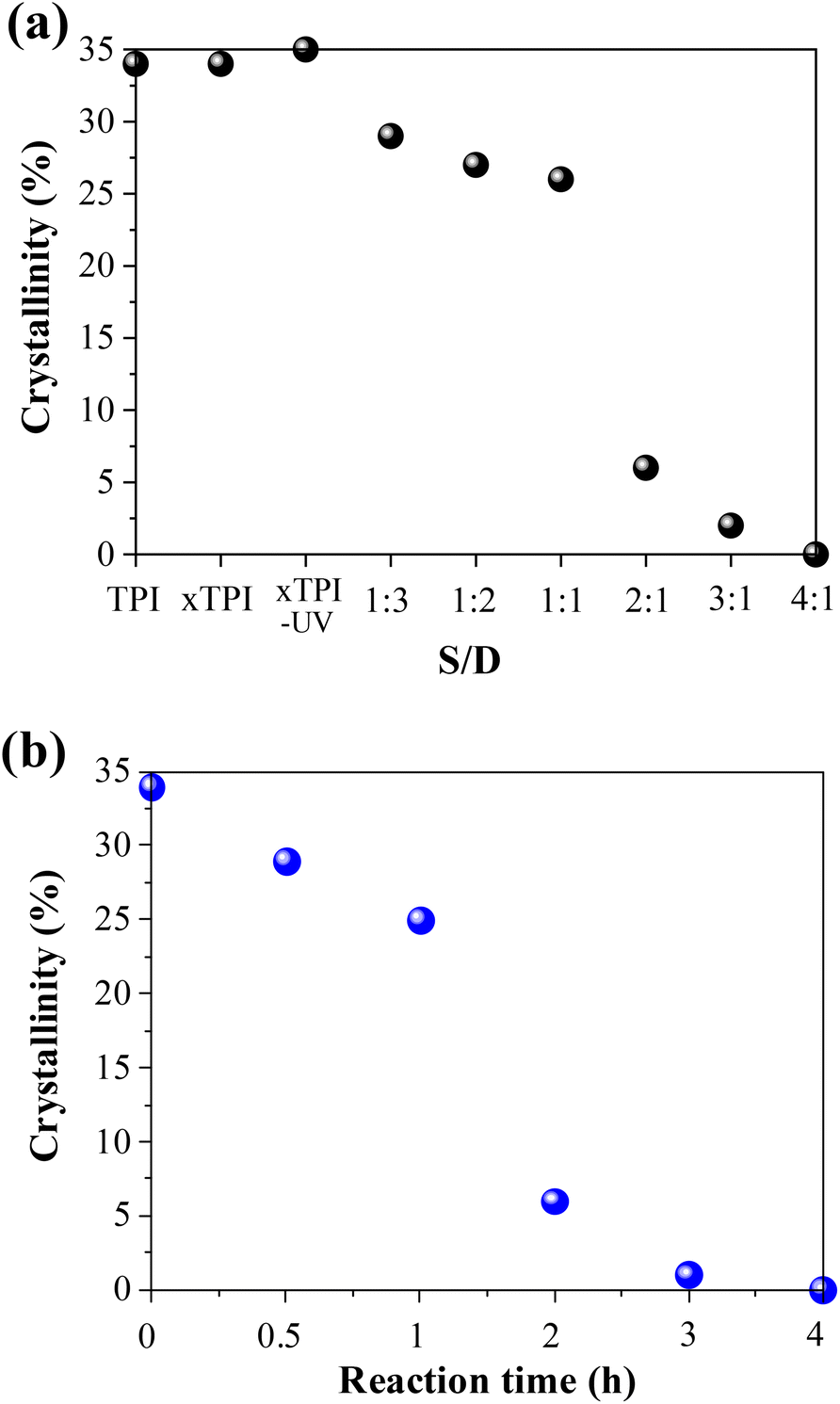
In order to further study the change of crystallinity of modified xTPI with S/D, we calculated the crystallinity by integrating the crystal peaks. When the grafting degree is low (S/D = 1:3–1:1) and (reaction times 0–1 h), the crystallinity changes little, as shown in Fig. 8. When the S/D ratio exceeds 1:1 or reaction times more than 1 h, the crystallinity drops remarkably, and this result is consistent with that shown in Fig. 5 and 6. When the S/D ratio exceeds 2:1, the crystallinity was close to 0. The results also indicate that the grafting degree for the S/D ratio = 2:1 is a critical value, for a transition from crystalline to amorphous.
