
Reducing defect density in UiO-68–CHO is key for its efficient and reliable post-synthetic modification†
Marcin
Wiszniewski
 and
Michał J.
Chmielewski
*
and
Michał J.
Chmielewski
*
Faculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: mchmielewski@chem.uw.edu.pl
First published on 6th July 2023
Abstract
Post-synthetic modification (PSM) is a powerful tool for introducing complex functionalities into metal–organic frameworks (MOFs). Aldehyde-tagged MOFs are particularly appealing platforms for covalent PSM due to the high reactivity of aldehyde groups, but the same feature also makes their solvothermal synthesis challenging. In this work, we show that while lowering the temperature during the synthesis of aldehyde-tagged UiO-68 avoids aldehyde group degradation and yields a highly porous and crystalline material, the resulting UiO-68–CHO contains a large fraction of missing linker defects and, as a result, its PSM is both inefficient and non-repeatable. However, we also show that this problem could be solved by 1) using an excess of linker during the synthesis of the MOF and 2) by soaking the crude material in the solution of the linker, which together reduce the density of defects enough to yield an excellent substrate for PSM. Treatment of the ‘healed’ material with model amines gives nearly quantitative conversions of aldehydes into imines, even if no excess of reagents is used. Importantly, the PSM of the ‘healed’ UiO-68–CHO gives repeatable results over many days, unlike the PSM of the highly defective MOF. Owing to these developments, various functionalities, such as new coordination sites, drug cargo, chirality, and hydrophobicity, were successfully introduced into the UiO-68 framework. The deleterious influence of defects on the PSM of MOFs and the solution to this problem proposed herein are likely to be of general nature and hence might help in developing new and versatile platforms for covalent PSMs.
Design, System, ApplicationIdeal MOFs for PSM should combine highly stable frameworks with highly reactive functional groups. With the aim to reconcile these seemingly contradictory requirements, efforts are made to synthesize these materials under mild conditions, wherein the reactive functional groups remain unchanged. Using UiO-68–CHO as an example, we show here that although the resulting MOFs may seem to be nicely crystalline by PXRD, they may also contain large amounts of missing linker defects, which make them unstable during PSM. As a result, PSMs of such materials give low yields and are non-repeatable. We show also that this problem could be easily solved by using an excess of linkers during the solvothermal syntheses of the MOFs, followed by a simple healing procedure, wherein the as-synthesized MOF is soaked in solution of ligand to fill its missing linker defects. Using this approach, we obtained an almost perfect substrate for PSM, i.e. the UiO-68–CHO MOF that gives nearly quantitative yields in model PSMs, even if no excess of reagents is used. Moreover, the PSM of the ‘healed’ UiO-68–CHO gives repeatable results over a long time, in contrast to the substrate with high defect density. Owing to these developments, various complex functionalities were successfully introduced into the UiO-68 framework. |
Introduction
Metal–organic frameworks are an exceptionally versatile class of porous materials owing mainly to their well-defined crystalline structures, permanent porosity and easy tunability.1,2 This unique combination of properties makes them attractive for numerous applications, such as gas storage and separation,3–5 catalysis,6–8 sensing,9–11 electronics,12–14 photonics,15–17 drug delivery,18–20 and many others.21–26 Some of these applications require functionalisation of the MOF's pores with complex functional groups, which are either too delicate to survive harsh conditions of typical MOF syntheses, or preclude the formation of the targeted MOF structure. Such functionalities may be introduced by covalent post-synthetic modification (PSM) of MOFs having linkers with reactive groups, such as –NH2, –N3, alkynyl, or –CHO.27–29 Unfortunately, however, the availability of such MOFs is limited, mainly because the high reactivity that makes a functional group attractive for PSM makes it also more prone to side reactions during the synthesis of MOFs.Thus far, the most popular platforms for covalent PSM have been amine-tagged MOFs.27,29 Aromatic amines are sufficiently stable to survive the harsh solvothermal conditions of many MOFs' synthesis, and yet sufficiently reactive to give high conversions in reactions with acyl halides, acid anhydrides, aldehydes, and other reagents. However, due to the rather poor nucleophilicity of aromatic amines, large, even several-fold excess of reagents is often required to reach a satisfactory degree of PSM.30–39 This is problematic for economic and environmental reasons, especially when complex, difficult to make, or very expensive reagents have to be used.
