
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP1 Glutamine isosteres in the design of inhibitors of 3C/3CL protease of human viruses of the Pisoniviricetes class
Louise A.
Stubbing
ab,
Jonathan G.
Hubert
a,
Joseph
Bell-Tyrer
a,
Yann O.
Hermant
 ab,
Sung Hyun
Yang
a,
Alice M.
McSweeney
bc,
Geena M.
McKenzie-Goldsmith
bc,
Vernon K.
Ward
bc,
Daniel P.
Furkert
ab and
Margaret A.
Brimble
*ab
ab,
Sung Hyun
Yang
a,
Alice M.
McSweeney
bc,
Geena M.
McKenzie-Goldsmith
bc,
Vernon K.
Ward
bc,
Daniel P.
Furkert
ab and
Margaret A.
Brimble
*ab
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street and 3b Symonds Street, Auckland 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3b Symonds Street, Auckland 1142, New Zealand
cDepartment of Microbiology and Immunology, School of Biomedical Sciences, University of Otago, PO Box 56, 720 Cumberland Street, Dunedin 9054, New Zealand
First published on 21st June 2023
Abstract
Viral infections are one of the leading causes of acute morbidity in humans and much endeavour has been made by the synthetic community for the development of drugs to treat associated diseases. Peptide-based enzyme inhibitors, usually short sequences of three or four residues, are one of the classes of compounds currently under development for enhancement of their activity and pharmaceutical properties. This review reports the advances made in the design of inhibitors targeting the family of highly conserved viral proteases 3C/3CLpro, which play a key role in viral replication and present minimal homology with mammalian proteases. Particular focus is put on the reported development of P1 glutamine isosteres to generate potent inhibitors mimicking the natural substrate sequence at the site of recognition.’
Introduction
The Pisoniviricetes class encompasses a diverse range of positive-sense single-stranded RNA viruses infecting eukaryotic organisms. Importantly to humans, this class includes the Picornaviridae, Coronaviridae and Caliciviridae families which represent some of the leading causes of acute morbidity in humans,1 and are among the most prevalent infectious agents. Picornaviruses are a large family of viruses, infecting both humans and animals, and their pathology ranges from mild infections such as the common cold and hepatitis A to more severe disease including meningitis and paralysis. In some instances viral infections by picornaviruses have been linked to autoimmune diseases such as myocarditis, diabetes, and multiple sclerosis.2–6 Coronavirus infections also span a range of severity,2,7 while caliciviruses are the leading cause of acute gastroenteritis but can have more severe symptoms in immunocompromised individuals.8 One of the unifying features of the Pisoniviricetes class is a highly structurally conserved cysteine protease belonging to the PA clan (proteases of mixed nucleophile, superfamily A), commonly referred to as 3Cpro (picornaviruses) or 3CLpro (coronaviruses and caliciviruses, also known as Mpro). 3Cpro and 3CLpro enzymes play a critical role in viral replication, initially catalysing their autocleavage from the nascent polypeptide chain, before performing secondary cleavages leading to maturation of the viral replication machinery.9–113Cpro and 3CLpro are chymotrypsin-like cysteine proteases, which have characteristic folding of two equivalent β-barrels to form the binding pocket and a catalytic triad or dyad.12–14 Substrate recognition is typically dictated by a hexapeptide sequence (P4–P3–P2–P1–P1′–P2′), each amino acid interacting with its corresponding binding site (Sx) of the protease enzyme (Fig. 1A). The scissile bond is between the P1–P1′ residues which are typically glutamine (Gln)–glycine (Gly), or Gln–other small amino acid. Though cleavage occurs less commonly, glutamic acid (Glu)–glycine (Gly) is another peptide bond of interest.15,16 Crystal structures of 3Cpro and 3CLpro proteases along with substrate specificity studies have demonstrated subtle variation in the binding pockets and orientation of the catalytic triad/dyad, however all are remarkably conserved to recognise glutamine at P1 position. Specificity for P1 glutamine is attributed to conserved histidine (His) residue located at the end of the S1 binding pocket which form important hydrogen bonds with the glutamine side chain of the natural substrate. Due to the critical roles that 3C and 3CLpro play in viral replication and minimal homology with mammalian proteases, 3Cpro and 3CLpro have emerged as promising targets for drug development. A commonly employed approach in developing 3C/3CLpro inhibitors is to mimic the natural substrate sequence, replacing the scissile bond with an electrophilic warhead to capture the catalytic cysteine residue, either via a reversible or irreversible mechanism. Peptidyl inhibitors targeting 3C/3CLpro typically consist of a two to four peptide sequence (P4–P1), with glutamine occupying the P1 position and an electrophilic “warhead” at P1′ while the N-terminal is protected (N-cap) (Fig. 1B).
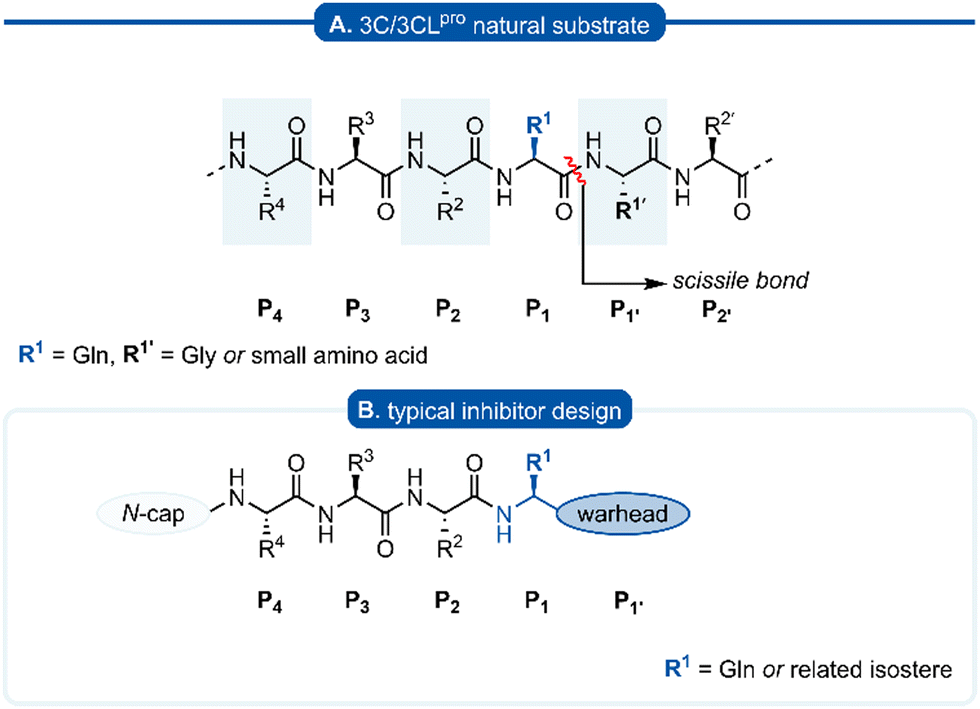 | ||
| Fig. 1 (A) Hexapeptide recognition sequence of 3C/3CLpro, where the scissile bond is located between P1 (Gln) and P1′. (B) Typical inhibitor design targeting 3C/3CLpro. | ||
Following this blueprint, a number of low micromolar inhibitors targeting 3C/3CLpro have been developed (representative examples, Fig. 2),17–26 equipped with various electrophilic warheads demonstrating their effectiveness at trapping the catalytic cysteine residue. A drawback of employing a P1 glutamine residue, however, is the high propensity for the amide side chain (Scheme 1A) to cyclise onto the electrophilic warhead, forming the hemiaminal tautomer (Scheme 1B). This reactivity has been observed with aldehyde,17,27 α-haloketone,23,28 and acyloxymethyl ketone25 warheads containing P1 glutamine or N-monoalkylated glutamine isosteres (see Section 1). Although the effects of tautomerisation to the hemiaminal are not clearly delineated, this is generally considered to be detrimental to antiviral activity while also causing complications during the chemical synthesis and purification. To avoid complications caused by the P1 glutamine residue in the design of 3C/3CLpro inhibitors, incorporation of a glutamine isostere which binds specifically in the S1 pocket while also preventing cyclisation onto the electrophilic warhead is desirable. This review reports the advances made in the design of inhibitors targeting viral 3C/3CLpro of human viruses in the Pisoniviricetes class and focusses on the reported development of P1 glutamine isosteres.
