
Effective pH-regulated release of covalently conjugated antibiotics from antibacterial hydrogels
Rita Das
Mahapatra†
a,
Ara
Jo†
b,
Kusuma Betha Cahaya
Imani
a,
Jin-Woong
Chung
*c and
Jinhwan
Yoon
 *a
*a
aGraduate Department of Chemical Materials, Institute for Plastic Information and Energy Materials, Sustainable Utilization of Photovoltaic Energy Research Center, Pusan National University, Busan, 46241, Republic of Korea. E-mail: jinhwan@pusan.ac.kr
bDepartment of Molecular Biology and Immunology, College of Medicine, Kosin University, Busan, 49267, Republic of Korea
cDepartment of Biomedical Science, Dong-A University, 37 Nakdong-Daero 550 beon-gil, Saha-gu, Busan, 49315, Republic of Korea. E-mail: jwchung@dau.ac.kr
First published on 22nd August 2022
Abstract
This paper describes the development of a pH-sensitive ampicillin-loaded double-network hydrogel for the local delivery of antibiotics against Gram-positive bacteria. Covalent polymerization of a cationic monomer constituted the first network, whereas imine bond formation between the aldehyde end group of Pluronic F127 (F127-CHO) and the amine of polyethyleneimine constituted the second network. The hydrogel was loaded with the antibiotic ampicillin. Ampicillin was conjugated by forming a reversible imine bond with F127-CHO, which allowed its sustained release over an extended period of time. The antibiotic's release was dependent on the initial amount of drug conjugated in the hydrogel and was greater at pH 5 than at pH 7. The hydrogel displayed excellent biocompatibility and antimicrobial activity against Gram-positive bacteria in vitro. Therefore, in the future, the proposed hydrogel could be used for in situ delivery of antibiotics in post-operative patients.
Introduction
Combating bacterial infections in vascular tissues (e.g., bone infection) and necrotic tissues (e.g., surgical site infections) is extremely challenging owing to the limited penetration of antibacterial drugs into the infection sites.1–3 Generally, treatment procedures include prolonged courses of antibacterial drugs administered orally or through intravenous injection over periods of weeks or months.4 Nevertheless, severe whole-body infection known as sepsis can emerge due to lack of effective antibiotic treatment at the infection site.5,6 Owing to the occurrence of bacterial resistance at the target site, a higher dose of antibiotic is sometimes required; however, this can have lethal side effects.7,8 Therefore, the development of biocompatible biomaterials with antibacterial properties could dramatically reduce the side effects of excessive drug loads at the target site by enabling instead their slow and sustained release. To this end, antibacterial hydrogels offer several advantages as their high-viscosity matrix allows the controlled and local release of therapeutics.9–26 Moreover, hydrogels are characterized by elevated biocompatibility, soft mechanical properties, high water content, and high capacity for absorption. In general, antibacterial hydrogels are fabricated from polymers with antibacterial properties,27,28 through incorporation of specific drugs, metal nanoparticles or a combination of the two.4,29,30 While conjugation of antibiotics in the hydrogel matrix is highly effective against bacteria, the rapid release of antibiotics limits their long-term effectiveness.Various strategies have been proposed for the long-lasting, sustained release of antibiotics, including manipulation of gel porosity and microstructure for slow diffusion,20 slow degradation for hydrogel matrix-assisted release,21 squeezing of antibiotics by a stimuli-responsive volume change of the hydrogels,22,23 and covalent conjugation of the hydrogel network with degradable linkers.24–26 Covalent conjugation of antibiotics in hydrogels has received growing attention owing to its long-lasting and controlled release of antibiotic through the dissociation of covalent bonds at a specific pH. Haldar et al. developed vancomycin-loaded injectable hydrogels to deliver antibiotics locally.4 The hydrogel was developed by mixing polydextran aldehyde with amine-functionalized chitosan through reversible imine bond formation. Vancomycin was conjugated covalently with the hydrogel network and showed sustained release over an extended period in a pH-dependent manner. Stewart et al. reported the sustained release of tobramycin, a polycationic aminoglycoside antibiotic, from a hydrogel over a period of 50 days at pH 6–8.19 Because infection sites are generally acidic in nature,4 efficient antimicrobial hydrogels should be designed for the controlled release of antibiotics under acidic conditions. While there is a previous report regarding the pH-dependent release of antibiotics for only injectable hydrogels,4 to the best of our knowledge, no one reports mechanical robust double network hydrogel which show the controlled release of antibiotics in the acidic pH.
The present study aimed to develop efficient, long-lasting antibacterial hydrogels, whose mechanical robustness was based on a double-network structure that enabled the covalent conjugation of antibiotics. The first network originated from the covalent polymerization of positively charged monomers, whereas the second network was attributed to imine bond formation between the aldehyde end of modified Pluronic F127 (F127-CHO) and the primary amine of polyethyleneimine (PEI). Accordingly, the first network maintained the mechanical properties of the hydrogel over time, which is particularly important in infections associated with wound healing, whereas the second network helped release the antibiotic at the target site in a pH-dependent manner. Ampicillin (Amp), a highly effective broad-spectrum antibiotic, was selected for this study. Because it contains a free primary amine group, it can be covalently conjugated in the hydrogel matrix through imine bond formation. Covalent conjugation allowed the sustained release of the antibiotic in a pH-dependent manner and was highly effective at killing bacteria in vitro.