A promising alternative to the amine-tagged MOFs is MOFs with aldehyde groups. In contrast to amines, which react with electrophiles, aldehydes react with nucleophiles, such as amines, hydrazines, hydroxylamines, hydrazides, alcohols, various carbanions, etc., and therefore offer complementary reactivity. Moreover, aldehydes can be easily reduced to alcohols or oxidised to carboxylic acids, as well as subjected to reductive amination and other reactions. Therefore, the incorporation of aldehyde groups into robust MOFs with large pores seems to be a very attractive strategy for obtaining versatile platforms for covalent PSM.
Unfortunately, however, the high reactivity of aldehydes makes the synthesis of such materials challenging and, in fact, only a few aldehyde-tagged MOFs have been described in the literature thus far.40–48 Besides, most of these MOFs have limited utility for PSM due to narrow pores or pore windows (ZIFs),40–43 framework catenation (IRMOF),44,45 and/or poor chemical stability (IRMOF, bioMOF).44–46 In 2016, Gao and co-workers developed the synthesis of the first aldehyde-tagged zirconium MOF, UiO-67–CHO.47 Zirconium MOFs from the UiO family are known for their high thermal and hydrolytic stability and large pores.49,50 They are also known to require high temperatures for their synthesis, and lowering the temperature results in large amounts of defects, giving materials with lower stability.51 Unfortunately, however, during the high temperature solvothermal synthesis of zirconium MOFs, aldehyde groups undergo side reactions with DMF.52,53 In order to avoid aldehyde degradation during the synthesis of UiO-67–CHO, Gao and co-workers lowered the reaction temperature from the typical 120 °C to 80 °C. The obtained material was crystalline and highly porous, and its aldehyde groups were successfully post-synthetically functionalised by means of various C–N and C–C coupling reactions. However, each PSM required a large excess of reagents and the repeatability of the functionalisation was not investigated. More recently, Manna and co-workers described the synthesis of UiO-68–CHO.48 With even larger pores and more expeditious linker synthesis, this material seemed to be an even more attractive platform for PSMs, and in particular for our planned studies on reversible, thermodynamically controlled PSM of MOFs via imine formation. However, in the original report, this material has been functionalised with only one amine, valinol, which was furthermore used in large excess. Also, our preliminary experiments with the PSM of UiO-68–CHO with stoichiometric amounts of amines gave disappointingly low and non-repeatable yields.
In this contribution, we link the low yields and non-repeatability of UiO-68–CHO post-synthetic modification to the large defect density in its structure, caused by the low temperature during the synthesis of this material. Furthermore, we develop an improved synthetic protocol for the synthesis of UiO-68–CHO, which reduces its defect density to a level that allows for nearly quantitative and highly repeatable PSM, without using a large excess of reagents. Finally, we show that the “defect-free” UiO-68–CHO is an excellent platform for PSM, by reacting it with various functional amines. In this way, new coordination sites, chirality, hydrophobicity, and even drug molecules were successfully introduced into the UiO-68 framework.
Results and discussion
Optimization of the synthesis of UiO-68–CHO
Thus, we reacted zirconium chloride with 1 equiv. of H2L in N,N-dimethylformamide, in the presence of water (3 equiv.) and benzoic acid (3 equiv.) as a modulator, at temperatures ranging from 100 °C to 75 °C. After 70 h, the resulting MOFs were washed with DMF and DCM, dried under vacuum, and digested in a mixture of DMSO-d6 and HF(aq) for 1H NMR studies.