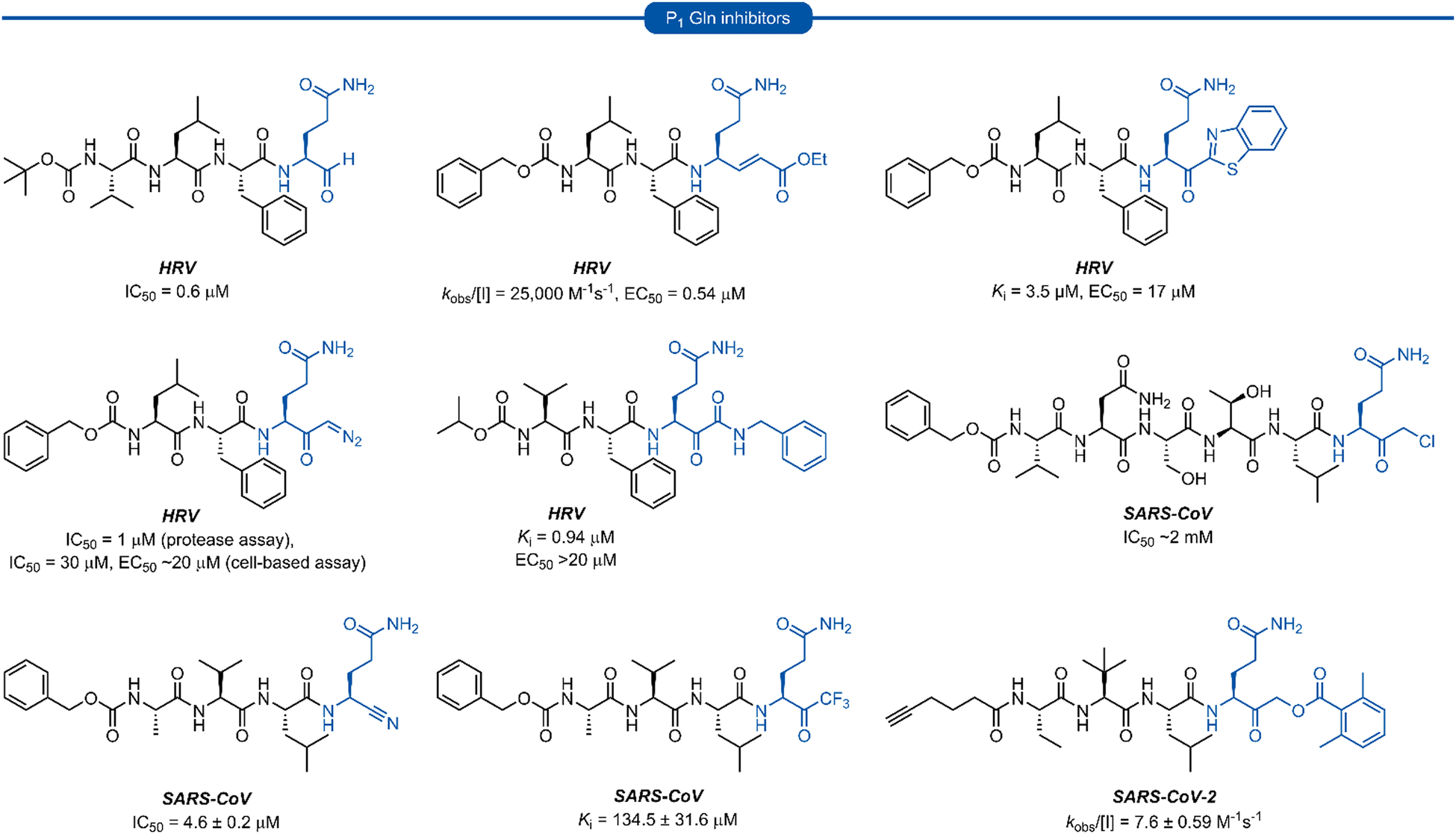 | ||
| Fig. 2 Representative 3C/3CLpro inhibitors possessing a P1 (L)-glutamine residue with various electrophilic warheads. | ||
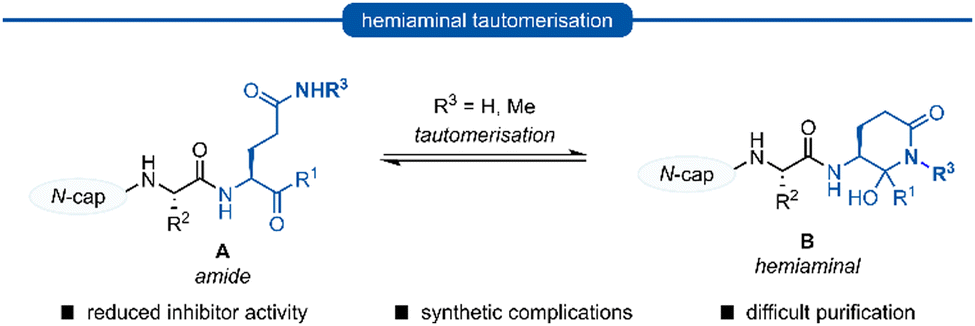 | ||
| Scheme 1 Generalised formation of hemiaminal B via tautomerisation of P1 glutamine inhibitors A onto the electrophilic warhead. | ||
Section 1. P1N-alkyl glutamine inhibitors
Early work in the development of P1 glutamine isosteres focused on alkylation of the glutamine side chain amide to retard its nucleophilicity and prevent cyclisation onto the electrophilic warhead. Selected examples of inhibitors possessing a mono- or di-substituted amide side-chain at P1 position and equipped with various warhead units are presented in Fig. 3. Malcolm et al. explored tetrapeptide inhibitors with N,N-dimethyl-Gln at the P1 position.29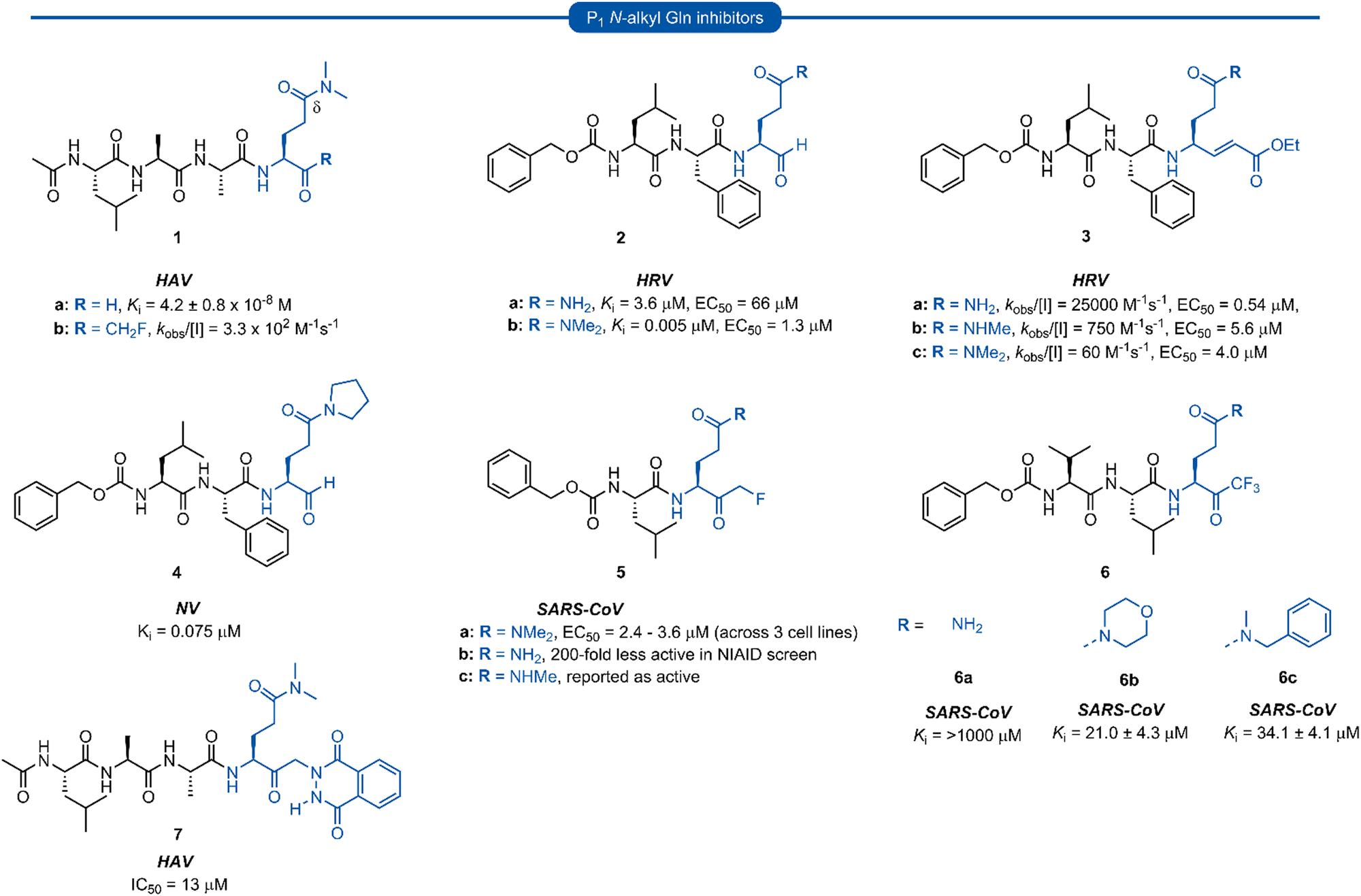 | ||
| Fig. 3 Representative examples of P1N-alkyl-Gln inhibitors targeting 3C/3CLpro. | ||
This was proposed to prevent cyclisation onto the aldehyde warhead while also maintaining hydrogen bonding between the glutamine δ-carbonyl oxygen and His160 of hepatitis A virus (HAV) 3Cpro.12,29 Inhibitor 1a was based on the preferred substrate sequence and was found to be a reversible, slow binding inhibitor of HAV 3Cpro, while also being 50-fold less active against human rhinovirus (HRV) 3Cpro. Investigation of a series of P1 glutaminal inhibitors targeting HRV found N,N-dimethyl-Gln containing inhibitor 2b was ∼50-fold more active compared with the corresponding P1 glutamine analogue 2a.27 The low potency of 2a was postulated to be due to the propensity for cyclisation to the less active hemiaminal tautomer (see Scheme 1). A similar study was conducted investigating Michael acceptor tetrapeptides for anti-HRV activity. In this study however, unmodified glutamine inhibitor 3a was found to have superior activity (EC50 = 0.54 μM) compared with both N-methyl-Gln 3b and N,N-dimethyl-Gln 3c containing analogues (EC50 = 5.6 μM and 4.0 μM, respectively).19 The discrepancies in these studies likely arise from the propensity of the unprotected glutamine side chain to cyclise onto the aldehyde warhead, while Michael acceptors have not been observed to undergo cyclisation with the P1 glutamine residue.18 A related series of N-alkylated glutaminal inhibitors, cyclic tertiary amide exemplified by 4 were also shown to be effective in targeting norovirus (NV) 3CLpro.30
A glutamine N-alkylation strategy has also been employed to generate inhibitors equipped with an α-fluoromethyl ketone (FMK) warhead and targeting HAV31 and severe acute respiratory syndrome coronavirus (SARS-CoV)23,28,32 Compound 1b was found to inhibit HAV 3Cpro and displayed a second order rate constant of kobs/[I] = 3.3 × 102 M−1 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 while N,N-dimethyl-Gln analogue 5a was shown to be a low micromolar inhibitor of SARS-CoV replication.28 The analogous inhibitor 5b containing an unmodified Gln-FMK unit was found to be predominantly cyclised in solution (as observed by 1H NMR) and 200-fold less active against SARS-CoV in a National Institute of Allergy and Infectious Diseases (NIAID) screen. An inhibitor containing an N-methyl-Gln-FMK unit 5c was on the other hand reported as active according to a personal communication in a footnote; however partial cyclisation in solution was observed by 1H NMR and a series containing this N-methyl-Gln-FMK unit does not appear to have been pursued further. A series of glutamine-trifluoromethyl ketone inhibitors, represented by inhibitor 6a (Ki >1000 μM), were all found to be at least partially cyclised in solution (as determined by 19F NMR in CDCl3), while some existed solely in their cyclic form. These exhibited only moderate anti-SARS-CoV activity23 while later work from the same laboratory showed N,N-dialkyl analogues 6b and 6c to have improved, albeit still low activity (Ki = 21.0 μM, 34.1 μM, respectively).32
31 while N,N-dimethyl-Gln analogue 5a was shown to be a low micromolar inhibitor of SARS-CoV replication.28 The analogous inhibitor 5b containing an unmodified Gln-FMK unit was found to be predominantly cyclised in solution (as observed by 1H NMR) and 200-fold less active against SARS-CoV in a National Institute of Allergy and Infectious Diseases (NIAID) screen. An inhibitor containing an N-methyl-Gln-FMK unit 5c was on the other hand reported as active according to a personal communication in a footnote; however partial cyclisation in solution was observed by 1H NMR and a series containing this N-methyl-Gln-FMK unit does not appear to have been pursued further. A series of glutamine-trifluoromethyl ketone inhibitors, represented by inhibitor 6a (Ki >1000 μM), were all found to be at least partially cyclised in solution (as determined by 19F NMR in CDCl3), while some existed solely in their cyclic form. These exhibited only moderate anti-SARS-CoV activity23 while later work from the same laboratory showed N,N-dialkyl analogues 6b and 6c to have improved, albeit still low activity (Ki = 21.0 μM, 34.1 μM, respectively).32
α-Ketophthalahydrazide warheads with P1N,N-dimethyl-Gln isosteres, as illustrated by 7, were investigated for anti-HAV 3Cpro activity.13,33 Intermolecular hydrogen bonding of the ketone and proximal hydrazide NH was postulated to restrict conformational mobility, thereby decreasing the entropic penalty upon binding, while also mimicking the P2′ phenylalanine (Phe) of the natural substrate.33 Compound 7 was found to be a competitive reversible inhibitor against HAV (IC50 = 13 μM), and no loss of activity was observed in the presence of a 100-fold excess of dithiothreitol (DTT), indicating its stability to extracellular thiols.
Section 2. P1 methionine derived inhibitors
Methionine (Met) and its oxidised sulfoxide and sulfone derivatives have also been investigated as non-nucleophilic glutamine P1 isosteres due to their ease of synthesis and H-bonding ability (Fig. 4). Methionine sulfone analogue 8a was found to be a good reversible inhibitor of HRV 3Cpro (Ki = 0.47 μM) and also displayed good antiviral activity in tissue culture assays (IC50 = 3.4 μM) with no observable cytotoxicity.34 Additionally, inhibitor 8a was found to be more potent than inhibitors 8b and 8c containing a P1 methionine or glutamine residue, respectively (IC50 = 9.7 μM and IC50 = 42 μM).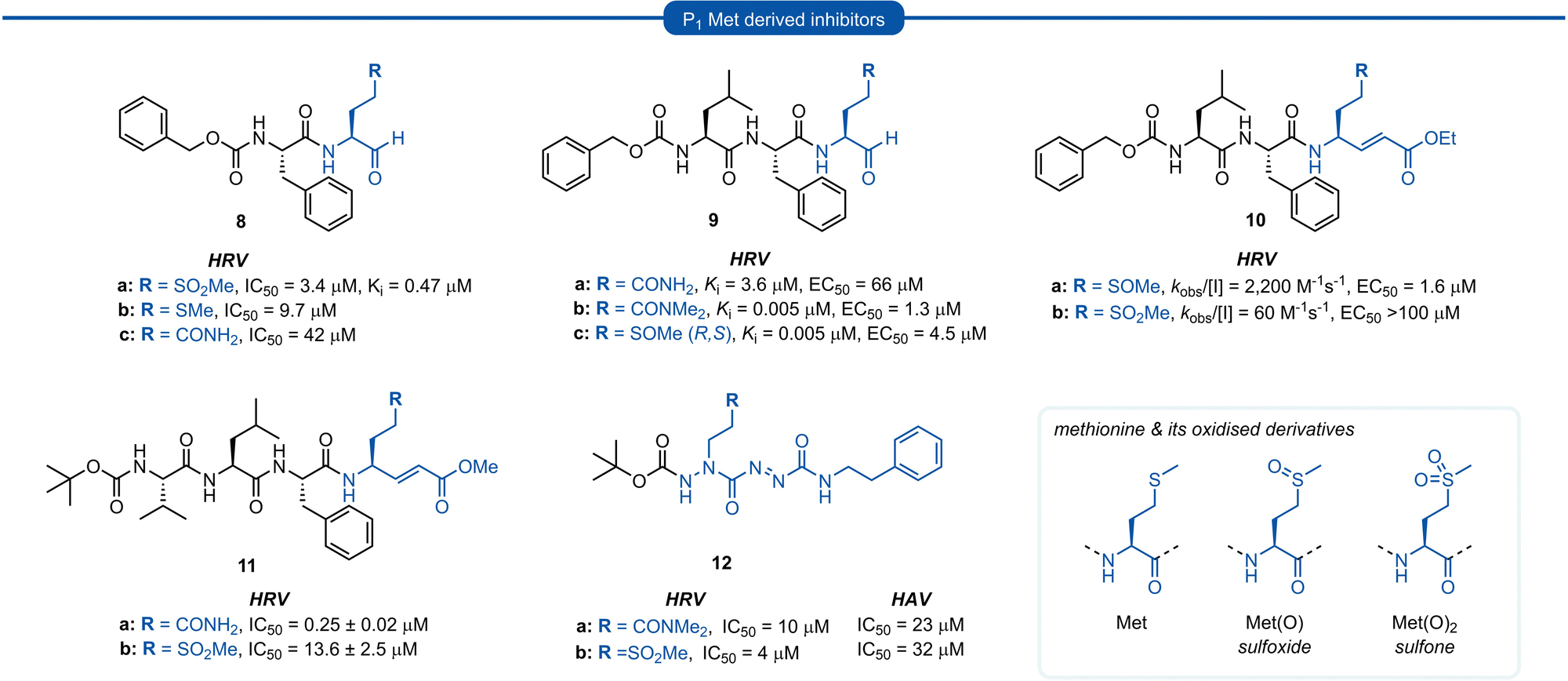 | ||