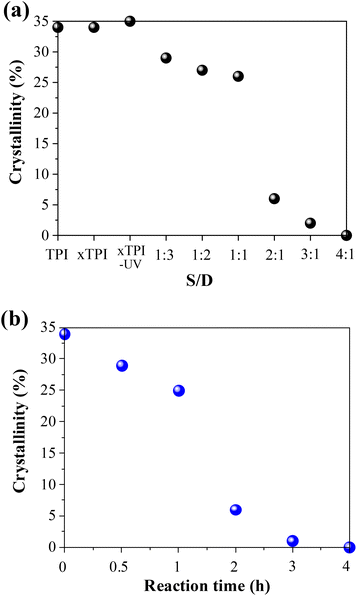 |
| | Fig. 8 The crystallinity of the modified xTPI (without heat treatment): (a) samples with the varied S/D ratio and fixed reaction time (2 h); (b) samples with different reaction times and a fixed S/D ratio (2:1). | |
3.6 Mechanical properties of g-xTPI
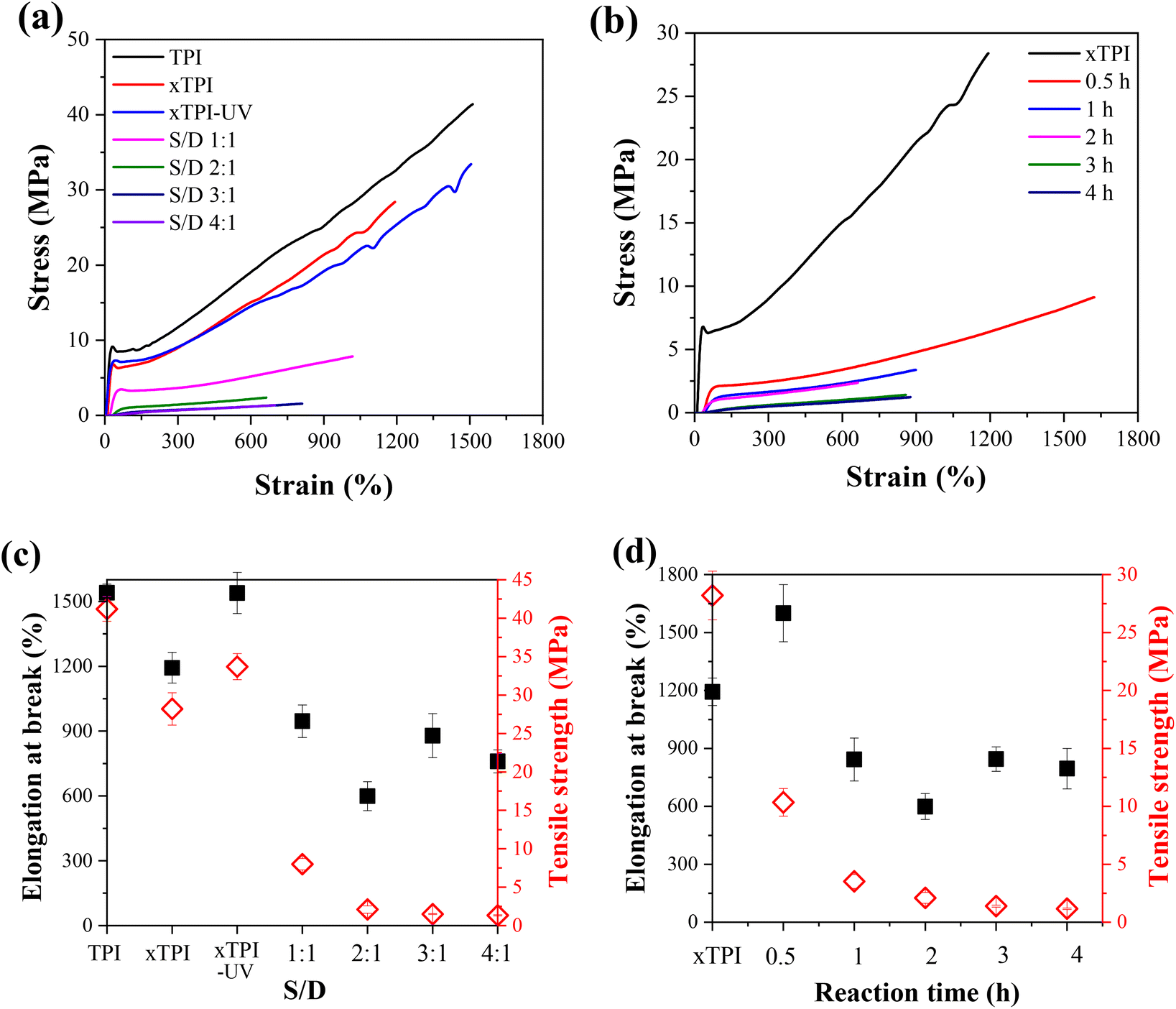
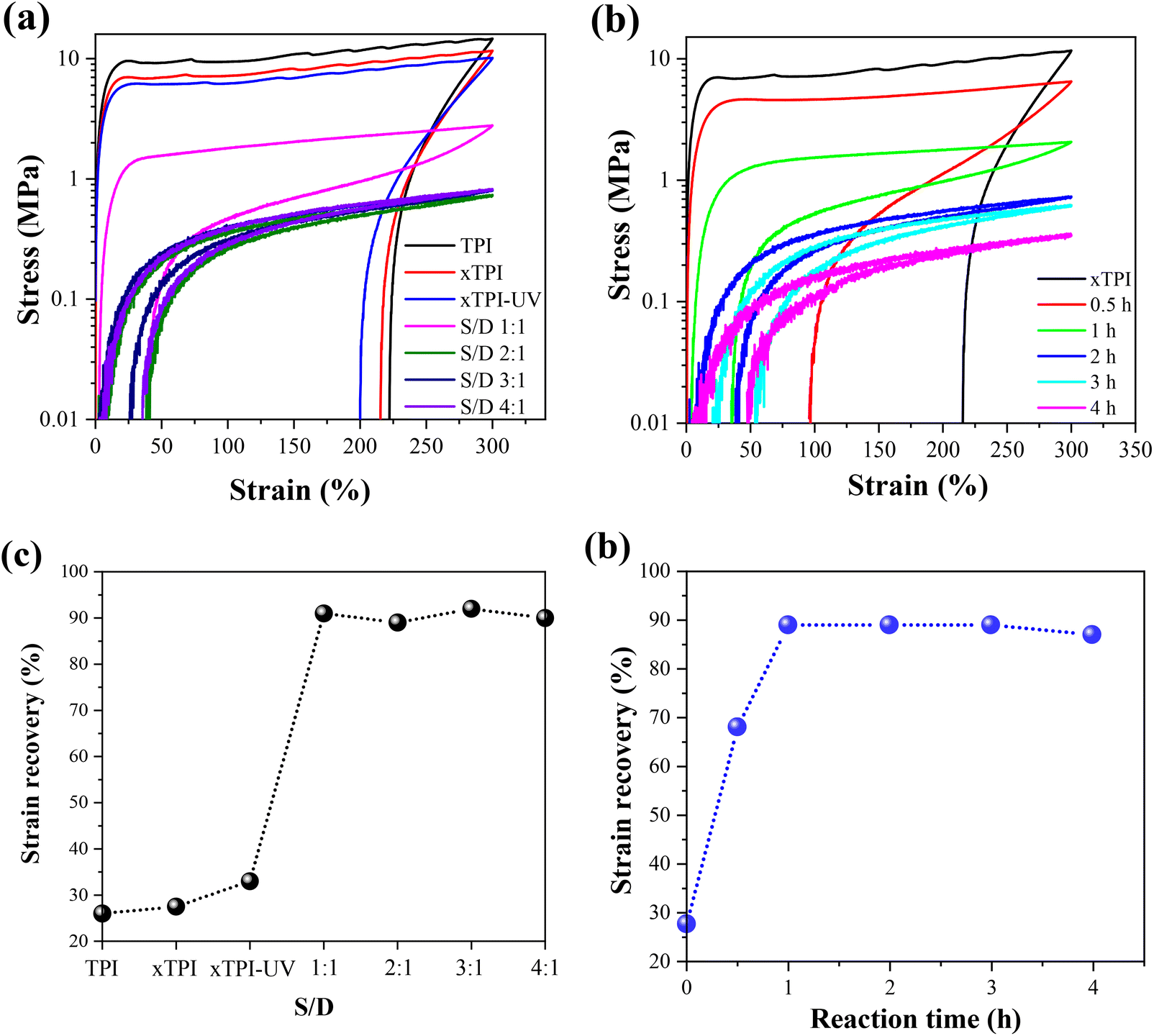
Fig. 9a and b show the engineering stress–strain curves of the modified xTPI. The tested samples are in their as-prepared state; therefore, the crystalline structures are reflected in Fig. 11a and b. With the increase of the S/D ratio, the tensile strength of g-xTPI decreased from 28.3 MPa to 1.3 MPa (Fig. 9c). xTPI and g-xTPI with a S/D ratio of 1:1 exhibit clear yielding behavior. With the increase of the S/D ratio, no yield point appears, and g-xTPI exhibits a rubber-like behavior. The decrease in stiffness with the grafting degree is in line with the decrease in crystallinity. When the S/D ratios were 3:1 