At 100 °C, an almost quantitative conversion of the aldehyde groups was observed, accompanied by clean formation of a single product [Fig. 1]. Based on the 1H NMR, 13C NMR and MS spectra, we identified the product as the tertiary amine formed by the reductive amination of aldehyde with dimethylamine coming from DMF decomposition (the Leuckart reaction, Fig. 1, see the ESI† for more details). This is in agreement with the results described by Gao and co-workers, who obtained analogous products from similar aldehyde linkers.53 Thus, the solvothermal synthesis at 100 °C yielded a new MOF, UiO-68–CH2NHMe2+Cl−, fully functionalised with aliphatic tertiary amines (in the form of hydrochloride). The material was fully characterised (see the ESI† for details) and shown to be crystalline and porous (BET surface area = 2215 m2 g−1). The lower than expected surface area might be due to the presence of HCl in the structure. Such aliphatic amine-substituted MOFs are appealing, among others, as heterogeneous catalysts, as shown by Gao and co-workers for UiO-66–CH2NMe2.53
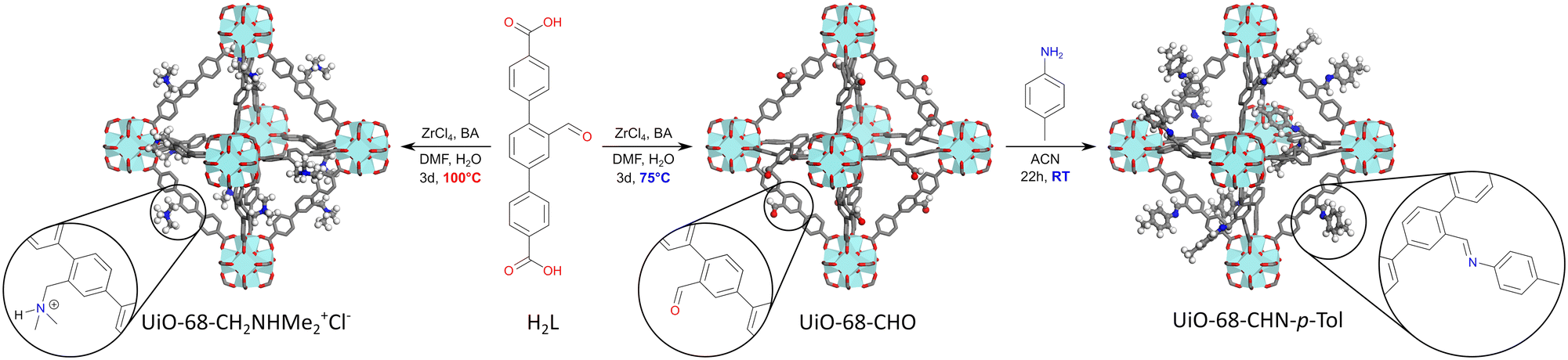 | ||
| Fig. 1 Solvothermal synthesis of UiO-68–CHO and UiO-68–CH2NHMe2+Cl−, and post-synthetic modification of UiO-68–CHO with 4-methylaniline. BA = benzoic acid, DMF = N,N-dimethylformamide, ACN = acetonitrile. | ||
When the solvothermal synthesis was performed at 90 °C, the aldehyde linker was still partially converted to the tertiary amine (in approx. 45%), but the degree of the conversion quickly decreased by lowering the reaction temperature and, at 75 °C, it was already imperceptible (Fig. 2). To our delight, the MOF obtained at this temperature was still crystalline and highly porous (BET = 3540 m2 g−1), although the presence of defects was evident from the N2 adsorption isotherm (Fig. S26;† type IV isotherm indicative of the presence of mesopores in the framework). Thus, for the preliminary PSM studies of UiO-68–CHO, we used the material synthesized at 75 °C.