Experimental
Materials
Acrylamide (AAm) and N,N′-methylenebisacrylamide (BisAA) were purchased from Bio Basic Inc. (Markham, ON, Canada). All other chemicals, including PEI (Mw = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), Pluronic F127 (Mw = 12600 g mol−1), ammonium persulfate (APS), N,N,N,N′-tetramethylethylenediamine (TEMED), and 2-(dimethylamino)ethyl methacrylate (DMAEMA), were purchased from Sigma-Aldrich (St. Louis, MO, USA).
000 g mol−1), Pluronic F127 (Mw = 12600 g mol−1), ammonium persulfate (APS), N,N,N,N′-tetramethylethylenediamine (TEMED), and 2-(dimethylamino)ethyl methacrylate (DMAEMA), were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Synthesis of cationic dimethylamino methacrylate monomer (DMA-6)
Briefly, 1 g DMAEMA (1 eq., 6.36 mmol) in 3.49 mL 1-chlorohexane (4 eq.) was mixed with 5 mL dimethyl formamide (DMF) in a single-neck round-bottom flask. The reaction mixture was refluxed for 24 h at 160 °C. The reaction was stopped, and once the mixture cooled to room temperature, ethyl acetate was added to obtain a white precipitate. The precipitate was collected via filtration and washed with hexane three times to remove unreacted 1-chlorohexane. The product was then transferred to a vacuum oven and dried.The chemical shifts (δ) obtained from the proton nuclear magnetic resonance (1H NMR) spectrum (400 MHz, CDCl3) of the synthesized DMA-6 was as follows: 0.86 ppm (3H, CH3CH2CH2CH2), 1.33 ppm (6H, CH3CH2CH2CH2), 1.74 ppm (2H, CH2CH2N(CH3)2), 1.96 ppm (3H, CH3C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 3.59 ppm (6H, N(CH3)2), 3.63 ppm (2H,CH2CH2N(CH3)2), 4.16 ppm (2H, N(CH3)2CH2CH2CO), 4.64 ppm (2H, N(CH3)2CH2CH2CO), as well as 5.66 and 6.13 ppm (2H, CH2 C(CH3).
CH2), 3.59 ppm (6H, N(CH3)2), 3.63 ppm (2H,CH2CH2N(CH3)2), 4.16 ppm (2H, N(CH3)2CH2CH2CO), 4.64 ppm (2H, N(CH3)2CH2CH2CO), as well as 5.66 and 6.13 ppm (2H, CH2 C(CH3).
Synthesis of F127-CHO
First, 6 g Pluronic F127 was dissolved in 40 mL dry dichloromethane (DCM) in a two-neck round-bottom flask under a N2 atmosphere. Then, an excess of triethylamine (10-fold molar ratio) was added to the mixture, and the temperature of the flask was brought to 0–5 °C using an ice bath. After 10 min, methanesulfonyl chloride (four-fold molar ratio) dissolved in a small amount of dry DCM was added in a dropwise fashion. The ice bath was removed, and the mixture was stirred at room temperature for an additional 24 h. Next, the reaction mixture was washed with water five times to remove side products, and the organic fraction was collected and concentrated under vacuum. The concentrated mixture was precipitated over chilled diethyl ether; the product was collected by centrifugation and then dried in vacuum for 1 day at 40 °C.The obtained white product was dissolved in dry DMF with K2CO3 (5-fold molar ratio) and 4-hydroxybenzaldehyde (three-fold molar ratio). The reaction mixture was refluxed at 80 °C for 48 h and washed with water and DCM five times to remove side products, and the organic fraction was collected and concentrated under vacuum. This concentrated mixture was then precipitated over chilled diethyl ether, and the product was collected by centrifugation. Based on 1H NMR spectra (400 MHz, CDCl3), the degree of end-functionalization was calculated to be greater than 90%: δ = 9.88 ppm (2H, CHO), δ = 7.81 ppm (4H, aromatic ring), δ = 7.03 ppm (4H, aromatic ring), and δ = 1.14 ppm (183H, PPG methyl protons).
Preparation of hydrogels
To prepare hydrogels, F127-CHO (55 mg mL−1) and BisAA (5.88 mg mL−1) were dissolved in buffer at pH 7. Separately, AAm (54 mg mL−1), DMA-6 (63 mg mL−1), and PEI (17.64 mg mL−1) were also mixed in buffer at pH 7. Both solutions were degassed to remove any dissolved oxygen and then mixed under N2 atmosphere with concomitant addition of APS and TEMED. The solution was quickly transferred into a mold and kept under N2 atmosphere for 1 h to obtain transparent hydrogels. To prepare the ampicillin-loaded hydrogels, ampicillin at different concentrations (5 or 10 mg mL−1) was mixed with F127-CHO (F127-CHO : AMP = 1:3 for 5 mg AMP and 1:6 for 10 mg AMP), and the solution was left for 15 min at 37 °C to ensure complete conjugation of ampicillin with F127-CHO. After gel formation, the hydrogel was immersed in water for 3–4 days to remove any unreacted monomers.
Characterization
The equilibrium swelling ratio (SR) was measured for lyophilized hydrogel and determined by the following equation:| SR = meq – m0/m0, |
Fourier-transform infrared (FT-IR) spectra of the hydrogels were obtained using an Agilent Cary 630 FT-IR spectrometer (Agilent Technologies, Santa Clara, CA, USA). The samples were freeze-dried prior to the measurement. NMR measurements were conducted in CDCl3 using a Bruker Avance Neo 500 MHz spectrometer (Bruker, Billerica, MA, USA). Field emission scanning electron microscopy (FE-SEM) images of the dried hydrogels were captured using a Sigma microscope (Zeiss, Oberkochen, Germany). The samples were sputtered with gold, and images were captured. A rotational rheometer AR G2 (TA instrument, New Castle, USA) having a parallel plate geometry with a diameter and gap of 20 mm 0.55 mm respectively was used to perform the rheological measurements. A solvent trap was used to prevent water evaporation during rheological measurements.