and 4:1, the mechanical properties of the samples no longer changed. Fig. 9b shows the engineering stress–strain curves of g-xTPI with a fixed S/D ratio and different reaction times. The results show that with the increase of the reaction time, the tensile strength of the sample decreases from 28.3 MPa to 1.2 MPa (Fig. 9d), similar to the effect of the increasing S/D ratio. When the reaction time was longer than 1 h, the yield point disappeared and the stiffness decreased significantly. No change in tensile strength or elongation at break was observed when the reaction time was longer than 3 h.
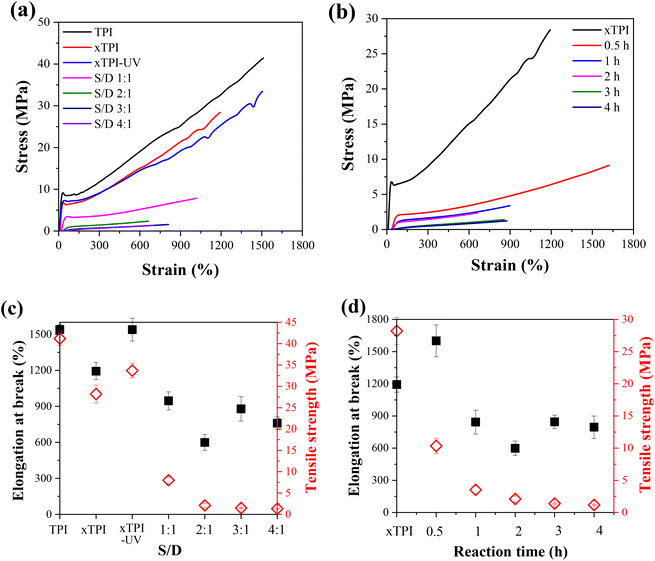 |
| | Fig. 9 The engineering stress–strain curves of g-xTPI with (a) different S/D ratios with a fixed reaction time (2 h) and (b) different reaction times with a fixed S/D ratio (2:1). The plots of elongation at break and tensile strength as a function of (c) S/D ratio and (d) reaction time. | |
Tensile recovery is a key property of elastomers. As shown in Fig. 10a and c, the initial elastic recovery of xTPI was ∼27.5%. The elastic recovery of g-xTPI significantly increased. When the S/D ratio was 1:1, the elastic recovery already reached 90%. Interestingly, the strain recovery of the samples does not change with further increasing the S/D ratio. Similar results were observed in Fig. 10b and d, where the strain recovery increases with the reaction time. The strain recovery of g-xTPI was about 90%, which is a relatively high value compared to other elastomers such as olefin block copolymers (OBCs, ∼75%)50 and polybutadiene elastomer (∼87%),34 and is close to styrene–butadiene rubber (∼92%),59 natural rubber (∼94%)60–62 and polyolefin elastomers (POEs, ∼92%).63 However, the tensile recovery of the sample shows a slight decrease, from 90% to 87%, when the reaction time was increased from 2 to 4 h. According to Carstensen,64 this may suggest that the UV reaction caused the degradation of xTPI, causing a decrease in strain recovery.