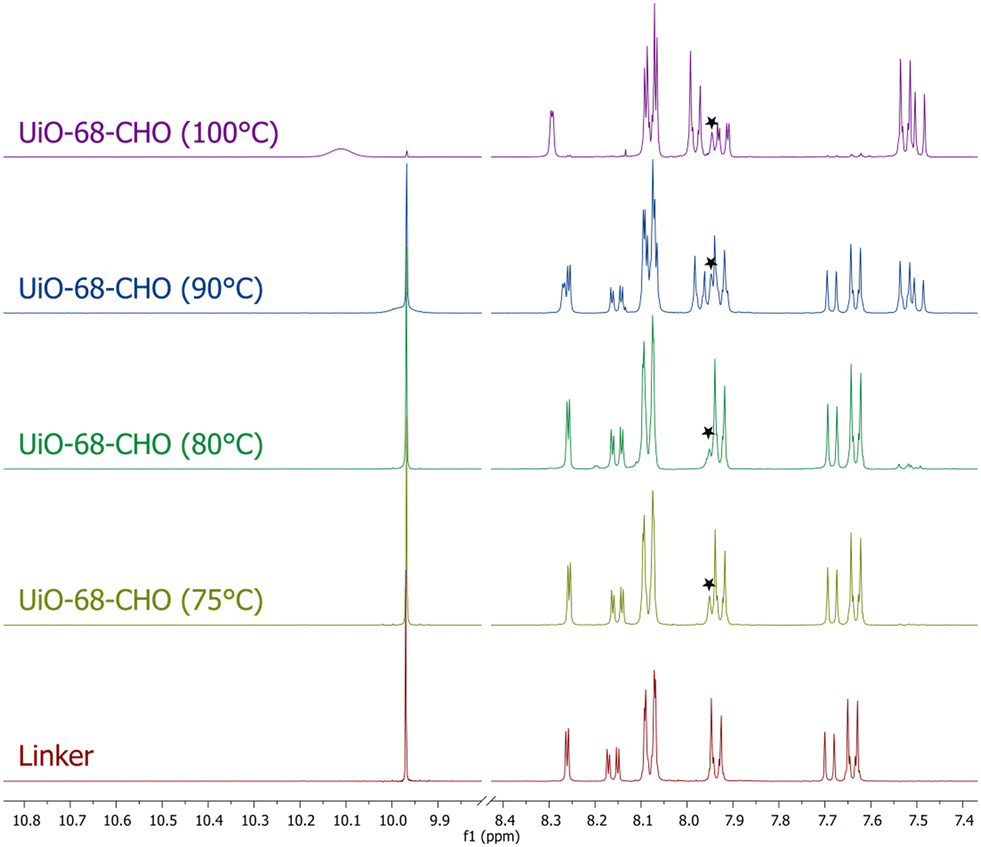 | ||
| Fig. 2 1H NMR spectra of linker H2L (bottom) and the digested samples of UiO-68–CHO synthesized at temperatures ranging from 75 °C to 100 °C. Signals from DMF are marked with stars. | ||
The molar mass of UiO-68–CHO was determined by quantitative 1H NMR, with respect to durene (1,2,4,5-tetramethylbenzene) as a standard. The obtained value – 533 g mol−1 – differs significantly from the theoretical value of 458 g mol−1, calculated for the defect-free material. Traces of DMF (2% w/w) in the MOF account for less than 12 g mol−1 difference, so the remaining difference must be due to excessive missing linkers.
As a model reaction for PSM, we chose imine formation with 4-methylaniline [Fig. 1]. Using freshly synthesized UiO-68–CHO and 2.2 equivalents of 4-methylaniline, a fair degree of PSM (69%) was achieved after 22 h in acetonitrile (ACN). Surprisingly, however, when the same procedure was repeated for the same batch of the starting material over the next few days, the yields dropped systematically [Table 1, entry 1]. Moreover, PXRD studies revealed that the crystallinity of UiO-68–CHO also decreased in time [Fig. S20†]. Such a rapid deterioration of the MOF properties significantly limits its practical utility. We hypothesized that the instability of the material might be due to the large number of defects in its structure and therefore resolved to further optimise its synthesis in order to reduce its defect density.
