
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Early-stage particle fragmentation behavior of a commercial silica-supported metallocene catalyst†
Silvia
Zanoni
 a,
Nikolaos
Nikolopoulos
a,
Alexandre
Welle
b,
Aurélien
Vantomme
b and
Bert M.
Weckhuysen
*a
a,
Nikolaos
Nikolopoulos
a,
Alexandre
Welle
b,
Aurélien
Vantomme
b and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterial Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: B.M.Weckhuysen@uu.nl
bCatalysis and Products R&D, Total Research & Technology, Zone Industrielle C, 7181 Feluy, Belgium
First published on 29th June 2021
Abstract
Efficient fragmentation of catalyst particles during the polymerization of olefins is a necessary process to maintain catalyst activity and obtain discrete polymer particles with ideal density and morphology. Collecting experimental evidence on the fragmentation process is essential for a fundamental understanding of this phenomenon. However, visualizing the very early onset of fragmentation within catalyst particles has been proven challenging. This study investigates the development of fragmentation at different reaction conditions in an industrial SiO2-supported metallocene catalyst of the type Zr/MAO/SiO2 (MAO = methylaluminoxane) early-stage of ethylene polymerization, i.e., in the range 1–20 g polyethylene per g catalyst. Focused ion beam-scanning electron microscopy was employed to visualize particle cross-sections with high 2-D resolution (i.e., a few tens of nm), showing that ethylene polymerization starts at two main fronts: one at the outer surface and one at the walls of the macropores within the particle. The relative rate of polymerization at these fronts was assessed by estimating the extent of polyethylene, catalyst, and macroporosity within each cross-section via image segmentation. When polymerization at these fronts proceeds at similar rates, fragmentation is extensive and a combination of both layer-by-layer and bi-sectioning modes; conversely, an imbalance between the two due to unideal reaction conditions, non-homogeneous active site distribution, or insufficient macroporosity leads to a sub-optimal fragmentation. This methodology revealed a detailed understanding of catalyst fragmentation for this commercial catalyst. However, it can be applied to a broader range of polymerization catalysts and parameters, amplifying the knowledge on such an intricated phenomenon.
1. Introduction
Containing only carbon and hydrogen, polyolefins are considered sustainable products thanks to their energy effectiveness and favorable cost/performance ratio, provided they can be recycled and made from more sustainable resources, including municipal and agricultural waste.1–3 In 2018, they reached a yearly global production of almost 180 million tonnes, making them the materials with the highest production volume in the plastics market.4,5 Metallocene-based catalysts, particularly zirconocenes, are highly active for the polymerization of olefins when combined with a co-catalyst, such as methylaluminoxane (MAO).6–9 Thanks to their tunable and well-defined active site nature, metallocene-based olefin polymerization catalysts are used for the production of high-grade polyolefins with narrow molecular weight distribution and controlled microstructure, presenting an essential alternative to the more commonly used Ziegler–Natta (TiCl4/MgCl2) and Phillips (CrO3/SiO2) olefin polymerization catalysts.10–16However, polyolefins need to be synthesized as discrete particles with homogeneous bulk density, narrow size distribution, and, ideally, spherical morphology to favor their processability and avoid reactor fouling.17–19 This is achieved by immobilizing metallocenes on inorganic supports, most commonly silica, which provide a template for the growing polymer. A crucial phenomenon during polymerization reactions is the fragmentation of the support material, which occurs due to the polymer accumulation and consequent stress build-up within the support pores. This is a necessary process, and its efficiency is essential to ensure adequate particle morphology development and allow sufficient catalyst activity. When the support material fragments, new diffusion pathways for the olefin are created, and active sites that were at first precluded from participating in the polymerization reaction become accessible. However, catalyst particle fragmentation cannot happen too rapidly; otherwise, control over particle morphology is lost. At industrial polymerization pressures and temperatures, particle fragmentation happens rapidly, making it challenging to control its course. For this reason, a pre-polymerization step at mild temperatures and pressures is often performed to control the initial fragmentation of the catalyst before introducing it into the main olefin polymerization reactor.20–22 In some cases, pre-polymerization is carried out in slurry phase conditions, as it allows for better heat dissipation, preventing temperature runaway and formation of sticky particles, which would occur in the gas phase. If fully gas-phase processes are required, (pre)polymerization is generally carried in fluidized bed reactor (FBR) systems.20,21
Several efforts have been made in the literature to investigate the fragmentation phenomenon, the vast majority focusing on developing single-particle models to predict particle morphology evolution under specific conditions.23–28 In general, two simplified limiting cases have been proposed: the shrinking core model, where fragmentation starts at the outer surface of the catalyst particle by peeling off small fractions and advancing layer-by-layer towards its center; and the bi-sectioning model, in which longer cracks occur across the catalyst core, followed by subsequent breakage of each section, and so on until the catalyst is finely dispersed into the growing polymer (see Fig. S1†).29–31 Advanced mathematical models implement the findings of experimental data to develop more accurate predictive systems that include the effect of a broad range of parameters, showing that fragmentation in real-life olefin polymerization catalysts is more likely a combination of these two modes.19,20,25,27,28,31–33 The factors influencing catalyst particle fragmentation can be classified under three categories: (a) catalyst properties (i.e., support morphology, mechanical strength, active sites distribution, and intrinsic activity), (b) polymer properties (i.e., polymer crystallinity and viscoelasticity, hence its ability to absorb stress) and (c) reaction conditions (which affect the local environment of each active site, i.e., monomer concentration across the particle, and heat accumulation during the exothermic reaction, viscosity and crystallization rate of the nascent polymer). Therefore, due to the extent and complexity of these factors that govern catalyst particle fragmentation, the development of a fully prognostic tool is not a straightforward task, as extensively reported in the review article by Alizadeh and McKenna.34
Furthermore, collecting reliable experimental data on catalyst particle fragmentation presents its challenges. Besides the difficulty of obtaining particles at the initial stage of polymerization,35 visualizing the fragments within the nascent polymer is not trivial. The strategy most reported in the literature is scanning electron microscopy (SEM), mainly used to observe the external morphology of polymerized particles which, however, reveal only that much about their internal fragmentation.36–39 Alternatively, melt microscopy has been reported to visualize catalyst fragments within the particle while melting the polymer phase under a microscope, though modifying the location of the fragments considerably.37,40 Recent developments by Bossers et al. have shown the combination of X-ray ptychography and X-ray fluorescence tomography (XRF) as a new powerful tool to study the fragmentation behavior of particles in 3D volume with submicron spatial resolution,41,42 a significant improvement from the resolution achieved in the past by X-ray computed microtomography.34,43–47 However, this methodology requires synchrotron radiation and time-demanding data collection, making it a less accessible and practical tool for fragmentation analysis of a broader range of catalyst particles. The current most effective lab-scale method to observe fragmentation is cutting particle slices (with a razor blade or diamond knife after embedding them in epoxy, i.e., micro-toming) and observing the obtained cross-sections with SEM or TEM.29,30,48–53 This method also has its limitations, mainly a possible deformation of the catalyst particle and the difficulty in distinguishing epoxy, polymer, and catalyst phases within the cross-section made.
Thus, it has not been possible so far to visualize the very early onset of catalyst particle fragmentation and polymerization fronts with sufficient spatial resolution as well as accuracy within real-life industrial metallocene-based olefin polymerization catalysts.
This study focuses on the influence of the reaction conditions on the fragmentation of an industrial silica-supported metallocene catalyst (i.e., Zr/MAO/SiO2 with methylaluminoxane, MAO, as co-catalyst) in the very early stages of ethylene polymerization (i.e., in the range of 1–20 g polyethylene (PE) per gram catalyst). To overcome the downsides of particle visualization methodologies, we have used a high-resolution focused ion beam-scanning electron microscopy (FIB–SEM) dual beam system. This allowed us to visualize the interior morphology of pre-polymerized catalyst particles and to distinguish catalyst fragments and pores a few tens of nm in size (Fig. S2†).54 Segmentation of the micrograms was then performed, allowing us to visualize PE accumulation and the catalyst particle fragmentation patterns at specific locations within the particle. The schematic representation of catalyst pre-polymerization, cross-section visualization via the dual-beam FIB–SEM instrument, and segmentation process are depicted in Fig. 1.
 | ||
| Fig. 1 a) Silica-supported metallocene-based olefin polymerization catalysts were pre-polymerized under a series of different reaction conditions and analysed by b) focused ion beam-scanning electron microscopy (FIB–SEM). The cross-sections are obtained with a FIB, which is capable of removing half of the particle and subsequently clean the cross-section surface; the cross-sections are then imaged in high-resolution by collecting the back-scattered electrons (BSE), which result in a greyscale image where catalyst and polymer can be distinguished due to the different atomic number (Z) of their main constituents (C and Si), making the polymer darker than the catalyst material. c) Schematic representation of the segmentation analysis of the SEM cross-section. First, the top half of the particle with the least artefacts is selected and subsequently, pixels belonging to the background and materials in the pores beyond the cross-section plane are removed. Then, k-means clustering is used to assign each pixel to labels corresponding to different materials. The areas of each label are calculated to estimate the extent of catalyst material (orange), polymer in the core (blue), polymer in the shell (light blue), and macropores (green) for each particle cross-section collected. | ||
The commercial silica-supported metallocene catalyst investigated in this work showed two main ethylene polymerization fronts: one on the outer surface and one at the macropores running across its core. Here, polymer formation in ideal reaction conditions induced fragmentation both in layer-by-layer and bi-sectioning mode, while mass transfer limitation phenomena and inhomogeneous active site distribution lead to sub-optimal catalyst particle fragmentation.
2. Experimental
2.1. Catalyst synthesis
Commercial SiO2 (ES70W, PQ Corporation) was first dried at 120 °C for 1 h, to remove any physisorbed water. The dry silica was then dehydroxylated at 450 °C for 4 h, under N2 atmosphere. MAO impregnation was performed by mixing the support to a solution of 16% MAO (Akzo-Nobel) in toluene for 4 h under reflux and in an inert atmosphere, with the target loading of 14.5 wt% Al. The so-obtained SiO2-supported MAO was then impregnated with a toluene solution of the metallocene catalyst Cp2ZrMe2 (bis(cyclopentadienyl)dimethyl-zirconium(IV) 97%, Sigma Aldrich) for 2 h at room temperature to reach the desired metallocene loading of 1.2 wt%. After impregnation, the catalyst was filtered and rinsed with dry toluene and pentane (both anhydrous, dried over molecular sieves, Ar, ChemSeal) and then further dried under vacuum to remove the remaining solvent traces.2.2. Catalyst testing
Pre-polymerized catalyst particles were prepared both in the gas-phase and in slurry-phase in a batch autoclave reactor, equipped with an inner quartz vial, which could be easily removed and thoroughly cleaned between reactions. The reactor was first loaded in N2 atmosphere with a few mg of catalyst material and then pressurized with ethylene (i.e., C2H4 4.0, Linde). For gas-phase ethylene polymerization experiments, the catalyst was finely dispersed across the quartz vial walls to avoid minimum contact among each particle to avoid catalyst deactivation due to over-heating and allow even ethylene access across the sample. Fig. S3† shows an example of sample over-heating and polymer melt when catalyst particles are reacted in the gas-phase if this expedient is not employed. For slurry-phase ethylene polymerization experiments, heptane (n-heptane, anhydrous, over molecular sieves, Ar, ChemSeal) was used as a diluent and tri-isobutyl aluminum (TiBA, 1.0 M in hexanes, Sigma-Aldrich) as impurities scavenger. All gas-phase reactions were performed at room temperature, while the slurry-phase reactions were done at temperatures up to 60 °C with a heating mantel around the autoclave body. Gas lines were flushed with N2 and vacuum cycles before pressurization of the autoclave reactor. Ethylene pressures were varied from 1 bar up to 15 bar. Reactions were stopped by depressurizing the reactor, and polymerized particles were quenched by adding ethanol. The so-obtained particles were then left drying at room temperature overnight to remove any solvent before weighting the polymer yield and perform FIB–SEM analysis. The fresh catalyst was also tested for ethylene polymerization at 9 bar in slurry-phase with heptane at 55 °C for 1 h, leading to a yield of 95 g of PE per g of catalyst, equal to 1990 kg PE per mol Zr h.2.3. Catalyst characterization
Samples were loaded on Al stubs with carbon tape and sputter-coated with 10–20 nm of Pt, before the measurement. Beam currents of 0.1 nA and 2 kV were used to image particles with dwell times varying between 10 and 20 μs. External morphologies were imaged by collecting secondary electrons (SE) with an Everhart–Thornley detector (ETD), while cross-sections were imaged by collecting back scattered electrons (BSE) with a through-the-lens detector (TLD) with a dual scanning electron microscope/focused-ion beam (FEI Helios NanoLab G3 UC). An extra layer of Pt (a rectangular area a few μm broad and 3 μm in thickness) was deposited over the top of the particle, at the height where a cross-section was planned to be exposed, via FIB assisted Pt deposition. Cross-sections were obtained with a Ga FIB, removing half of the particle material and then cleaning with precision milling the exposed cross-section. Discrete, isolated spherical particles were selected for cross-section analysis, based on the observation of a wide range of particles across the sample batch and after estimating average particle size and morphology, to choose a representative particle for each batch. Micrograms are measured by collecting back-scattered electrons (BSE); hence the contrast difference will depend on the atomic number (Z) of the elements constituting the material they have interacted with. The main constituents of catalyst and polymer are Si and C, respectively; therefore, these two materials will show different BSE intensities resulting in a greyscale SEM image with high contrast between the two phases. Thanks to this feature, cross-sections were then analyzed by k-means clustering, assigning each pixel to either catalyst, macropores, or polymer (in the core or accumulated at the outer surface). For a more detailed description of the k-means clustering segmentation and pixel labeling of the 2D cross-section images, the reader is referred to Fig. S4 and S5.† The area extension of each label was then calculated as a ratio to the overall cross-section area.3. Results & discussion
3.1. Pristine catalyst particles morphology
The fragmentation of polymerization catalysts is affected by several parameters, most notably the morphology of the catalyst particle itself. Each particle of the metallocene-based olefin catalyst material under study is made of an agglomeration of highly mesoporous domains, encapsulated by a layer of less porous silica a few micrometers thick (hereafter referred to as “dense silica shell”) and interconnected by macropores that run across the core of the particle, as shown and schematically represented in Fig. 2. The macropores are connected to the outer surface, as evident from the pristine particle SEM shown in Fig. S6.† The two different silica phases can be seen as an agglomeration of micrograins, forming interstitial mesopores of ∼15–20 nm (as evidenced by N2-physisorption reported in Table S1 and Fig. S7†), although coalescing more in the dense silica shell. The distribution of the MAO co-catalyst is homogeneous throughout the support oxide (Fig. S8†), and the intrinsic activity of each Zr+ active site is assumed to be comparable.7,9,55 Therefore, the rate of olefin polymerization at any active site in the catalyst particle will mainly depend on ethylene accessibility, i.e., ethylene diffusion through such support morphology. We can then predict that the active sites at the outer surface of the catalyst particle and those at the macropores will be the first to be reached, as diffusion of ethylene through the macropores network is expected to be fast. Then, the monomer will diffuse through the mesopores, making its way through their tortuous paths. However, this diffusion pattern will evolve as the morphology of the particle changes due to the accumulation of polymer and catalyst fragmentation. Polymers are semi-crystalline materials, i.e., a mixture of entangled chains (amorphous phase) and ordered compact chains (crystalline phase). The monomer can diffuse through the amorphous polymer phase, although with different diffusion times depending on its compactness/entanglement, while it cannot permeate through the very compact crystallites.56,57 Therefore, the level of entanglement and crystallinity of the nascent polymer will affect the diffusion of monomer to the active sites. The crystallinity of the nascent polymer might be different from the bulk crystallinity of the final polymer, and it will be dependent on the local environment of the growing chain, which is highly affected by the reaction conditions.19,58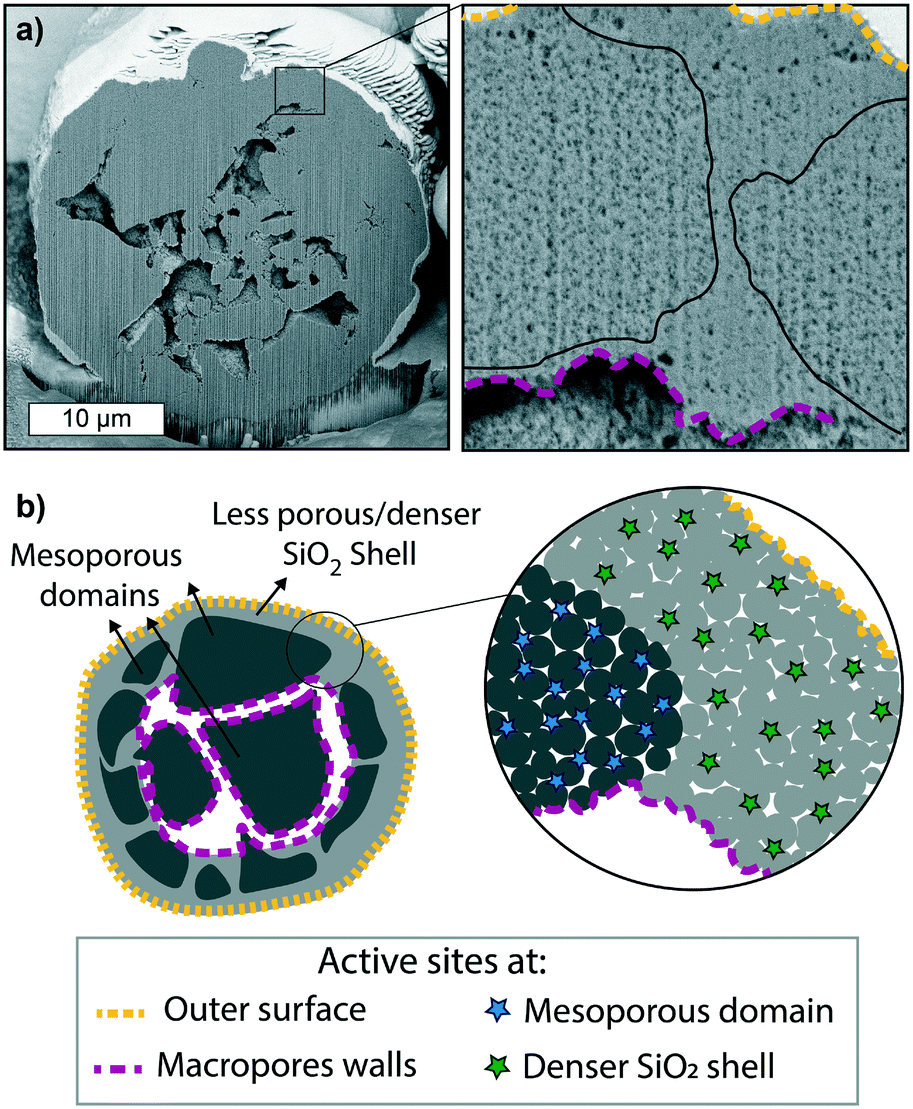 | ||
Fig. 2 a) Cross-section of a pristine supported metallocene-based olefin polymerization catalyst, showing macropores a few μm in diameter, extending across the core of the particle. The zoom-in reveals the presence of two different mesoporous SiO2 areas with different porosity present in the shell of the particle, one being less porous than the other (black lines are placed to highlight the physical border between these two phases – see also Fig. S6c†). b) Schematic representation of the particle cross-section, showing the highly porous silica domains being encapsulated by a shell of the denser silica material and interconnected by macropores, with a zoom-in showing the different porosity of the two. Active sites at different locations are also indicated: on the outer surface of the particle (yellow dashed-line), at the macropores walls (purple dashed-line), within the mesoporous domains (green ![[*]](https://www.rsc.org/images/entities/char_e103.gif) ) and in the pores of the dense silica shell (blue ). ) and in the pores of the dense silica shell (blue ). | ||
3.2. Polymerized particles morphology and fragmentation behavior at different reaction conditions
Ethylene polymerization reactions were performed with this commercial silica-based metallocene catalyst at a series of reaction conditions and polymerization times (see Table S2† for the yields obtained). The resulting morphologies and fragmentation behaviors were observed via FIB–SEM analysis.When exposed to 1 bar pressure of ethylene under gas-phase conditions, within the first 5 minutes, PE primarily forms at the outer surface and within the macropores (Fig. 3a), causing the silica at these two polymerization fronts to fragment in a concentrical layer-by-layer manner, as also schematically depicted in Fig. 3c. After 15 minutes (Fig. 3b), both polymerization fronts have proceeded further, as more polymer has accumulated on the particle's outer surface and within the macropores. The contrast between the dense silica shell and the mesoporous domains is now more marked than in the pristine catalyst. This further confirms the higher density/lower porosity of the silica shell, where less polymer has formed, making the silica/polymer ratio higher, resulting in a lighter grey. Some mesopores are still visible in areas further away from both polymerization fronts (Fig. 3b zoom-in), indicating that the monomer slowly diffuses through the mesoporous domains.
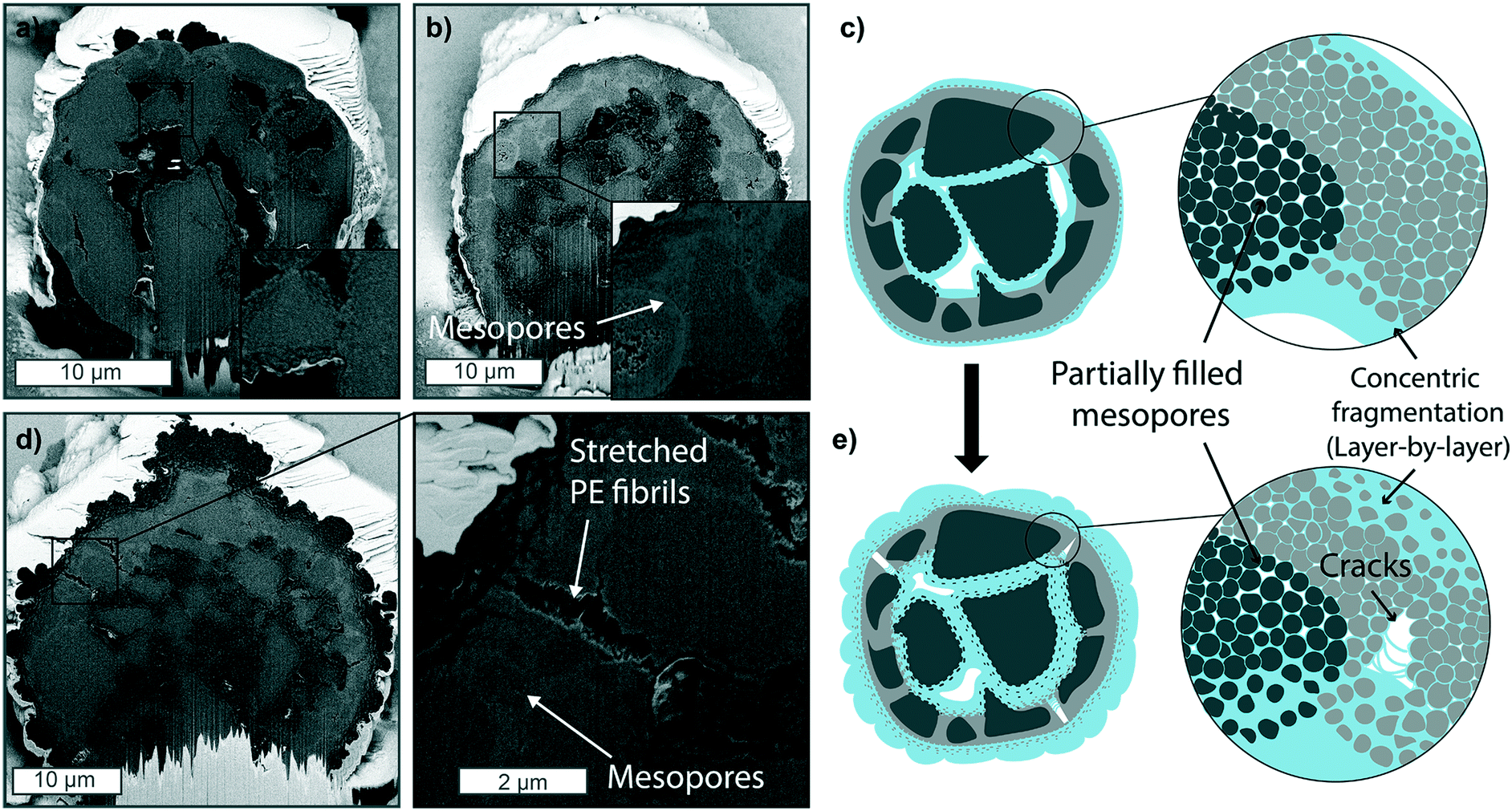 | ||
| Fig. 3 Focused-ion beam-scanning electron microscopy (FIB-SEM) cross-sections of a metallocene-based catalyst supported on silica polymerized at 1 bar ethylene pressure for a) 5 and b) 15 min and c) schematic representation of the particle fragmentation at this stage, where the catalyst is peeled-off both at the outer surface at the macropores where PE is forming. d) Metallocene catalyst supported on silica polymerized at 1 bar ethylene pressure for 60 min. e) Schematic representation of catalyst particle fragmentation at this stage, where pressure build-up has caused the formation of cracks with stretched PE fibrils in the dense silica shell. | ||
The two fronts proceed further, continuing the peeling of catalyst fragments over time (see Fig. S9c and d†). After 60 min (Fig. 3d), the PE on the particle's outer surface has increased considerably in thickness, folding onto itself in bump-like protrusions (see also Fig. S9e†). On the other hand, the dense silica shell is still clearly distinguishable. However, it is starting to fracture as pressure has built up in the expanding core, pushing it outwards and leading to the formation of cracks in this area (zoom-in of Fig. 3d). The fracturing via the formation of cracks could be due to the different mechanical strength of the dense (and less polymerized) silica shell compared to the highly mesoporous domains. On the other hand, the formation of PE stretched fibrils within such cracks is a consequence of PE being pulled apart as the distance between the two catalyst fragments increases faster than the formation of PE itself.
Increasing the ethylene pressure to 9 and 15 bar (Fig. 4a and b, respectively) results in catalyst particles with thicker PE layers on their outer surface, which considerably hinders ethylene diffusion to the core substantially less PE is formed. This is evident both by the lower amounts of PE being present in the core macropores as well as the clear presence of non-filled mesopores (Fig. 4d). Fragmentation at the two main fronts is still occurring in a layer-by-layer manner, as clearly visible in the zoom-in of Fig. 4b and schematically depicted in Fig. 4c. However, the accumulation of PE in the macropores at the core of the particle is less extensive than what was found in the sample polymerized at lower pressures of Fig. 3. To be noted, these two particles show a particularly thick silica dense shell and very low macroporosity (Fig. 4a and b), contributing further to slowing the diffusion of monomer to the core, while other particles polymerized at these pressures in the gas-phase showed more polymer to be accumulated in the macropores (Fig. S10†). Nonetheless, the very low polymer yields (below 4 g PE per g catalyst after 15 min of reaction at 15 bar – see Table S2†) are an indication that the overall fast accumulation of polymer on the surface of the particle leads to a slower pressure build-up in the core and consequent incomplete fragmentation of the catalyst. The morphology of this external PE layer (Fig. 4d) is also an indication of the fast accumulation of PE not being accompanied by an equally rapid core expansion, causing the PE to be corrugated in this manner.
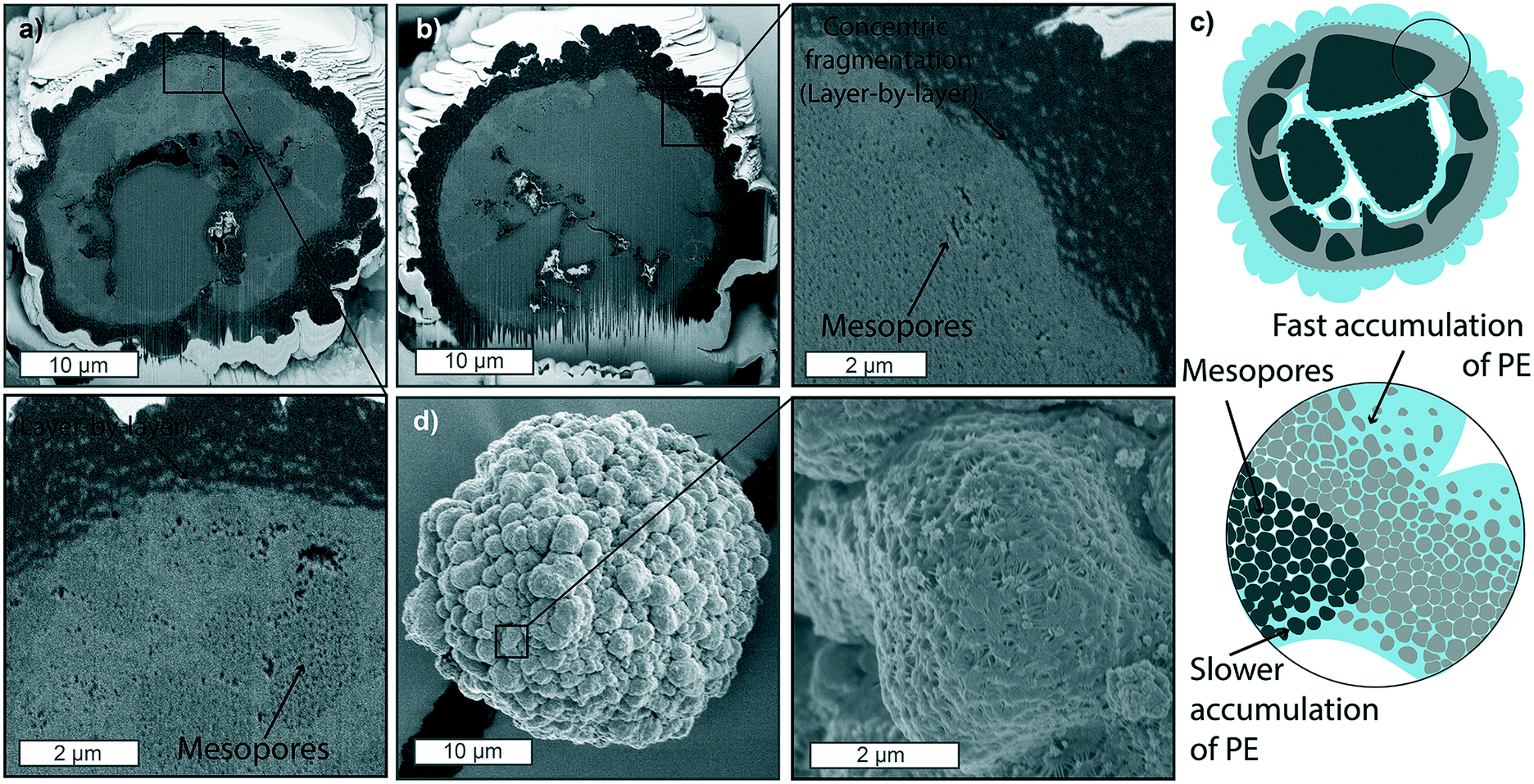 | ||
| Fig. 4 Focused-ion beam-scanning electron microscopy (FIB-SEM) cross-sections of supported metallocene-based catalyst polymerized for 15 min at a) 9 bar and b) 15 bar of ethylene pressure. c) Schematic representation of catalyst particle fragmentation at conditions of high pressure in the gas-phase, where polymer is quickly formed at the outer surface, folding onto itself in bump-like protrusions, slowing down the diffusion of monomer to the core. d) Outside morphology of the catalyst particle polymerized at 15 bar for 15 minutes in the gas-phase. | ||
Introducing a diluent such as heptane to the reaction mixture leads to changes in the catalyst fragmentation behavior for the catalyst under study (Fig. 5, S10 and S11†). As shown in Fig. 5, at 9 bar and room temperature, a thin layer of PE is formed on the outer surface of the particles and at the macropore walls within the first minute (Fig. 5a), similarly to what was found in gas-phase polymerized particles. However, mesopores appear to be filled entirely with PE across the entire particle, and fractures filled with PE are also found across the mesoporous domains (zoom-in of Fig. 5a). Within 2 minutes of reaction (Fig. 5b), it is even more clear that fragmentation is occurring both via concentric peeling-off of catalyst fragments as well as via the formation of longer fractures across the mesoporous domains, indicating that a combination of the layer-by-layer and bisection models is present. This process is continued over the following stages at 5 and 10 minutes (Fig. 5c and d), where small fragments of catalyst are finely dispersed in the polymer phase, with some bigger domains still seen undergoing concentrical layer-by-layer fragmentation (zoom-in Fig. 5c). The external morphology (Fig. 5e) shows to be smoother than that of the gas-phase polymerized particles, and macropores never appear to be filled with polymer. Both observations can be ascribed to the more homogeneous polymerization across the particle diameter, leading to particle expansion proceeding at comparable rates to the polymer growth. Increasing the pressure to 15 bar in the slurry-phase reaction (Fig. S11†) does not lead to the polymer accumulation on the outer shell found in the gas-phase. On the contrary, extensive fragmentation has already occurred within the first 5 minutes of reaction (Fig. S11a†), predominantly in a layer-by-layer mode, with fragments being peeled-off from the catalyst surface and mesoporous domains in a concentric manner, eventually leading to a polymer particle with a myriad of catalyst grains a few tens of nm in size, homogeneously dispersed in the PE phase (Fig. S11b†). The extensive macroporosity still found in particles after 15 min of reaction (Fig. S11b†) and the smooth morphology of the outer surface of the polymer particle (Fig. S11c†) are again evidence of the expansion of the particle occurring at similar or faster rates than polymer accumulation in the core. The effect of fast particle expansions is also evident from a close-up on the outer surface morphology (zoom-in Fig. S11c†), where PE stretched fibrils connect small catalyst fragments as they are being pulled apart from the growing polymer beneath. Such expansion maintains a level of mesoporosity and macroporosity, which ensures continuous diffusion of monomer to the active sites and avoids the accumulation of a thick shell of polymer on the outer surface, leading to a smoother external morphology without the protuberances which were occurring in the gas-phase.
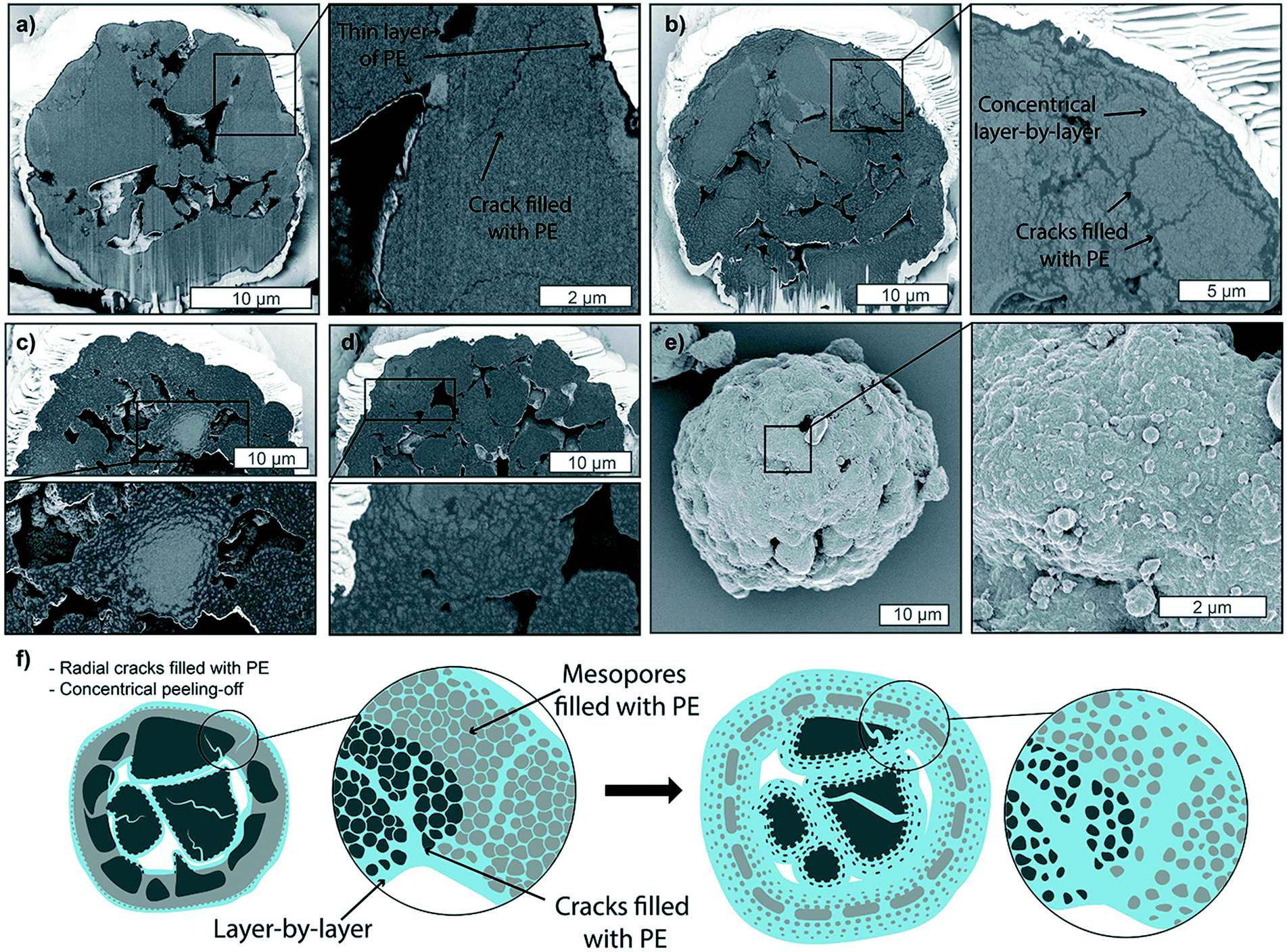 | ||
| Fig. 5 Focused-ion beam-scanning electron microscopy (FIB-SEM) cross-sections of supported metallocene-based catalyst polymerized in heptane at 9 bar of ethylene pressure and room temperature (RT) with 400 rotations per minute (RPM) stirring performed for a) 1 min, b) 2 min c) 5 min and d) 10 min. e) Outside morphology of the particle in d). f) Schematic representation of catalyst particle fragmentation in slurry-phase, where fragmentation starts in a layer by-layer mode at the outer surface and at the macropores, while simultaneously developing thin radial fractures filled with PE across the different domains within the particle. The catalyst is eventually extensively and homogeneously fragmented, while still presenting a level of macroporosity. | ||
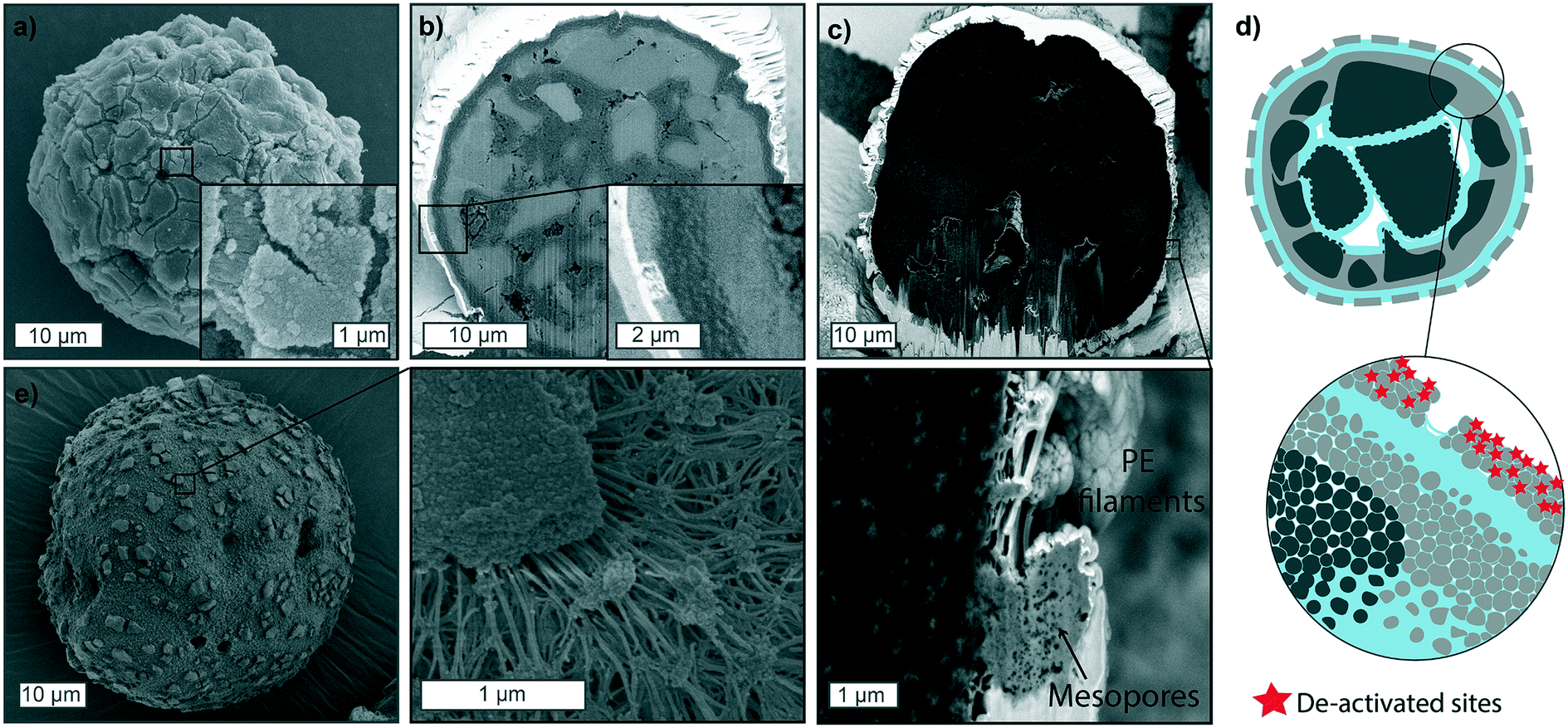 | ||
| Fig. 6 a) Scanning electron microscopy (SEM) images of the particle surface morphology and b) cross-section of a polymerized particle (gas-phase, 9 bar, 5 min) that suffered from mild deactivation, mainly located on a thin layer on its outer surface. c) FIB cross-section of a particle polymerized in the slurry-phase with similar deactivation phenomena and its outside morphology in e). d) Schematic of the ethylene particle fragmentation when partial deactivation of the outer surface has occurred. The non-active catalyst layer presents big cracks connected by polyethylene (PE) filaments, as a result of pressure build-up in the core, where polymer is forming at faster rates. To be noted that the schematic shows the case of gas-phase reactions. For slurry-phase reaction the behaviour of the deactivated shell fraction is comparable, while the core will polymerize more extensively. | ||
The effect of slowing the stirring speed was investigated by reducing it from 400 to 60 rpm, which lead to lower polymer yields (i.e., 2.3 g PE per g catalyst vs. 22 g PE per g catalyst obtained with fast stirring at same ethylene pressure and time). Decreasing the stirring speed reduces the mixing of the gas in the diluent, thus a lower concentration of monomer is reaching the catalyst particle. Yields are comparable to particles polymerized in the gas-phase at low pressure (i.e., 60 min at 1 bar of Fig. 3d). However, the polymer produced in the slurry-phase with slow stirring is more homogeneously distributed (Fig. S11d†), forming a smoother layer of PE around the particle and filling the mesopores of the catalyst completely, proving that the presence of a diluent in the reaction mixture is the main factor contributing to a more homogeneous accumulation of polymer across the particles of the catalyst under analysis.
Finally, as a diluent in the reaction mixture consents for a better temperature control over the sample, slurry-phase reactions were used to investigate the influence of reaction temperature.20–22 Polymerization reactions are exothermic processes; therefore, too elevated temperatures would not benefit the reaction kinetics, while milder temperatures can be used to overcome the activation energy barrier.59 Hence, temperatures up to 60 °C were considered (Fig. S12†). Similar to the progress observed at room temperature (RT) over time, PE yields increased as a function of temperature, catalyst particle fragmentation proceeded further, while macropores are never completely filled at the temperatures and polymerization time investigated. From Fig. S12c,† it is visible that at 60 °C, macropores contain thick PE “worm-like” threads connecting different domains. Similar polymer morphology has been reported before and ascribed to different reasons, such as the presence of a cluster of active sites, an extrusion-like formation of polymer, a rapid crystallization of the polymer chains, or the occurrence of a rapid axial extension.60 It is plausible that at these conditions, PE growth vs. its crystallization rate is causing such polymer conformation to form and to pull strain within the particle, contributing to the particle expansion.
3.3. Fragmentation of partially deactivated samples
One final case worth reporting is the catalyst behavior when partial deactivation has occurred. Fig. 6 shows polymerized particles in the gas-phase (Fig. 6a and b) and slurry-phase (Fig. 6c and e), where deactivation had affected a thin fraction of the outer particle shell (up to 1 μm in thickness), due to exposure to very low amounts of air. In this scenario, active sites on the immediate outer surface of the catalyst are not active, preventing the formation of a thick PE shell and facilitating the diffusion of monomer through the macropores and mesopores. This generally results in more polyethylene being formed in the core and unreacted catalyst fragments on the external portion of the particle, separated by cracks visible on the outer surface, which increase in distance as the polymerization proceeds in the core (schematically shown in Fig. 6d). Besides the loss of some active sites, this scenario is highly undesirable as the unreacted fragments and residual polymer can detach from the catalyst particle and create fines in the reactor which would lead to fouling. The presence of external cracks across the particle surface has been shown before in literature, but the causes are generally different than what experienced in this study and mainly ascribed to fragmentation primarily occurring in the continuous bisection mode, where cracks form across the particle, extending to its surface.38,39,60,613.4. Semi-quantitative analysis of particle fragmentation via image segmentation
Thanks to the high resolution of the SEM and contrast between catalyst and polymer phase, each cross-section image collected was segmented in different areas to calculate the extent of catalyst, macropores, and polymer, as reported in Fig. 7. This analysis gives a semi-quantitative evaluation of catalyst particle fragmentation trends for each reaction parameter investigated. As two main polymerization fronts were identified (at the outer surface and at the macropores), the polymer phase was also divided into two sub-regions when possible, that is, PE accumulated outside the catalyst particle and PE in the core. The rate of propagation during ethylene polymerization at a specific location (i) within the catalyst particle can be described by the following equation:26,35,59| ri = ki [C2H4]i[Zr+]i |
| ki = Ae−Ea/RTi |
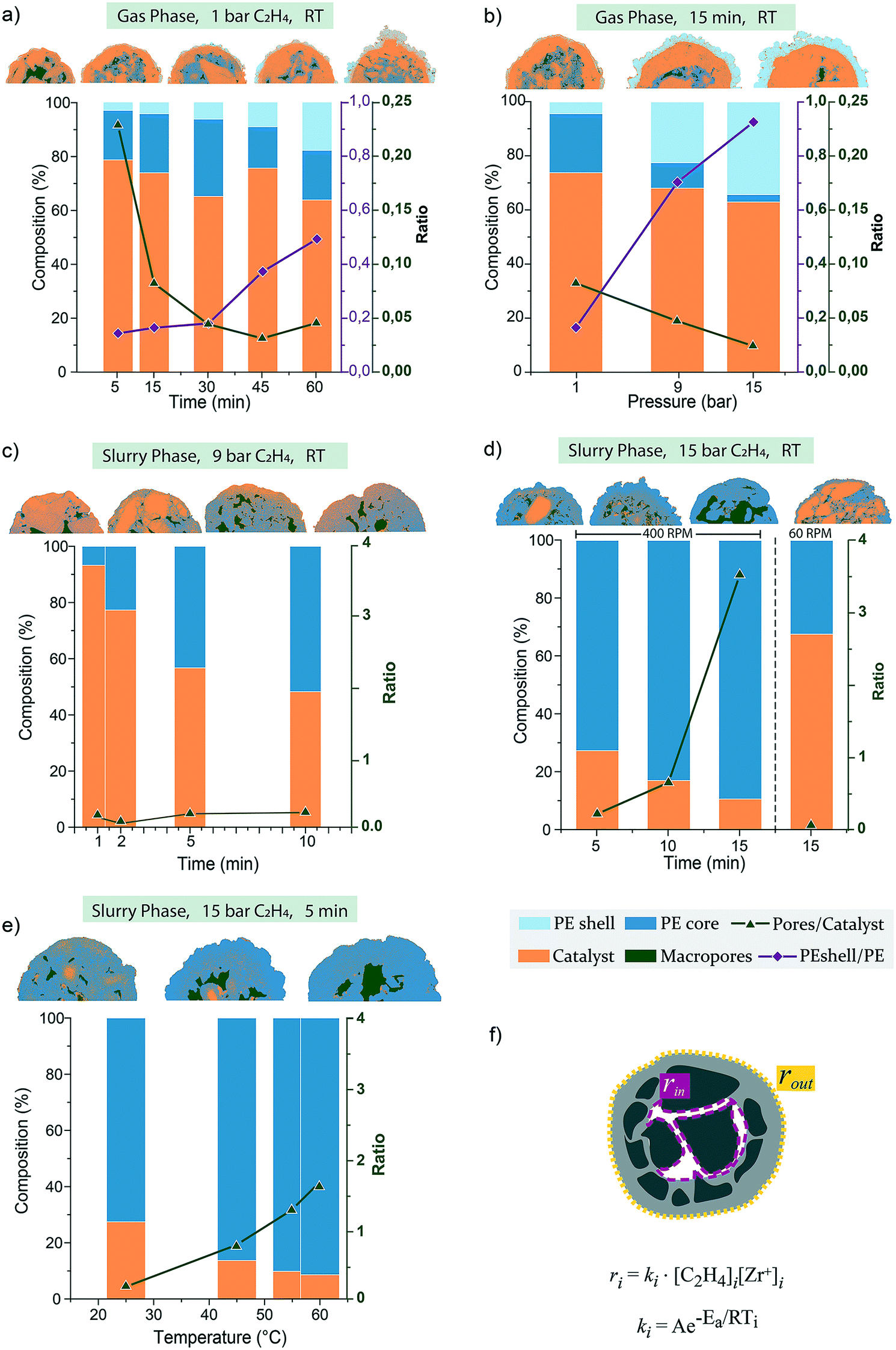 | ||
| Fig. 7 Segmentation of the cross-sections of the silica-based metallocene catalyst polymerized a) in the gas-phase at 1 bar of ethylene and RT from 5 to 60 min; b) in the gas-phase, for 15 min and at RT, for increasing pressures of 1, 9 and 15 bar of ethylene; c) in slurry-phase at 9 bar of ethylene and at RT, 400 RPM stirring speed, from 1 to 10 min; d) in slurry-phase at 15 bar of ethylene and RT with 400 and 60 RPM stirring speed and increasing polymerization time; e) in slurry-phase at 15 bar of ethylene and for 5 min, with increasing temperature up to 60 °C. f) Schematic representation of the metallocene-based ethylene polymerization catalyst under study, with the two main polymerization fronts highlighted, at the outer surface (yellow dashed line) and at the macropores (purple dashed line). The polymerization propagation rates at these two frons are dependent on the concentration of monomer [C2H4]i, concentration of active sites [Zr+]i and temperature Ti at the specific location i. | ||
When the catalyst is exposed to low pressures of ethylene at RT in the gas-phase, polymerization rates ri will be slow due to low [C2H4] and T. However, the relative rate of polymerization between the two fronts (rinvs. rout) will depend on the local concentration of C2H4, which is dependent on its diffusion times through the particle, τd (equal to L2/Deff where L is the characteristic length and Deff is the diffusivity). Macropores are generally connected to the outside, thus sites on the outer surface and at the walls of the macropores are reached almost simultaneously by the monomer; however, from the segmentation analysis (Fig. 7a), we can observe that at the beginning of the reaction, PE is predominantly formed in the core (PEshell/PE ratio < 0.2 up to 30 min of reaction). This is because the surface area (and therefore the relative [Zr+]) of the exterior of the particle is lower than that of the macropores.
After 60 min of reaction, the PE shell thickness has increased considerably, at the expense of PE in the core (PEshell/PE ratio ∼ 0.5), meaning that rout has increased and become comparable to rin. This is a result of monomer diffusion being hindered by the thickening of the PE shell itself, lowering [C2H4] in the core.20,33 As polymer fills the macropores, pores/catalyst ratio values decrease over time up to 45 min of reaction (from 0.25 to 0.035) until pressure build-up is sufficient to cause sectioning of the dense silica shell, which restores some macroporosity (pore/catalyst = 0.05) and allows for better monomer diffusion and continuation of the polymerization process. Mesopores further away from the two main polymerization fronts are the last to be filled with polymer, indicating that in the gas-phase the rate of polymerization appears to be considerably lower in the mesopores than at the outer surface (rout) and at the macropore walls (rin). This is likely a consequence of the slow diffusion of monomer to the core of the mesoporous domains, leading to lower [C2H4] in the mesopores and consequent lower polymerization rates at those active sites. Higher ethylene pressures mean an increased monomer concentration in the gas-phase, [C2H4]bulk, which should lead to proportionally faster polymerization rates. However, polymer yields only reached 3.1 and 3.6 g PE per g catalyst after 15 min of reaction at 9 and 15 bar, respectively, with most of the polymer being synthesized by sites at the outer surface, as indicated by PEshell/PE ratio of 0.7–0.8 (Fig. 7b). The high concentration of ethylene in the gas-phase and or the low macroporosity of the catalyst could cause a gradient of monomer concentration across the particle ([C2H4]out > [C2H4]in), leading to initial faster rates at sites on the outer surface where [C2H4] is highest. Furthermore, as the polymerization reactions is exothermic, poor heat removal can lead to a local increase in temperature, which could in turn increase the reaction rate. Gas velocities in this batch reactor is nil, making heat removal from the catalyst very inefficient. Overlap of multiple catalyst particles would lead to severe over-heating with catalyst deactivation and polymer melts (see Fig. S3†), and such effect was minimized by dispersing the catalyst powder across the reactor walls. Although this expedient prevents serious over-heating, a local increase in temperature on each particle can still be expected when high ethylene pressures are employed. Nonetheless, the temperature across the particle is expected to be homogeneous. Hence the faster rate at the outer polymerization front (rout ≫ rin) is likely due to the initial high [C2H4]out and accumulation of a thicker layer of polymer which hinders further diffusion of monomer to the core of the particle, explaining the overall low yields and incomplete fragmentation.56,62
The segmentation analysis in Fig. 7c–e shows that the extent and homogeneity of fragmentation in these particles makes the distinction between polymer formed in the core from that in the shell unfeasible. Yet, it also implies that rates of ethylene polymerization across the particle must be fast and comparable (rin = rout). Hence, the monomer concentration is also expected to be homogeneous across the particle ([C2H4]out = [C2H4]in). On the other hand, this was also concluded to be the case for the gas-phase reactions at low pressure and high macroporosity, but where fragmentation behavior was however different. The heat capacity of liquids is much higher than that of gasses, which is why slurry-phase processes are more efficient at dissipating the heat produced during olefin polymerization reactions therefore preventing a local increase of temperature at the active sites. This could explain the absence of a thick PE layer on the outer shell of the catalysts and/or in the macropores in the slurry-phase polymerized samples of this study. However, the role of a diluent in polymerization reactions is more complex and goes beyond its capacity of preventing overheating. In fact, both PE yield and catalyst particle fragmentation benefit from adding a diluent to the reaction mixture in our lab-scale autoclave.
We must consider that the presence of a third (liquid) phase to the reaction mixture affects the overall diffusion of the monomer: a) from the gas-phase to the liquid-phase, b) from the liquid diluent through the pores of the catalyst and c) from the swelled polymer to the active sites. To address a), fast stirring of the reaction mixture ensures quick saturation of the diluent with the monomer. This means that [C2H4] at the outer surface and within the macropores is comparable to that of the bulk gas-phase. When stirring speed is reduced to 60 RPM, [C2H4] in the liquid-phase is much lower than in the bulk gas, with consequent polymer yields equal to those in the gas-phase at low pressure (∼2 g PE per g catalyst). Segmentation analysis confirms that the PE% area is comparable between the slow-stirring sample and that of the sample polymerized in gas-phase at low pressure (∼35%). However, the polymer formed in the slurry-phase is more evenly distributed between macropores and the external layer. No external folding of polymer occurs, resulting in smooth outside morphologies. This again confirms that accumulation of PE and consequent fragmentation on the mesoporous domain scale is different between the gas- and slurry-phase, and the difference between monomer diffusion through the gas-phase and the bulk diluent is not at the root of these differences. As for (b), the time of diffusion of monomer through (meso)pores in the slurry-phase is calculated to be several factors shorter than in the gas-phase (Deff_gas = 10−8–10−7 m2 s−1 and Deff_slurry = 10−10–10−8 m2 s−1),20,33 which again does not explain faster filling of mesopores and more extensive catalyst particle fragmentation seen for these slurry-phase samples. However, the fast expansion of the catalyst particle in the slurry-phase, maintains a level of macro- and mesoporosity that facilitates diffusion compared to the more compact polymer layer which forms in the gas-phase. This is evident by the increasing macroporosity both at RT over time and with increasing temperature (Fig. 7d and e) and by the presence of segregated micrograins on the external layer of these particles being connected by an intricate web of PE stretched fibrils (Fig. S11†). As for (c), a diluent changes the environment of the active site by decreasing the local micro-viscosity of the swelled polymer. This could improve monomer accessibility at first, but as soon as the chain length reaches a critical length, it also facilitates its crystallization process, leading to a less permeable polymer matrix.19,63
Nonetheless, three main differences can be highlighted when a diluent is introduced: first, PE does not accumulate as a thick layer at the outer shell and/or at the macropore walls, while it forms in the mesopores from the very beginning of the reaction; second, fragmentation is more extensive, with a higher contribution of the bisectioning mode; finally, a certain level of macro- and mesoporosity is maintained throughout the course of the reaction as particles expand. Clearly, the monomer diffuses more slowly through the polymer phase formed in the gas-phase than that of the slurry-phase. In the initial stages, this could be ascribed to the monomer being more permeable through the heptane-swelled nascent polymer, as well as the better heat dissipation which prevented the formation of a thicker PE shell. In contrast, in the lack of a diluent, the polymer formed both at the outer surface and at the macropore walls seems more compact (although not necessarily more crystalline), thicker, and less permeable to the monomer, hindering its diffusion to the mesopores of the catalyst particles. Because mesopores are filled very quickly in the slurry-phase, stress-build-up within the silica domains leads to bisectioning-type fragmentation, which exposes more active sites and results in an overall more extensive fragmentation.
In summary, a series of catalyst particles pre-polymerized under a broad range of experimental conditions were studied by visualizing with a high spatial resolution both their outside and inner morphology using a dual-beam FIB–SEM system. In the gas-phase, PE forms both at the sites on the outer surface and at the macropores walls, where the catalyst slowly fragmented in a layer-by-layer manner. Once PE has filled the macropores, internal pressure build-up causes the denser silica shell to release stress via the formation of cracks. If the catalyst is exposed to higher pressures of ethylene and/or macroporosity of the particle is low, a monomer concentration gradient occurs; the outer side of the particle polymerizes faster, forming a thick and dense PE layer which further hinders the diffusion of monomer, leading to a less extensive polymerization of the particle core. When polymerized in the slurry-phase, monomer diffuses more quickly through the diluent-swelled polymer and resulting in more homogeneous monomer concentrations and polymerization rates across the particle core, with higher polymer yields and more extensive fragmentation, which occurs both with concentric layer-by-layer peeling of catalyst fragments and the formation of radial fractures in a sectioning mode. Finally, if the catalyst is exposed to low amounts of air, sites on the outer surface of the polymer are quenched, causing a lack of polymer production at these sites and resulting in the formation of cracks on the outer portion of the catalyst, as unreacted silica plate-like sections drift apart while the polymer is expanding beneath, in the core of the catalyst particle.
Hence, two main polymerization fronts could be discerned for this specific commercial silica-based catalyst: one at the active sites located on the outer surface of the catalyst particle and one at the active sites on the walls of the macropores. Fragmentation at these fronts occurs in the concentric layer-by-layer mode, forming radial or surface cracks, depending on reaction conditions and active site distribution. In general, the fragmentation behavior can better be explained by the relative rate of ethylene polymerization at the two main fronts (rinvs. rout), as schematically depicted in Fig. 8.
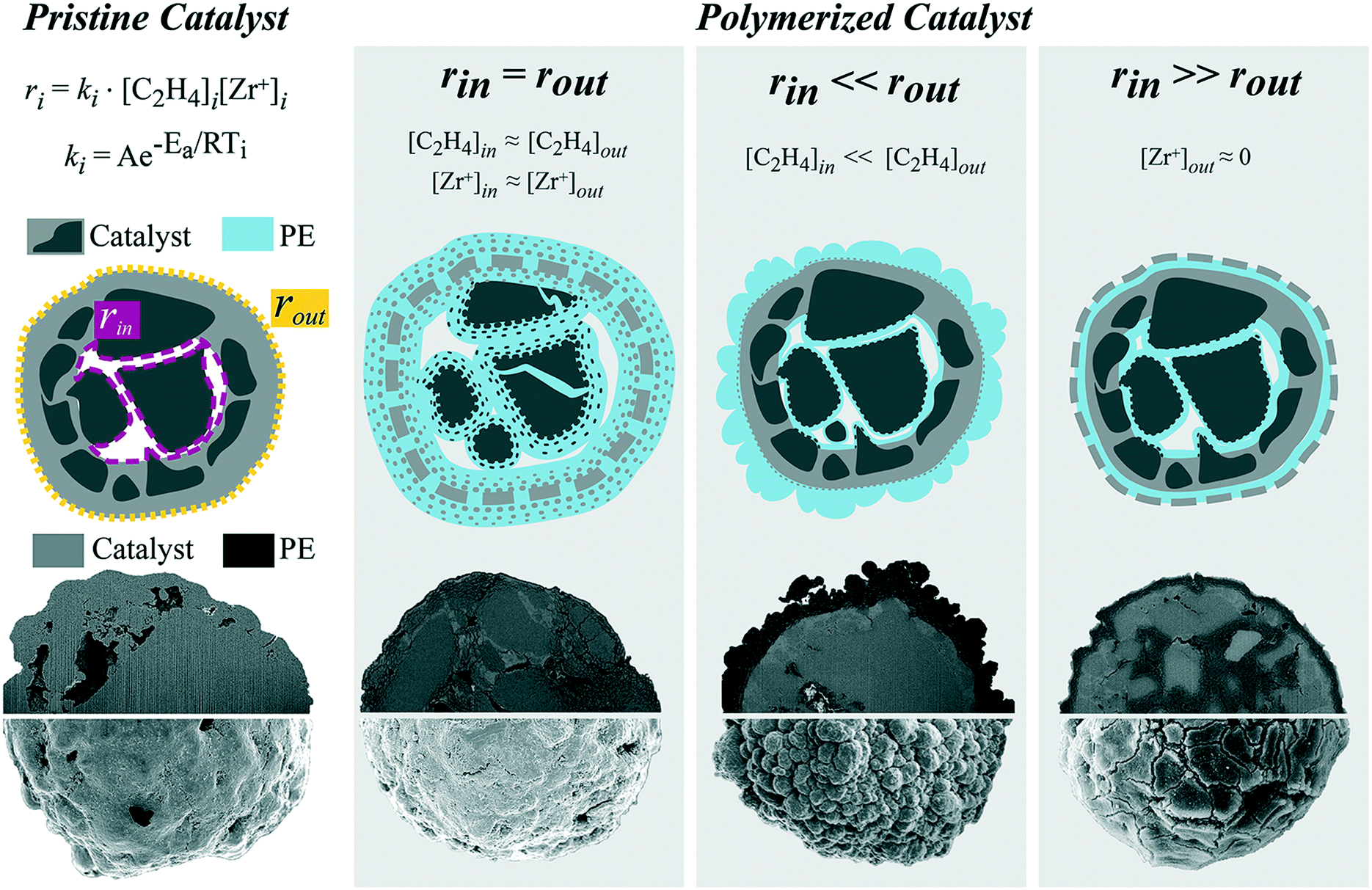 | ||
| Fig. 8 Schematic of fragmentation modes experimentally observed for a metallocene-based ethylene polymerization catalyst supported on a silica support. Every experimentally observed catalyst particle fragmentation pattern can be described by the ratio between the rate of ethylene polymerization at the sites in the macropore walls in the core of the catalyst particle (rin) and the rate at the sites on the outer surface of the catalyst particle (rout). | ||
When the two rates are comparable (rin = rout), ethylene polymerization is uniform across the catalyst particle as monomer concentration and active site distribution are sufficiently homogeneous within the particle. This occurred when the catalyst was polymerized in the gas-phase at low pressures and in the slurry-phase. The outer surface and the inner silica domains polymerize in a shrinking core manner for both gas- and slurry-phase, while the formation of cracks differs in the two cases. Particles reacted in the gas-phase only seldomly present cracks interconnected by PE fibrils. In the slurry-phase several radial thin cracks filled with PE are formed both across the dense shell and the mesoporous domains, indicating a continuous bisection type of fragmentation. This scenario is also the most desirable as it allows for complete and homogeneous fragmentation of the catalyst while maintaining adequate particle morphology.
The rate of ethylene polymerization is considerably faster on the outer surface than in the macropores (rout ≫ rin) when a gradient of monomer concentration is present. This was found to be the case for high-pressure gas-phase reactions and within particles with low macroporosity. In this situation, the outside of the catalyst particle is quickly covered by a thick and dense PE layer, hindering the diffusion of ethylene to the particle core, where, therefore, polymerization occurs more slowly. The outer surface peels off in a layer-by-layer mode, but the overall PE yield is low due to incomplete catalyst fragmentation.
Finally, if the catalyst outer surface has been partially deactivated ([Zr+]out ≈ 0), the inner rate will be faster than the outer rate (rin ≫ rout). The silica domains in the core polymerize in the shrinking core manner, expanding and creating a pressure build-up inside the catalyst particle. This causes the less active (or inactive) silica layer to fragment, developing cracks visible on the outer surface of the catalyst particle connected by PE stretched fibrils.
4. Conclusions
Early fragmentation stages of supported ethylene polymerization catalysts affect both the polymer product's catalyst activity and final particle morphology. As they are of great relevance for industrial olefin polymerization processes, a great effort has been made in the scientific community to develop predictive mathematical models. However, obtaining reliable experimental data on catalyst particle fragmentation presents even more challenges, making them less available in the literature and generally reporting later olefin polymerization stages as well as imaging techniques with low spatial resolution. The employment of a high-resolution FIB–SEM dual beam system allowed us to visualize the early onset of fragmentation within an industrial silica-supported metallocene-based ethylene polymerization catalyst under a broad range of reaction conditions. Two main ethylene polymerization fronts were identified: one at the outer surface and one at the macropores walls. Image processing via segmentation of the cross-section micrograms resulted in a semi-quantitative analysis of the catalyst fragmentation behavior, enabling us to evaluate relative kinetic rates at the two different fronts within the olefin polymerization catalyst particle. In particular, the rates were found to be affected by the efficiency of mass transfer or active site distribution. For ideal reaction conditions, fragmentation at these two fronts occurs by peeling off the catalyst in a layer-by-layer mode, with the simultaneous formation of bisection-type more extended fractures, which ensure together ensure a faster but controlled and complete fragmentation.The results obtained provide a detailed understanding of catalyst fragmentation for the specific catalyst particles and experimental conditions analyzed. This type of analysis could be applied to a broader range of supported olefin polymerization catalysts and add valuable information to how fragmentation fronts develop and proceed for various parameters, amplifying the knowledge on such intricated phenomenon.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received funding from Total Research and Technologies, Feluy. The authors would like to thank Koen Bossers (Utrecht University) for the useful discussions on image processing and Guusje Delen (Utrecht University) for the valuable feedback.Notes and references
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- M. A. Alma and I. Krupa, Polyolefin Compounds and Materials, Springer International Publishing, Cham, 2016 Search PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- PlasticsEurope, Plastics - the Facts 2019, 2019.
- PlasticsEurope, Annual Review 2017–2018, 2018, vol. 15.
- H. Sinn, W. Kaminsky, H.-J. Vollmer and R. Woldt, Angew. Chem., 1980, 92, 396–402 CrossRef CAS.
- J. C. W. Chien and B.-P. Wang, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 3089–3102 CrossRef CAS.
- J. Ewen and M. Elder, US005387568A, 1995.
- A. R. Barron, Macromol. Symp., 1995, 97, 15–25 CrossRef CAS.
- W. Kaminsky, J. Chem. Soc., Dalton Trans., 1998, 1413–1418 RSC.
- W. Kaminsky, Macromol. Symp., 1998, 134, 63–71 CrossRef CAS.
- W. Kaminsky, in Macromolecular Symposia, 2016, vol. 360 Search PubMed.
- W. Kaminsky, Rend. Fis. Acc. Lincei, 2017, 28, 87–95 CrossRef.
- M. R. Ribeiro, A. Deffieux and M. F. Portela, Ind. Eng. Chem. Res., 1997, 36, 1224–1237 CrossRef CAS.
- G. G. Hlatky, Coord. Chem. Rev., 1999, 181, 243–296 CrossRef CAS.
- W. Kaminsky, Metalorganic Catalysts for Synthesis and Polymerization, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999 Search PubMed.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS PubMed.
- F. Ciardelli, A. Altomare and M. Michelotti, Catal. Today, 1998, 41, 149–157 CrossRef CAS.
- T. F. L. McKenna, A. Di Martino, G. Weickert and J. B. P. Soares, Macromol. React. Eng., 2010, 4, 40–64 CrossRef CAS.
- J. B. P. Soares and T. F. L. McKenna, Polyolefin Reaction Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- J. R. Severn and J. C. Chadwick, Tailor-Made Polymers, Wiley, 2008 Search PubMed.
- M. Ahsan Bashir and T. F. L. McKenna, in Advances in Polymer Science, Springer New York LLC, 2017, vol. 280, pp. 19–63 Search PubMed.
- T. McKenna and V. Mattioli, Macromol. Symp., 2001, 173, 149–162 CrossRef CAS.
- A. G. Fisch, J. H. Z. dos Santos, A. R. Secchi and N. S. M. Cardozo, Ind. Eng. Chem. Res., 2015, 54, 11997–12010 CrossRef CAS.
- P. Kittilsen, H. F. Svendsen and T. F. L. McKenna, AIChE J., 2003, 49, 1495–1507 CrossRef CAS.
- T. F. L. McKenna and J. B. P. Soares, Chem. Eng. Sci., 2001, 56, 3931–3949 CrossRef CAS.
- D. M. Merquior, E. L. Lima and J. C. Pinto, Polym. React. Eng., 2003, 11, 133–154 CrossRef CAS.
- P. Kittilsen, T. F. L. McKenna, H. Svendsen, H. A. Jakobsen and S. B. Fredriksen, Chem. Eng. Sci., 2001, 56, 4015–4028 CrossRef CAS.
- J. T. M. Pater, G. Weickert, J. Loos and W. P. M. Van Swaaij, Chem. Eng. Sci., 2001, 56, 4107–4120 CrossRef CAS.
- X. Zheng and J. Loos, Macromol. Symp., 2006, 236, 249–258 CrossRef CAS.
- B. Horáčková, Z. Grof and J. Kosek, Chem. Eng. Sci., 2007, 62, 5264–5270 CrossRef.
- V. F. Tisse, R. M. Briquel and T. F. L. McKenna, Macromol. Symp., 2009, 285, 45–51 CrossRef CAS.
- M. G. Chiovetta and D. A. Estenoz, Macromol. Mater. Eng., 2004, 289, 1012–1026 CrossRef CAS.
- A. Alizadeh and T. F. L. L. McKenna, Macromol. React. Eng., 2018, 12, 1700027 CrossRef.
- T. F. L. Mckenna, E. Tioni, M. M. Ranieri, A. Alizadeh, C. Boisson and V. Monteil, Can. J. Chem. Eng., 2013, 91, 669–686 CrossRef CAS.
- F. Machado, E. L. Lima, J. C. Pinto and T. F. L. McKenna, Polym. Eng. Sci., 2011, 51, 302–310 CrossRef CAS.
- M. Emami, M. Parvazinia and H. Abedini, Iran. Polym. J., 2017, 26, 871–883 CrossRef CAS.
- E. L. Weist, A. H. Ali, B. G. Naik and W. C. Conner, Macromolecules, 1989, 22, 3244–3250 CrossRef CAS.
- H.-L. L. Rönkkö, T. Korpela, H. Knuuttila, T. T. Pakkanen, P. Denifl, T. Leinonen, M. Kemell and M. Leskelä, J. Mol. Catal. A: Chem., 2009, 309, 40–49 CrossRef.
- M. Abboud, P. Denifl and K.-H. Reichert, Macromol. Mater. Eng., 2005, 290, 558–564 CrossRef.
- K. W. Bossers, R. Valadian, S. Zanoni, R. Smeets, N. Friederichs, J. Garrevoet, F. Meirer and B. M. Weckhuysen, J. Am. Chem. Soc., 2020, 142, 3691–3695 CrossRef CAS PubMed.
- K. W. Bossers, R. Valadian, J. Garrevoet, S. van Malderen, R. Chan, N. Friederichs, J. Severn, A. Wilbers, S. Zanoni, M. K. Jongkind, B. M. Weckhuysen and F. Meirer, JACS Au, 2021, 1(6), 852–864 CrossRef CAS PubMed.
- W. Curtis Conner, S. W. Webb, P. Spanne and K. W. Jones, Macromolecules, 1990, 23, 4742–4747 CrossRef.
- K. W. Jones, P. Spanne, W. B. Lindquist, W. C. Conner and M. Ferrero, Nucl. Instrum. Methods Phys. Res., Sect. B, 1992, 68, 105–110 CrossRef.
- S. Boden, M. Bieberle, G. Weickert and U. Hampel, Powder Technol., 2008, 188, 81–88 CrossRef CAS.
- L. Seda, A. Zubov, M. Bobak, J. Kosek and A. Kantzas, Macromol. React. Eng., 2008, 2, 495–512 CrossRef CAS.
- L. Meisterová, A. Zubov, K. Smolná, F. Štěpánek and J. Kosek, Macromol. React. Eng., 2013, 7, 277–288 CrossRef.
- X. Zheng, M. S. Pimplapure, G. Weickert and J. Loos, e-Polym., 2006, 6, 1–10 Search PubMed.
- G. Fink, B. Steinmetz, J. Zechlin, C. Przybyla and B. Tesche, Chem. Rev., 2000, 100, 1377–1390 CrossRef CAS PubMed.
- S. Knoke, F. Korber, G. Fink and B. Tesche, Macromol. Chem. Phys., 2003, 204, 607–617 CrossRef CAS.
- H. Hammawa and S. E. Wanke, J. Appl. Polym. Sci., 2007, 104, 514–527 CrossRef CAS.
- J. Zhou, N. Li, N. Bu, D. T. Lynch and S. E. Wanke, J. Appl. Polym. Sci., 2003, 90, 1319–1330 CrossRef CAS.
- T. Vestberg, P. Denifl, C.-E. Wilen and C.-E. Wilén, J. Appl. Polym. Sci., 2008, 110, 2021–2029 CrossRef CAS.
- J. I. Goldstein, D. E. Newbury, J. R. Michael, N. W. M. Ritchie, J. H. J. Scott and D. C. Joy, Scanning Electron Microscopy and X-Ray Microanalysis, Springer New York, New York, NY, 2018 Search PubMed.
- M. E. Z. Velthoen, J. D. Meeldijk, F. Meirer and B. M. Weckhuysen, Chem. – Eur. J., 2018, 24, 11944–11953 CrossRef CAS PubMed.
- A. S. Michaels and H. J. Bixler, J. Polym. Sci., 1961, 50, 393–412 CrossRef CAS.
- S. Ebnesajjad, in Chemical Resistance of Commodity Thermoplastics, Elsevier, 2016, pp. xxvii–xli Search PubMed.
- M. Daftaribesheli, PhD Thesis, University of Twente, 2009 Search PubMed.
- M. F. Bergstra and G. Weickert, Macromol. Mater. Eng., 2005, 290, 610–620 CrossRef CAS.
- A. Di Martino, G. Weickert and T. F. L. McKenna, Macromol. React. Eng., 2007, 1, 165–184 CrossRef CAS.
- T. F. L. McKenna, Macromol. Symp., 2007, 260, 65–73 CrossRef CAS.
- A. Yiagopoulos, H. Yiannoulakis, V. Dimos and C. Kiparissides, Chem. Eng. Sci., 2001, 56, 3979–3995 CrossRef CAS.
- P. Smith, H. D. Chanzy and B. P. Rotzinger, J. Mater. Sci., 1987, 22, 523–531 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic of fragmentation modes, smallest fragment detectable size, N2-physisorption and EDX of pristine catalyst, over-heating during reaction, segmentation methods, polymer yields and additional SEM cross-sections and segmented images. See DOI: 10.1039/d1cy00930c |
| This journal is © The Royal Society of Chemistry 2021 |