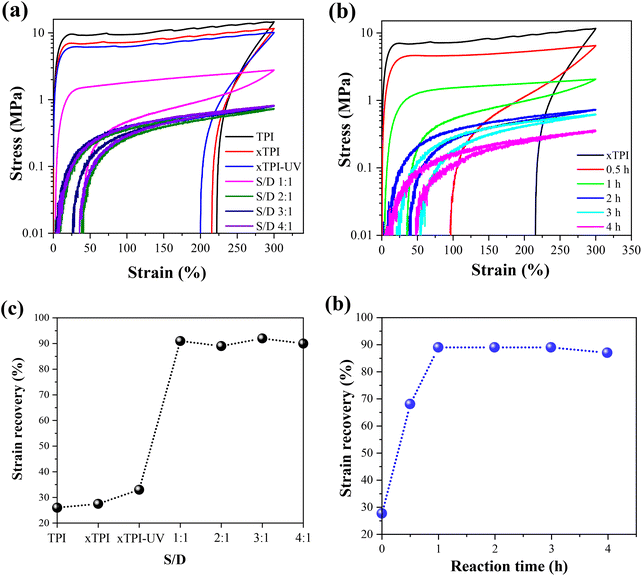 |
| | Fig. 10 Stress–strain curves of strain recovery tests (a) TPI samples with different S/D ratios (reaction time: 2 h); (b) g-xTPI with different reaction times (S/D = 2:1). The strain recovery of the TPI samples (c) with different S/D ratios and (d) with different reaction times. | |
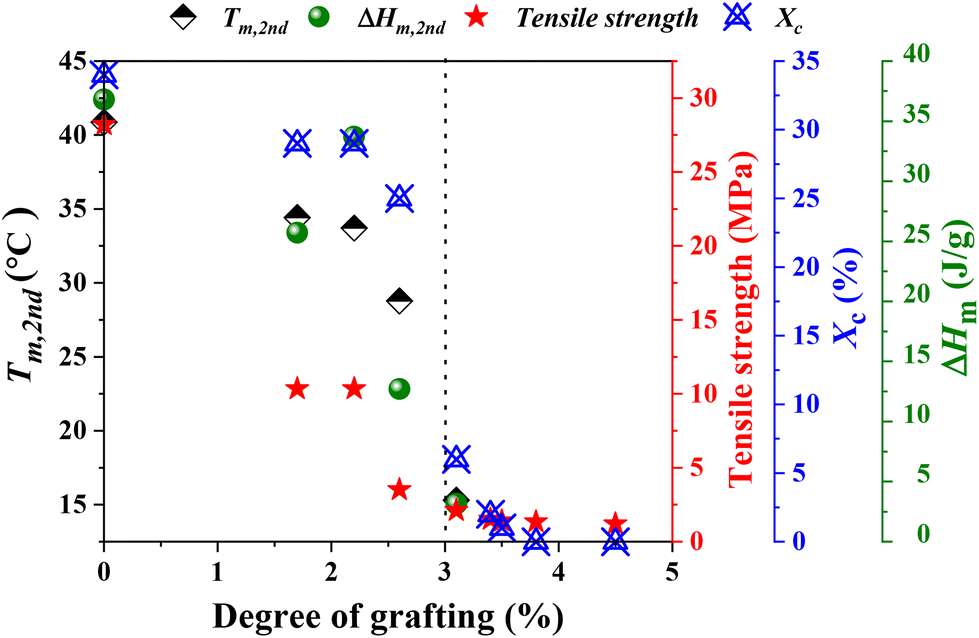
The physical properties of g-xTPI are closely related to its microstructures. The crosslinks provide a permanent network, where the junctions are defects for crystallization. As shown in the monodisperse polyethylene glycol network, the melting temperature decreases with the length of strands.65 A similar effect is known in ethylene copolymers where commoners such as butene or octane act as defects that shorten the crystallizable sequence length.66 In analogies to the comonomer content, the grafting degree is the fundamental parameter for the physical properties of g-xTPI. Tm, ΔHm, tensile strength and Xc were plotted as a function of grafting degree, as shown in Fig. 11. Those physical quantities decreased in a roughly similar trend. As compared to the crosslinks, the advantage of grafting chains is that they shorten the crystallizable sequences while keeping the density of the network to maintain stretchability.