| Entry | Equiv. of linker for synthesis | Equiv. of linker for healing | Experimental molar massb [g mol−1] | Yield of PSMb | |||
|---|---|---|---|---|---|---|---|
| 1 d | 2 d | 3 d | 8 d | ||||
| a ZrCl4 was reacted with 1 or 1.5 equiv. of H2L (as specified in column 2) in DMF, in the presence of 3 equiv. of H2O and 3 equiv. of benzoic acid. T = 75 °C, reaction time = 70 h; see the ESI† for further details. b Every molar mass and every yield of PSM were averaged from 3 measurements; for further details see Tables S1 and S2 in the ESI.† | |||||||
| 1a | 1.0 | — | 533 | 69% | 58% | 60% | 50% |
| 2a | 1.5 | — | 513 | 93% | 95% | 94% | 94% |
| 3a | 1.5 | 1.0 | 493 | 100% | 100% | 100% | 100% |
In order to reduce the density of defects even further, we applied a ‘healing’ procedure, which consisted of soaking the MOF with a fresh portion of the linker in DMF. Thus, immediately after the solvothermal synthesis, UiO-68–CHO was shortly washed with DMF twice, mixed with a suspension of the linker in DMF, and heated at 75 °C for 22 h. After washing with copious amounts of DMF and DCM, we obtained a material with a molar mass of 493 g mol−1, i.e. even closer to the ideal value. BET analysis gave a specific surface area of 3629 m2 g−1, and SEM micrographs showed that the ‘healed’ UiO-68–CHO exists in the form of nicely shaped octahedral crystals, ca. 1–4 μm in size. Also, as previously, PXRD did not show any significant deterioration of its crystallinity over time [Fig. S22†]. The PSM of the ‘healed’ UiO-68–CHO gave quantitative conversion of aldehyde groups, meaning that virtually every linker in its structure is accessible to amine. Satisfyingly, the same quantitative conversion was repeatedly obtained over the next 8 days. IR spectra confirmed that the post-synthetically modified materials contained no aldehyde groups, and PXRD – that the MOF retained its crystallinity after the reaction [Fig. S53 and S62†].
To confirm our conclusions regarding defect density with an independent measurement, we also performed thermogravimetric analysis (TGA) of the synthesised MOFs in an oxygen-containing atmosphere. Missing linker defects lower the carbon-to-Zr ratio in MOFs, reducing the initial weight of a MOF sample with respect to the residual ZrO2 – the only solid product of complete material oxidation. The obtained TGA curves show that indeed the initial sample weight (expressed in % with respect to ZrO2) drops with the increasing density of missing linker defects, estimated by experimental molar mass determination [Fig. 3].
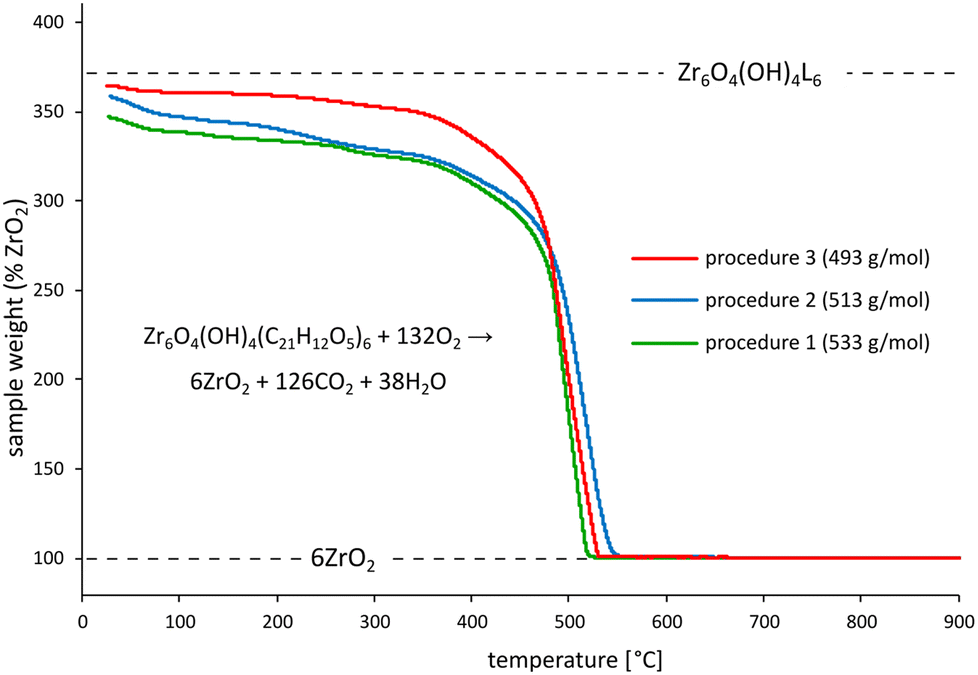 | ||
| Fig. 3 TGA curves (relative to ZrO2) obtained from UiO-68–CHO samples synthesized according to procedures 1, 2 and 3. The position of the theoretical TGA plateau for defect-free UiO-68–CHO is marked with the dashed line labelled ‘Zr6O4(OH)4L6’. | ||
| Amine | Equiv. used for PSM | Yield of PSM | Amine | Equiv. used for PSM | Yield of PSM |
|---|---|---|---|---|---|
| Conditions: UiO-68–CHO was stirred with an appropriate amine solution (0.05 M) at rt for 22 h, centrifuged, washed three times with DCM and dried under vacuum. See the ESI† for details. *Due to the poor solubility of 8 in ACN, THF was used as a solvent. | |||||
 1
1
|
1.0 | 92% |
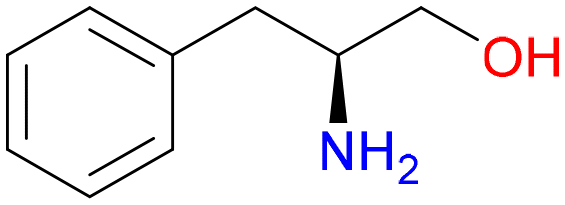 5
5
|
1.0 | 96% |
| 2.2 | 100% | ||||
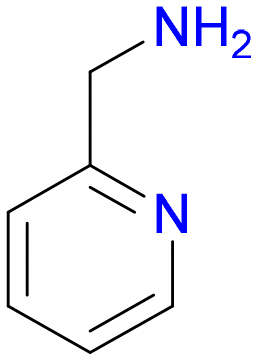 2
2
|
1.0 | 94% |
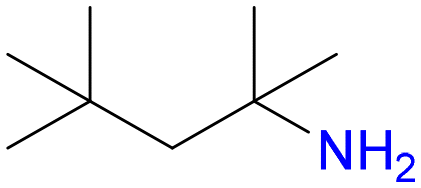 6
6
|
1.0 | 86% |
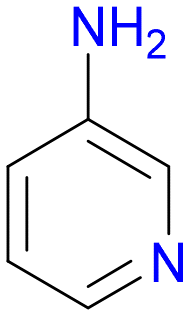 3
3
|
1.0 | 26% |
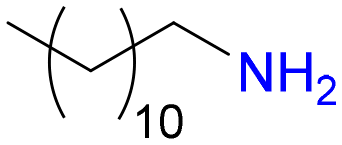 7
7
|
1.0 | 97% |
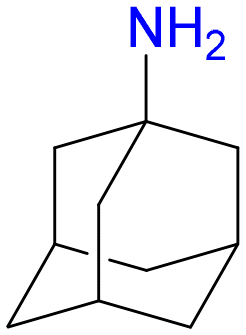 4
4
|
1.0 | 95% |
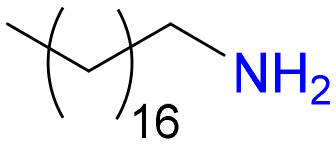 8
8
|
1.0* | 46% |
PXRD and nitrogen adsorption studies show that the ‘healed’ UiO-68–CHO retains its crystallinity and porosity after all the above-mentioned post-synthetic modifications. The IR spectra of the modified materials show greatly decreased aldehyde bands at around 1700 cm−1, suggesting that most of the aldehyde groups were consumed in these reactions [Fig. S52†]. Thus, while the ‘healing’ procedure developed in this work is certainly unable to fix the missing cluster defects and cannot even replenish all of the missing linkers, it strengthens the framework enough for very efficient and reproducible post-synthetic modification.
Conclusion
In conclusion, the results presented in this paper point to the strong and detrimental impact of missing linker defects on the yield and repeatability of covalent PSM of UiO-68–CHO. This phenomenon is likely to be of a more general nature, because the synthesis of ‘defect-free’ and chemically stable MOFs – i.e. the MOFs which are the most attractive for PSM – necessitates high temperature solvothermal conditions, which are not, in turn, compatible with the typically very reactive functional groups required for PSM. Nevertheless, this possibility seems to have been largely overlooked in the previous literature on PSM; in part perhaps because the repeatability of PSM has seldom been studied, and in part because the density of defects is difficult to measure. As a result, the often unsatisfactory yields of PSM have been traditionally ascribed to other factors, such as diffusion problems, overcrowding, or insufficient reactivity, and addressed by using a large excess of reagents and harsher conditions during PSM. In contrast, our results suggest that the low degree of PSM might be also due to instability of highly defective MOFs during PSM, and hence circumvented by strengthening the framework, for example by reducing the number of missing linker defects. Accordingly, we have developed a protocol for the synthesis of zirconium UiO-68–CHO with a reduced number of missing linker defects, and the resulting material has been shown to give nearly quantitative yields in PSM with model amines, even if no excess of reagents was used. The thus-obtained ‘healed’ UiO-68–CHO thus became one of the most appealing MOFs for PSM, owing to its very spacious pores, large pore windows, 3D pore structure, chemical stability and, especially, the ease and reliability of PSM. We believe that the findings described in this work will help in developing even more versatile platforms for covalent PSM in the future, by showing the way to combine the two highly desirable, but seemingly contradictory features: high reactivity and high stability.Author contributions
M. W. carried out the synthesis and characterization of MOFs and their ligands, performed the PSM of MOFs, collected and analysed the data, prepared graphical materials and contributed to manuscript writing and editing. M. J. C. was responsible for conceptualization, supervision, funding acquisition, project administration, and manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland through the OPUS grant 2017/27/B/ST5/00941 to M. J. C. This study was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within a project co-financed by the European Union through the European Regional Development Fund under the Operational Programme Innovative Economy 2007–2013. The authors thank Dr. Artur Chołuj for his expertise and assistance during this project.Notes and references
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, Wiley, 2019 Search PubMed.
- S. Kaskel, The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, Wiley, 2016 Search PubMed.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. M. Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- D. N. Dybtsev and K. P. Bryliakov, Coord. Chem. Rev., 2021, 437, 213845 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ChemCatChem, 2020, 12, 4732–4753 CrossRef CAS.
- Z. Zhai, X. Zhang, X. Hao, B. Niu and C. Li, Adv. Mater. Technol., 2021, 6, 2100127 CrossRef CAS.
- E. Moumen, L. Bazzi and S. El Hankari, Coord. Chem. Rev., 2022, 455, 214376 CrossRef CAS.
- L. Sun, L. Yang, J.-H. Dou, J. Li, G. Skorupskii, M. Mardini, K. O. Tan, T. Chen, C. Sun, J. J. Oppenheim, R. G. Griffin, M. Dincă and T. Rajh, J. Am. Chem. Soc., 2022, 144, 19008–19016 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. Usman, S. Mendiratta and K.-L. Lu, Adv. Mater., 2017, 29, 1605071 CrossRef PubMed.
- Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC.
- C. Cong and H. Ma, Adv. Opt. Mater., 2021, 9, 2100733 CrossRef CAS.
- X. Yang, X. Lin, Y. S. Zhao and D. Yan, Chem. – Eur. J., 2018, 24, 6484–6493 CrossRef CAS PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS PubMed.
- J. Yang and Y. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- J. W. M. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2021, 16, 2000005 CrossRef CAS PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130–8160 CrossRef CAS PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
- S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- D.-W. Lim and H. Kitagawa, Chem. Rev., 2020, 120, 8416–8467 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Adv. Funct. Mater., 2021, 31, 2006291 CrossRef CAS.
- S. Jeoung, S. Kim, M. Kim and H. R. Moon, Coord. Chem. Rev., 2020, 420, 213377 CrossRef CAS.
- L. Garzón-Tovar, S. Rodríguez-Hermida, I. Imaz and D. Maspoch, J. Am. Chem. Soc., 2017, 139, 897–903 CrossRef PubMed.