Agar diffusion test
Antimicrobial activity was tested in accordance with the Clinical Laboratory Standards Institute manual using disk diffusion and growth curve measurements. Bacillus subtilis Gram-positive bacteria were grown on cation-adjusted Mueller-Hinton broth (CAMHB) agar, from which bacterial colonies were selected using a sterile inoculating loop and suspended in 10 mL sterile broth medium for overnight incubation at 35 °C. On the next day, when the cultures’ optical density at 600 nm (OD600) reached 0.4–0.5, bacteria were spread on dried agar plates. Hydrogels were placed on the surface of the agar plates, and they were incubated for 24 h at 35 °C. Antibacterial activity was measured by evaluating the bacterial growth inhibition zone around the hydrogel.Bacterial growth curve and colony forming unit (CFU) determination
Bacteria were grown in 100 mL CAMHB medium at 35 °C starting at OD600 = 0.1. The inoculum density was determined every 3 h for a total of 24 h by measuring OD600 using a UV/visible spectrophotometer. All experiments were performed in triplicates.For CFU determination, bacteria were cultured overnight at 35 °C in a continuous shaking incubator at 180 rpm. The next day, hydrogel samples were added at a ratio of 1 g per 50 mL bacterial solution. Samples were incubated at 35 °C with shaking for 24 h. Every 3 h, OD600 was measured using a microplate reader. The bacterial solution (100 μL) was diluted with 900 μL culture medium in serial 10-fold dilutions from 10-1 to 10-7. The diluted bacterial suspension (100 μL) was spread onto broth agar medium, and colonies were counted after incubation for 24 h at 35 °C. The following equation was used: CFU/mL = (number of colonies/dilution factor)/volume of culture solution. The dilution factor was calculated as the ratio of the bacterial suspension's volume after dilution vs. its volume before dilution.
Cell culture and proliferation assay
NIH3T3 mouse embryonic fibroblasts, human dermal fibroblasts, and newborn human foreskin fibroblasts were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (100 U mL−1) and maintained at 37 °C in a humidified atmosphere containing 5% CO2.The cytotoxicity of the hydrogel was measured using the methyl thiazolyl tetrazolium (MTT) cell proliferation assay. The hydrogel was under the same conditions as those used for the disk diffusion experiments. Cells were seeded in 12-well plates at a density of 1 × 105 cells per well. The following day, the hydrogel was added to the medium, and cells were cultured for 24 h at 37 °C in a humidified atmosphere in a CO2 incubator. After incubation, the cells were observed under a microscope, the hydrogel was removed, the medium was replaced with 10 μL MTT solution at 5 mg mL−1 in 1× phosphate-buffered saline, and the cells were incubated for 4 h at 37 °C. MTT–formazan was dissolved in 100 μL dimethyl sulfoxide, and absorbance at 550 nm (A550) was measured using a microplate reader. Cell viability (%) was calculated as (A550 of treated cells/A550 untreated cells) × 100.
Results and discussion
Fig. 1 illustrates the synthetic procedure for the various components of double-network antibacterial hydrogels. The resulting hydrogel comprised a positively charged cross-linked polymer chain as the first network and covalently-linked Pluronic F127 and PEI as the second network. The positively charged DMA-6 monomer was chosen because of its inherent antibacterial properties, whereas F127-CHO and PEI were selected because of their biocompatibility and amine functionality, respectively. Branched PEI has been applied in biomedicine as a nanoparticle coating for drug and gene delivery.27 As PEI contains many free amines that remain in the protonated form at physiological pH, it can bind to the negatively charged bacterial cell wall, resulting in cell membrane disruption and lysis.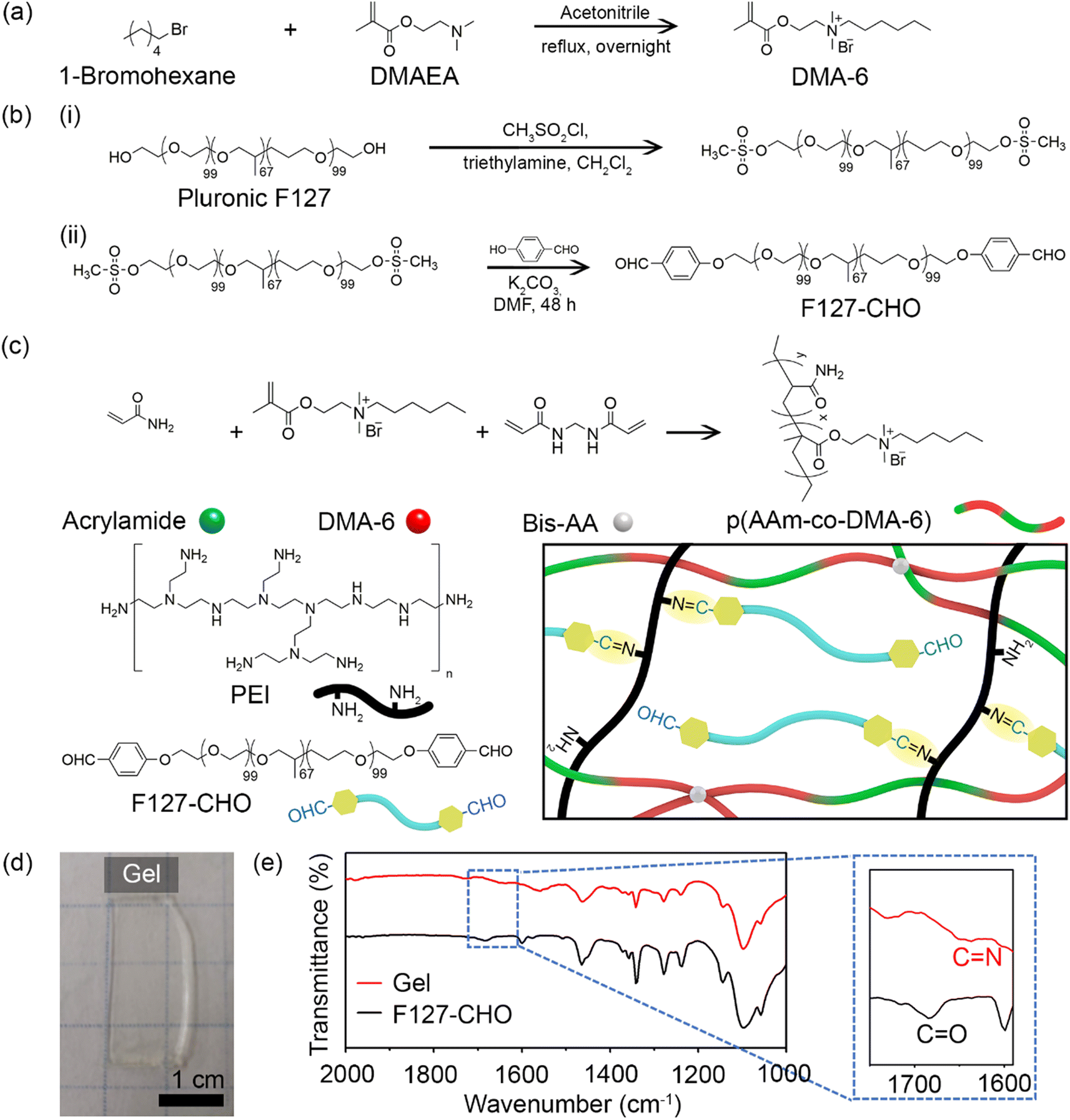 | ||