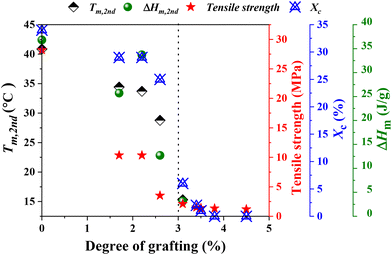 |
| | Fig. 11 A plot of Tm, ΔHm, tensile strength, and Xc as a function of grafting degree. | |
4. Conclusion
In this work, we studied the modification of TPI for potential applications as elastomers. TPI was crosslinked first by γ-ray radiation to form a permanent network. Then, the chains between crosslinks were grafted by a thiol–ene reaction. In this way, the regularity of TPI was destroyed, while maintaining an appropriate crosslinking density that enables the polymer to exhibit good stretchability. The influence of reagent concentration and reaction time was systematically studied. The results showed that the crystallinity of the modified TPI decreased significantly with the grafting ratio. For TPI with an absorbed dose of 300 kGy, a critical grafting degree of 3% was identified for a transition from a semicrystalline plastic to an amorphous rubber. Interestingly, the strain recovery increases from 27.5% to 90%, indicating a promising improvement in elasticity.
Author contributions
Hui Zhao: investigation, formal analysis, and writing – original draft. Chunbo Zhang: validation and formal analysis. Bo Yang: investigation. Xiuqin Zhang: validation. Xia Dong: validation. Dujin Wang: writing – review & editing. Guoming Liu: conceptualization, supervision, and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21922308). G. L. is grateful to the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Grant No. Y201908). X. Z. is grateful to the Beijing Scholars Program (RCQJ20303). We thank Qingdao Junxiang Technology Co., Ltd for providing the TPI samples, Prof. Wei You (ICCAS) and Prof. Yang Wang (UCAS) for helpful discussions.
References
- J. Wang, H. Kang, F. Yang and Q. Fang, China Synth. Rubber Ind., 2017, 40, 315–319 CAS.
- J. C. Zhang, Z. H. Xue and R. F. Yan, Chin. J. Polym. Sci., 2011, 29, 157–163 CrossRef CAS.
- J. Zhang and Z. Xue, Polym. Bull., 2011, 67, 511–525 CrossRef CAS.
- H. Moon, J. Lee, J. Ahn, H. Song and Y. Park, Int. Endod. J., 2015, 48, 556–563 CrossRef CAS.
- X. Wei, P. Peng, F. Peng and J. Dong, J. Agric. Food Chem., 2021, 69, 3797–3821 CrossRef CAS PubMed.
- A. Du, Y. Liu, B. Huang and W. Yao, J. Macromol. Sci., Part B: Phys., 2010, 49, 479–486 CrossRef CAS.
- Z. N. Liu, L. I. Xu Dong, B. C. Huang, W. Yao, Y. X. Zhao and D. Q. Wang, China Elastomerics, 2006, 16, 8–12 CAS.
- J. Fu, S. Zhang, J. Zhao and Y. Feng, Mod. Plast. Process. Appl., 2014, 24, 18–21 Search PubMed.
- X. Qi, X. Zhao, Y. Li, J. Zhang, L. Zhang and D. Yue, J. Appl. Polym. Sci., 2021, 138, 50007 CrossRef CAS.
- D. R. Burfield and A. H. Eng, Polymer, 1989, 30, 2019–2022 CrossRef CAS.
- F. Yang, Q. Liu, X. Li, L. Yao and Q. Fang, Polym. Bull., 2017, 74, 3657–3672 CrossRef CAS.
- Y. Zhao, B. Huang, W. Yao, H. Cong, H. Shao and A. Du, J. Appl. Polym. Sci., 2008, 107, 2986–2993 CrossRef CAS.
- T. Tsujimoto, K. Toshimitsu, H. Uyama, S. Takeno and Y. Nakazawa, Polymer, 2014, 55, 6488–6493 CrossRef CAS.
- H. Kang, M. Xu, H. Wang, L. Li, J. Li, Q. Fang and J. Zhang, J. Appl. Polym. Sci., 2020, 137, 49133 CrossRef CAS.
- H. Zhang, C. Ma, R. Sun, X. Liao, J. Wu and M. Xie, Polymer, 2019, 184, 121904 CrossRef CAS.
- H. Li, Q. Ren, J. Chen, H. Zhang, J. Wu and M. Xie, React. Funct. Polym., 2018, 132, 104–111 CrossRef CAS.
- Y. Wei, J. Song, A. He, W. Zuo, J. He and B. Huang, China Elastomerics, 1995, 4, 1–7 Search PubMed.
- W. Fu, A. Liu, J. Li and L. Wang, Asian – J. Chem., 2014, 26, 5546–5550 CrossRef CAS.
- D. Saunders, Trans. Faraday Soc., 1956, 52, 1414–1425 RSC.
- D. Saunders, Trans. Faraday Soc., 1957, 53, 860–870 RSC.
- Y. Ikeda, N. Higashitani, K. Hijikata, Y. Kokubo, Y. Morita, M. Shibayama, N. Osaka, T. Suzuki, H. Endo and S. Kohjiya, Macromolecules, 2009, 42, 2741–2748 CrossRef CAS.
- M. Shibayama, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS.
- J. Valentín, P. Posadas, A. Fernández-Torres, M. Malmierca, L. González, W. Chassé and K. Saalwachter, Macromolecules, 2010, 43, 4210–4222 CrossRef.
- H. Zhao, C. Zhang, G. Liu, J. Li, B. Yang, H. Ma, X. Zhang and D. Wang, Polym. Degrad. Stab., 2022, 197, 109869 CrossRef CAS.
- M. Baboo, K. Sharma, N. Saxena and M. Kumar, Polym. Eng. Sci., 2013, 53, 443–451 CrossRef CAS.
- Z. Zheng, W. M. Kwok and W. M. Lau, Chem. Commun., 2006, 29, 3122–3124 RSC.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- T. Zhao, R. Yu, S. Li, X. Li, Y. Zhang, X. Yang, X. Zhao, C. Wang, Z. Liu and R. Dou, ACS Appl. Mater. Interfaces, 2019, 11, 14391–14398 CrossRef CAS.
- R. Fraser, A. Carletto, M. Wilson and J. Badyal, ACS Appl. Mater. Interfaces, 2016, 8, 21832–21838 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- D. Wang, H. Zhang, B. Cheng, Z. Qian, W. Liu, N. Zhao and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1357–1366 CrossRef CAS.
- K. D. Nguyen, W. V. Megone, D. Kong and J. E. Gautrot, Polym. Chem., 2016, 7, 5281–5293 RSC.
- D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC.
- J.-X. Lei, Q.-Y. Wang, F.-S. Du and Z.-C. Li, Chin. J. Polym. Sci., 2021, 39, 1146–1154 CrossRef CAS.
- W. B. Zhang, J. Luo, Y. M. Wang, X. Z. Zhu, C. Zhang, J. Liu, M. L. Ni and G. H. Zhang, Chin. J. Polym. Sci., 2021, 39, 994–1003 CrossRef CAS.
- S. Deuri and P. Phukan, Int. J. Quantum Chem., 2012, 112, 801–808 CrossRef CAS.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag and U. S. Schubert, Macromol. Chem. Phys., 2016, 217, 2541–2550 CrossRef CAS.
- R. Steer and A. Knight, Can. J. Chem., 1969, 47, 1335–1345 CrossRef CAS.
- C. Resetco, B. Hendriks, N. Badi and F. Du Prez, Mater. Horiz., 2017, 4, 1041–1053 RSC.
- C. Gao, S. Wang, J. Zhang, Y. Liu and C. Wang, Polym. Degrad. Stab., 2020, 181, 109336 CrossRef CAS.
- P.
J. Flory, J. Chem. Phys., 1950, 18, 108–111 CrossRef CAS.
- J. Zhang, T. Zhang, M. Dong, G. Liu and Y. Dong, Polym. Test., 2017, 63, 511–520 CrossRef CAS.
- R. Qian, Z. Wu, Z. Xue and R. Yan, Macromol. Rapid Commun., 1995, 16, 19–23 CrossRef CAS.
- P. Bala, B. Samantaray, S. Srivastava and G. Nando, J. Appl. Polym. Sci., 2004, 92, 3583–3592 CrossRef CAS.
- M. Dong, T. Zhang, J. Zhang, G. Hou, M. Yu and L. Liu, Polym. Test., 2020, 87, 106539 CrossRef CAS.
- H. Wang, S. Chum, A. Hiltner and E. Baer, J. Appl. Polym. Sci., 2009, 113, 3236–3244 CrossRef CAS.
- S. Trabelsi, P. A. Albouy and J. Rault, Macromolecules, 2003, 36, 7624–7639 CrossRef CAS.
- Q. Niu, X. Jiang and A. He, Polymer, 2014, 55, 2146–2152 CrossRef CAS.
- H. Y. Chen, Anal. Chem., 1962, 34, 1134–1136 CrossRef CAS.
- K. Dong, J. Zhang and A. He, Polymer, 2021, 235, 124231 CrossRef CAS.
- J. Lu, H. Yang, Y. Ji, X. Li, Y. Lv, F. Su and L. Li, Macromol. Chem. Phys., 2017, 218, 1700235 CrossRef.
- C. W. Bunn, Proc. R. Soc. London, Ser. A, 1942, 180, 40–66 CAS.
- P. J. Ratri, K. Tashiro and M. Iguchi, Polymer, 2012, 53, 3548–3558 CrossRef CAS.
- C. B. Zhang, L. Wang, B. Yang, H. Zhao, G. M. Liu and D. J. Wang, Chin. J. Polym. Sci., 2022, 40, 256–265 CrossRef CAS.
- J. B. Le Cam and E. Toussaint, Mech. Mater., 2009, 41, 898–901 CrossRef.
- S. Amnuaypornsri, S. Toki, B. S. Hsiao and J. Sakdapipanich, Polymer, 2012, 53, 3325–3330 CrossRef CAS.
- L. Mullins, Rubber Chem. Technol., 1969, 42, 339–362 CrossRef CAS.
- X. Qi, L. Wang, Y. Zhang, M. Jia, L. Zhang and D. Yue, Macromolecules, 2022, 55, 2758–2767 CrossRef CAS.
- F. Yang, L. Pan, Z. Ma, Y. Lou, Y. Li and Y. Li, Polym. Chem., 2020, 11, 3285–3295 RSC.
- P. Carstensen, Makromol. Chem., 1970, 135, 219–234 CrossRef CAS.
- C. Zhang, C. Liu, L. Wang, Y. Zhao, G. Liu and D. Wang, Chem. Commun., 2022, 58, 286–289 RSC.
- J. Isasi, J. Haigh, J. Graham, L. Mandelkern and R. Alamo, Polymer, 2000, 41, 8813–8823 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |

 b,
Xia
Dong
b,
Xia
Dong