- T. Maity, P. Ghosh, S. Das, D. Saha and S. Koner, New J. Chem., 2021, 45, 5568–5575 RSC.
- S. Daliran, A. Santiago-Portillo, S. Navalón, A. R. Oveisi, M. Álvaro, R. Ghorbani-Vaghei, D. Azarifar and H. García, J. Colloid Interface Sci., 2018, 532, 700–710 CrossRef CAS PubMed.
- Y.-P. Wei, Y. Liu, F. Guo, X.-Y. Dao and W.-Y. Sun, Dalton Trans., 2019, 48, 8221–8226 RSC.
- Y.-P. Wei, S. Yang, P. Wang, J.-H. Guo, J. Huang and W.-Y. Sun, Dalton Trans., 2021, 50, 384–390 RSC.
- Q. Yang and H. Jiang, Small Methods, 2018, 2, 1800216 CrossRef.
- C. Yildiz, K. Kutonova, S. Oßwald, A. Titze-Alonso, J. Bitzer, S. Bräse and W. Kleist, ChemCatChem, 2020, 12, 1134–1142 CrossRef CAS.
- A. Xu, Z. Chen, L. Jin, B. Chu, J. Lu, X. He, Y. Yao, B. Li, L. Dong and M. Fan, Appl. Catal., A, 2021, 624, 118307 CrossRef CAS.
- F. Al-Ghazzawi, L. Conte, K. K. Wagner, C. Richardson and P. Wagner, Chem. Commun., 2021, 57, 4706–4709 RSC.
- H. Alinezhad, S. Ghasemi and M. Cheraghian, J. Organomet. Chem., 2019, 898, 120867 CrossRef.
- M. Baias, A. Lesage, S. Aguado, J. Canivet, V. Moizan-Basle, N. Audebrand, D. Farrusseng and L. Emsley, Angew. Chem., Int. Ed., 2015, 54, 5971–5976 CrossRef CAS PubMed.
- F. Cacho-Bailo, M. Etxeberría-Benavides, O. Karvan, C. Téllez and J. Coronas, CrystEngComm, 2017, 19, 1545–1554 RSC.
- S. Aguado, J. Canivet and D. Farrusseng, J. Mater. Chem., 2011, 21, 7582 RSC.
- F.-M. Zhang, H. Dong, X. Zhang, X.-J. Sun, M. Liu, D.-D. Yang, X. Liu and J.-Z. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 27332–27337 CrossRef CAS PubMed.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482–8486 CrossRef CAS PubMed.
- D. E. Williams, E. A. Dolgopolova, P. J. Pellechia, A. Palukoshka, T. J. Wilson, R. Tan, J. M. Maier, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2015, 137, 2223–2226 CrossRef CAS PubMed.
- C. Liu, T.-Y. Luo, E. S. Feura, C. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2015, 137, 10508–10511 CrossRef CAS PubMed.
- F.-G. Xi, H. Liu, N.-N. Yang and E.-Q. Gao, Inorg. Chem., 2016, 55, 4701–4703 CrossRef CAS PubMed.
- N. Antil, N. Akhtar, R. Newar, W. Begum, A. Kumar, M. Chauhan and K. Manna, ACS Catal., 2021, 11, 10450–10459 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- M. A. Hussein, A. H. Dinh, V. T. Huynh and T. V. Nguyen, Chem. Commun., 2020, 56, 8691–8694 RSC.
- F.-G. Xi, W. Sun, Z. Dong, N.-N. Yang, T. Gong and E.-Q. Gao, Chem. Commun., 2020, 56, 13177–13180 RSC.
- R. Pallach, J. Keupp, K. Terlinden, L. Frentzel-Beyme, M. Kloß, A. Machalica, J. Kotschy, S. K. Vasa, P. A. Chater, C. Sternemann, M. T. Wharmby, R. Linser, R. Schmid and S. Henke, Nat. Commun., 2021, 12, 4097 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00071k |
| This journal is © The Royal Society of Chemistry 2023 |