| Fig. 4 Representative examples of P1 Met derived inhibitors and their P1 Gln/N-alkyl-Gln analogues targeting 3C/3CLpro. | ||
Similar observations were made in a separate study investigating aldehyde HRV 3Cpro inhibitors. Sulfoxide 9c displayed superior potency compared to analogue 9a containing an unmodified Gln residue (IC50 = 0.005 μM vs. 3.6 μM), and was approximately equipotent to the N,N′-dimethyl-Gln analogue 9b. The potency of sulfoxide 9c was further supported by X-ray crystal structure data of the enzyme-inhibitor complex, which revealed the P1 sulfoxide moiety fits snugly into the S1 pocket and the H-bonding pattern of the active site His161 and Thr142 residues was analogous to that of the natural substrate.27 The use of sulfoxide and sulfone P1 isosteres in conjunction with Michael acceptor warheads on the other hand has met with less success. Two separate studies both reported that peptidyl inhibitors of HRV 3Cpro containing a P1 sulfoxide 10a or sulfone 10b and 11b residue showed a significant reduction in potency compared with their P1 glutamine counterparts 3a and 11a, respectively.19 Monopeptidyl aza-carboxamides, exemplified by 12a/b (For aza-backbone modifications, see Section 5) have also been investigated for anti HRV and HAV activity. P1 sulfone 12b was found to have improved activity against HRV 3Cpro compared to N,N′-dimethyl-Gln analogue 12a. Conversely, 12a was found to exhibit slightly higher potency against HAV 3Cpro over 12b.35,36
Section 3. P1 inhibitors derived from non-proteogenic amino acids
A number of P1 glutamine isosteres derived from unnatural amino acids were investigated during the early development of 3C/3CLpro inhibitors. Most notably, publications by Dragovich et al. and Webber et al. explored a diverse array of P1 derivatives including glutamine related P1 substrates (Section 1) and non-glutamine derived P1 residues such as sulfones/sulfoxides (Section 2), as well as a number of glutamine isosteres (representative examples, Fig. 5A). During these studies however, none of these analogues 13–15 or 16–18 showed increased potency over inhibitors containing the unmodified side chain 3a19 or the N,N′-dimethyl-Gln 9b27 derivatives, respectively. Alternatively, peptidyl inhibitors bearing a hydrophobic amino acid in the P1 position have also been investigated (Fig. 5B). The substitution of P1 glutamine with non-nucleophilic residues circumvented synthetic complications caused by the reactivity of the electrophilic warhead. Representative examples 19 and 20 possess a norvaline (Nva) or phenylalanine hydrophobic amino acid at the P1 position, respectively. Norvaline P1 tetrapeptide inhibitor 19 was found to have low micromolar activity against HRV 3Cpro (Ki = 0.17) and also having good activity in tissue culture (EC50 = 0.85 μM),24 while P1-phenylalanine inhibitor 20 was found to exhibit moderate time-dependent activity against SARS-CoV 3CLpro (IC50 = 10 μM).37 A recent development in the inhibitor design has been made using self-masked aldehyde inhibitors (SMAIs) methodology for targeting 3CL protease.38 Originally developed by Meek and co-workers to target cruzain, SMAIs was then applied to the development of SARS-CoV-2 inhibitor 21.39 SMAIs methodology is designed to mask the reactive aldehyde warhead through the spontaneous formation of a δ-lactol via nucleophilic attack by the P1 2-pyridone side-chain on the aldehyde warhead, which would closely mimic the γ-lactam P1 moiety that is now commonly employed in the design of 3C/3CL protease inhibitors (see Section 7). Upon binding to the protease active site, acid catalysed ring-opening by an acidic residue, revealing the reactive aldehyde warhead, which captures the cysteine residue to afford the enzyme-bound moiety (Scheme 2).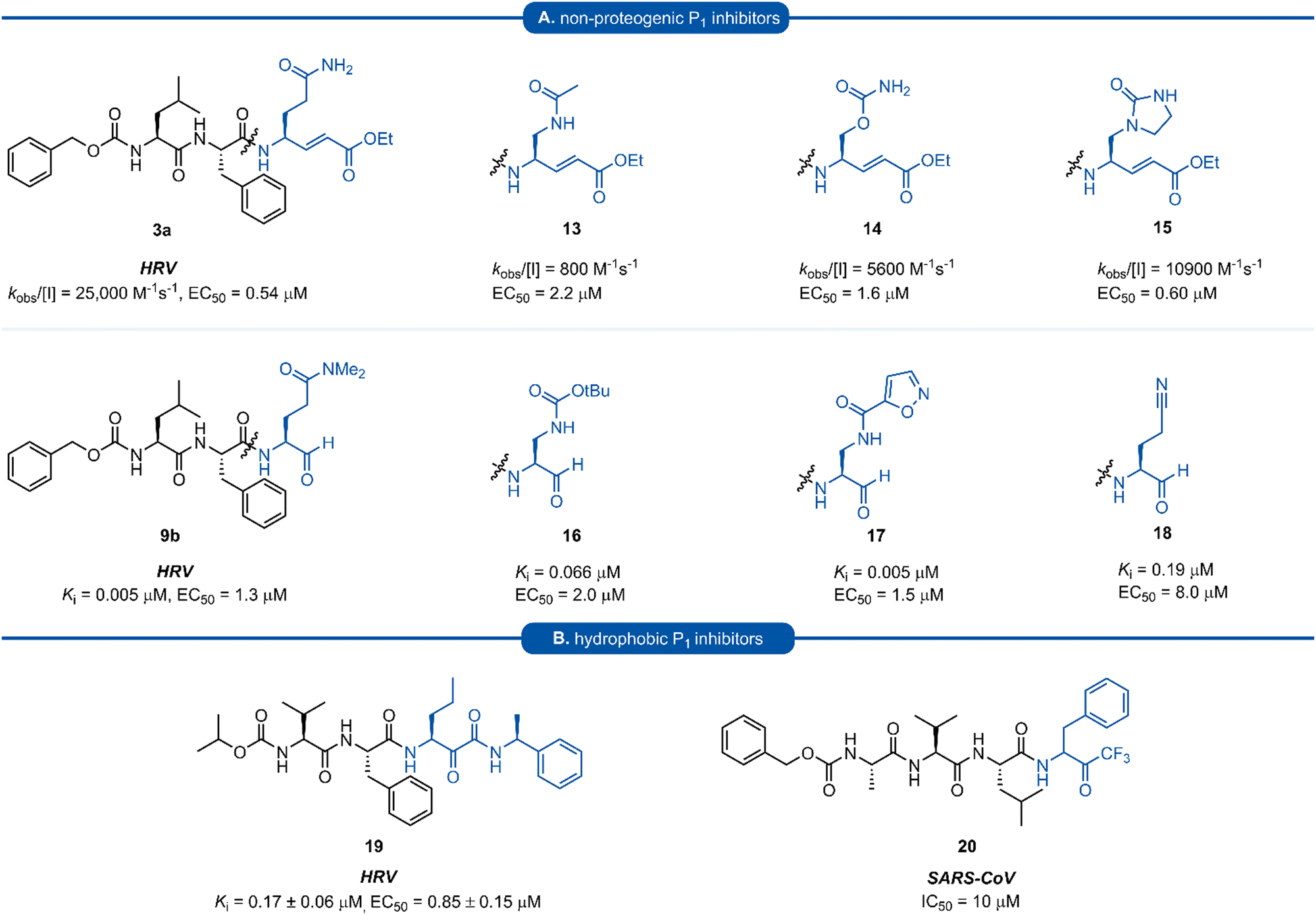 | ||
| Fig. 5 (A) Inhibitors targeting 3C/3CLpro containing non-proteogenic P1 isosteres. (B) Hydrophobic P1 residue inhibitors targeting 3C/3CLpro. | ||
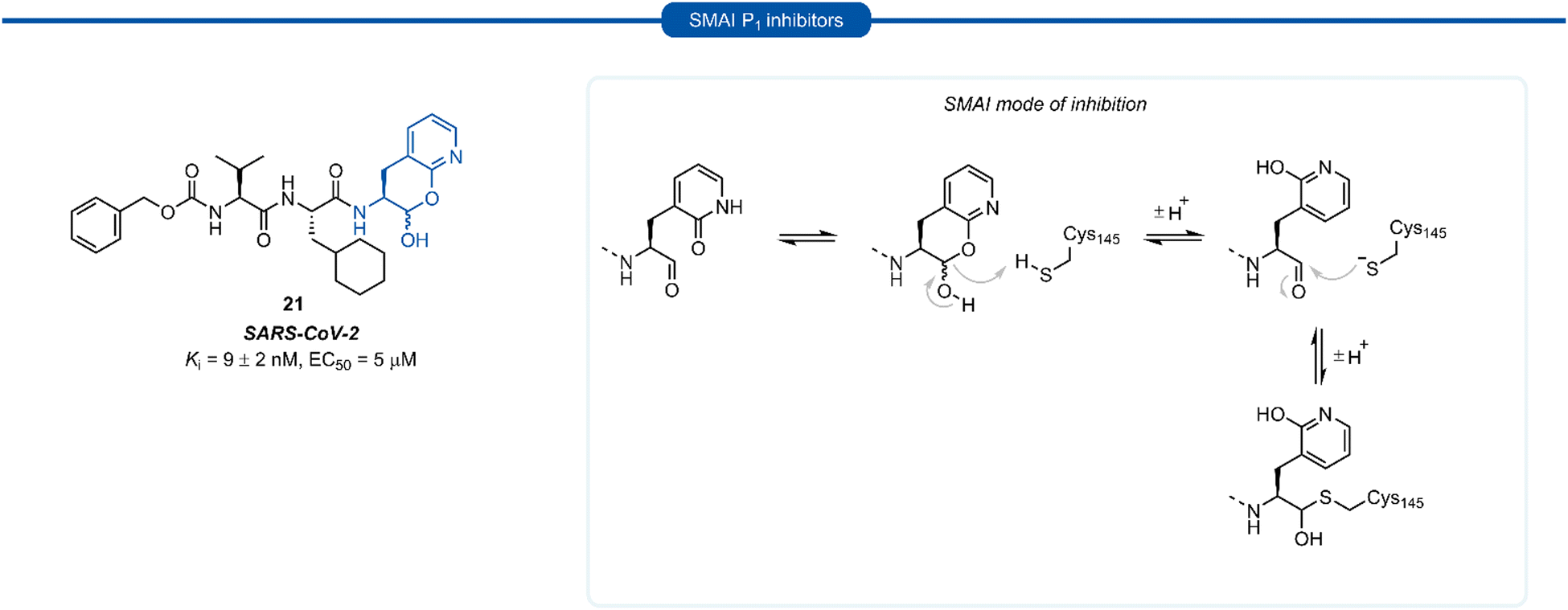 | ||
| Scheme 2 Putative mechanism of reversible covalent inhibition of a cysteine protease by SMAIs. | ||
Formation of the lactol moiety was confirmed by NMR in organic solvents, however, unambiguous confirmation of the major species in aqueous solutions has not been made.38 Compound 21 was found to be a competent inhibitor of SARS-CoV-2 with EC50 value of 5 μM. A high-resolution X-ray crystal structure of 21 bound in the active site of SARS-CoV-2 3CLpro confirmed the formation of the anticipated hemithioacetal between the aldehyde group and Cys145, thereby confirming the inhibitor's mechanism of action. The P1 2-pyridone residue also establishes the expected H-bonding pattern, with the 2-pyridone carbonyl group accepting H-bonding from His163 and Ser144 while the amide proton donates an H-bond to Glu166 and Phe140.38
Section 4. P1 histidine inhibitors
During work towards the development of SARS-CoV 3CLpro inhibitors, investigations into the substrate preference for SARS-CoV 3CLprovia positional scanning of synthetic recombinant libraries based on the 7-amino-4-carbamoyl-coumarin (ACC) fluorogenic leaving group were undertaken.A surprising preference for histidine at P1 was revealed which was further confirmed by synthesising single substrates and comparing their kinetic constraints (Fig. 6).40 Early investigations of histidine P1 analogues for anti SARS-CoV activity initially led to the discovery of the potent tetrapeptide inhibitor 22 (IC50 = 65 nM).41 Analysis of the binding pocket of the inhibitor 22-bound X-ray crystal structure showed that the cyclohexyl ring of the P2 cyclohexylalanine (Cha) was proximal to the α-nitrogen of the corresponding amino acid residue. A novel decahydroisoquinoline inhibitor scaffold was designed by introduction of a methyl linker between the P2 cyclohexyl ring and the P2 backbone nitrogen and a peptide chain extending off the 3-position of the ring system of compound 23. Although compound 23 represents a novel class of 3CLpro inhibitors, improved potency against SARS-CoV 3CLpro compared with its linear predecessor 22, was not achieved.42–44 Histidine P1 residues were also employed by Hayashi et al. against 3CLpro of SARS-CoV using α-keto-thiazole warheads 24, however, these did not show superior activity to analogous P1 lactam inhibitors (see Section 7) and were not further explored.45
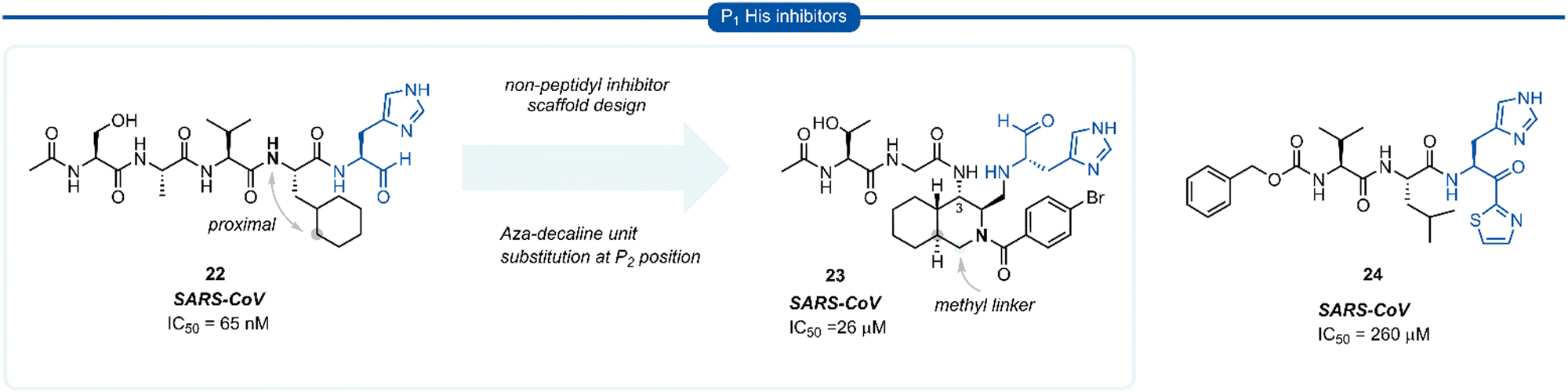 | ||
| Fig. 6 Representative examples of P1 His inhibitors targeting SARS-CoV 3CLpro. | ||
Section 5. P1 aza-glutamine inhibitors
Azapeptides are peptide chains where one of the amino acid residues are replaced by a semicarbazide which conformationally restricts the peptide, bending the amino acid away from the natural linear geometry. Incorporation of semicarbazides into peptidyl inhibitor manifolds has been shown to improve activity and selectivity while also showing improved pharmacokinetic properties such as metabolic stability and duration of action.46 The success of aza-backbone modifications at the P1 position of papain inhibitors initially led to their incorporation in the design of inhibitors for the closely related 3C/3CLpro (Fig. 7).47,48 Norbeck et al. developed P1 aza-glutamine bromomethylketone 25 for anti-HRV activity as a time-dependent irreversible inhibitor (kinact/Kinact = 23400 M−1 s−1).49,50 In a separate study, a series of aza-glutamine inhibitors such as 26 were found to have moderate activity against both HRV and HAV 3Cpro (IC50 = 12 μM and IC50 = 10 μM) respectively.35 Although a number of P1 aza-glutamine inhibitors have been investigated, generally only moderate potency has been achieved against HRV and HAV.48,51 P1 aza-glutamine inhibitors have also been developed against SARS-CoV. Notably, James et al. demonstrated through X-ray crystal structure elucidation that aza-glutamine epoxide 27 irreversibly binds via an induced-fit model.52,53 Müller and co-workers have also developed a potent inhibitor of 3CLpro SARS CoV-2 containing a N-methyl-azanitrile warhead unit 28 (Ki = 24 nM).54
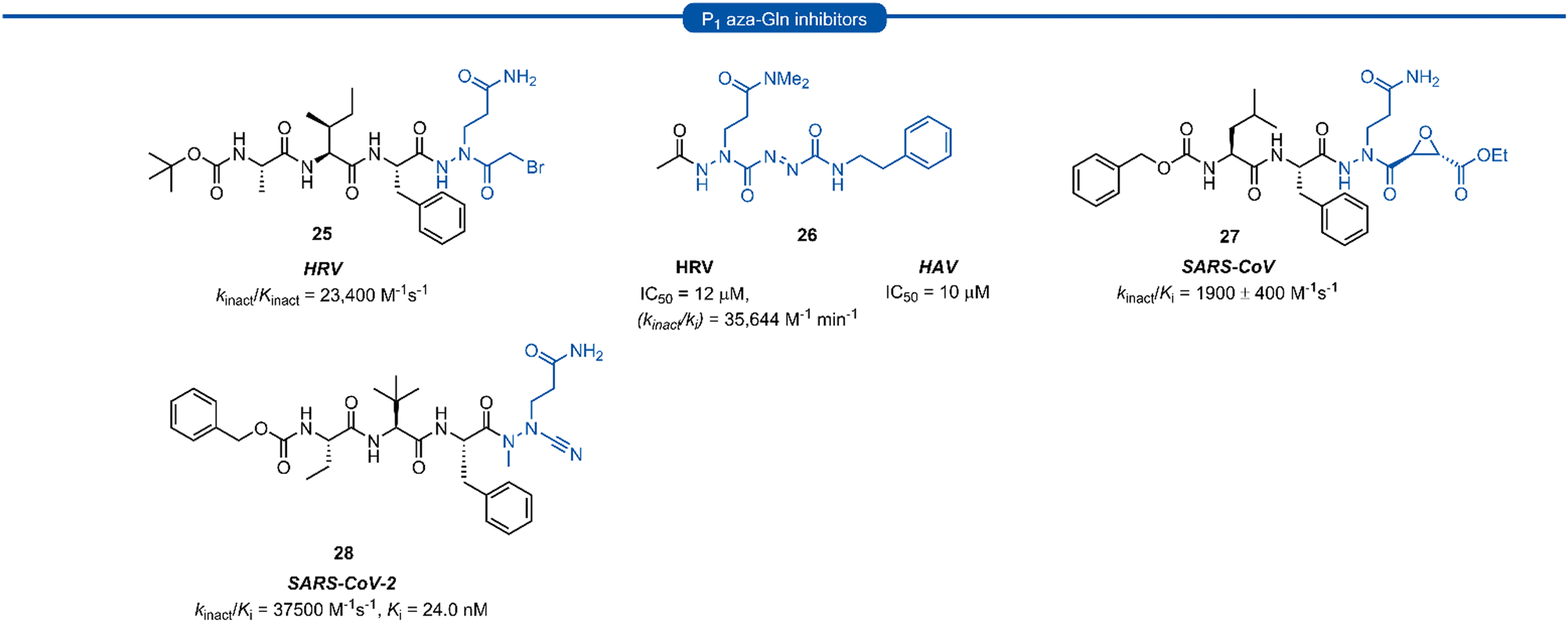 | ||
| Fig. 7 Representative examples of P1 aza-Gln inhibitors targeting 3C/3CLpro. | ||
Section 6. P1 Gln macrocyclic inhibitors
Macrocyclic inhibitors have been shown to display improved properties over their acyclic counterparts for the inhibition of protease enzymes. Protease enzymes recognise their ligands in a β-strand conformation, and macrocyclization can help facilitate the backbone hydrogen bonding pattern of peptide inhibitors to mimic that of an extended antiparallel β-sheet of the natural substrate.55–57 Macrocyclization may also increase the structural rigidity of peptide-based inhibitors, restricting the conformational interchange and thereby enhancing the affinity for the receptor by reducing the loss of entropy upon binding, as well as exhibiting improved receptor selectivity and favourable drug-like characteristics including increased cell permeability and proteolytic stability.55,58–60 The first investigations into macrocyclic 3Cpro and 3CLpro inhibitors arose from analysis of the crystal structure of inhibitors bound to HRV 3Cpro, revealing close spatial proximity between P1 and P3 side-chains. Modelling studies yielded tripeptidyl macrocyclic inhibitor 29, where a triazole-containing linker adjoined the P1 and P3 residues (Fig. 8). The inhibitor activity was evaluated against norovirus, enterovirus and SARS-CoV, and in all cases 29 was found to inhibit the protease in the low micromolar range.61 Further investigation showed poor correlation between 3CLpro enzyme inhibition assays (IC50) and inhibition of norovirus replication in a cell-based replicon system assays (EC50) for 29 and derivatives thereof.62 This result indicates that compounds such as 29 likely suffer from poor cellular permeability due to the triazole ring increasing the polar surface area. An X-ray crystal structure of NV 3CLpro with bound inhibitor 29 revealed the expected antiparallel β-sheet backbone binding conformation, and the tetrahedral adduct between the aldehyde warhead and Cys139 residue was also present. It was noted however, that binding induced suboptimal structural changes of the protease binding pocket and more importantly, the S1 pocket, resulting in loss of the key Thr134 and His157 H-bonds to P1 Gln and thus maximal binding efficiency was not obtained.62,63 Further publications explored related macrocyclic inhibitors with alkyl linkers which generally gave increased cell permeability, with inhibitor 30 found to have good activity in both enzyme and cell-based assays. Co-crystallisation of 30 and NV 3Cpro showed the expected backbone H-bonding pattern indicating the expected bonding orientation. The key H-bonding interaction between the P1 amide of 30 and His157 and Thr134 of NV 3Cpro however was again absent; instead, a new H-bond between the oxygen of the tetrahedral thioacetal adduct and Pro136 was evident.64 Rupintrivir 33 (AG-7088), a potent anti-rhinoviral agent that became the subject of extensive preclinical and clinical investigation, was used as a scaffold for macrocyclic inhibitors, with the goal of improving rupintrivir's pharmacokinetic properties.65 The crystal structure of rupintrivir/HRV2 3Cpro showed only 4.6 Å between the P1 and P3 side chains, providing an opportunity for the introduction of a macrocyclic linkage.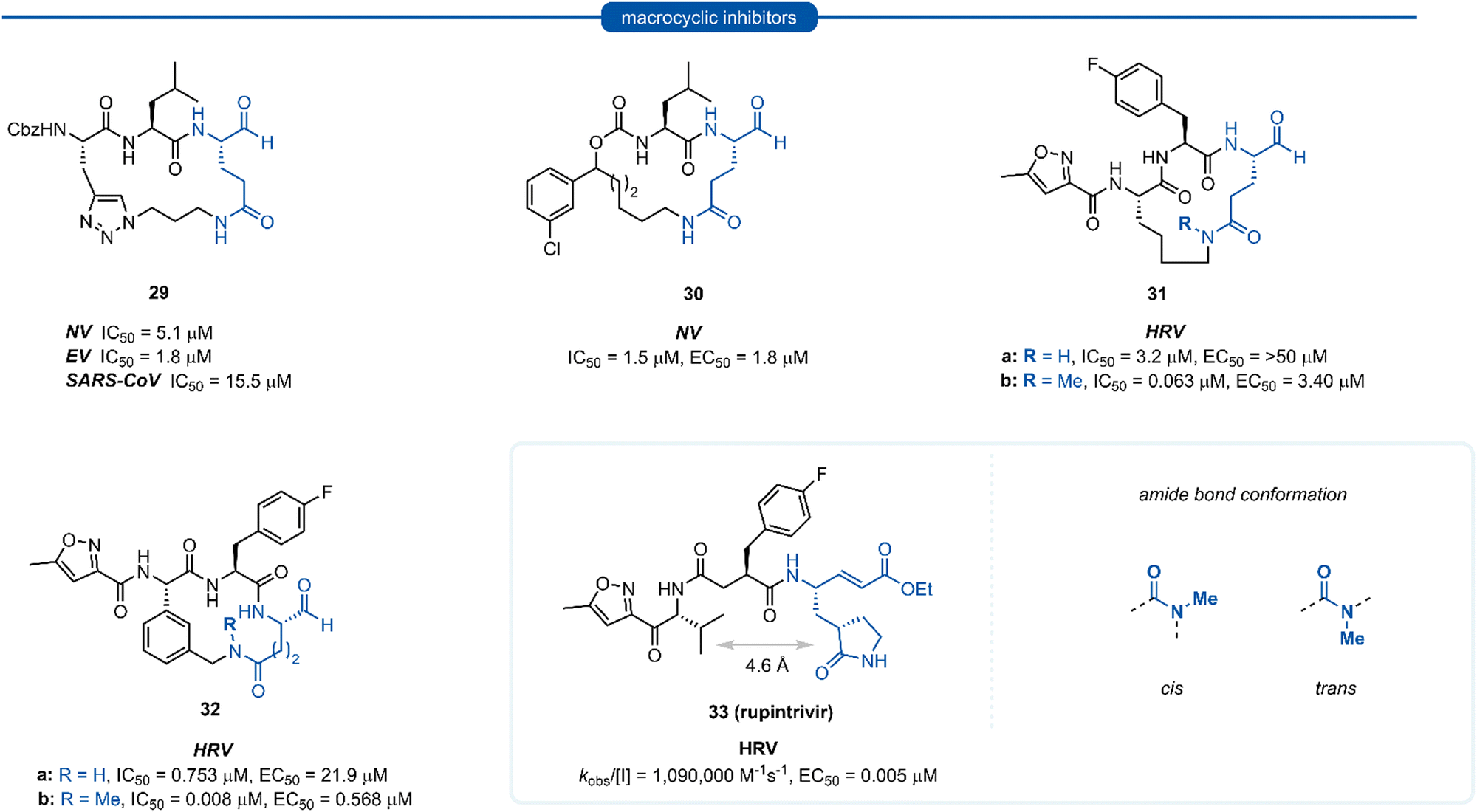 | ||
| Fig. 8 Representative examples of macrocyclic lactam inhibitors containing a P1 Gln residue. | ||
The resulting macrocycle 31a was tested against HRV 3Cpro and showed decreased potency compared to rupintrivir (EC50 > 50 μM) in HeLa cells infection model, albeit conserving moderate protease inhibition activity with IC50 values in the low micromolar range (IC50 = 3.2 μM). Modelling of the macrocyclic peptide in the binding pocket predicted the glutamine-derived amide bond was required to be in the cis conformation for efficient binding. Despite the cis conformation being higher energy, methylation of the amide nitrogen allows increased population of the cis conformer. Inhibitor 31b containing an N-methyl moiety showed a 50-fold increase in potency against HRV 3Cpro (IC50 = 0.063 μM) despite losing an H-bonding interaction between the P1 amide proton and backbone carbonyl of Thr142. Alternatively, incorporation of a phenyl ring into the macrocyclic linkage gave inhibitor 32a (IC50 = 0.753 μM), which displayed a 4-fold increase in activity over 31a. Additional incorporation of the N-methyl moiety afforded 32b and further increased the activity by 100-fold (IC50 = 0.008 μM).
Section 7. P1 lactam inhibitors
Pioneering work by Dragovich and co-workers first led to the use of γ-lactams as P1 glutamine isosteres in a series of publications developing tripeptide inhibitors of HRV 3Cpro.18,19,66,67 Early investigations (see Sections 3 and 4) found that none of the P1 non-glutamine inhibitors showed improved potency over the inhibitor containing an unmodified glutamine side chain 3a (Fig. 9A). Inspection of the X-ray crystal structure of 3a bound to HRV 3Cpro revealed that while the P1trans amide proton is engaged in a H-bond to the backbone carbonyl of Thr142, the cis amide proton is solvent exposed, providing a vector for further optimisation of the P1 side chain. N-Monomethyl inhibitor 3b displayed significantly weaker protease inhibition, presumably due to the amide preferentially adopting a trans conformation, interrupting the key P1 amide proton-Thr142 H-bond (Fig. 9A). In order to lock in the desired cis amide conformation, an (S)-γ-lactam was incorporated into the P1 side chain.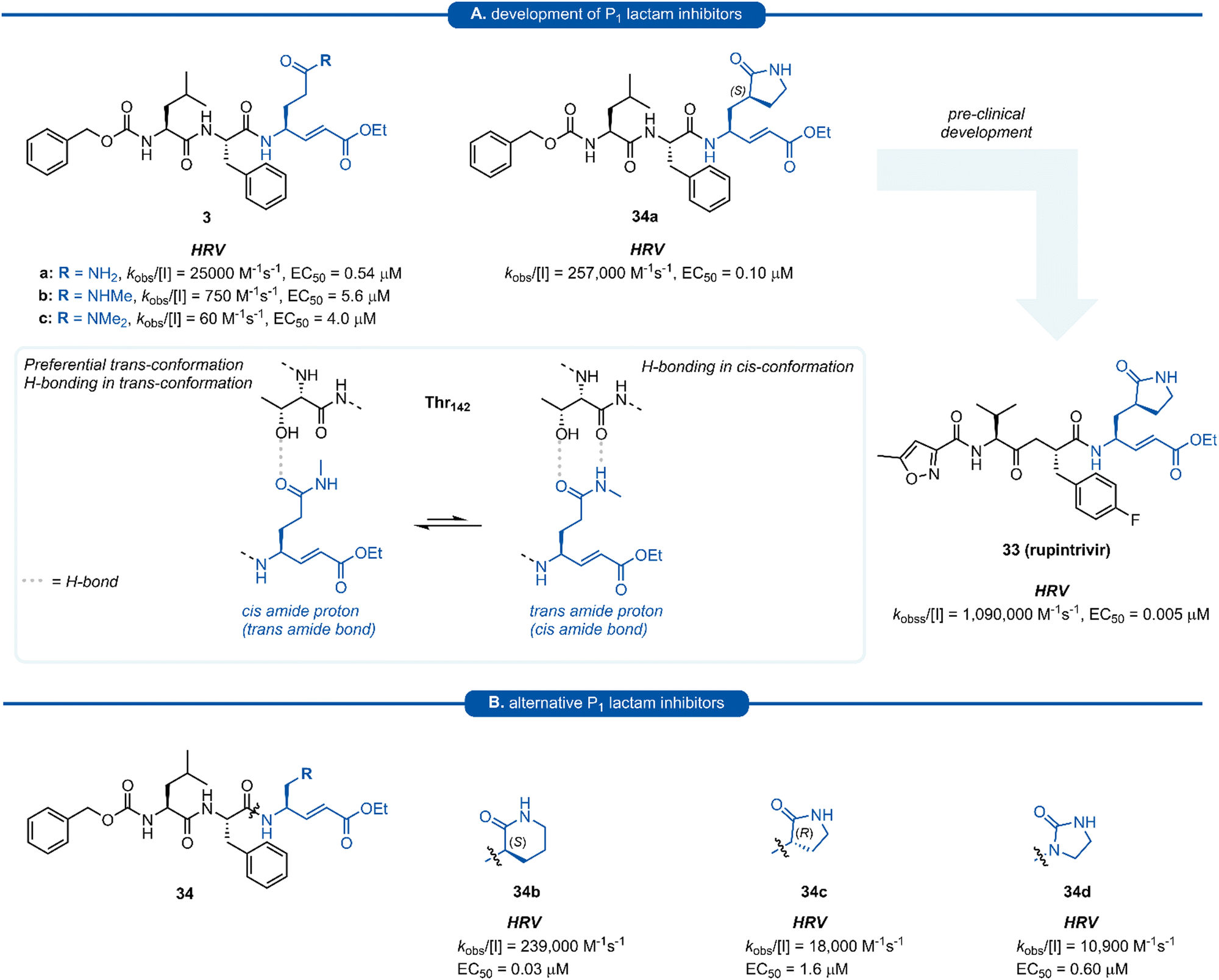 | ||
| Fig. 9 (A) Development of HRV inhibitors possessing a P1 (S)-γ-lactam moiety. (B) Alternative P1 lactam isosteres. | ||
The resulting tripeptide 34a displayed a remarkable increase in protease inhibition activity against HRV 3Cpro as well as sub-micromolar antiviral activity in cell culture.67 The combination of this newly optimised P1 side chain with favourable P2–P4 modifications discovered in related work gave rise to rupintrivir 33 (AG-7088). During the initial investigation into P1 lactam-based glutamine isosteres, it was found that while a (S)-δ-lactam moiety 34b was equally effective as the (S)-γ-lactam 34a, switching the stereochemistry of the lactam ring 34c or replacing the lactam moiety with a 5-membered cyclic urea 34d resulted in a significant reduction in activity (Fig. 9B).67 Following the work of Dragovich et al. and guided by crystal structure studies revealing the high degree of structural homology among 3Cpro and 3CLpro enzymes, P1 (S)-lactam isosteres have been integrated into the design of numerous inhibitors targeting 3C/3CLpro. The first example outside of the preliminary work on HRV protease was for the development of inhibitors of hepatitis A virus (HAV) 3Cpro. From a small series of compounds bearing an α-ketophthalahydrazide, compound 35 proved to be the most effective (IC50 = 1.6 μM).68 Representative examples of 3C/3CLpro inhibitors bearing a P1 (S)-lactam isostere are shown in Fig. 10, showcasing the compatibility of the P1 (S)-lactam with a range of electrophilic warhead functionalities. The (S)-lactam P1 unit has been shown to be effective in targeting numerous viral 3C/3CLpro including compounds 36–38 for enterovirus,69–7439 for norovirus,75–7940–41 for SARS-CoV,32,45,80–8942 for MERS-CoV,90–92 and 43–51 for SARS-CoV-2 (Fig. 11). A handful of inhibitors demonstrating broad spectrum activity have also been developed.93–96 As represented in Fig. 10, 3C/3CLpro inhibitors typically employ a (S)-γ-lactam P1 isostere, however enterovirus has been shown to prefer the (S)-δ-lactam isostere at the P1 position. This was first demonstrated through docking studies of EV71 3Cpro, revealing the δ-lactam had shorter H-bond distances with His161 and Thr142 compared to the γ-lactam, while also adopting a preferable binding orientation in the S1 pocket.70,71 This model was validated through the design of inhibitor 36c, which was 7–10 fold more active than the corresponding γ-lactam 36b. In a separate study, screening of a compound library revealed compound 37 containing a chiral cyanohydrin warhead.72 Each epimer was tested separately and exhibited good activity against EV71 with little effect of the warhead stereochemistry on the potency of the inhibitor. The same library screening also revealed an inhibitor scaffold analogous to that of 37 with an aldehyde warhead 38, to be the most potent compound tested against EV71 in both 3Cpro protease inhibition assay and human rhabdomyosarcoma cells viral infection model.70–72
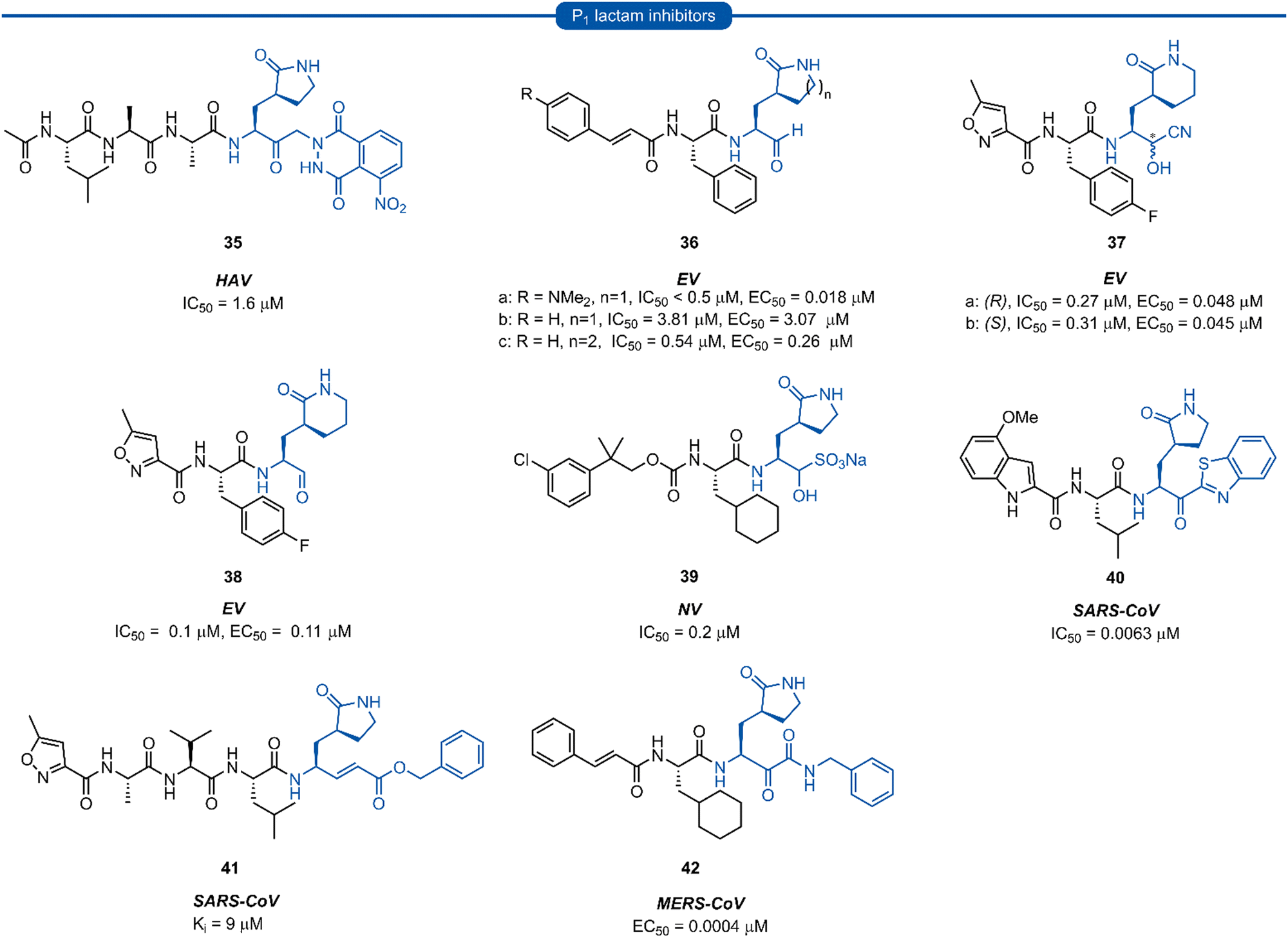 | ||
| Fig. 10 Representative examples of inhibitors targeting 3C/3CLpro containing a P1 (S)-γ/δ-lactam isostere. | ||
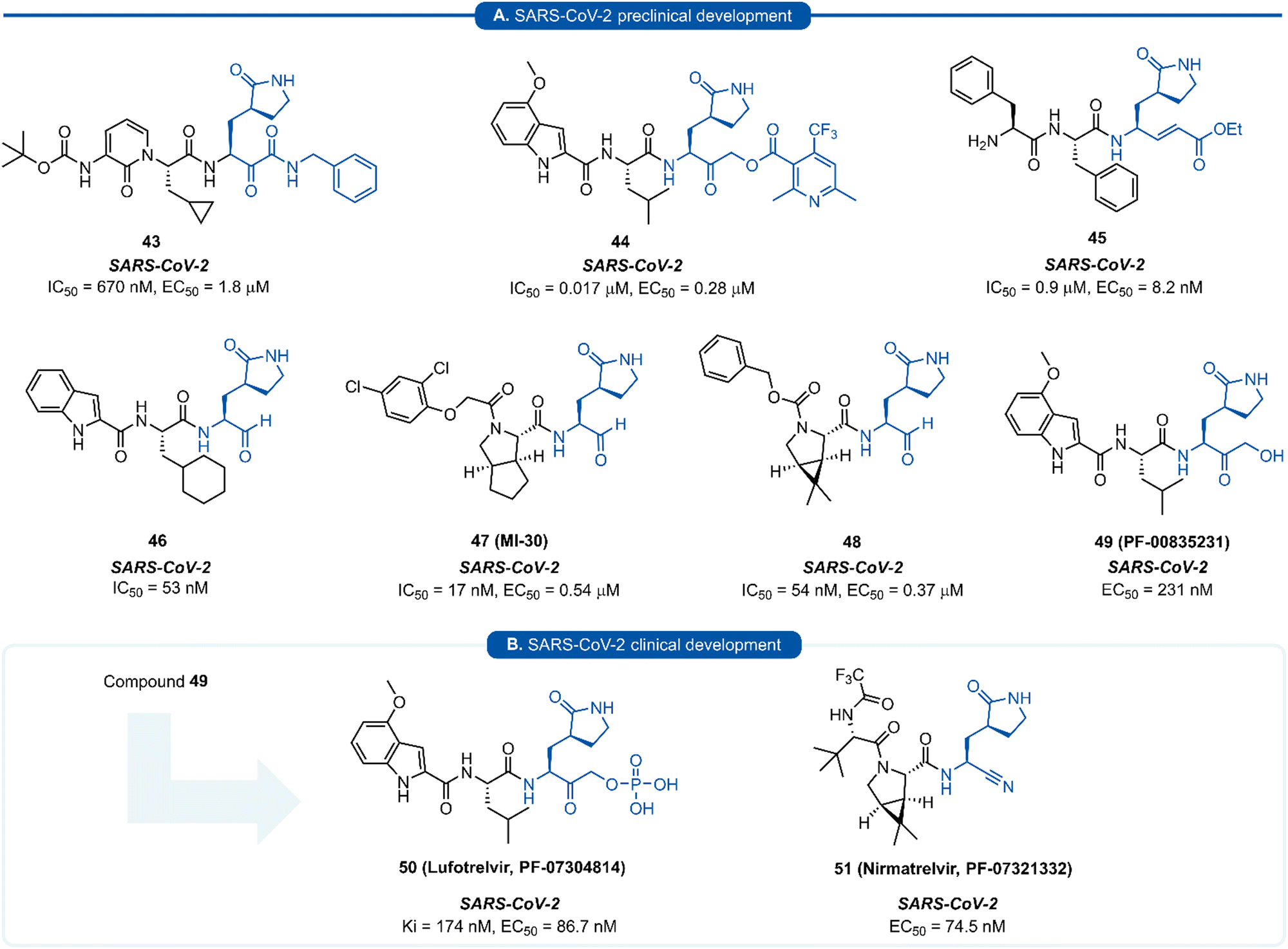 | ||
| Fig. 11 Representative examples of SARS-CoV-2 inhibitors in preclinical (A) or clinical (B) development. | ||
Since the SARS-CoV-2 outbreak in 2019, new inhibitors of the main protease 3CLpro (aka Mpro, Nsp5) have almost exclusively been developed using P1 (S)-γ-lactam isostere. In early work, it was realised that a high degree of homology existed between 3CLpro of SARS-CoV and SARS-CoV-2.97 In addition to their 96% sequence identity, X-ray crystal data has demonstrated there to be a high structural similarity between SARS-CoV and SARS-CoV-2 3CLpro with a root mean square deviation of 0.53 Å across all Cα positions.98 This suggested that structural features from known coronaviral protease inhibitors are likely to be effective when incorporated into the design of SARS-CoV-2 3CLpro inhibitors. This was demonstrated in an early report by Hilgenfeld and co-workers in the development of α-ketoamide inhibitor 43 from known inhibitor 42. The (S)-γ-lactam moiety was found to occupy the 3CLpro S1 pocket, with the crystal structure of SARS-CoV-2 3CLpro with 43 highlighting the crucial H-bonding between the lactam carbonyl oxygen with His163 side-chain imidazole, and the lactam nitrogen forming a three-centred hydrogen bond with Phe140 and Glu166.98 Various electrophilic warhead moieties have been demonstrated as effective in combination with a P1 (S)-γ-lactam group targeting SARS-CoV-2 3CLpro. Dipeptides bearing a heterocyclic acyloxymethylketone warhead were shown to be excellent irreversible inhibitors of SARS-CoV-2 3CLpro, exemplified by 44.99 Compound 44 also exhibited excellent plasma stability, glutathione stability, and selectivity over human proteases cathepsin B and cathepsin S. Inhibitor 45, which possesses a Michael acceptor warhead unit and inhibits SARS-CoV-2 3CLpro in the low micromolar range (IC50 = 0.9 μM), was recently demonstrated to exhibit significantly superior antiviral activity in hACE2 cell assays (EC50 = 8.2 nM).100 Upon further investigation, this enhanced potency was attributed to a dual mode of action of compound 45 in targeting the SARS-CoV-2 virus lifecycle. In addition to inhibiting 3CLpro, 45 was found to also inhibit cathepsin L (CatL), a protease enzyme responsible for cleaving the viral Spike protein, promoting cell entry. Intraperitoneal administration of 45 in SARS-CoV-2 infected transgenic mice also showed to cause reduced viral load in the lungs and enhanced survival rate. In line with the development of inhibitors of other viral proteases, the introduction of an aldehyde C-terminal warhead has also proved effective for the discovery of SARS-CoV-2 3CLpro inhibitors (46–48). Structure-based design utilising the crystal structure of SARS-CoV 3CLpro gave rise to dipeptide aldehyde 46, which demonstrates good anti-SARS-CoV-2 activity (EC50 = 0.53 nM) and was selected for further preclinical development.101 Compounds 47 (aka MI-30)102 and 48 (aka UAWJ9-36-3)103 represent a series of aldehyde-based hybrid inhibitors which combine the P1 (S)-γ-lactam group with a bicycloproline P2 residue derived from the hepatitis C virus (HCV) inhibitors, telaprevir104 (P2 5,5-fused ring) or boceprevir105 (P2 3,5-fused ring), respectively. Unlike work towards other viral 3C/3CLpro inhibitors highlighted in this review, SARS-CoV-2 inhibitors are now at various stages of clinical development (Fig. 11). Compound 49 (aka PF-00835231), containing a P1 (S)-γ-lactam group in conjunction with a hydroxymethylketone C-terminal electrophilic warhead, was identified as a potent inhibitor of SARS-CoV-2 3CLpro (IC50 = 4 nM) and was selected as the basis for the development of a protease inhibitor for the treatment of COVID-19.106 One drawback of compound 49 that became apparent during preclinical characterisation was its relatively poor intrinsic aqueous solubility (<0.1 mg mL−1), which was insufficient to formulate for intravenous (IV) infusion. Accordingly, IV administration was enabled by utilizing a prodrug strategy, resulting in the phosphate prodrug lufotrelvir (compound 50) which displays vastly superior aqueous solubility of >200 mg mL−1 over a pH range suitable for IV infusion.107 Lufotrelvir (50) is rapidly converted to the active free hydroxymethyl ketone (PF-00835231, 44) following administration. Work towards development of an orally bioavailable SARS-CoV-2 inhibitor has been undertaken by Owen and co-workers (Pfizer). Again using 49 as a lead compound, several modifications were made in order to reduce the hydrogen bond donor (HBD) count which has been shown to correlate with poor oral bioavailability.108 Changing the electrophilic warhead from a hydroxymethyl ketone (HMK) to a nitrile group significantly increased its oral bioavailability (Oral F = 1.4% vs. 7.6% for otherwise identical HMK and nitrile inhibitors, respectively). It was also noted that compounds containing a nitrile warhead were less likely to undergo epimerization at the P1 group during chemical synthesis compared to those equipped with heterocyclic ketone warheads that were also being investigated.108 Incorporation of a P2 bicycloproline derivative and N-terminal cap modifications afforded nirmatrelvir (51), which displays superior activity (EC50 = 74.5 nM) and oral bioavailability (F = 50%) over its predecessor 49 (EC50 = 231 nM, oral F = 1.4%).109
A drawback of the P1 (S)-γ-lactam glutamine isostere which became apparent through preclinical development of nirmatrelvir (51), is its susceptibility to undergo cytochrome P450 (CYP450) oxidative metabolism. The primary route of inactivation of nirmatrelvir in human liver microsomes occurs via CYP3A4-mediated oxidation. The major metabolite 52 resulting from hydroxylation of the pyrrolidine ring and which, albeit exhibiting similar inhibition potency against 3CLpro and repressing viral replication, was 10-fold less active against SARS-CoV-2 compared to 51 in epithelial Vero E6 cells infection model (Scheme 3). In the same study, other minor metabolites of nirmatrelvir, produced by CYP3A4 via alternative oxidation patterns at the P1 and P2 position, were detected in trace amounts, but their individual inhibitory potencies were not measured due to their low relative abundance.109 This has ultimately led to nirmatrelvir being co-administered with the CYP3A4 inhibitor, ritonavir, to increase its bioavailability. This combined formulation, PaxlovidTM has proven to be a safe and effective treatment for COVID-19 for patients with high risk of disease progression, resulting in an 89% risk reduction in treated patients relative to a placebo group.110
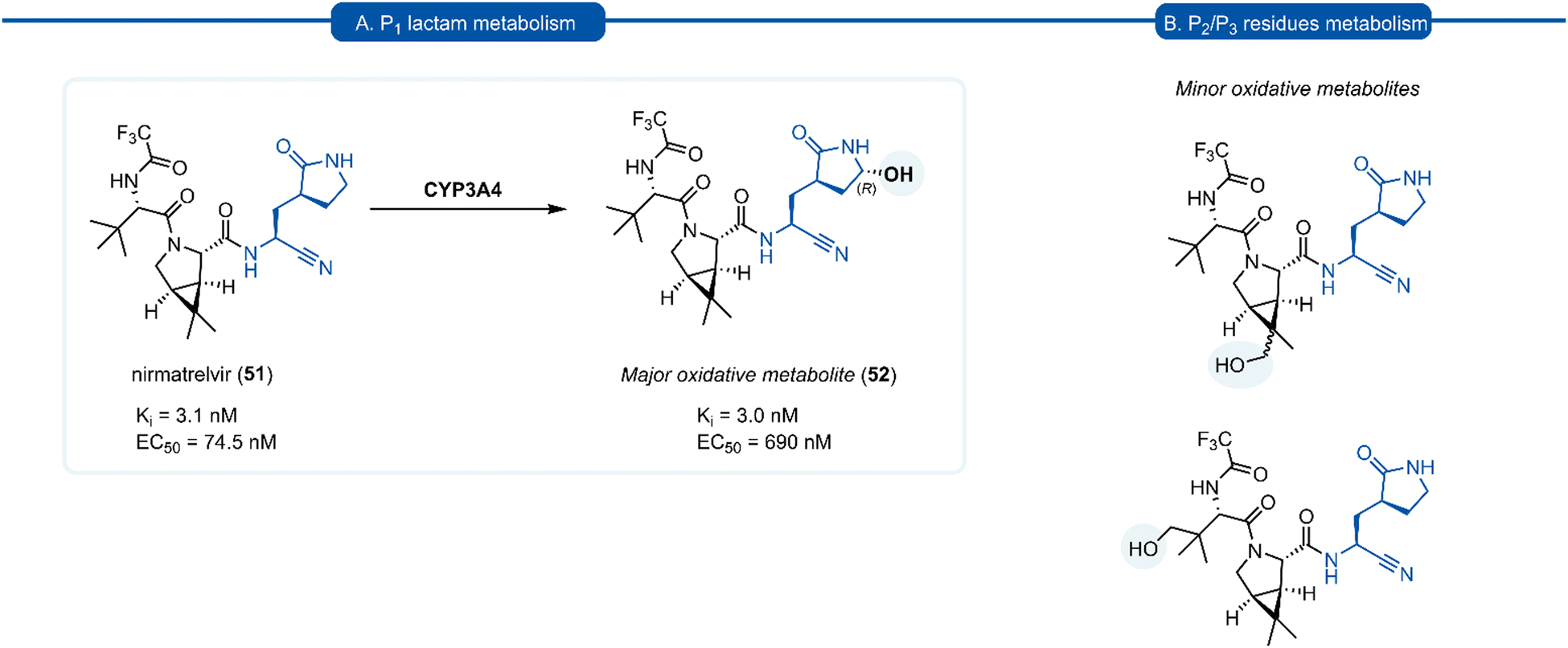 | ||
| Scheme 3 (A) The primary route of metabolism of nirmatrelvir 51via CYP3A4 hydroxylation of the P1 (S)-γ-lactam moiety. (B) Minor metabolites products of nirmatrelvir 51via CYP3A4 hydroxylation of the P2 and P3 moiety. | ||
Conclusion
Over the last 25 years, considerable effort has been invested into the development of P1 glutamine isosteres for incorporation into inhibitors targeting 3Cpro and 3CLpro of viruses in the Pisoniviricetes class. Early efforts exploring P1N-alkyl glutamine inhibitors were met with mixed success, while more structurally diverse methionine and carbonyl derived isosteres of glutamine, were generally found to be detrimental to inhibitor activity. Macrocyclization and aza group backbone modifications at the P1 position, although delivering low micromolar inhibitors, also did not demonstrate any significant improvements over their linear and unmodified counterparts. Incorporation of the P1 lactam isostere resulted in significantly more potent inhibitors and the development of the (S)-γ-lactam moiety marked the first real progress towards potent 3C/3CLpro inhibitors. The lactam was found to conformationally restrict the P1 side chain amide bond in the cis conformation, providing optimal H-bonding interactions in the S1 binding pocket and reducing the entropic cost of binding. Moreover, the inhibitor inactivation by intramolecular cyclisation of the nucleophilic P1 amide moiety onto the electrophilic warhead, was not observed for the (S)-lactam. The P1 (S)-lactam moiety has now found widespread use in targeting 3C/3CLpro due to its optimal binding interactions and broad compatibility with electrophilic warheads. Recent metabolic studies on nirmatrelvir, however, have now revealed the lactam group to be the primary metabolic hotspot. Although the P1 lactam moiety has provided one of the major milestones in targeting 3Cpro and 3CLpro, further structural optimisation of the lactam side chain is desirable to improve the pharmacokinetics of P1 lactam containing inhibitors.Author contributions
L. A. Stubbing: concept and original version. J. Bell-Tyrer: writing – original draft. Y. O. Hermant, J. G. Hubert, S. H. Yang, A. M. McSweeney, G. M. McKenzie-Goldsmith, D. P. Furkert, V. K. Ward, M. A. Brimble: writing – review and editing. V. K. Ward, M. A. Brimble: project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Ministry of Business, Innovation and Employment (MBIE), contract number U00X1904 and Auckland Medical Research Foundation, grant 1720013 for financial support.References
- World Health Organization, The top 10 causes of death, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death, 2020, accessed 13/03/2023.
- T. Heikkinen and A. Jarvinen, Lancet, 2003, 361, 51 CrossRef PubMed.
- X. Wang, J. Ren, Q. Gao, Z. Hu, Y. Sun, X. Li, D. J. Rowlands, W. Yin, J. Wang, D. I. Stuart, Z. Rao and E. E. Fry, Nature, 2015, 517, 85 CrossRef CAS PubMed.
- A. Bouin and B. L. Semler, Curr. Clin. Microbiol. Rep., 2020, 7, 31 CrossRef PubMed.
- T. M. Szopa, P. A. Titchener, N. D. Portwood and K. W. Taylor, Diabetologia, 1993, 36, 687 CrossRef CAS PubMed.
- J. D. Kriesel, A. White, F. G. Hayden, S. L. Spruance and J. Petajan, Mult. Scler., 2004, 10, 145 CrossRef PubMed.
- S. Payne, Viruses, 2017, 149 Search PubMed.
- S. M. Ahmed, A. J. Hall, A. E. Robinson, L. Verhoef, P. Premkumar, U. D. Parashar, M. Koopmans and B. A. Lopman, Lancet Infect. Dis., 2014, 14, 725 CrossRef PubMed.
- S. C. Baker, Encyclopedia of Virology, Oxford Academic Press, Oxford, Third edn, 2008, p. 554 Search PubMed.
- D. Sun, S. Chen, A. Cheng and M. Wang, Viruses, 2016, 8, 82 CrossRef PubMed.
- C. P. Campillay-Veliz, J. J. Carvajal, A. M. Avellaneda, D. Escobar, C. Covian, A. M. Kalergis and M. K. Lay, Front. Immunol., 2020, 11, 961 CrossRef CAS PubMed.
- D. A. Matthews, W. W. Smith, R. A. Ferre, B. Condon, G. Budahazi, W. Sisson, J. E. Villafranca, C. A. Janson, H. E. McElroy and C. L. Gribskov, et al. , Cell, 1994, 77, 761 CrossRef CAS PubMed.
- J. Yin, M. M. Cherney, E. M. Bergmann, J. Zhang, C. Huitema, H. Pettersson, L. D. Eltis, J. C. Vederas and M. N. James, J. Mol. Biol., 2006, 361, 673 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763 CrossRef CAS PubMed.
- X. Fan, X. Li, Y. Zhou, M. Mei, P. Liu, J. Zhao, W. Peng, Z. B. Jiang, S. Yang, B. L. Iverson, G. Zhang and L. Yi, ACS Chem. Biol., 2020, 15, 63 CrossRef CAS PubMed.
- H. Chen, Z. Zhu, Y. Qiu, X. Ge, H. Zheng and Y. Peng, Virol. Sin., 2022, 37, 437 CrossRef CAS PubMed.
- S. W. Kaldor, M. Hammond, B. A. Dressman, J. M. Labus, F. W. Chadwell, A. D. Kline and B. A. Heinz, Bioorg. Med. Chem. Lett., 1995, 5, 2021 CrossRef CAS.
- P. S. Dragovich, S. E. Webber, R. E. Babine, S. A. Fuhrman, A. K. Patick, D. A. Matthews, C. A. Lee, S. H. Reich, T. J. Prins, J. T. Marakovits, E. S. Littlefield, R. Zhou, J. Tikhe, C. E. Ford, M. B. Wallace, J. W. Meador, 3rd, R. A. Ferre, E. L. Brown, S. L. Binford, J. E. Harr, D. M. DeLisle and S. T. Worland, J. Med. Chem., 1998, 41, 2806 CrossRef CAS PubMed.
- P. S. Dragovich, S. E. Webber, R. E. Babine, S. A. Fuhrman, A. K. Patick, D. A. Matthews, S. H. Reich, J. T. Marakovits, T. J. Prins, R. Zhou, J. Tikhe, E. S. Littlefield, T. M. Bleckman, M. B. Wallace, T. L. Little, C. E. Ford, J. W. Meador, 3rd, R. A. Ferre, E. L. Brown, S. L. Binford, D. M. DeLisle and S. T. Worland, J. Med. Chem., 1998, 41, 2819 CrossRef CAS PubMed.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld and Z. Rao, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190 CrossRef CAS PubMed.
- P. S. Dragovich, R. Zhou, S. E. Webber, T. J. Prins, A. K. Kwok, K. Okano, S. A. Fuhrman, L. S. Zalman, F. C. Maldonado, E. L. Brown, J. W. Meador, 3rd, A. K. Patick, C. E. Ford, M. A. Brothers, S. L. Binford, D. A. Matthews, R. A. Ferre and S. T. Worland, Bioorg. Med. Chem. Lett., 2000, 10, 45 CrossRef CAS PubMed.
- M. A. Murray, J. W. Janc, S. Venkatraman and L. M. Babe, Antivir. Chem. Chemother., 2001, 12, 273 CrossRef CAS PubMed.
- M. O. Sydnes, Y. Hayashi, V. K. Sharma, T. Hamada, U. Bacha, J. Barrila, E. Freire and Y. Kiso, Tetrahedron, 2006, 62, 8601 CrossRef CAS PubMed.
- S.-H. Chen, J. Lamar, F. Victor, N. Snyder, R. Johnson, B. A. Heinz, M. Wakulchik and Q. M. Wang, Bioorg. Med. Chem. Lett., 2003, 13, 3531 CrossRef CAS PubMed.
- M. A. van de Plassche, M. Barniol-Xicota and S. H. Verhelst, ChemBioChem, 2020, 21, 3383 CrossRef CAS PubMed.
- C. P. Chuck, C. Chen, Z. Ke, D. C. Wan, H. F. Chow and K. B. Wong, Eur. J. Med. Chem., 2013, 59, 1 CrossRef CAS PubMed.
- S. E. Webber, K. Okano, T. L. Little, S. H. Reich, Y. Xin, S. A. Fuhrman, D. A. Matthews, R. A. Love, T. F. Hendrickson, A. K. Patick, J. W. Meador, 3rd, R. A. Ferre, E. L. Brown, C. E. Ford, S. L. Binford and S. T. Worland, J. Med. Chem., 1998, 41, 2786 CrossRef CAS PubMed.
- H. Z. Zhang, H. Zhang, W. Kemnitzer, B. Tseng, J. Cinatl, M. Michaelis, H. W. Doerr and S. X. Cai, J. Med. Chem., 2006, 49, 1198 CrossRef CAS PubMed.
- B. A. Malcolm, C. Lowe, S. Shechosky, R. T. McKay, C. C. Yang, V. J. Shah, R. J. Simon, J. C. Vederas and D. V. Santi, Biochemistry, 1995, 34, 8172 CrossRef CAS PubMed.
- L. S. Deng, Z. Muhaxhiri, M. K. Estes, T. Palzkill, B. V. V. Prasad and Y. C. Song, MedChemComm, 2013, 4, 1354 RSC.
- T. S. Morris, S. Frormann, S. Shechosky, C. Lowe, M. S. Lall, V. Gauss-Muller, R. H. Purcell, S. U. Emerson, J. C. Vederas and B. A. Malcolm, Bioorg. Med. Chem., 1997, 5, 797 CrossRef CAS PubMed.
- T. Regnier, D. Sarma, K. Hidaka, U. Bacha, E. Freire, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2009, 19, 2722 CrossRef CAS PubMed.
- Y. K. Ramtohul, M. N. G. James and J. C. Vederas, J. Org. Chem., 2002, 67, 3169 CrossRef CAS PubMed.
- T. A. Shepherd, G. A. Cox, E. McKinney, J. Tang, M. Wakulchik, R. E. Zimmerman and E. C. Villarreal, Bioorg. Med. Chem. Lett., 1996, 6, 2893 CrossRef CAS.
- R. D. Hill and J. C. Vederas, J. Org. Chem., 1999, 64, 9538 CrossRef CAS.
- J. S. Kong, S. Venkatraman, K. Furness, S. Nimkar, T. A. Shepherd, Q. M. Wang, J. Aube and R. P. Hanzlik, J. Med. Chem., 1998, 41, 2579 CrossRef CAS PubMed.
- Y. M. Shao, W. B. Yang, T. H. Kuo, K. C. Tsai, C. H. Lin, A. S. Yang, P. H. Liang and C. H. Wong, Bioorg. Med. Chem., 2008, 16, 4652 CrossRef CAS PubMed.
- L. Li, B. C. Chenna, K. S. Yang, T. R. Cole, Z. T. Goodall, M. Giardini, Z. Moghadamchargari, E. A. Hernandez, J. Gomez, C. M. Calvet, J. A. Bernatchez, D. M. Mellott, J. Zhu, A. Rademacher, D. Thomas, L. R. Blankenship, A. Drelich, A. Laganowsky, C. K. Tseng, W. R. Liu, A. J. Wand, J. Cruz-Reyes, J. L. Siqueira-Neto and T. D. Meek, J. Med. Chem., 2021, 64, 11267 CrossRef CAS PubMed.
- J. Zhu, L. Li, A. Drelich, B. C. Chenna, D. M. Mellott, Z. W. Taylor, V. Tat, C. Z. Garcia, A. Katzfuss, C. K. Tseng and T. D. Meek, Front. Chem., 2022, 10, 867928 CrossRef CAS PubMed.
- D. H. Goetz, Y. Choe, E. Hansell, Y. T. Chen, M. McDowell, C. B. Jonsson, W. R. Roush, J. McKerrow and C. S. Craik, Biochemistry, 2007, 46, 8744 CrossRef CAS PubMed.
- K. Akaji, H. Konno, H. Mitsui, K. Teruya, Y. Shimamoto, Y. Hattori, T. Ozaki, M. Kusunoki and A. Sanjoh, J. Med. Chem., 2011, 54, 7962 CrossRef CAS PubMed.
- K. Teruya, Y. Hattori, Y. Shimamoto, K. Kobayashi, A. Sanjoh, A. Nakagawa, E. Yamashita and K. Akaji, Biopolymers, 2016, 106, 391 CrossRef CAS PubMed.
- Y. Shimamoto, Y. Hattori, K. Kobayashi, K. Teruya, A. Sanjoh, A. Nakagawa, E. Yamashita and K. Akaji, Bioorg. Med. Chem., 2015, 23, 876 CrossRef CAS PubMed.
- K. Ohnishi, Y. Hattori, K. Kobayashi and K. Akaji, Bioorg. Med. Chem., 2019, 27, 425 CrossRef CAS PubMed.
- S. Konno, P. Thanigaimalai, T. Yamamoto, K. Nakada, R. Kakiuchi, K. Takayama, Y. Yamazaki, F. Yakushiji, K. Akaji, Y. Kiso, Y. Kawasaki, S. E. Chen, E. Freire and Y. Hayashi, Bioorg. Med. Chem., 2013, 21, 412 CrossRef CAS PubMed.
- C. Proulx, D. Sabatino, R. Hopewell, J. Spiegel, Y. Garcia Ramos and W. D. Lubell, Future Med. Chem., 2011, 3, 1139 CrossRef CAS PubMed.
- J. Magrath and R. H. Abeles, J. Med. Chem., 1992, 35, 4279 CrossRef CAS PubMed.
- S. Venkatraman, J. S. Kong, S. Nimkar, Q. M. Wang, J. Aube and R. P. Hanzlik, Bioorg. Med. Chem. Lett., 1999, 9, 577 CrossRef CAS PubMed.
- W. M. Kati, H. L. Sham, J. O. McCall, D. A. Montgomery, G. T. Wang, W. Rosenbrook, L. Miesbauer, A. Buko and D. W. Norbeck, Arch. Biochem. Biophys., 1999, 362, 363 CrossRef CAS PubMed.
- H. L. Sham, W. Rosenbrook, W. Kati, D. A. Betebenner, N. E. Wideburg, A. Saldivar, J. J. Plattner and D. W. Norbeck, J. Chem. Soc., Perkin Trans. 1, 1995, 1081 RSC.
- Y. T. Huang, B. A. Malcolm and J. C. Vederas, Bioorg. Med. Chem., 1999, 7, 607 CrossRef CAS PubMed.
- T. W. Lee, M. M. Cherney, C. Huitema, J. Liu, K. E. James, J. C. Powers, L. D. Eltis and M. N. James, J. Mol. Biol., 2005, 353, 1137 CrossRef CAS PubMed.
- T. W. Lee, M. M. Cherney, J. Liu, K. E. James, J. C. Powers, L. D. Eltis and M. N. James, J. Mol. Biol., 2007, 366, 916 CrossRef CAS PubMed.
- J. Breidenbach, C. Lemke, T. Pillaiyar, L. Schakel, G. Al Hamwi, M. Diett, R. Gedschold, N. Geiger, V. Lopez, S. Mirza, V. Namasivayam, A. C. Schiedel, K. Sylvester, D. Thimm, C. Vielmuth, L. Phuong Vu, M. Zyulina, J. Bodem, M. Gutschow and C. E. Muller, Angew. Chem., Int. Ed., 2021, 60, 10423 CrossRef CAS PubMed.
- C. Gilon, D. Halle, M. Chorev, Z. Selinger and G. Byk, Biopolymers, 1991, 31, 745 CrossRef CAS PubMed.
- J. D. Tyndall and D. P. Fairlie, Curr. Med. Chem., 2001, 8, 893 CrossRef CAS PubMed.
- M. P. Glenn, L. K. Pattenden, R. C. Reid, D. P. Tyssen, J. D. Tyndall, C. J. Birch and D. P. Fairlie, J. Med. Chem., 2002, 45, 371 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef CAS PubMed.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961 CrossRef CAS PubMed.
- F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278 CrossRef CAS PubMed.
- S. R. Mandadapu, P. M. Weerawarna, A. M. Prior, R. A. Uy, S. Aravapalli, K. R. Alliston, G. H. Lushington, Y. Kim, D. H. Hua, K. O. Chang and W. C. Groutas, Bioorg. Med. Chem. Lett., 2013, 23, 3709 CrossRef CAS PubMed.
- P. M. Weerawarna, Y. Kim, A. C. Galasiti Kankanamalage, V. C. Damalanka, G. H. Lushington, K. R. Alliston, N. Mehzabeen, K. P. Battaile, S. Lovell, K. O. Chang and W. C. Groutas, Eur. J. Med. Chem., 2016, 119, 300 CrossRef CAS PubMed.
- A. C. Galasiti Kankanamalage, P. M. Weerawarna, A. D. Rathnayake, Y. Kim, N. Mehzabeen, K. P. Battaile, S. Lovell, K. O. Chang and W. C. Groutas, Proteins, 2019, 87, 579 CrossRef CAS PubMed.
- V. C. Damalanka, Y. Kim, A. C. Galasiti Kankanamalage, G. H. Lushington, N. Mehzabeen, K. P. Battaile, S. Lovell, K. O. Chang and W. C. Groutas, Eur. J. Med. Chem., 2017, 127, 41 CrossRef CAS PubMed.
- K. Namoto, F. Sirockin, H. Sellner, C. Wiesmann, F. Villard, R. J. Moreau, E. Valeur, S. C. Paulding, S. Schleeger, K. Schipp, J. Loup, L. Andrews, R. Swale, M. Robinson and C. J. Farady, Bioorg. Med. Chem. Lett., 2018, 28, 906 CrossRef CAS PubMed.
- P. S. Dragovich, T. J. Prins, R. Zhou, S. A. Fuhrman, A. K. Patick, D. A. Matthews, C. E. Ford, J. W. Meador, 3rd, R. A. Ferre and S. T. Worland, J. Med. Chem., 1999, 42, 1203 CrossRef CAS PubMed.
- P. S. Dragovich, T. J. Prins, R. Zhou, S. E. Webber, J. T. Marakovits, S. A. Fuhrman, A. K. Patick, D. A. Matthews, C. A. Lee and C. E. Ford, J. Med. Chem., 1999, 42, 1213 CrossRef CAS PubMed.
- R. P. Jain and J. C. Vederas, Bioorg. Med. Chem. Lett., 2004, 14, 3655 CrossRef CAS PubMed.
- C. J. Kuo, J. J. Shie, J. M. Fang, G. R. Yen, J. T. A. Hsu, H. G. Liu, S. N. Tseng, S. C. Chang, C. Y. Lee, S. R. Shih and P. H. Liang, Bioorg. Med. Chem., 2008, 16, 7388 CrossRef CAS PubMed.
- Y. Wang, B. Yang, Y. Zhai, Z. Yin, Y. Sun and Z. Rao, Antimicrob. Agents Chemother., 2015, 59, 2636 CrossRef CAS PubMed.
- Y. Wang, L. Cao, Y. Zhai, Z. Yin, Y. Sun and L. Shang, Antimicrob. Agents Chemother., 2017, 61, e00298 CAS.
- Y. Y. Zhai, X. S. Zhao, Z. J. Cui, M. Wang, Y. X. Wang, L. F. Li, Q. Sun, X. Yang, D. B. Zeng, Y. Liu, Y. N. Sun, Z. Y. Lou, L. Q. Shang and Z. Yin, J. Med. Chem., 2015, 58, 9414 CrossRef CAS PubMed.
- D. Zeng, Y. Ma, R. Zhang, Q. Nie, Z. Cui, Y. Wang, L. Shang and Z. Yin, Bioorg. Med. Chem. Lett., 2016, 26, 1762 CrossRef CAS PubMed.
- Y. Y. Zhai, Y. Y. Ma, F. Ma, Q. D. Nie, X. J. Ren, Y. X. Wang, L. Q. Shang and Z. Yin, Eur. J. Med. Chem., 2016, 124, 559 CrossRef CAS PubMed.
- K. C. Tiew, G. He, S. Aravapalli, S. R. Mandadapu, M. R. Gunnam, K. R. Alliston, G. H. Lushington, Y. Kim, K. O. Chang and W. C. Groutas, Bioorg. Med. Chem. Lett., 2011, 21, 5315 CrossRef CAS PubMed.
- A. C. Galasiti Kankanamalage, Y. Kim, P. M. Weerawarna, R. A. Z. Uy, V. C. Damalanka, S. R. Mandadapu, K. R. Alliston, N. Mehzabeen, K. P. Battaile and S. Lovell, J. Med. Chem., 2015, 58, 3144 CrossRef CAS PubMed.
- A. D. Rathnayake, Y. Kim, C. S. Dampalla, H. N. Nguyen, A. M. Jesri, M. M. Kashipathy, G. H. Lushington, K. P. Battaile, S. Lovell, K. O. Chang and W. C. Groutas, J. Med. Chem., 2020, 63, 11945 CrossRef CAS PubMed.
- A. C. Galasiti Kankanamalage, Y. Kim, A. D. Rathnayake, K. R. Alliston, M. M. Butler, S. C. Cardinale, T. L. Bowlin, W. C. Groutas and K. O. Chang, J. Med. Chem., 2017, 60, 6239 CrossRef CAS PubMed.
- F. Amblard, S. M. Zhou, P. Liu, J. Yoon, B. Cox, K. Muzzarelli, B. D. Kuiper, L. C. Kovari and R. F. Schinazi, Bioorg. Med. Chem. Lett., 2018, 28, 2165 CrossRef CAS PubMed.
- R. P. Jain, H. I. Pettersson, J. Zhang, K. D. Aull, P. D. Fortin, C. Huitema, L. D. Eltis, J. C. Parrish, M. N. James and D. S. Wishart, J. Med. Chem., 2004, 47, 6113 CrossRef CAS PubMed.
- A. K. Ghosh, K. Xi, K. Ratia, B. D. Santarsiero, W. Fu, B. H. Harcourt, P. A. Rota, S. C. Baker, M. E. Johnson and A. D. Mesecar, J. Med. Chem., 2005, 48, 6767 CrossRef CAS PubMed.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam and Z. Rao, PLoS Biol., 2005, 3, e324 CrossRef PubMed.
- S. Yang, S. J. Chen, M. F. Hsu, J. D. Wu, C. T. Tseng, Y. F. Liu, H. C. Chen, C. W. Kuo, C. S. Wu, L. W. Chang, W. C. Chen, S. Y. Liao, T. Y. Chang, H. H. Hung, H. L. Shr, C. Y. Liu, Y. A. Huang, L. Y. Chang, J. C. Hsu, C. J. Peters, A. H. Wang and M. C. Hsu, J. Med. Chem., 2006, 49, 4971 CrossRef CAS PubMed.
- P. Thanigaimalai, S. Konno, T. Yamamoto, Y. Koiwai, A. Taguchi, K. Takayama, F. Yakushiji, K. Akaji, S. E. Chen, A. Naser-Tavakolian, A. Schon, E. Freire and Y. Hayashi, Eur. J. Med. Chem., 2013, 68, 372 CrossRef CAS PubMed.
- P. Thanigaimalai, S. Konno, T. Yamamoto, Y. Koiwai, A. Taguchi, K. Takayama, F. Yakushiji, K. Akaji, Y. Kiso, Y. Kawasaki, S. E. Chen, A. Naser-Tavakolian, A. Schon, E. Freire and Y. Hayashi, Eur. J. Med. Chem., 2013, 65, 436 CrossRef CAS PubMed.
- C. S. B. Chia, W. Xu and P. Shuyi Ng, ChemMedChem, 2022, 17, e202100576 CAS.
- K. Akaji, Amino Acids Pept. Proteins, 2017, 228 Search PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S. H. Jung, J. Med. Chem., 2016, 59, 6595 CrossRef CAS PubMed.
- T. Pillaiyar, L. L. Wendt, M. Manickam and M. Easwaran, Med. Res. Rev., 2021, 41, 72 CrossRef CAS PubMed.
- V. Kumar, J. S. Shin, J. J. Shie, K. B. Ku, C. Kim, Y. Y. Go, K. F. Huang, M. Kim and P. H. Liang, Antiviral Res., 2017, 141, 101 CrossRef CAS PubMed.
- A. C. G. Kankanamalage, Y. Kim, V. C. Damalanka, A. D. Rathnayake, A. R. Fehr, N. Mehzabeen, K. P. Battaile, S. Lovell, G. H. Lushington, S. Perlman, K. O. Chang and W. C. Groutas, Eur. J. Med. Chem., 2018, 150, 334 CrossRef PubMed.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. Von Brunn, P. Leyssen, K. Lanko and J. Neyts, J. Med. Chem., 2020, 63, 4562 CrossRef CAS PubMed.
- S. L. Binford, F. Maldonado, M. A. Brothers, P. T. Weady, L. S. Zalman, J. W. Meador, 3rd, D. A. Matthews and A. K. Patick, Antimicrob. Agents Chemother., 2005, 49, 619 CrossRef CAS PubMed.
- H. M. Wang and P. H. Liang, Expert Opin. Ther. Pat., 2010, 20, 59 CrossRef CAS PubMed.
- R. Ramajayam, K. P. Tan and P. H. Liang, Biochem. Soc. Trans., 2011, 39, 1371 CrossRef CAS PubMed.
- Y. Kim, S. Lovell, K. C. Tiew, S. R. Mandadapu, K. R. Alliston, K. P. Battaile, W. C. Groutas and K. O. Chang, J. Virol., 2012, 86, 11754 CrossRef CAS PubMed.
- J. Tan, K. H. Verschueren, K. Anand, J. Shen, M. Yang, Y. Xu, Z. Rao, J. Bigalke, B. Heisen, J. R. Mesters, K. Chen, X. Shen, H. Jiang and R. Hilgenfeld, J. Mol. Biol., 2005, 354, 25 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409 CrossRef CAS PubMed.
- B. Bai, A. Belovodskiy, M. Hena, A. S. Kandadai, M. A. Joyce, H. A. Saffran, J. A. Shields, M. B. Khan, E. Arutyunova, J. Lu, S. K. Bajwa, D. Hockman, C. Fischer, T. Lamer, W. Vuong, M. J. van Belkum, Z. Gu, F. Lin, Y. Du, J. Xu, M. Rahim, H. S. Young, J. C. Vederas, D. L. Tyrrell, M. J. Lemieux and J. A. Nieman, J. Med. Chem., 2022, 65, 2905 CrossRef CAS PubMed.
- S. Mondal, Y. Chen, G. J. Lockbaum, S. Sen, S. Chaudhuri, A. C. Reyes, J. M. Lee, A. N. Kaur, N. Sultana and M. D. Cameron, J. Am. Chem. Soc., 2022, 144, 21035 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331 CrossRef CAS PubMed.
- J. Qiao, Y. S. Li, R. Zeng, F. L. Liu, R. H. Luo, C. Huang, Y. F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z. L. Duan, Q. C. Zou, H. L. Zhang, W. Qu, Y. H. Long, M. H. Li, R. C. Yang, X. Liu, J. You, Y. Zhou, R. Yao, W. P. Li, J. M. Liu, P. Chen, Y. Liu, G. F. Lin, X. Yang, J. Zou, L. Li, Y. Hu, G. W. Lu, W. M. Li, Y. Q. Wei, Y. T. Zheng, J. Lei and S. Yang, Science, 2021, 371, 1374 CrossRef CAS PubMed.
- Z. Xia, M. Sacco, Y. Hu, C. Ma, X. Meng, F. Zhang, T. Szeto, Y. Xiang, Y. Chen and J. Wang, ACS Pharmacol. Transl. Sci., 2021, 4, 1408 CrossRef CAS PubMed.
- C. Lin, K. Lin, Y. P. Luong, B. G. Rao, Y. Y. Wei, D. L. Brennan, J. R. Fulghum, H. M. Hsiao, S. Ma, J. P. Maxwell, K. M. Cottrell, R. B. Perni, C. A. Gates and A. D. Kwong, J. Biol. Chem., 2004, 279, 17508 CrossRef CAS PubMed.
- S. Venkatraman, S. L. Bogen, A. Arasappan, F. Bennett, K. Chen, E. Jao, Y. T. Liu, R. Lovey, S. Hendrata, Y. Huang, W. Pan, T. Parekh, P. Pinto, V. Popov, R. Pike, S. Ruan, B. Santhanam, B. Vibulbhan, W. Wu, W. Yang, J. Kong, X. Liang, J. Wong, R. Liu, N. Butkiewicz, R. Chase, A. Hart, S. Agrawal, P. Ingravallo, J. Pichardo, R. Kong, B. Baroudy, B. Malcolm, Z. Guo, A. Prongay, V. Madison, L. Broske, X. Cui, K. C. Cheng, Y. Hsieh, J. M. Brisson, D. Prelusky, W. Korfmacher, R. White, S. Bogdanowich-Knipp, A. Pavlovsky, P. Bradley, A. K. Saksena, A. Ganguly, J. Piwinski, V. Girijavallabhan and F. G. Njoroge, J. Med. Chem., 2006, 49, 6074 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, Jr., B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, 3rd, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725 CrossRef CAS PubMed.
- B. Boras, R. M. Jones, B. J. Anson, D. Arenson, L. Aschenbrenner, M. A. Bakowski, N. Beutler, J. Binder, E. Chen, H. Eng, H. Hammond, J. Hammond, R. E. Haupt, R. Hoffman, E. P. Kadar, R. Kania, E. Kimoto, M. G. Kirkpatrick, L. Lanyon, E. K. Lendy, J. R. Lillis, J. Logue, S. A. Luthra, C. Ma, S. W. Mason, M. E. McGrath, S. Noell, R. S. Obach, O. B. Mn, R. O'Connor, K. Ogilvie, D. Owen, M. Pettersson, M. R. Reese, T. F. Rogers, R. Rosales, M. I. Rossulek, J. G. Sathish, N. Shirai, C. Steppan, M. Ticehurst, L. W. Updyke, S. Weston, Y. Zhu, K. M. White, A. Garcia-Sastre, J. Wang, A. K. Chatterjee, A. D. Mesecar, M. B. Frieman, A. S. Anderson and C. Allerton, Nat. Commun., 2021, 12, 6055 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586 CrossRef CAS PubMed.
- H. Eng, A. L. Dantonio, E. P. Kadar, R. S. Obach, L. Di, J. Lin, N. C. Patel, B. Boras, G. S. Walker, J. J. Novak, E. Kimoto, R. S. P. Singh and A. S. Kalgutkar, Drug Metab. Dispos., 2022, 50, 576 CrossRef CAS PubMed.
- J. Hammond, H. Leister-Tebbe, A. Gardner, P. Abreu, W. Bao, W. Wisemandle, M. Baniecki, V. M. Hendrick, B. Damle, A. Simon-Campos, R. Pypstra, J. M. Rusnak and E.-H. Investigators, N. Engl. J. Med, 2022, 386, 1397 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |