
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface composition of size-selected sea salt particles under the influence of organic acids studied in situ using synchrotron radiation X-ray photoelectron spectroscopy†
Minna
Patanen
 *a,
Isaak
Unger
bc,
Clara-Magdalena
Saak
bd,
Geethanjali
Gopakumar
b,
Rebecka
Lexelius
e,
Olle
Björneholm
b,
Matthew
Salter
fg and
Paul
Zieger
fg
*a,
Isaak
Unger
bc,
Clara-Magdalena
Saak
bd,
Geethanjali
Gopakumar
b,
Rebecka
Lexelius
e,
Olle
Björneholm
b,
Matthew
Salter
fg and
Paul
Zieger
fg
aNano and Molecular Systems Research Unit, Faculty of Science, University of Oulu, P. O. Box 3000, 90014, Finland. E-mail: minna.patanen@oulu.fi; Tel: +358 294481326
bDepartment of Physics and Astronomy, Uppsala University, Box 516, 75120 Uppsala, Sweden
cDeutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22763 Hamburg, Germany
dUniversity of Vienna, Department of Physical Chemistry, Währinger Straße 42, 1090 Vienna, Austria
eDepartment of Cell and Molecular Biology, Uppsala University, Box 596, 75124 Uppsala, Sweden
fDepartment of Environmental Science, Stockholm University, 10691 Stockholm, Sweden
gBolin Centre for Climate Research, 10691 Stockholm, Sweden
First published on 30th July 2022
Abstract
Sea spray aerosols play a key role in the climate system by scattering solar radiation and by serving as cloud condensation nuclei. Despite their importance, the impact of sea spray aerosols on global climate remains highly uncertain. One of the key knowledge gaps in our understanding of sea spray aerosol is the chemical composition of the particle surface, important for various atmospheric chemical processes, as a function of size and bulk composition. Here, we have applied X-ray photoelectron spectroscopy (XPS) to determine the surface composition of both pure inorganic sea salt aerosols and sea salt aerosols spiked with an amino acid (phenylalanine) and a straight chain fatty acid (octanoic acid). Importantly, the use of a differential mobility analyser allowed size-selection of 150, 250 and 350 nm monodisperse aerosol particles for comparison to polydisperse aerosol particles. We observed enrichment of magnesium at the particle surfaces relative to chloride in all aerosols tested, across all particle sizes. Interestingly, the magnitude of this enrichment was dependent on the type of organic present in the solution as well as the particle size. Our results suggest that the observed enrichment in magnesium is an inorganic effect which can be either enhanced or diminished by the addition of organic substances.
Environmental significanceSea spray aerosol is a complex mixture of organic matter and inorganic salts, and is one of the most numerous natural aerosols in the atmosphere. Sea spray aerosol particles are involved in various atmospheric chemical processes. They play an important role in the Earth's radiation budget, especially due their hygroscopicity and thus capacity to serve as cloud condensation nuclei. Many aerosol particle characterisation techniques probe integrated particle properties such as size and total elemental content. In contrast, the interaction of aerosol particles with their environment is often determined by their surface chemical composition. Through the use of high brilliance synchrotron radiation, we have carried out surface characterisation of size-resolved laboratory generated free flying sea spray aerosol particles. |
1 Introduction
Bubble bursting processes at the ocean surface eject sea spray aerosol particles to the atmosphere, where they are involved in a multitude of physical and chemical processes.1,2 Bubble bursting produces mainly two types of droplets, film and jet droplets, which have their own typical size distributions and compositions.3–6 Film droplets are formed when the bubble cap bursts, and they contain typically more organics and are smaller than the jet droplets produced when the bubble cavity collapses.4,7 Thus, sea spray aerosols are a complex mixture of organic matter and inorganic salts and their representation in atmospheric models is challenging.8,9 Several studies have reported that certain ions or compounds are enriched in sea spray aerosol particles compared to bulk seawater.10,11 Further, this enrichment has been shown to depend on particle size11 which also affects key properties such as particle hygroscopicity.12Considering that interactions with gas phase species like water pickup or heterogeneous chemical reactions depend on the composition of the particle surface adds another layer of difficulty.13,14 However, it also highlights that obtaining a better understanding about the factors shaping the surface of sea spray aerosol is imperative in order to further our understanding of atmospherical processes. We opted to use X-ray photoelectron spectroscopy (XPS) in the present study due to its high chemical and surface sensitivity. We studied in situ generated aerosol particles as a free-flying stream in order to avoid any X-ray induced charging and damage, or substrate and deposition effects. This also implies that the particle density in the interaction region was very small (order of 106 particles per cm3) compared to studies where particles have been deposited on substrates. Thus, these types of experiments benefit from high brilliance light sources, like synchrotron radiation facilities and free-electron lasers (FELs), which have revolutionised studies of dilute matter. The experimental techniques available at these facilities have a great potential to provide highly accurate data on atmospheric aerosols and can shed light on fundamental particle formation processes, particle composition and morphology. For example, today it is possible to carry out high resolution experiments in the gas phase on atmospheric precursor molecules and clusters15,16 and size-selected cluster ions,17 follow reactant formation isomers selectively,18 and image single aerosol particles.19 Synchrotron radiation XPS studies of atmospheric aerosols span the range from organic droplets20 to soot particles21 and inorganic salt systems.22–25 Recently, we reported an XPS study on free-flying nanoparticles generated with an atomiser from a model sea salt aqueous solution.26 The results indicated that dried particles had a core–shell structure, which can be modified through the addition of an organic acid.
Here, we have carried out the first size-selected core-level XPS measurements of atomiser-generated sea salt nanoparticles and studied the effect of two organic compounds, octanoic acid and phenylalanine, on them. The aim was to determine whether there was a size-dependence in the surface chemical composition of pure inorganic sea salt particles, and whether the surface propensity of inorganic ions could be influenced by these organic substances11,27 in a similar way to that observed on liquid surfaces.28 We have concentrated on size-selected particles with dry diameters in the 150–350 nm range. Based on the studied 0.6 M sea water salt concentration, we estimate these particles to originate from droplets with their wet diameters in the 1–2 μm range. This is an interesting size range for SSA where both sea salt and organic content are important.10 Smaller SSA droplets typically contain a higher fraction of organics.10,29 However, in our experiments theaerodynamic lens setup used limited the particles we could study to those with dry diameters larger than 100.
2 Materials and methods
2.1 Aerosol generation
We have atomised three aqueous solutions using an aerosol generator (model 3076, TSI Inc., USA) operated with nitrogen gas at 2.8 bar (Air Liquide, purity ≥ 99.999%). The first solution was pure Sigma sea salt aqueous solution made by dissolving 17.5 g of sea salt (ACS reagent grade, Sigma-Aldrich, S9883) in 500 ml of MilliQ water (specific resistance of 18.2 MΩ cm). In the second experiment, we mixed Sigma sea salt with 0.05 M phenylalanine (Phe) (2.06 g Phe and 8.75 g sea salt in 250 ml MilliQ), which led to a solution with a pH of 7.8. In the third experiment, we added ∼0.003 M octanoic acid (Oct) to the aqueous sea salt solution (0.205 g octanoic acid + 17.5 g Sigma sea salt in 500 ml MilliQ water). The pH of this solution was 5.9.2.2 Measurement of monodisperse aerosols using a differential mobility analyser
Fig. 1 shows the setup of the monodisperse aerosol experiments. Atomiser generated aerosol was dried using two silica diffusion driers after which the particle stream entered a differential mobility analyser (DMA, TSI Inc., USA, type 3008003 equipped with a long column 3081 and a soft X-ray neutralizer source 3088) via an impactor with a cutoff of approximately 1 μm. The DMA was operated with a sheath to sample flow ratio of approximately 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (sheath flow: 2 l min−1, sample flow 0.58 l min−1). With the internal software, quasi-monodisperse particles with aerodynamic diameters of 150 nm, 250 nm and 350 nm were selected by varying the voltage applied to the DMA. The size-selected aerosol was directed from the outlet of the DMA into the flow limiting orifice (270 μm in diameter) of the aerodynamic lens. To inititally determine the particle number size distribution a condensation particle counter (CPC, model 3786, TSI Inc., USA) was connected after the DMA.
:1 (sheath flow: 2 l min−1, sample flow 0.58 l min−1). With the internal software, quasi-monodisperse particles with aerodynamic diameters of 150 nm, 250 nm and 350 nm were selected by varying the voltage applied to the DMA. The size-selected aerosol was directed from the outlet of the DMA into the flow limiting orifice (270 μm in diameter) of the aerodynamic lens. To inititally determine the particle number size distribution a condensation particle counter (CPC, model 3786, TSI Inc., USA) was connected after the DMA.
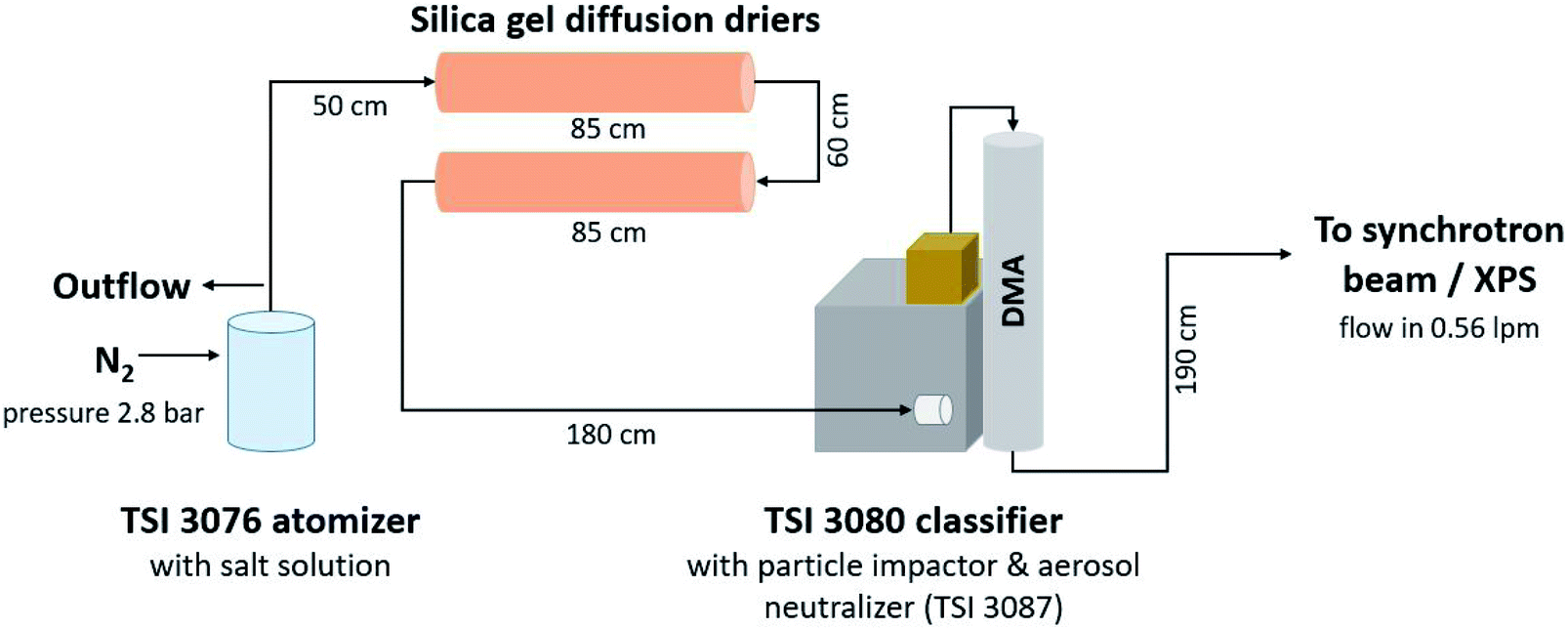 | ||
| Fig. 1 Schematic of the experimental set-up used to probe polydisperse and monodisperse particles at 350 nm, 250 nm and 150 nm using a differential mobility analyzer (DMA). For the polydisperse measurements, the DMA was bypassed. | ||
2.3 Measurement of polydisperse aerosols
For pure inorganic sea salt aerosol, three different schemes to measure polydisperse particles were tested. First, the particles were directed to the aerodynamic lens directly after the drying stage. Second, an impactor with a cutoff of about 1 μm (aerodynamic diameter) was placed before a flow limiting orifice of the aerodynamic lens. Third, in addition to the impactor, the particle stream also flew through a soft X-ray neutraliser. As discussed below, these three schemes resulted in closely similar enrichment values, and the results obtained with the soft X-ray neutralised and the impactor are shown in the elemental ratio and enrichment graphs. Polydisperse aerosol particles generated from a solution with phenylalanine were measured directly after they pass through the diffusion driers. Both the X-ray neutraliser and the impactor were used in the polydisperse measurements of inorganic sea salt and octanoic acid. The polydisperse size distributions were processed and corrected for multiple-charge effects using the manufacturer's software (AIM Ver. 9, TSI Inc., USA).2.4 Measurements of the photoelectron spectra
The experiments were carried out at the French national synchrotron radiation laboratory SOLEIL (Saint-Aubin, France) at the soft X-ray beamline PLEIADES.30 An Apple II-type permanent magnet HU80 (80 mm period) undulator produced linearly polarised light with a 55° angle between the polarisation vector and detection axis of the VG-Scienta R4000 hemispherical electron energy analyser. This configuration ensured that the recorded partial photoionisation cross-sections are independent of the photoionisation asymmetry parameter β.The spectrometer's contribution to instrumental broadening was always 400 meV during the experiments, which was determined by a pass energy of 200 eV and 0.8 mm curved entrance slits of the electron analyser. These settings offered a reasonable compromise between the resolution and the transmission. The exit slit of the monochromator was kept constant at 600 μm, and resulted in a broadening of 400–1000 meV depending on the photon energies. For each core level XPS measurement, the photon energy was set approximately 70 eV above the ionisation threshold of that level to ensure that the electron escape depth remained similar to all experiments. This means that for Mg 2p, Cl 2p, and C 1s experiments, photon energies of 135 eV, 273 eV, and 360 eV were used. The inelastic mean free path is approximately 0.9 nm at 70 eV in NaCl.31
A collimated beam of isolated, dry sea salt nanoparticles was focused to the interaction volume with the synchrotron light using an aerodynamic lens installed on the so-called multi purpose source chamber (MPSC).32 A low limiting orifice of 270 μm was used, and the aerosol flow was approximately 0.6 l min−1 before the orifice. A 2 mm skimmer between the MPSC and the experimental chamber housing the spectrometer was used to skim away most of the N2 carrier gas. The pressure in the experimental chamber was around 2.5 × 10−6 mbar during the experiments.
The photoelectron spectra were recorded in short sequences (2–12 sweeps per level in one sequence) so that possible fluctuations in the intensity of the nanoparticle beam would not affect the final intensity ratios between the levels. The nanoparticle beam intensity was monitored using a Faraday cup installed on the axis to the beam. The spectra were normalised with respect to the number of sweeps and photon flux measured using an AXUV 100 photodiode (IRD Inc., USA).
2.5 Calculation of the surface enrichment
In order to present the composition of the particle surface in a simple numerical form, we have defined two parameters from the experimental spectroscopic data: ratio R and enrichment ER. Ratio R is defined as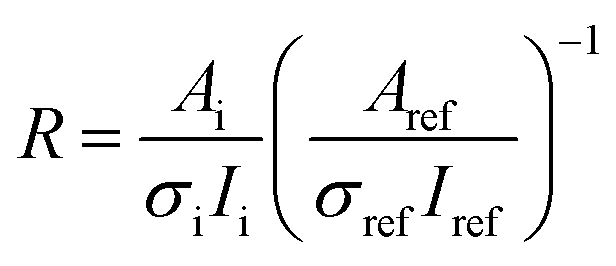 | (1) |
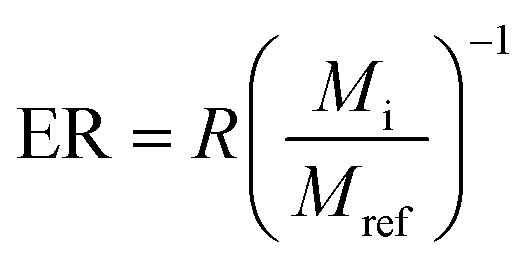 | (2) |
Error bars represent the estimated statistical error of the XPS measurement. It is assumed that the measurement follows Poisson statistics and that the error in the area A of the given photoelectron peak can be taken as the square root of the normalised peak area (total number of counts divided by the number of acquisitions). Generally, the error bars are about ±6–11% in the case of monodisperse aerosol experiments. As the count rate was much higher during experiments using polydisperse aerosol particles, the statistical errors are smaller, 1–2%. It should be noted that these error bars present only the uncertainty in the spectral acquisition. In general, the atomiser provided a stable flow of particles. The effect of possible fluctuations in particle density in the interaction region was further minimised by taking short acquisitions of Mg 2p, Cl 2p, and C 1s XPS in a repetitive manner. Thus, slow variations in particle density affected recording of all levels in the same way.
3 Results
3.1 Aerosol size distribution measurements
Fig. 2 presents the surface size distributions of the three sea salt solutions used during the experiments along with single lognormal fits to these data. The corresponding number size distributions are presented in the ESI.† We observed an interesting increase in the mean of the mode (Dmode) between particles generated from pure sea salt solution and the two solutions with added organics. This effect has been observed previously by McNeill et al.35 An explanation for this observation may be that long-chain acids inhibit the evaporation of the particle's water film causing generally larger particles that may still contain water. Corresponding to the increase in Dmode, the increase in the particle volume is so large (160% and 110% for phenylalanine and octanoic acid solutions, respectively), that the additional organic layer with the used concentrations is unlikely to be the only contributing factor to the increased particle size. The relative humidity of the particle stream was not measured during this experiment, but in a previous experiment with the same drying setup it remained constant at 3%, measured before the flow limiting orifice of the aerodynamic lens.26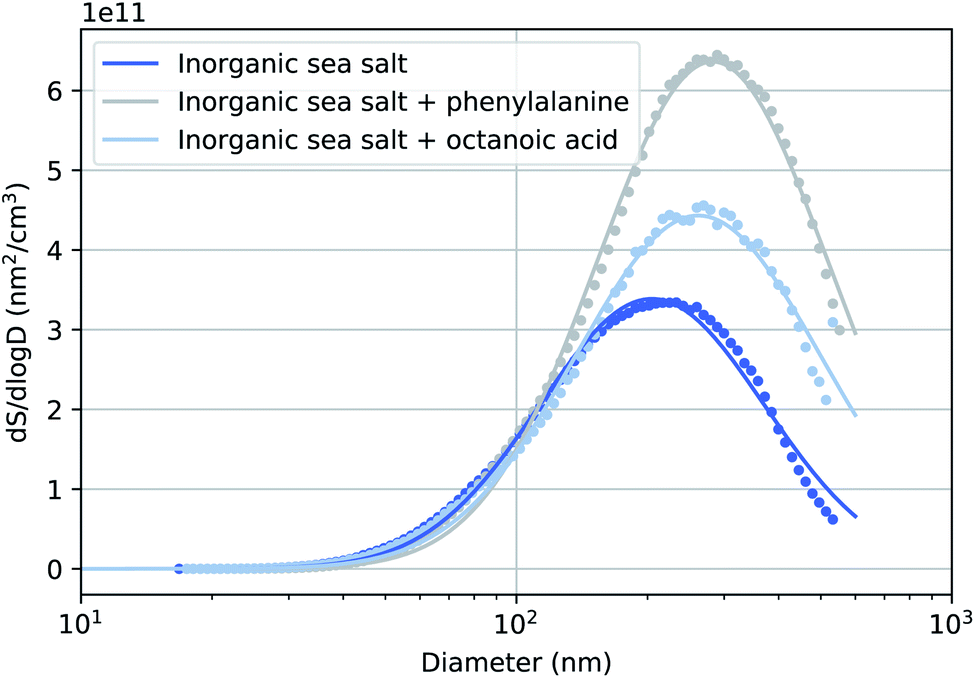 | ||
| Fig. 2 Surface size distribution vs. electrical mobility diameter measurement using a scanning mobility particle sizer (SMPS) for sea spray particles generated using a TSI 3076 aerosol generator. Single lognormal fits to these data are also presented (fit coefficients: total particle surface S = 2.2 × 1011 nm2 cm−3, 4.2 × 1011 nm2 cm−3, 3.1 × 1011 nm2 cm−3, mode diameter Dmode = 205 nm, 282 nm, and 262 nm; geometric standard deviation σ = 1.8, 1.8, and 1.9 for inorganic sea salt, inorganic sea salt + phenylalanine and inorganic sea salt + octanoic acid, respectively). | ||
3.2 Relative abundance of elements on the particle surface
Fig. 3 presents the cross-section and photon flux normalised Mg-to-Cl and C-to-Cl spectral intensity ratios from eqn (1) in the studied mixtures obtained from the XPS measurements. The photoelectron spectra and examples of peak fits used to estimate the peak areas can be found in the ESI.† The C-to-Cl and C-to-Mg spectral intensity ratios are not shown for the pure inorganic sea salt due to the very low carbon signal.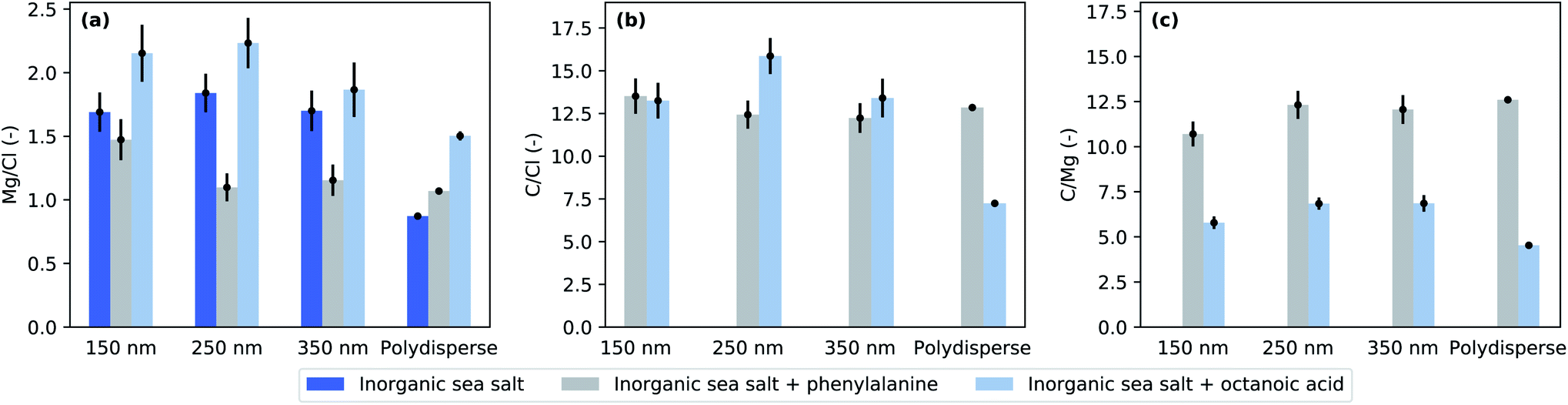 | ||
| Fig. 3 Elemental ratios at the particle surface as defined by eqn (1) for (a) magnesium to chloride, (b) carbon to chloride, and (c) carbon to magnesium. | ||
Fig. 4 presents enrichment factors calculated using Eq. 2. It is evident that Mg is highly enriched on the nanoaerosol surface at each selected size as well as in polydisperse particles. This observation is in accordance with the literature.36 However, in the case of pure inorganic sea salt and pure inorganic sea salt + octanoic acid, the polydisperse particles show the smallest enrichment. This is also true for pure inorganic sea salt + phenylalanine, but in this case the ER values of monodisperse 250 and 350 nm particles are also very close to those of polydisperse particles. The three different polydisperse measurements performed for pure inorganic particles gave slightly different enrichment factors depending on the setup used. When the aerosol was measured without the impactor and neutraliser in the path of the particle stream, an Mg-to-Cl enrichment of about 7.8 ± 0.1 was obtained. The measurement using an X-ray neutraliser resulted in an ER of 8.7 ± 0.1 and when both the impactor and neutraliser were used, the ER was 9.2 ± 0.2. All of these ER values are clearly smaller than the values obtained for size-selected particles. While the error bars for size-selected measurements are large, the observed trend indicates that the surface enrichment decreases towards larger particles, since the polydisperse particle stream contains a significant amount of surface area from micron-size particles. The used aerodynamic lens follows the design by Zhang et al.,37 and simulations predict an inlet transmission efficiency of 100% for spherical particles with diameters 80–600 nm,32 dropping gradually37 until the used impactor cuts off super-micron particles.
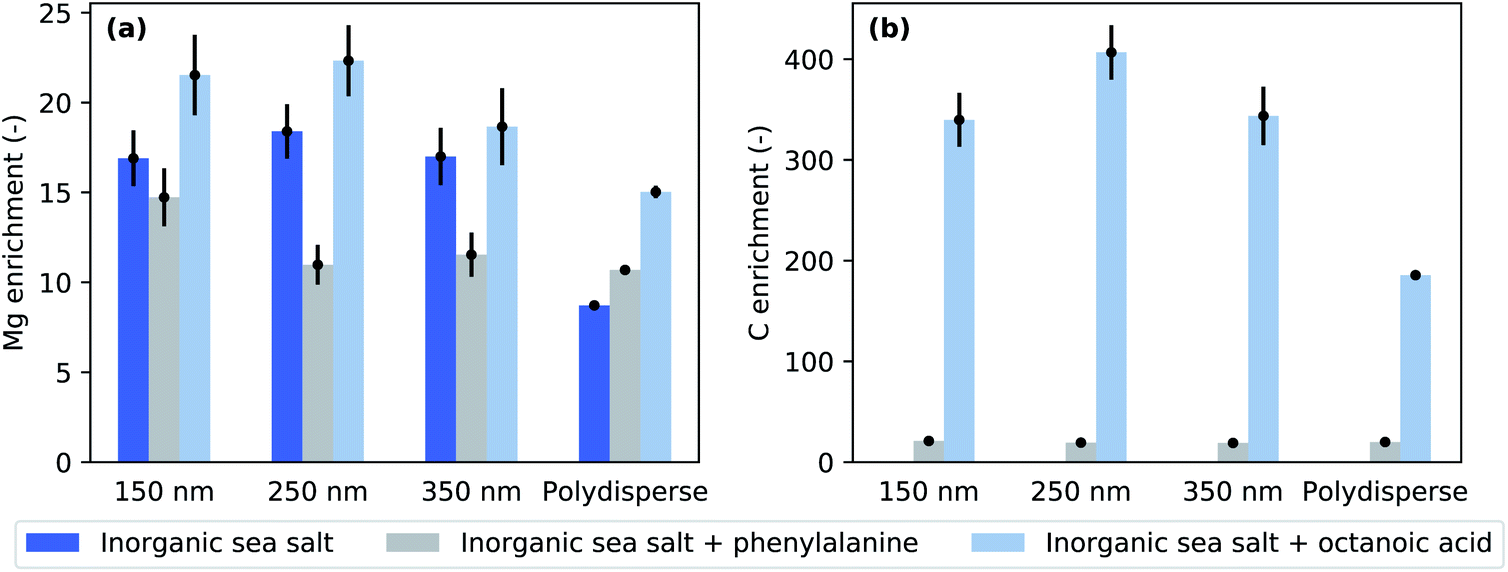 | ||
| Fig. 4 (a) Magnesium and (b) carbon enrichment factors for three probed chemical compositions and different particle sizes and polydisperse particles. | ||
Given the photon energies used, this XPS measurement is especially sensitive to the surface composition of the particles. The addition of organic compounds to the inorganic sea salt water solution modified the Mg-to-Cl ratio and consequently the Mg enrichment observed in the aerosol particles. Compared to pure sea salt particles, the addition of octanoic acid increased the Mg enrichment while the addition of phenylalanine decreased it. This effect can be achieved in two ways: in the case of octanoic acid, Mg is enriched and/or Cl depleted from the particle surface, and in the case of phenylalanine, either Mg was depleted or Cl enriched. Interestingly, the C-to-Cl ratios (Fig. 3b) for both phenylalanine and octanoic acid enriched inorganic sea salt aerosols were very close to each other, while for the polydisperse measurements the C-to-Cl ratio was lower for aerosol containing octanoic acid. As the amount of octanoic acid added was much lower than that of phenylalanine (0.003 M vs. 0.05 M), the C enrichment was respectively much higher for particles containing octanoic acid (Fig. 4b). While particles formed from phenylalanine spiked solutions did not show any size-dependence, the polydisperse particles generated from octanoic acid spiked solutions clearly exhibited a smaller enrichment than corresponding size-selected particles.
Concerning the particles formed upon addition of acids, we can use binding energy differences between the aliphatic or phenyl carbons and carboxyl carbons to deduce whether they were in acid or acetate form in the aerosol particles. In polydisperse sea salt particles with phenylalanine, we found that the α and carboxyl carbons were shifted 1.3 eV and 3.9 eV towards higher binding energies compared to phenyl carbons. These binding energy shifts are smaller than those observed for gas phase phenylalanine (1.6 and 4.55 eV (ref. 38)) and closer to the measured values of solid state zwitterionic form (approximately 1.2 and 3.4 eV (ref. 39)). Similarly, the binding energy difference between aliphatic and carboxyl carbons in sea salt particles with octanoic acid was about 3.8 eV, which is lower than expected for acid indicating a carboxylate form.34 Thus, we have an indication that it was mostly COO− groups that were present on the particle surface. However, it has to be noted that the signal of the carboxyl carbon is very small, and the integrated intensity of the fitted peak is only about 4% from the aromatic carbon signal, whereas 17% would be expected based on purely stoichiometric arguments. Thus, it is possible that the carboxyl peak is very broad since the group can have many different chemical environments and our fits do not reflect this variety of carbon species to the fullest extent. On the other hand, variation from the stoichiometry has been observed before in different systems40–44 and can be explained by various factors, e.g. a preferred orientation of molecules combined with scattering effects. Radiation damage which can similarly alter the stoichiometry39 can be ruled out in our study due to a continuously renewing beam of particles which spend less than 1 μs in the photon beam (approximately 4 × 107 photons per s per μm2)45 so that the probability for multiple ionization of the same molecule is very low.
4 Discussion
4.1 Inorganic particles
The pure inorganic sea salt solution was prepared with a concentration of 35 g l−1, and according to the specification sheet provided by the supplier, the concentration of Mg was approximately 1.2 g l−1, i.e. 0.05 M. However, the photon flux and cross-section corrected Mg XPS signal was either comparable to or up to twice the signal of Cl, even though the Cl concentration in the sea salt solution was about ten times higher (0.5 M). This is interpreted as a 10–20-fold surface enrichment of Mg, since at these photoelectron kinetic energies XPS gives information only from the depth of first 1–2 nm from the particle surface. In a similar experiment by Pelimanni et al. on particles produced via atomisation of aqueous binary salt mixtures of MgBr2 and NaBr, Mg was observed to be highly enriched over Na.25 When they atomised an aqueous solution of MgCl2 and CaCl2, the particles produced did not show any surface enrichment, but the Mg-to-Ca corresponded well to the bulk ratio at all relative concentrations of MgCl2 and CaCl2 salts studied. This behaviour showed a better correlation with differences in efflorescence dynamics than solubilities of these salts, which has also been discussed to be one factor driving the surface segregation. The efflorescence and deliquescence RHs for MgCl2 and CaCl2 are very similar, but differ for MgBr2 and NaBr. The sea salt studied here is a complex mixture of ions. However, Mg and Na are the most abundant cations, and their chloride salts are likely to be the most abundant species in the dried sea salt particles. The solubilities of NaCl and MgCl2 are very similar, being 6.1 and 5.8 mol kg−1, respectively.46 The efflorescence relative humidity (ERH) of NaCl particles has been reported to be in the range of 45–48%,47,48 whereas MgCl2 effloresces only below 2% RH48 (as a hydrate MgCl2·6H2O at 9–11% RH49). Gupta et al.50 studied 1–10 μm deposited NaCl–MgCl2 particles as a function of RH and mixing ratio. At the seawater mixing ratio (XNa = 0.9) they observed two efflorescence points, at 45.6% and 5.1%. Electron microscopy with X-ray emission elemental analysis of effloresced particles revealed a cubical Na-rich core with a Mg-rich layer on the top. The surface layer also contained oxygen, and based on Raman micro-spectrometry, it was concluded that the formed phase was MgCl2·4H2O. Our observations of in-flight analysed particles are in line with Gupta et al. Furthermore, we observed increased enrichment in size-selected particles compared to polydisperse particles. As the aerodynamic lens used in our study has better transmission for larger particles, the polydisperse particle experiment probed preferentially these larger particles. Thus, there is an indication that the surface chemical composition depends on the particle size. This is further supported by the trend observed in ER values of three different measurements on inorganic polydisperse particles. The dried aerosol measured directly after the atomiser gave the smallest ER, and the measurement carried out with both the neutraliser and the impactor gave the largest ER. When the impactor was not used, also the super-micron particles were probed. The introduction of a neutraliser may have caused particle losses especially on large particles. Thus these three experiments probed on average slightly different size distributions, and the trend in ER values is consistent with the result that larger particles show lower Mg enrichment. The slightly reduced enrichment of Mg in the polydisperse particles may be due to island growth on the surface of large particles, whereas smaller size-selected particles could be completely covered. This speculation needs verification by experiments using electron microscopy or preferably in-flight single particle imaging available at FELs. Furthermore, increasing the electron escape depth by using a substantially higher photon energy would be beneficial to clarify the composition of surface and bulk layers. This requires a very high photon flux and high particle densities to compensate the quickly decreasing photoionisation cross-section as a function of photon energy. We look forward to future development of high brilliance hard X-ray beamlines to accommodate gas-phase nanoparticle sample environments that would enable such depth-profiling experiments.4.2 Addition of organics
We have used octanoic acid as a proxy for the hydrophobic long-carbon chain organic molecules present in seawater due to decomposing marine organisms.51 Its addition to the sea salt solution further enhanced the Mg-enrichment over Cl (Fig. 4a). The acid itself was also highly enriched on the particle surfaces (Fig. 4b). Cochran et al. observed that the addition of surface active organics (including octanoic acid) to the aerosolised solution leads to the enrichment of certain inorganic species (especially Ca) and the depletion of chloride and sulfate in the aerosol phase. They observed that this effect was especially pronounced in smaller aerosol particles (diameter < 200 nm).10 They did not observe enrichment of Mg.However, their method of aerosol generation differs from our experiment, since they used a bubble-chamber which is likely to preferentially produce film drops, and thus they probed more the surface of the solution. Furthermore, their mass spectrometric elemental analysis probes the composition of the aerosol particle as a whole, while we have information only about the particle surface. In our previous study,26 we observed that in the liquid phase, Ca was present in excess on the surface of NaCl/CaCl2 solutions with and without acetic acid, but this surface enrichment in the liquid phase was not reflected in the surface chemical composition of dried sea salt aerosol particles. In contrast, upon addition of acetic acid, the Ca signal disappeared from the particle surface. The reason for this behaviour is the efflorescence points of the various salts contained in the aerosol particles. Thus, one has to be careful when comparing enrichment between liquid surfaces probed in liquid jet experiments, aerosol particles extracted mostly from the liquid surface, and atomised dried particles. First of all, our method for aerosol generation (atomiser) extracts the liquid from the bulk solution. If there is strong surface–bulk partitioning in the solution, this may lead to different compositions already in the initial droplets compared to methods which extract preferentially droplets from the surface. In addition, our XPS probe is extremely surface sensitive, and thus a direct comparison with mass spectrometry experiments which probe the total amount of species in the aerosol particles is not straightforward, especially, when the dynamics upon particle drying drive some species towards the surface and other species to the bulk. In addition, our previous work suggested that drying aerosol in free flight instead of drying them on a hydrophobic substrate, as is often done, might lead to a different morphology of the dry aerosol.26
Aerosol particles with long-chain organic compounds have been suggested to be “inverted micelles”, consisting of a highly concentrated aqueous core surrounded by a layer of organics with their hydrophobic tail pointing outwards from the particle core.51 Such structures are expected here as well, and the octanoic acid layer may prohibit complete drying of the core. This is supported by the lower than expected signal intensity from COO− compared to aliphatic carbons. However, it should be noted that deduction of the orientation of the molecules using only XPS intensities is not straightforward.44 Mg and Na can form (di)octanoate salts with octanoic acid, which are sparingly soluble in water (solubilities are 0.004 mmol l−1 and 4.7 mmol l−1 for Mg dioctanoate and Na octanoate, respectively, as estimated using ALOGPS 2.1 (ref. 52)). One possible reason for increased Mg-enrichment upon addition of octanoic acid would be that Cl is expelled from the surface due to salt formation between cations and carboxylate. Cl can form HCl, which will leave the solution owing to its volatile nature. Thus, our method, where we compare the abundance of the ions to the Cl signal instead of the more commonly used Na reference,10,11 is not ideal. However, Na XPS signal is so small that taking it as a reference would introduce large errors in the analysis. Also, addition of organic acid was shown to change the relative cation ratio on the particle surface,26 and thus, it is difficult to find an absolute constant reference for the ER.
Phenylalanine is a common amino acid present in seawater and we have used it here as a model of surface active amino acids.53 The effect of phenylalanine seems to be exactly opposite to the effect of octanoic acid. Compared to pure inorganic particles, Mg-enrichment decreases upon phenylalanine addition. Phenylalanine has both carboxylic acid and amine groups, and thus it can be present in the aqueous environment in three ionized forms: cation, zwitterion, and anion. In a recent liquid microjet experiment, it was reported that under acidic conditions, phenylalanine increases the surface propensity of Cl− in aqueous KCl solution.28 Close to neutral pH, there was no enrichment of either of the ions, K+ or Cl− compared to the same solution without phenylalanine. However, Cl− exhibited overall surface propensity in all these solutions. In our experiment, the pH of the atomised solution was 7.8, in which the zwitterionic form should be prevalent. It is therefore difficult to explain the reduced Mg-enrichment by an increase in Cl surface abundance. Again, the dried aerosol particles show different behaviour from their aqueous solution, highlighting the importance of studying aerosol particles under realistic conditions. We would have liked to quantify the water content remaining in the aerosol particles but this was not possible in our experiment. The O 1s signal from organic acids would overlap with the water signal,38,54 and very high statistics would be needed for the quantification. In pure sea salt particles, the intensity of the O 1s signal is very low in the polydisperse particles (see ESI†), indicating a very small water content.
The ionic ratios in particles generated from phenylalanine spiked solutions (Fig. 3) do not seem to be size-dependent, with only the smallest size (d = 150 nm) exhibiting a higher Mg surface content than other sizes and polydisperse particles. This differs from inorganic particles and particles with octanoic acid, which both exhibited lower enrichment with polydisperse particles. When the particle diameter gets larger, the surface-to-bulk ratio decreases. Assuming that the very surface active octanoic acid forms a monolayer on the particle surface, it can be expected that in the experiment on polydisperse particles a larger proportion of Cl is observed. As explained earlier, the polydisperse experiment probes larger sizes, which have a smaller surface-to-bulk ratio compared to smaller sizes. Thus, while Cl is more preferentially in the bulk, the lower surface-to-bulk ratio reduces the C enrichment in polydisperse particles. A similar effect is not observed in phenylalanine spiked particles. This can be due to the much higher solubility of phenylalanine than octanoic acid, meaning that phenylalanine may not form a monolayer but is highly present under the immediate particle surface. XPS analysis carried out as a function of the concentration of the organic compound would be beneficial to shed light on the organisation of the organics on the particle surface. This is left for further study, in which also the photon polarization could be varied for additional information on the surface layer orientation.
5 Conclusions
Here, we report the first X-ray photoelectron study of laboratory generated, size-selected sea salt aerosol particles. We observed the enrichment of Mg in dry aerosol particles as a pure inorganic effect, which can be amplified or suppressed depending on the organic compound chosen. Addition of phenylalanine, a common amino acid present in seawater, decreased the Mg-enrichment. A very surface active organic acid, octanoic acid, in contrast, caused an enhancement of the Mg enrichment. The results are discussed in light of experiments carried out in aqueous solutions using liquid jets as well as experiments probing the total elemental content of particles. We conclude that the enrichment behaviour observed on dried particle surfaces cannot be satisfactorily explained based on previous studies of infinite surfaces or total elemental composition. Therefore, it is important to carry out chemical analysis of actual aerosols rather than aerosols sampled on filters or proxy liquid solutions. In our experiment, we especially investigate about 2 nm of the particle surface, which changes as a function of particle size and organic content. Since the surface is relevant for the chemical processes within the particle and its surrounding, it is not enough to probe only bulk chemical processes. In future, it would be interesting to collect the particles after the XPS analysis of grids to obtain complementary information on the total elemental composition and morphology of the exactly same particles probed with XPS. Brilliant light sources, especially FELs, also enable single particle imaging experiments for studies of the particle morphology, which can be combined with mass spectroscopic analysis of particle fragments. These experiments would give further complementary information about particle shape and total chemical composition. Furthermore, the recent development of ambient pressure XPS systems will provide opportunities to study free-flying particles under controlled relative humidity conditions, which are more realistic for atmospheric applications.Author contributions
Conceptualization & methodology – Matthew Salter, Paul Zieger, Minna Patanen, Isaak Unger, Clara-Magdalena Saak, Olle Björneholm. Formal analysis – Minna Patanen, Isaak Unger. Investigation – Minna Patanen, Clara-Magdalena Saak, Paul Zieger, Geethanjali Gopakumar, Matthew Salter, Rebecka Lexelius, Isaak Unger. Resources – Paul Zieger, Matthew Salter, Olle Björneholm. Writing the original draft – Minna Patanen. Editing the final manuscript – Minna Patanen, Isaak Unger, Matthew Salter, Paul Zieger. Visualization – Paul Zieger. Funding acquisition – Minna Patanen, Matthew Salter, Paul Zieger, Olle Björneholm. All the authors commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. acknowledges the Academy of Finland for financial support. M. S. was financed by the Swedish Research Council (project number 2016-05100). P. Z. acknowledges funding form the Swedish Research Council Starting Grant 2018-05045. O. B. acknowledges the support by the Swedish Research Council (grant 2017-04162). I. U. acknowledges the support from the Carl Tryggers Foundation and thanks Stephan Thürmer for his data analysis routines, which have been used for the data analysis. The research leading to this result has been supported by the project CALIPSOplus under Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. We acknowledge SOLEIL for provision of synchrotron radiation facilities (proposal 20170532) and we thank Aleksandar Milosavljević for assistance in using the PLÉIADES beamline.Notes and references
- A. H. Woodcock, J. Meteorol., 1953, 10, 362–371 CrossRef.
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chem. Rev., 2015, 115, 4383–4399 CrossRef CAS PubMed.
- F. Knelman, N. Dombrowski and D. M. Newitt, Nature, 1954, 173, 261 CrossRef.
- D. C. Blanchard, Estuaries, 1989, 12, 127–137 CrossRef CAS.
- D. E. Spiel, J. Geophys. Res.: Oceans, 1998, 103, 24907–24918 CrossRef.
- R. Lewis and E. Schwartz, Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models—A Critical Review, American Geophysical Union, Washington, D. C., 2004, vol. 152 Search PubMed.
- R.-S. Tseng, J. T. Viechnicki, R. A. Skop and J. W. Brown, J. Geophys. Res.: Oceans, 1992, 97, 5201–5206 CrossRef CAS.
- M. Schulz, C. Textor, S. Kinne, Y. Balkanski, S. Bauer, T. Berntsen, T. Berglen, O. Boucher, F. Dentener, S. Guibert, I. S. A. Isaksen, T. Iversen, D. Koch, A. Kirkevåg, X. Liu, V. Montanaro, G. Myhre, J. E. Penner, G. Pitari, S. Reddy, Ø. Seland, P. Stier and T. Takemura, Atmos. Chem. Phys., 2006, 6, 5225–5246 CrossRef CAS.
- H. Grythe, J. Ström, R. Krejci, P. Quinn and A. Stohl, Atmos. Chem. Phys., 2014, 14, 1277–1297 CrossRef.
- R. E. Cochran, T. Jayarathne, E. A. Stone and V. H. Grassian, J. Phys. Chem. Lett., 2016, 7, 1692–1696 CrossRef CAS PubMed.
- M. E. Salter, E. Hamacher-Barth, C. Leck, J. Werner, C. M. Johnson, I. Riipinen, E. D. Nilsson and P. Zieger, Geophys. Res. Lett., 2016, 43, 8277–8285 CrossRef CAS.
- P. Zieger, O. Väisänen, J. C. Corbin, D. G. Partridge, S. Bastelberger, M. Mousavi-Fard, B. Rosati, M. Gysel, U. K. Krieger and C. Leck, et al. , Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- C. Moreno and M. T. Baeza-Romero, Phys. Chem. Chem. Phys., 2019, 21, 19835–19856 RSC.
- C. Zhang, X. Lin, X. Tang, C. Fittschen, S. Hartweg, G. A. Garcia, B. Long, W. Zhang and L. Nahon, Phys. Chem. Chem. Phys., 2022, 24, 2015–2021 RSC.
- M. Ahmed and O. Kostko, Phys. Chem. Chem. Phys., 2020, 22, 2713–2737 RSC.
- M. J. Ryding, A. Giuliani, M. Patanen, J. Niskanen, G. Simões, G. B. S. Miller, E. Antonsson, T. Jokinen, C. Miron, O. Björneholm, K. Hansen, K. J. Børve and E. Uggerud, RSC Adv., 2014, 4, 47743–47751 RSC.
- M. T. Baeza-Romero, F. Gaie-Levrel, A. Mahjoub, V. López-Arza, G. A. Garcia and L. Nahon, Eur. Phys. J. D, 2016, 70, 154 CrossRef.
- N. D. Loh, C. Y. Hampton, A. V. Martin, D. Starodub, R. G. Sierra, A. Barty, A. Aquila, J. Schulz, L. Lomb, J. Steinbrener, R. L. Shoeman, S. Kassemeyer, C. Bostedt, J. Bozek, S. W. Epp, B. Erk, R. Hartmann, D. Rolles, A. Rudenko, B. Rudek, L. Foucar, N. Kimmel, G. Weidenspointner, G. Hauser, P. Holl, E. Pedersoli, M. Liang, M. S. Hunter, L. Gumprecht, N. Coppola, C. Wunderer, H. Graafsma, F. R. N. C. Maia, T. Ekeberg, M. Hantke, H. Fleckenstein, H. Hirsemann, K. Nass, T. A. White, H. J. Tobias, G. R. Farquar, W. H. Benner, S. P. Hau-Riege, C. Reich, A. Hartmann, H. Soltau, S. Marchesini, S. Bajt, M. Barthelmess, P. Bucksbaum, K. O. Hodgson, L. Strüder, J. Ullrich, M. Frank, I. Schlichting, H. N. Chapman and M. J. Bogan, Nature, 2012, 486, 513–517 CrossRef CAS PubMed.
- M. I. Jacobs, B. Xu, O. Kostko, A. A. Wiegel, F. A. Houle, M. Ahmed and K. R. Wilson, J. Phys. Chem. A, 2019, 123, 6034–6044 CrossRef CAS PubMed.
- F.-X. Ouf, P. Parent, C. Laffon, I. Marhaba, D. Ferry, B. Marcillaud, E. Antonsson, S. Benkoula, X.-J. Liu and C. Nicolas, et al. , Sci. Rep., 2016, 6, 1–12 CrossRef.
- E. Antonsson, M. Patanen, C. Nicolas, J. J. Neville, S. Benkoula, A. Goel and C. Miron, Phys. Rev. X, 2015, 5, 011025 Search PubMed.
- E. Antonsson, C. Raschpichler, B. Langer, D. Marchenko and E. Rühl, J. Phys. Chem. A, 2018, 122, 2695–2702 CrossRef CAS PubMed.
- O. Kostko, B. Xu, M. Jacobs and M. Ahmed, J. Chem. Phys., 2017, 147, 013931 CrossRef CAS PubMed.
- E. Pelimanni, C.-M. Saak, G. Michailoudi, N. Prisle, M. Huttula and M. Patanen, Phys. Chem. Chem. Phys., 2022, 24, 2934–2943 RSC.
- I. Unger, C.-M. Saak, M. Salter, P. Zieger, M. Patanen and O. Björneholm, J. Phys. Chem. A, 2020, 124, 422–429 CrossRef CAS PubMed.
- P. Zieger, O. Väisänen, J. Corbin, D. Partridge, S. Bastelberger, M. Mousavi-Fard, B. Rosati, M. Gysel, U. Krieger, C. Leck, A. Nenes, I. Riipinen, A. Virtanen and M. Salter, Nat. Commun., 2017, 8, 15883 CrossRef CAS PubMed.
- G. Gopakumar, I. Unger, C.-M. Saak, G. Öhrwall, A. Naves de Brito, T. C. Rizuti da Rocha, C. Nicolas, C. Caleman and O. Björneholm, Environ. Sci.: Atmos., 2022, 2, 441–448 CAS.
- C. Oppo, S. Bellandi, N. Degli Innocenti, A. M. Stortini, G. Loglio, E. Schiavuta and R. Cini, Mar. Chem., 1999, 63, 235–253 CrossRef CAS.
- PLÉIADES: French National Synchrotron Facility (SOLEIL), https://www.synchrotron-soleil.fr/en/beamlines/pleiades.
- T. Boutboul, A. Akkerman, A. Breskin and R. Chechik, J. Appl. Phys., 1996, 79, 6714–6721 CrossRef CAS.
- A. Lindblad, J. Söderström, C. Nicolas, E. Robert and C. Miron, Rev. Sci. Instrum., 2013, 84, 113105 CrossRef PubMed.
- R. A. Duce, W. Stumm and J. M. Prospero, J. Geophys. Res., 1972, 77, 5059–5061 CrossRef.
- N. Ottosson, E. Wernersson, J. Söderström, W. Pokapanich, S. Kaufmann, S. Svensson, I. Persson, G. Öhrwall and O. Björneholm, Phys. Chem. Chem. Phys., 2011, 13, 12261–12267 RSC.
- V. F. McNeill, J. Patterson, G. M. Wolfe and J. A. Thornton, Atmos. Chem. Phys., 2006, 6, 1635–1644 CrossRef CAS.
- H.-S. Xiao, J.-L. Dong, L.-Y. Wang, L.-J. Zhao, F. Wang and Y.-H. Zhang, Environ. Sci. Technol., 2008, 42, 8698–8702 CrossRef CAS.
- X. Zhang, K. A. Smith, D. R. Worsnop, J. L. Jimenez, J. T. Jayne, C. E. Kolb, J. Morris and P. Davidovits, Aerosol Sci. Technol., 2004, 38, 619–638 CrossRef CAS.
- W. Zhang, V. Carravetta, O. Plekan, V. Feyer, R. Richter, M. Coreno and K. C. Prince, J. Chem. Phys., 2009, 131, 035103 CrossRef PubMed.
- Y. Zubavichus, M. Zharnikov, A. Shaporenko, O. Fuchs, L. Weinhardt, C. Heske, E. Umbach, J. D. Denlinger and M. Grunze, J. Phys. Chem. A, 2004, 108, 4557–4565 CrossRef CAS.
- M.-M. Walz, C. Caleman, J. Werner, V. Ekholm, D. Lundberg, N. L. Prisle, G. Öhrwall and O. Björneholm, Phys. Chem. Chem. Phys., 2015, 17, 14036–14044 RSC.
- M.-T. Lee, F. Orlando, L. Artiglia, S. Chen and M. Ammann, J. Phys. Chem. A, 2016, 120, 9749–9758 CrossRef CAS.
- J. Söderström, N. Mårtensson, O. Travnikova, M. Patanen, C. Miron, L. J. Sæthre, K. J. Børve, J. J. Rehr, J. J. Kas, F. D. Vila, T. D. Thomas and S. Svensson, Phys. Rev. Lett., 2012, 108, 193005 CrossRef PubMed.
- O. Björneholm, J. Werner, N. Ottosson, G. Öhrwall, V. Ekholm, B. Winter, I. Unger and J. Söderström, J. Phys. Chem. C, 2014, 118, 29333–29339 CrossRef.
- R. Dupuy, J. Filser, C. Richter, R. Seidel, F. Trinter, T. Buttersack, C. Nicolas, J. Bozek, U. Hergenhahn, H. Oberhofer, B. Winter, K. Reuter and H. Bluhm, Phys. Chem. Chem. Phys., 2022, 24, 4796–4808 RSC.
- O. Sublemontier, C. Nicolas, D. Aureau, M. Patanen, H. Kintz, X. Liu, M.-A. Gaveau, J.-L. Le Garrec, E. Robert, F.-A. Barreda, A. Etcheberry, C. Reynaud, J. B. Mitchell and C. Miron, J. Phys. Chem. Lett., 2014, 5, 3399–3403 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press LLC, 85th edn, 2004 Search PubMed.
- X. Li, D. Gupta, H.-J. Eom, H. Kim and C.-U. Ro, Atmos. Environ., 2014, 82, 36–43 CrossRef CAS.
- D. J. Cziczo and J. P. D. Abbatt, J. Phys. Chem. A, 2000, 104, 2038–2047 CrossRef CAS.
- C. Peng, L. Chen and M. Tang, Fundam. Res., 2022, 2, 578–587 CrossRef.
- D. Gupta, H.-J. Eom, H.-R. Cho and C.-U. Ro, Atmos. Chem. Phys., 2015, 15, 11273–11290 CrossRef CAS.
- G. B. Ellison, A. F. Tuck and V. Vaida, J. Geophys. Res.: Atmos., 1999, 104, 11633–11641 CrossRef CAS.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed.
- H. B. Bull and K. Breese, Arch. Biochem. Biophys., 1974, 161, 665–670 CrossRef CAS PubMed.
- R. Sankari, M. Ehara, H. Nakatsuji, Y. Senba, K. Hosokawa, H. Yoshida, A. D. Fanis, Y. Tamenori, S. Aksela and K. Ueda, Chem. Phys. Lett., 2003, 380, 647–653 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ea00035k |
| This journal is © The Royal Society of Chemistry 2022 |