| Fig. 1 Schematic representation of the synthesis of (a) DMA-6 and (b) F127-CHO. (c) Illustration of the double-network structure of the gel. (d) Photograph of the prepared gel. (e) FT-IR spectra of the gel and F127-CHO. | ||
DMA-6 was synthesized by reacting DMAEMA with 1-chlorohexane under reflux condition for 24 h in DMF (Fig. 1a). F127-CHO was synthesized from commercially available Pluronic F127 using a two-step method (Fig. 1b). First, Pluronic F127 was treated with methanesulfonyl chloride, followed by reaction with 4-hydroxybenzaldehyde in DMF. Second, it was reacted with 4-hydroxybenzaldehyde in dry DMF in the presence of K2CO3.
The double-network resulted from the covalent polymerization of DMA-6, AAm, and BisAA as the first network and imine bond formation between the aldehyde of F127-CHO with primary amines of PEI as the second network (Fig. 1c). The total polymer concentration was 30% (w/w), and the first to second network ratio was 70:30. To prepare the hydrogels, a solution containing DMA-6, AAm, and PEI was mixed with a second solution containing F127-CHO and BisAA. After APS and TEMED were added to initiate free radical polymerization, the mixture was transferred to a mold and maintained in a N2 atmosphere for 1 h until optically transparent hydrogels were obtained (Fig. 1d). Because DMA-6 alone formed a weak hydrogel, 8% (mol mol−1) AAm was introduced to fabricate a much stronger hydrogel capable of maintaining structural integrity for several months. The swelling ratio of the hydrogel was measured to be 5.6.
To confirm imine bond formation between the aldehyde and amine groups, the F127-CHO solution and hydrogel were analyzed by FT-IR. As can be seen in Fig. 1e, the F127-CHO solution showed a peak at 1684 cm−1, corresponding to the CO in aldehyde group, but was shifted to 1650 cm−1 after gel formation with PEI. The peak at 1650 cm−1 corresponded to the CN imine bond formed during hydrogel cross-linking.
As the final goal of this study was to develop effective long-lasting hydrogels with antibacterial properties, the free primary amine group of ampicillin was allowed to form a reversible covalent imine bond with F127-CHO. This bond led to covalent conjugation of the drug in the gel network. Two different ampicillin concentrations (5 and 10 mg mL−1) were used, and ampicillin was dissolved in DMA-6 during hydrogel formation.
To form the covalent imine conjugate, F127-CHO (50 mg mL−1) and ampicillin (5 mg mL−1) were mixed and incubated at 37 °C for 1 h. The solution was then dialyzed for 24 h at 4 °C in deionized water, with water changes at regular intervals to remove any non-conjugated ampicillin. The polymer-antibiotic solution was then lyophilized, and 1H NMR spectra were recorded with D2O to confirm the covalent coupling of ampicillin with F127-CHO. The 1H NMR spectrum of ampicillin was recorded for comparison, revealing that the peaks in the aromatic region (5.0–8.0 ppm) exhibited by the polymer–drug corresponded to ampicillin peaks along with other protons from F127-CHO (Fig. 2a). This result clearly indicated that ampicillin was bound to F127-CHO via imine conjugation. The covalent conjugation of ampicillin with the hydrogel network was further confirmed by FT-IR spectra, which revealed a stronger peak at 1652 cm−1 (CN stretching) for the drug-polymer (Fig. 2b).
 | ||
| Fig. 2 (a) 1H-NMR spectra of free ampicillin (AMP), F127-CHO and ampicillin-contained F127-CHO recorded using D2O as the solvent. (b) FT-IR spectra of the hydrogel and ampicillin-containing hydrogel. (c, d) Storage modulus (G′) and loss modulus (G′′) of hydrogels as a function of (c) mechanical stress and (d) frequency. (e) SEM images of hydrogel and ampicillin-containing hydrogel. | ||
Given that some double-network hydrogels are chemically cross-linked, permanent damage to the network causes irreversible softening of the gels and poor fatigue resistance.28,29,31 To overcome this issue, non-covalent, reversible bonds are introduced. Here, a reversible imine bond was introduced in the second network, and the viscoelastic properties of the hydrogels were determined by rheological experiments. As shown in Fig. 2c, the hydrogel without ampicillin exhibited a G′ value of 750 ± 50 Pa; whereas addition of 5 and 10 mg ampicillin increased the G′ value to 1100 ± 100 Pa and 2000 ± 180 Pa, respectively. Similarly, measurements of the storage modulus (G′) and loss modulus (G′′) indicated that the G′ and G′′ values were higher for ampicillin-loaded hydrogels than unloaded hydrogels (Fig. 2c). This behavior can be attributed to the increased density in ampicillin-loaded hydrogels. Notably, the G′ value was much higher than the G′′ value throughout the frequency range (1–100 rad s−1) for all hydrogel formulations, indicating strong hydrogel formation (Fig. 2d). These results clearly suggest that the hydrogel network tends to become stronger with increasing ampicillin concentration. Moreover, ampicillin owing to the presence of amide and carboxylic acid functionality in its chemical structure can undergo H-bonding bonding interactions with the hydrogel network.32,33 This additional H-bonding interactions can affect the overall mechanical properties of the hydrogels.
The surface morphology of hydrogels with and without ampicillin was observed by FE-SEM. Following air-drying, the hydrogels without ampicillin showed a porous structure with large pores, which became smaller and denser in the case of ampicillin-loaded hydrogels (Fig. 2e). This denser network can be attributed to the conjugation between the free amines of ampicillin and the aldehyde of F127-CHO, explaining also the results obtained in the rheological experiments, whereby hydrogels became stronger as the amount of ampicillin was increased. Overall, these findings clearly indicated that ampicillin was successfully conjugated in the hydrogel network.
As previously mentioned, the antibiotic was covalently bound to the hydrogel network to allow its release over time and thereby, ensure long-lasting antibacterial activity. As illustrated in Fig. 3a, in an acidic medium (pH 5), the imine bonds between ampicillin and F127-CHO, as well as between PEI and F127-CHO, broke down, resulting in antibiotic release. Moreover, this is to notify that the pKa values for carboxylic acid and the amines are 3.24 and 7.44 respectively hence, ampicillin will exist in zwitterionic state in the pH range of pH 3.0 and 7.5. Hence, the drug release would remain same in our experimental pH values.34
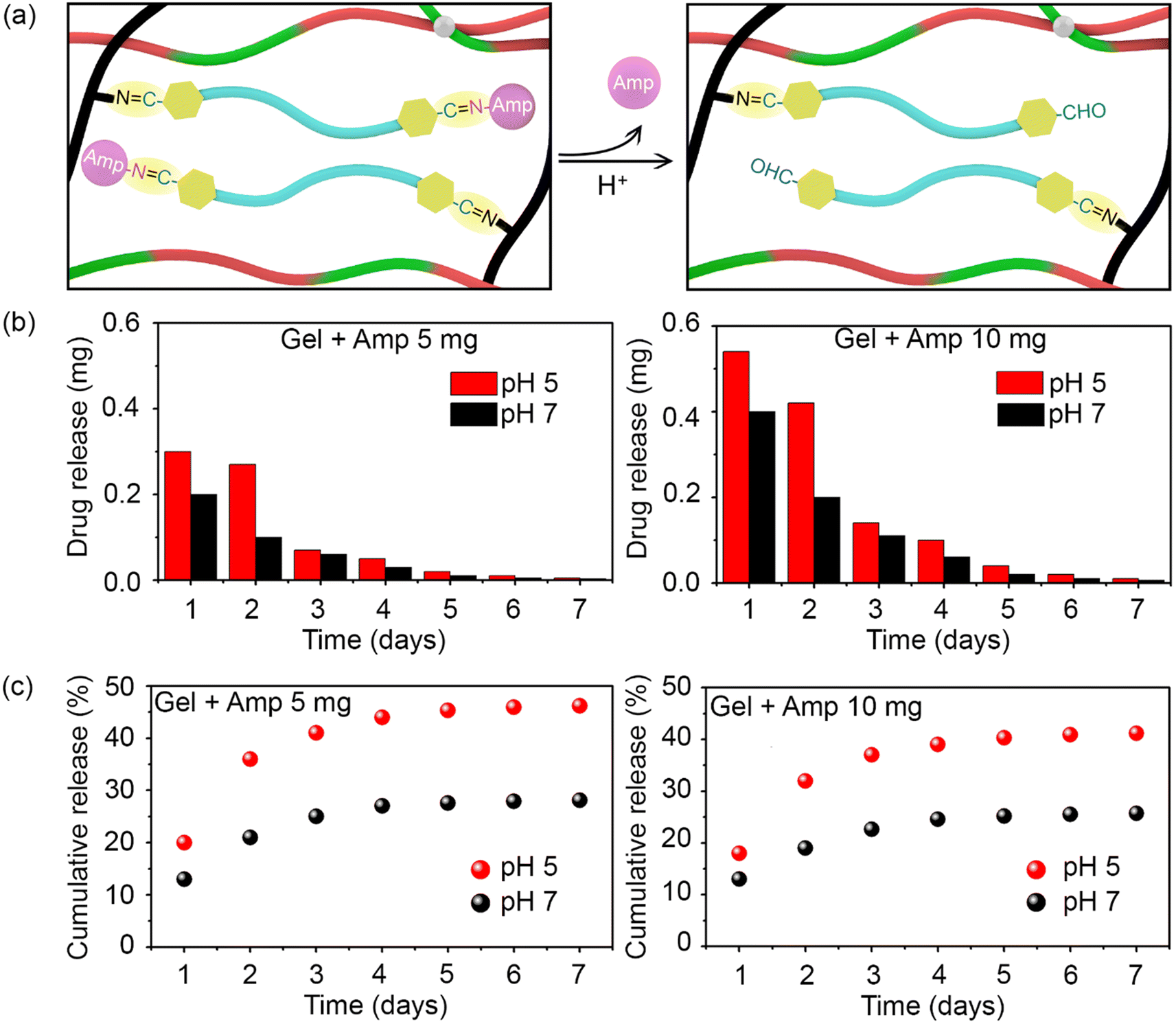 | ||
| Fig. 3 (a) Illustration of ampicillin release from encapsulation in the hydrogel network upon acidification of the medium. (b) Ampicillin release rate from the hydrogel with respect to antibiotic concentration and medium pH. (c) Cumulative release of ampicillin from the hydrogel with respect to antibiotic concentration and medium pH. | ||
To attain the controlled release of antibiotic under acidic conditions, we determined the release kinetics of the prepared hydrogels loaded with different amounts of drug (5 and 10 mg) and at pH 5 and pH 7. Hydrogels (0.4 mL) were placed in Eppendorf tubes and covered with 1 mL buffer of different pH.
The hydrogels with the buffer were incubated at 37 °C with constant shaking for 24 h, after which the upper part of the buffer was collected for UV-visible spectroscopy measurements and replaced with fresh buffer. This process was repeated every day for 7 days, and the amount of ampicillin released was quantified based on absorbance at 257 nm.
As shown in Fig. 3b, ampicillin was successfully released from the hydrogels for over 7 days in both pH groups. Ampicillin release was rapid at the initial stage and slowed down over time for all samples in a manner that depended on the initial ampicillin concentration and medium pH. The release was always higher at pH 5 than at pH 7 because the acidic buffer broke the imine bonds between the antibiotic and F127-CHO in the hydrogel network. Interestingly, the ampicillin release rate was much higher during the first and second days, tapering off after that at both pH values.
Based on the detected amount of ampicillin, its cumulative release was plotted. A higher cumulative release was measured at pH 5 than at pH 7 for both concentrations of ampicillin (Fig. 3c). Over 7 days, almost 45% and 40% of overall ampicillin present in the hydrogels at 10 mg and 5 mg, respectively, was released. In comparison, only half this amount of drug was released at pH 7. These results showcase how hydrogels in which ampicillin is conjugated through imine bonds can be used for the controlled and prolonged release of antibiotic under acidic conditions.
To confirm the antibacterial activity of the prepared hydrogels, an agar diffusion test was carried out with the Gram-positive bacterium B. subtilis. When material with antibacterial activity is placed on an agar-grown bacterial culture, bacterial growth is inhibited by the diffused antibiotic. The antibiotic activity was determined by measuring the diameter of the inhibition zone around the hydrogel. Unloaded hydrogels and hydrogels loaded with 5 and 10 mg ampicillin produced clear zones of 1.3, 2.6, and 4.3 mm in diameter, respectively (Fig. 4a). Even though a minor inhibitory effect was detected for the unloaded hydrogel, the antibacterial effect was more pronounced at a higher ampicillin concentration.
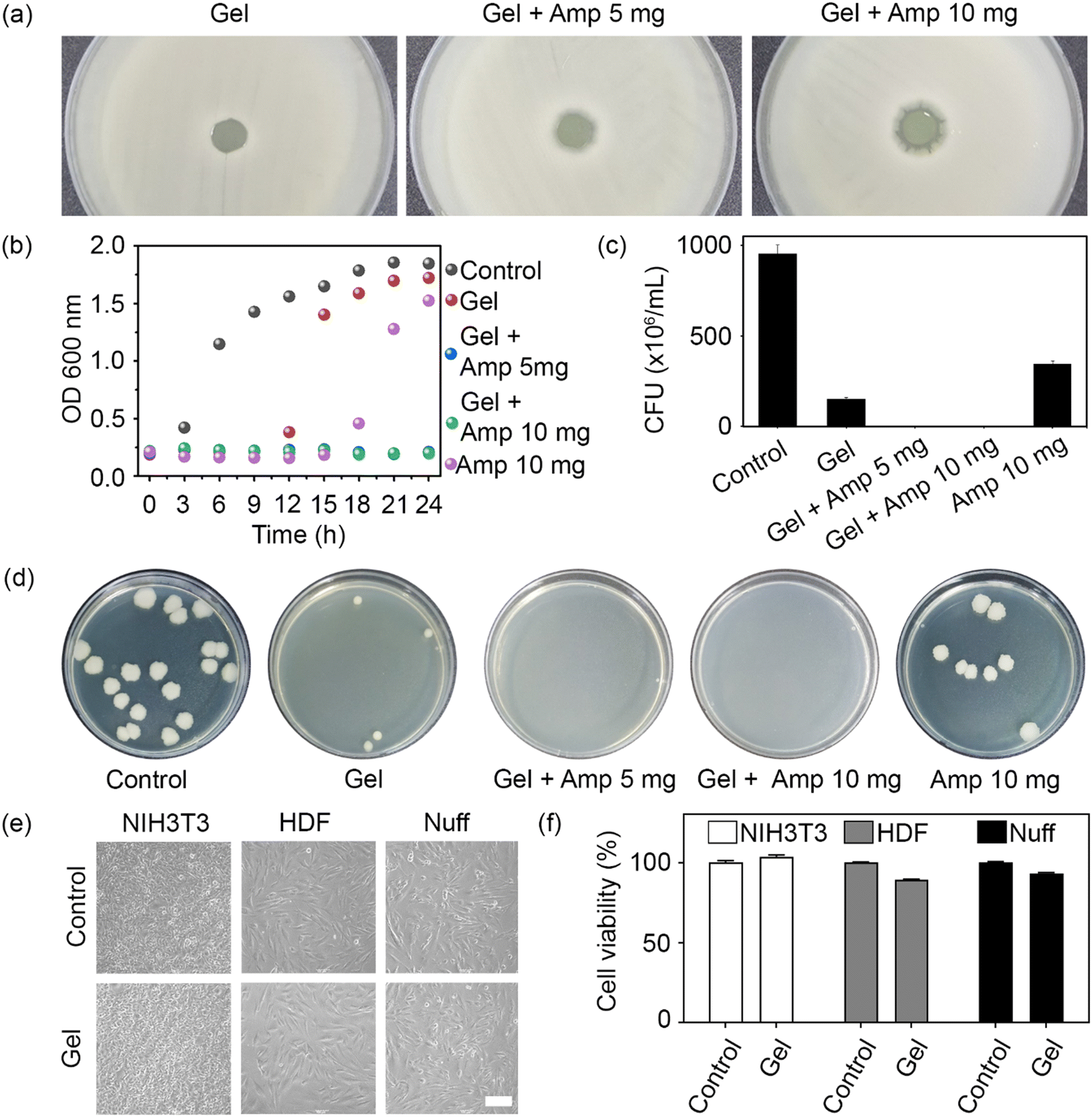 | ||
| Fig. 4 (a) Inhibition zone test using B. subtilis on agar plates; hydrogels with or without ampicillin were placed in the center of the bacterial culture, and the inhibitory halo was quantified. (b) Growth of B. subtilis in the presence of hydrogels containing ampicillin or not over a period of 24 h. (c) CFU measurement after incubation of B. subtilis with the prepared gels for 24 h. (d) Representative images of surviving B. subtilis colonies on agar plates. (e) Morphology of NIH3T3 mouse fibroblasts, human dermal fibroblasts (HDF), and newborn human foreskin fibroblasts (Nuff) exposed to the prepared hydrogels for 24 h. Scale bar = 200 μm. (f) Cytotoxicity of the prepared hydrogels toward the three types of fibroblasts. | ||
To assess the antibacterial activity of the prepared hydrogels in a three-dimensional environment, the number of live cells in a bacterial population exposed to the loaded or unloaded hydrogels over 24 h was recorded, both in the form of OD600 measurements (Fig. 4b) and CFU (Fig. 4c). While ampicillin alone (positive control) retarded bacterial growth for 15 h, no cell growth was observed in the ampicillin-loaded hydrogel groups over at least 24 h owing to the long-lasting antibacterial activity of the hydrogel (Fig. 4b). This finding was consistent with the outcome of the agar diffusion test. Similarly, CFU counts confirmed the absence of live bacteria when exposed to hydrogels loaded with either 5 or 10 mg ampicillin (Fig. 4c). It should be noted that the number of bacteria in the unloaded hydrogel group was lower than in the ampicillin control, suggesting that the positively charged chain and PEI backbone conferred some basic antibacterial activity to the hydrogel network. Therefore, a synergistic antibacterial effect was attributed to the chemical structure of the hydrogel and the loaded antibiotic, which endowed the prepared hydrogels with superior antimicrobial activity.
To evaluate the biocompatibility of the unloaded hydrogel, mouse and human fibroblasts were cultured with the hydrogel for 24 h. No morphological differences were observed between the control cells and those exposed to unloaded hydrogels (Fig. 4e). Finally, an MTT assay was performed to determine the cytotoxicity of the hydrogels toward mouse and human fibroblasts. The unloaded hydrogel did not exhibit any cytotoxicity toward the tested cell lines (Fig. 4f). Taken together, these results show that the unloaded hydrogel had a negative effect only on bacteria but displayed an otherwise excellent biocompatibility toward mammalian cells.
Conclusions
In summary, the present study describes the development of antibiotic-loaded antimicrobial hydrogels based on a double-network structure. The hydrogels integrated a first network composed of a positively charged monomer obtained via free radical polymerization, with a second network generated via a reversible imine bond between the free amines of PEI and the aldehyde of F127-CHO. The antibiotic ampicillin was loaded on the hydrogel through reversible imine bond formation. This pH-sensitive bond enabled the effective long-lasting release of antibiotic in an acidic environment. Importantly, the prepared hydrogels showed excellent antimicrobial activity against Gram-positive bacteria, while at the same time allowing for strong biocompatibility toward mammalian cells. Hence, the proposed hydrogels promise materials with long-lasting antibiotic activity and mechanical integrity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Ministry of Science and ICT, Ministry of Education, and the National Research Foundation of Korea through the Basic Science Research Program (grants 2019R1I1A2A01040856 and 2022R1A2C2008256).References
- E. S. R. Darley and A. P. MacGowan, J. Antimicrob. Chemother., 2004, 53, 928–935 CrossRef CAS PubMed.
- R. L. Smith, D. J. Schurman, G. Kajiyama, M. Mell and E. Gilkerson, J. Bone Joint Surg. Am., 1987, 69, 1063–1068 CrossRef CAS PubMed.
- C. D. Owens and K. Stoessel, J. Hosp. Infect., 2008, 70, 3–10 CrossRef PubMed.
- J. Hoque, B. Bhattacharjee, R. G. Prakash, K. Paramanandham and J. Haldar, Biomacromolecules, 2018, 19, 267–278 CrossRef CAS PubMed.
- D. E. Reichman and J. A. Greenberg, Rev. Obstet. Gynecol., 2009, 2, 212–221 Search PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- R. S. Howell-Jones, M. J. Wilson, K. E. Hill, A. J. Howard, P. E. Price and D. W. Thomas, J. Antimicrob. Chemother., 2005, 55, 143–149 CrossRef CAS PubMed.
- D. Stengel, K. Bauwens, J. Sehouli, A. Ekkernkamp and F. Porzsolt, Lancet Infect. Dis., 2001, 1, 175–188 CrossRef CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS PubMed.
- Y. Li, K. Fukushima, D. J. Coady, A. C. Engler, S. Liu, Y. Huang, J. S. Cho, Y. Guo, L. S. Miller, J. P. K. Tan, P. L. R. Ee, W. Fan, Y. Y. Yang and J. L. Hedrick, Angew. Chem., Int. Ed., 2013, 52, 674–678 CrossRef CAS PubMed.
- K. Vulic and M. S. Shoichet, J. Am. Chem. Soc., 2012, 134, 882–885 CrossRef CAS PubMed.
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Biomaterials, 2010, 31, 1444–1452 CrossRef CAS PubMed.
- J. Guo, W. Wang, J. Hu, D. Xie, E. Gerhard, M. Nisic, D. Shan, G. Qian, S. Zheng and J. Yang, Biomaterials, 2016, 85, 204–217 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, M. Chen, H. Gong, S. Thamphiwatana, L. Eckmann, W. Gao and L. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 18367–18374 CrossRef CAS PubMed.
- I. Stefanov, S. Pérez-Rafael, J. Hoyo, J. Cailloux, O. O. S. Pérez, D. Hinojosa-Caballero and T. Tzanov, Biomacromolecules, 2017, 18, 1544–1555 CrossRef CAS PubMed.
- S. Q. Liu, C. Yang, Y. Huang, X. Ding, Y. Li, W. M. Fan, J. L. Hedrick and Y.-Y. Yang, Adv. Mater., 2012, 24, 6484–6489 CrossRef CAS PubMed.
- M. C. Giano, Z. Ibrahim, S. H. Medina, K. A. Sarhane, J. M. Christensen, Y. Yamada, G. Brandacher and J. P. Schneider, Nat. Commun., 2014, 5, 4095 CrossRef CAS PubMed.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
- D. D. Lane, A. K. Fessler, S. Goo, D. L. Williams and R. J. Stewart, Acta Biomater., 2017, 50, 484–492 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- C. S. Kwok, T. A. Horbett and B. D. Ratner, J. Controlled Release, 1999, 62, 301–311 CrossRef CAS PubMed.
- W.-C. Liao, S. Lilienthal, J. S. Kahn, M. Riutin, Y. S. Sohn, R. Nechushtai and I. Willner, Chem. Sci., 2017, 8, 3362–3373 RSC.
- H. Liu, X. Shi, D. Wu, F. K. Khshen, L. Deng, A. Dong and J. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 19700–19711 CrossRef CAS PubMed.
- R. G. Schoenmakers, P. Van De Wetering, D. L. Elbert and J. A. Hubbell, J. Controlled Release, 2004, 95, 291–300 CrossRef CAS PubMed.
- Y. Yeo, T. Ito, E. Bellas, C. B. Highley, R. Marini and D. S. Kohane, Ann. Surg., 2007, 245, 819–824 CrossRef PubMed.
- K. Nakamae, T. Nishino, K. Kato, T. Miyata and A. S. Hoffman, J. Biomater. Sci. Polym. Ed., 2004, 15, 1435–1446 CrossRef PubMed.
- H. Du, G. Zha, L. Gao, H. Wang, X. Li, Z. Shen and W. Zhu, Polym. Chem., 2014, 5, 4002–4008 RSC.
- C. J. Waschinski, S. Barnert, A. Theobald, R. Schubert, F. Kleinschmidt, A. Hoffmann, K. Saalwächter and J. C. Tiller, Biomacromolecules, 2008, 9, 1764–1771 CrossRef CAS PubMed.
- A. GhavamiNejad, C. H. Park and C. S. Kim, Biomacromolecules, 2016, 17, 1213–1223 CrossRef CAS PubMed.
- V. W. L. Ng, J. M. W. Chan, H. Sardon, R. J. Ono, J. M. García, Y. Y. Yang and J. L. Hedrick, Adv. Drug Delivery Rev., 2014, 78, 46–62 CrossRef CAS PubMed.
- C. Maiti, K. B. C. Imani and J. Yoon, ChemPlusChem, 2021, 86, 601–611 CrossRef CAS PubMed.
- M. Catauro, M. G. Raucci, D. de Marco and L. Ambrosio, J. Biomed. Mater. Res., Part A, 2006, 77A, 340–350 CrossRef CAS PubMed.
- E. Khan, A. Shukla, A. Srivastava, Shweta and P. Tandon, New J. Chem., 2015, 39, 9800–9812 RSC.
- G. Malloci, G. Serra, A. Bosin and A. V. Vargiu, Computation, 2016, 4, 5 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |