
Hybrid particles derived from alendronate and bioactive glass for treatment of osteoporotic bone defects†
Mani
Diba‡
a,
Winston A.
Camargo§
a,
Tatiana
Zinkevich§
b,
Alina
Grünewald
c,
Rainer
Detsch
c,
Yoones
Kabiri
 d,
Arno P. M.
Kentgens
b,
Aldo R.
Boccaccini
c,
Jeroen J. J. P.
van den Beucken
a and
Sander C. G.
Leeuwenburgh
*a
d,
Arno P. M.
Kentgens
b,
Aldo R.
Boccaccini
c,
Jeroen J. J. P.
van den Beucken
a and
Sander C. G.
Leeuwenburgh
*a
aDepartment of Regenerative Biomaterials, Radboud University Medical Center, Philips van Leydenlaan 25, 6525 EX, Nijmegen, The Netherlands. E-mail: Sander.Leeuwenburgh@radboudumc.nl
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
cInstitute of Biomaterials, Department of Materials Science and Engineering, University of Erlangen-Nuremberg, Cauerstraße 6, 91058 Erlangen, Germany
dKavli Institute of Nanoscience Delft, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 4th January 2019
Abstract
Osteoporosis is the most widespread metabolic bone disease which represents a major public health burden. Consequently, novel biomaterials with a strong capacity to regenerate osteoporotic bone defects are urgently required. In view of the anti-osteoporotic and osteopromotive efficacy of alendronate and 45S5 bioactive glass, respectively, we investigated the feasibility to synthesize novel hybrid particles by exploiting the strong interactions between these two compounds. Herein, we demonstrate the facile preparation of a novel class of hybrid particles of tunable morphology, chemical composition and structure. These hybrid particles (i) release alendronate and various inorganic elements (Ca, Na, Si, and P) in a controlled manner, (ii) exhibit a strong anti-osteoclastic effect in vitro, and (iii) stimulate regeneration of osteoporotic bone in vivo. Consequently, this novel class of hybrid biomaterials opens up new avenues of research on the design of bone substitutes with specific activity to facilitate regeneration of bone defects in osteoporotic patients.
1. Introduction
Osteoporosis is the most widespread metabolic bone disease which increases the risk of bone fractures considerably by reducing bone mass.1 Each year around 9 million osteoporotic bone fractures occur worldwide,2 which represents a major public health burden. Biomaterials are frequently applied to support the process of bone healing, but conventional bone-substituting biomaterials lack the capacity to ensure successful healing of bone suffering from osteoporosis.3,4 Consequently, novel biomaterials specifically designed to regenerate osteoporotic bone defects are urgently required. Generally, catabolic processes dominate anabolic processes in osteoporotic bone, which compromises regeneration of bone defects.5 Bisphosphonates such as alendronate (alendronic acid; ALN) are common anti-catabolic drugs which inhibit osteoclastic bone resorption.6 These drugs are usually administered systemically, which results in low bioavailability, a lack of site specificity and increased risks of side effects. A major side effect of such high-dose and long-term bisphosphonate treatment is osteonecrosis of the jaw.7 Although bisphosphonates effectively decrease bone resorption, effective stimulation of anabolic bone formation is required to achieve successful regeneration of osteoporotic bone defects.5,8 Bioactive glasses (BGs) are typically amorphous calcium-containing silicates which are well-known for their osteopromotive capacity.9,10 The most frequently used type of BGs is the 45S5 composition, which contains 45 wt% SiO2, 24.5 wt% CaO, 24.5 wt% Na2O, and 6 wt% P2O5.9,11 This type of BG is osteoconductive, osteogenic, and biodegradable.10,12–14 Consequently, in view of the strong anti-osteoporotic and osteopromotive capacity of bisphosphonate drugs (such as ALN) and 45S5 BG, respectively, we hypothesize that hybrid formulations of both compounds facilitate highly effective regeneration of defects in osteoporotic bone. Nevertheless, only very few studies investigated the feasibility of combining BGs with bisphosphonate drugs. Srisubut et al.15 reported that local delivery of ALN with BG granules (Biogran®) enhanced the osteopromotive efficacy of BG granules when implanted in mandibular bone defects of healthy rats. Nevertheless, this study did not provide any details on the effects of ALN on the chemical structure of BG granules. Bui et al.16 developed zoledronate-46S6 BG composites by simple adsorption of zoledronate onto BG particles. In 2011, Lee et al.17 reported that osteoblastic differentiation of cells cultured in the presence of a bioactive glass-ceramic was increased when a specific concentration (10−7 M) of ALN was added to the culture media. In 2013, Rosenqvist et al.18 studied the interaction between S53P4 BG with clodronate and reported the formation of a calcium clodronate layer on the BG surface upon mixing of both substances in a saline solution. However, this precipitated layer could not be isolated and its chemical composition was not determined. Other studies have used physical adsorption19 or vacuum infiltration20 methods to achieve local delivery of bisphosphonate drugs from BG nanoparticles or coatings, respectively. Generally, despite the fact that promising indications have been reported for the strong affinity between BGs and BPs, these interactions have not been studied systematically as basis for the synthesis of a new class of hybrid BG–BP biomaterials.Herein, we demonstrate the facile preparation of a novel class of hybrid biomaterials by exploiting the strong interaction between ALN and 45S5 BG particles. By immersing BG particles in aqueous ALN solutions at different pH and ALN/BG ratios, we synthesize hybrid particles of tunable morphology, chemical composition and structure through a carefully controlled precipitation process. By monitoring the precipitation kinetics as a function of immersion time using inductively coupled plasma-optical emission spectroscopy and solid-state nuclear magnetic resonance spectroscopy, we obtain fundamental understanding of the interactions between BG and ALN and the resulting formation mechanism of hybrid particles. We demonstrate that these hybrid particles (i) release alendronate and various inorganic elements (Ca, Na, Si, and P) in a controlled manner, (ii) exhibit a strong anti-osteoclastic effect in vitro, and (iii) stimulate regeneration of osteoporotic bone in vivo. Consequently, we conclude that this novel class of hybrid biomaterials opens up new avenues of research on the design of bone substitutes with specific activity to induce regeneration of bone in osteoporotic patients.
2. Materials and methods
2.1 Materials
BG particles (45S5 composition) with a mean particle size of 2.0 ± 1.2 μm were obtained from Schott AG (Mainz, Germany). Alendronic acid (4-amino-1-hydroxybutane-1,1-diphosphonic acid) powder was purchased from AK Scientific (Union City, CA, USA). 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES; ≥99.5%) and 2-(N-morpholino)ethanesulfonic acid hydrate (MES hydrate; ≥99.5%) were obtained from Sigma-Aldrich. Sodium hyaluronate powder (1.01 MDa–1.8 MDa) was purchased from Lifecore Biomedical (Chaska, MN, USA). All other chemicals were reagent grade and used without any purification.2.2 Immersion procedure
Bisphosphonate solutions were prepared by dissolving 100 mM of ALN powder in Milli-Q water and adjusting their pH using NaOH solution. BG particles were immersed in bisphosphonate solutions (pH 5, 7 or 9) at concentrations of 11.5, 23, and 46 mg mL−1 corresponding to CaBG/ALN molar ratios of 0.5, 1, and 2, where CaBG and ALN correspond to Ca content of BG particles and ALN molecules in solution, respectively. The mixtures were kept at 37 °C for various time periods up to three days. Subsequently, the mixtures were centrifuged at 5000 rpm for 5 min. The supernatants were collected for further analysis and the solid sediments were rinsed with Milli-Q water through a redispersion and centrifugation (5000 rpm for 5 min) step. Thereafter, the sedimented hybrid particles were dried at 65 °C for 2 h, after which they were kept at room temperature for further analysis. The composition of hybrid particles in this paper is denoted by HPX–Y, where X and Y indicate the CaBG/ALN molar ratio and the pH of bisphosphonate solution used for the immersion procedure, respectively.2.3 Electron microscopy
A Field Emission Scanning Electron Microscope (FE-SEM; Sigma 300, Zeiss, Germany) equipped with an energy-dispersive X-ray detector (EDX; XFlash detector 610 M, Brucker Nano GmbH, Germany) was used for morphological evaluation of particles and EDX mapping of Si. For these experiments, sample preparation was carried out by dispersing the particles on a sticky carbon tape and sputter-coating with a conductive chromium layer with a thickness of ∼5 nm. The size of hybrid microparticles was determined by measuring the width and length of 50 particles in SEM images.A Transmission Electron Microscope (TEM; Titan cubed G2 60-300) equipped with an EDX detector was used for scanning transmission electron microscopy (STEM) and EDX mapping of single particles at 300 kV. For these experiments, sample preparation was carried out by dispersing 0.02 g of the particles in 2 mL of ethanol using a sonication bath (5 min). Thereafter, 4 μL of the dispersion was drop-casted onto a glow-discharged standard perforated carbon TEM grid (Quantifoil® 200 mesh) and incubated for 1 minute. Finally, the TEM grid was rinsed with Milli-Q water, dried and used for the imaging experiments.
2.4 Elemental analysis
The ALN content of the precipitated particles was determined by measuring their nitrogen content using a CN Elemental Analyzer (NA1500, Carlo Erba – Thermo Fisher Scientific, USA). Calcium (Ca), sodium (Na), phosphorus (P) and silicon (Si) contents of the precipitated particles were determined by elemental analysis of the supernatants using Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES, iCAP 6300, Thermo Fisher Scientific, USA). First, the total mass of each element in the mixture (ALN solution + BG particles) was calculated based on the elemental composition of the ALN solutions and BG particles. Subsequently, the composition of precipitated particles was determined by deducting the mass of each element in the supernatant from the total mass of that specific element in the mixture. Since both ALN and BG contain phosphorus, the mass of P derived from ALN (as measured using the CN Elemental Analyzer) was subtracted from the total mass of P present in the solid sediments to determine the mass of P derived from BG. Subsequently, the percentage of each element in the precipitated particles was calculated as a percentage of the total mass of dry precipitates.Sample preparation for the ICP-OES measurements was carried out by diluting all immersion solutions using 2 v/v% nitric acid solutions (both before and after precipitation of the particles). A similar ICP-OES method was used to determine the composition of supernatants at different stages of the immersion process.
2.5 Powder X-ray diffraction (XRD)
Powder XRD analysis of precipitated particles was carried out using a PANalytical X’Pert3 Powder X-ray diffractometer with Cu Kα radiation at a voltage of 45 kV and a current of 40 mA. The measurements were performed from 7° to 47° (2θ) using a step size of 0.05° and a counting time of 30 s per step.2.6 Solid-state nuclear magnetic resonance (NMR) spectroscopy
Solid-state NMR spectroscopy was used to study the chemical structure and the precipitation mechanism of hybrid particles. In view of the composition of BG and ALN, phosphorous, carbon, sodium, and proton spectra were collected as described below.Phosphorous spectra were acquired on a Varian NMR spectrometer operating at a proton frequency of 300 MHz. Experiments were performed at a Magic angle spinning (MAS) frequency of 10 kHz, which did not produce spinning sidebands in the region of real NMR-signals. Cross-polarization (CP) experiments were performed for qualitative evaluation of different samples to save experimental time. In these experiments, a proton 2.5 μs π/2 pulse was applied (ν1 = 100 kHz). The resulting phosphorous signal was acquired using 1H decoupling employing a radiofrequency field (rf-field) strength of 75 kHz. The CP contact-time was 1.75 ms. Direct-polarization (DP) measurements were conducted for a quantitative analysis of phosphonate and phosphate fractions in the various samples. The relaxation delay of 300 s was used after relaxation time measurements, which provided spin-lattice relaxation (T1) values in the range of 35–60 s. 31P chemical shifts were referenced using the known shifts of ALN as a secondary reference.21
Carbon spectra were acquired on a Varian NMR spectrometer operating at a proton frequency of 400 MHz. Experiments were performed at a MAS frequency of 15 kHz. Again the CP pulse sequence was used at a proton rf-field strength of 91 kHz, using a 2.75 μs proton π/2 pulse. The CP contact-time was optimized to 2 ms. 75 kHz decoupling power was used to remove dipole–dipole interactions between carbons and protons. 13C chemical shifts were referenced to the known chemical shifts of adamantane; 29 ppm and 38 ppm for methine and methylene carbons, respectively.
Sodium spectra were recorded on an 850 MHz Varian NMR spectrometer at a MAS frequency of 35 kHz. The 23Na signal was obtained by means of single pulse excitation using a 30° pulse to ensure quantitative evaluation of the spectra. The 23Na radiofrequency field strength of 135 kHz was calibrated using solid NaCl, which was also used as a secondary reference for the chemical shift.
Proton spectra were acquired on an 850 MHz Varian NMR spectrometer at a MAS frequency of 35 kHz. Single Pulse Excitation was employed using a hard 90° pulse (at an rf-field strength of 100 kHz). The acquisition time was 50 ms. These experiments were performed at high spinning frequencies and high magnetic field to average homonuclear proton–proton couplings and enhance the spectral resolution.
2.7 In vitro degradation and release behavior of hybrid particles
Degradation of hybrid particles and release of their content were evaluated upon immersion in aqueous solutions for up to 14 days. To investigate the effect of pH on the degradation of the particles, the immersion process was carried out at neutral (pH 7.4) or acidic (pH 5.5) conditions using HEPES- or MES-buffers, respectively. These two conditions were selected to mimic the local pH of healthy and inflamed tissues, respectively. The buffer solutions were prepared by dissolving 100 mM of HEPES or MES powder in Milli-Q water and adjusting the pH using NaOH solution. Thereafter, 20 mg of hybrid particles were added into 2 mL Eppendorf tubes. The tubes were filled with 2 mL of a buffered solution and incubated at 37 °C on a rotating plate at a rate of 120 rpm. At each time point, the tubes were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min. Afterwards, the solutions were collected for further analysis, fresh solutions were added into the tubes, and the tubes were put back in the incubator until the next time point. After the last time point (day 14), the remaining particles were dried at 65 °C for 2 h and their weight was measured using an analytical balance. The elemental composition of the collected solutions was analyzed by means of ICP-OES. The cumulative release of each element (Ca, Na, P and Si) was expressed relative to its original content in the hybrid powder. The expressed values for P include P derived from both ALN and BG constituents.
000 rpm for 2 min. Afterwards, the solutions were collected for further analysis, fresh solutions were added into the tubes, and the tubes were put back in the incubator until the next time point. After the last time point (day 14), the remaining particles were dried at 65 °C for 2 h and their weight was measured using an analytical balance. The elemental composition of the collected solutions was analyzed by means of ICP-OES. The cumulative release of each element (Ca, Na, P and Si) was expressed relative to its original content in the hybrid powder. The expressed values for P include P derived from both ALN and BG constituents.
2.8 Influence of hybrid particles on osteoclastic cells
To assess the biological effect of the particles on osteoclasts, the effects of different concentrations of the particles (n = 6 for each concentration) were evaluated using osteoclastic cells derived from a mouse leukaemic monocyte-macrophage (RAW 264.7) cell line.Actin/nucleus staining. The medium was removed from each well and the cells were washed with a solution of 1% bovine serum albumin (BSA) in PBS solution (BSA/PBS). Thereafter, a fixing solution (3.7% paraformaldehyde in PBS) was added to the samples for 15 min. The fixing solution was then removed and samples were washed with PBS/BSA for three times, each time for 5 minutes. Thereafter, cells were permeabilized with a buffer (0.1% Triton-X in PBS) for 5 min. The buffer was removed from each well and the samples were stained with rhodamine phalloidin (8 μL mL−1 solutions in PBS/BSA; Thermo Fisher Scientific, USA) for 1 h in the dark to stain F-actin. Thereafter, cells were washed with PBS/BSA for two times, each time for 5 minutes. Next, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, 1 μL mL−1 solutions in PBS/BSA, Thermo Fisher Scientific, USA) for 5 min. Finally, cells were washed once with PBS/BSA for 5 min and kept in PBS for microscopic analysis.
TRAP staining. The medium was removed from each well and the cells were washed with PBS. Thereafter, cells were fixed with a fixing solution of 4% paraformaldehyde in a citrate–acetone solution for 5 min. The fixing solution was removed and cells were washed with PBS. The staining solution (2% Sodium-Garnet solution, 1% naphtol, 4% acetate, and 2% tartrate in water) was added to the cells and left for 2 h in the dark at 37 °C. The staining solution was removed and cells were washed with distilled water. Finally, the well plate was air-dried for microscopic analysis.
2.9 Influence of hybrid particles on regeneration of osteoporotic bone
To assess the influence of hybrid particles on the regeneration of osteoporotic bone defects in vivo, HP1–7 and HP2–7 hybrid particles were selected for implantation into defects created in the femoral condyles of osteoporotic rats. In these experiments, pure BG particles were used as positive control.:12 h, temperature: 20–22 °C, relative humidity: 45–55%). After acclimatization, the animals underwent orchidectomy (ORX) surgery to induce an osteoporotic bone condition as described previously,27 and had free access to water and low calcium pelleted chow (0.01% Ca and 0.77% P; ssniff Spezialdiäten GmbH, Soest, Germany). After 6 weeks, osteoporotic conditions in the femoral condyle trabecular bone of the animals were confirmed using an in vivo micro-CT imaging system (Inveon; Siemens Medical Solutions, Knoxville, TN).27 Thereafter, bilateral frontal femoral condyle bone defects were made (n = 8 per experimental group) and the particulate pastes were injected in the left or right femur of each animal under inhalation anesthesia (2% Isoflurane® by volume) in a randomized manner (Table S1, ESI†). To perform the surgery, the rats were immobilized supine with the knee joint in a maximally flexed position and the hind limbs were shaved, washed and disinfected with povidone iodine (10%). After exposure of the knee joint, a 1 mm pilot hole was drilled and the defect was gradually widened using various drills of increasing size by means of a surgical motor (Elcomed 100, W&H Dentalwerk Burmoos, Austria) at low rotational drilling speed (800 rpm) and continuous external cooling with saline solution, until a cylindrical defect of 2.5 mm in diameter and 5 mm in depth was created. The surgery was carried out in both legs of the animals and one defect was created in each femoral condyle. Thereafter, the pastes were injected into the defects using 1 mL disposable syringes and the wounds were closed with metallic wound clips (Becton Dickinson, Franklin Lakes, NJ). For pain control, Temgesic® (0.02 mg kg−1) and Rimadyl® (5.0 mg kg−1) were used for 2 days postoperatively. During the implantation period, the rats were fed with the low calcium diet.
2.10 Statistics
All values were presented as average ± standard deviation. The statistical tests were carried out using GraphPad Prism® software. A two-way ANOVA with Tukey's multiple comparisons test was used for statistical analysis of the in vitro tests. Histomorphometrical results were statistically evaluated using a one-way ANOVA test, followed by Tukey's multiple comparisons test. In all tests, the significance threshold was set at P < 0.05.3. Results and discussion
Previous investigations18,29,30 revealed strong interactions between bisphosphonate molecules and BG particles (Fig. S1, ESI†). To explore the feasibility of exploiting these interactions for the synthesis of hybrid particles, we immersed the BG particles in aqueous solutions containing a high concentration of dissolved ALN (100 mM). BG particles rapidly shrank upon immersion in ALN solutions, and precipitates were formed within 3 days of incubation. However, immersion of BG particles in ALN-free solutions resulted into sedimentation of particles without visual changes during 3 days of incubation. To unravel the chemical nature of these precipitates, we immersed different amounts of BG particles in ALN solutions at different pH values, and studied the physicochemical properties of resulting precipitates using SEM, TEM, EDX, XRD, solid-state NMR and elemental analysis.3.1 Morphology, composition and structure
Fig. 1 shows SEM images of hybrid particles formed at different immersion conditions. The morphology of the precipitated particles varied depending on the respective CaBG/ALN ratio (R). While spherulitic particulate clusters were formed at a CaBG/ALN ratio of 0.5 (Fig. 1A and D), discrete needle-like microparticles were formed at ratios of 1 and 2 (Fig. 1B, C, E, and F). Importantly, particles synthesized at a ratio of 2 contained unreacted BG residues as confirmed by EDX mapping (Fig. S2, ESI†). The size and morphology of precipitated particles were also affected by the initial pH of the ALN solution. At a fixed CaBG/ALN ratio of 0.5, the precipitated clusters revealed a spherulitic structure (∼2 mm in diameter) composed of nanosheets or honeycomb-like morphologies (∼0.1 mm in diameter) composed of entangled nanofibers (Fig. 2) when prepared at pH 7 or 9, respectively. In general, at a basic pH of 9, smaller particles were formed than at a neutral pH of 7. For example, at a CaBG/ALN ratio of 1, the length of needle-like microparticles was about 9 ± 5 μm or 4 ± 3 μm when prepared at pH 7 or 9, respectively. However, the shape of the precipitated particles did not depend on the pH at CaBG/ALN ratios of 1 and 2. Importantly, when ALN solutions were acidic (pH 5), the precipitation of hybrid particles was inhibited due to the protonation of bisphosphonate groups at acidic conditions, reducing their binding affinity for BG surfaces.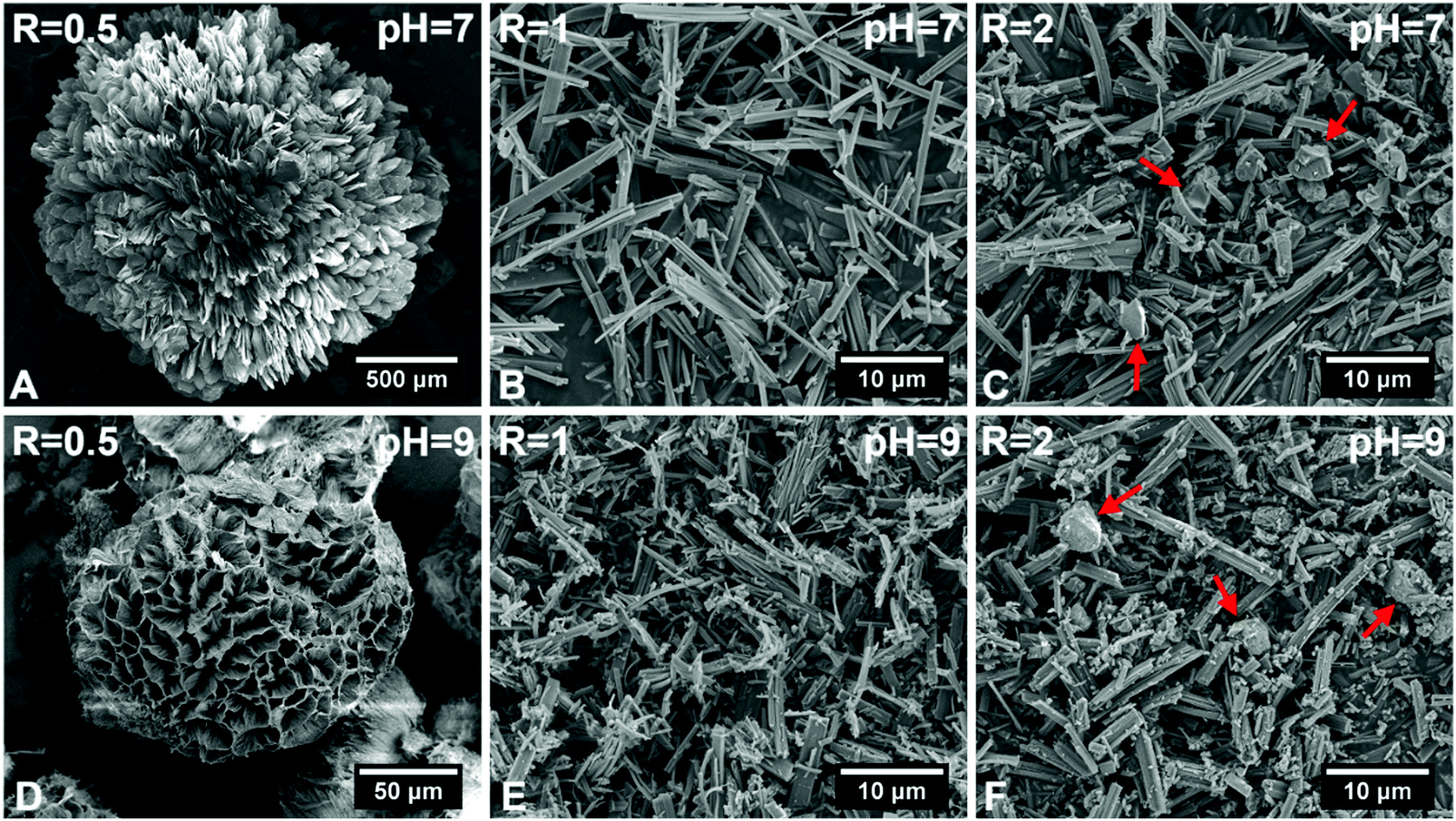 | ||
| Fig. 1 Scanning electron micrographs of hybrid particles precipitated at different CaBG/ALN ratios (R) and pH values after 3 days of incubation at 37 °C. Red arrows indicate bioactive glass residues. | ||
 | ||
| Fig. 2 High magnification scanning electron micrographs of hybrid particles precipitated at a CaBG/ALN ratio of 0.5 and a pH of 7 (A and B) or 9 (C and D). | ||
The multiple morphologies observed for these hybrid particles resemble the morphological variability as observed in natural structures (e.g. diatoms) where self-assembly occurs between inorganic ions and organic molecules.31 Indeed, similar processes have also been studied to enable rational design of precipitating microstructures such as carbonate-silica co-precipitates by controlling reaction conditions such as ionic concentration and solution pH.32,33
Variation of the immersion conditions allowed tuning of both the morphology of the precipitated hybrid particles as well as their chemical composition (Table S2, ESI†). While hybrid particles formed at CaBG/ALN ratios of 0.5 or 1 contained ∼55–70 wt% of ALN, the ALN content of particles was significantly lowered at a CaBG/ALN ratio of 2. At this ratio, the particles also contained higher amounts of Si and P (not derived from ALN; see Materials and methods) which can be partially attributed to BG residues. Due to the compositional similarity of samples prepared at pH 7 or 9, further investigations were focused on hybrid particles formed using ALN solutions at a pH of 7.
We used EDX elemental mapping combined with STEM imaging to determine the elemental distribution within single hybrid particles, which revealed a uniform distribution of elements throughout each particle (Fig. 3A). To assess the crystallinity of hybrid particles, we collected the XRD patterns of various particles types (Fig. 3B). The XRD spectra indicated that the ALN powder was highly crystalline, whereas the BG particles were fully amorphous. Interestingly, the hybrid particles revealed crystalline structures that depended on the CaBG/ALN ratio used during immersion. The particles precipitated at a ratio of 2 were less crystalline as compared to particles containing lower amounts of BG (prepared at ratios of 0.5 and 1). Nevertheless, XRD peaks could not be identified using available crystallographic databases and literature, indicating the formation of novel crystalline phases that were not reported before to the best of our knowledge. Similar to novel metal-organic frameworks,34 structural determination of these hybrid particles was challenging. Our attempts to use single-crystal XRD and electron diffraction techniques were not successful due to the small size and electron-beam sensitivity of the hybrid particles, respectively.
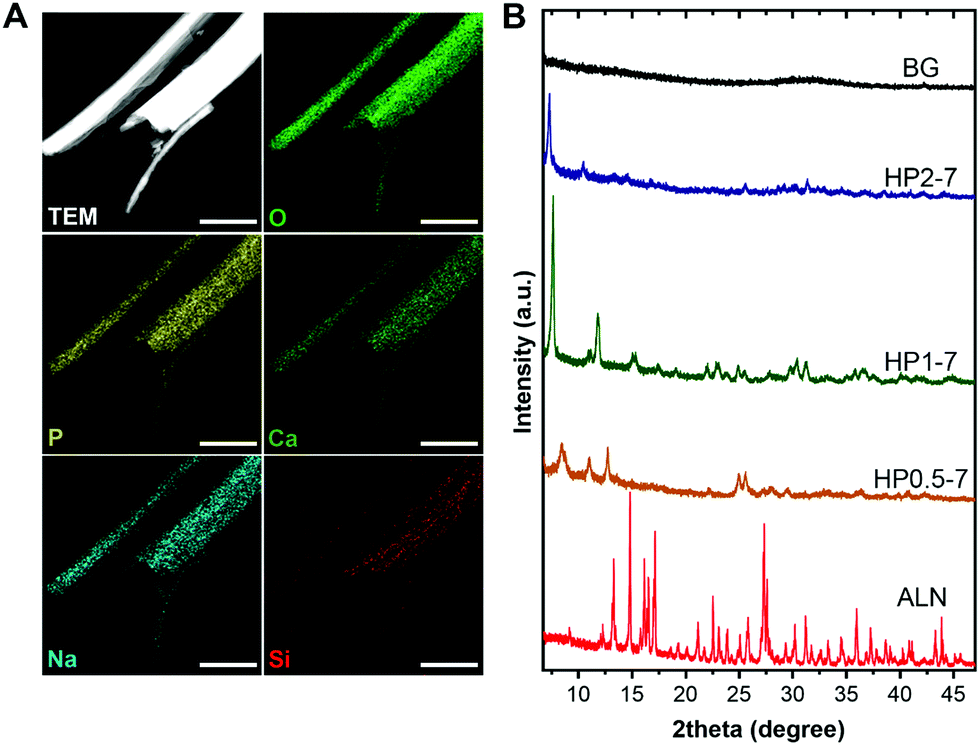 | ||
| Fig. 3 (A) Scanning TEM image and EDX elemental maps of single HP1–7 particles. (B) Powder XRD spectra of different particle types. Scale bars in images correspond to 1 μm. | ||
Therefore, we used solid-state NMR spectroscopy to gain fundamental understanding of the chemical structure of these hybrid particles (Fig. 4). As shown in Fig. 4A, the 13C spectra of hybrid particles showed pronounced peak broadening and shifts as compared to the peaks of pure ALN, indicating that hybrid particles were less crystalline and contain coordination complexes, respectively.21,35–37 The 13C spectrum of HP0.5–7 consisted of four broad signals. In this sample, the largest peak shift (Δδ = 4.6 ppm) occurred for the peak at ∼72 ppm corresponding to the carbon closest to the phosphonate groups of ALN (see Fig. S3, ESI† for the peak assignment). The two peaks at ∼24 and ∼32 ppm corresponded to the CH2 groups and shifted 3.2 ppm downfield, suggesting a large conformational change for ALN molecules.37 The smallest shift was observed for the peak located at ∼40 ppm (Δδ = −0.9 ppm), indicating that the amine-linked CH2 group was affected least. Interestingly, by increasing the CaBG/ALN ratio, the 13C spectra became narrower and revealed different features. Particularly, for HP1–7 and HP2–7, the amine-linked CH2 signal showed a larger downfield shift (Δδ ≈ −4 ppm) as compared to HP0.5–7, indicating that the amine group also participated in the complex formation when higher CaBG/ALN ratios were used. Similar trends were observed in the 31P spectra (Fig. 4B) of the hybrid particles. The 31P spectrum of HP0.5–7 showed a broad featureless peak positioned at 20 ppm, corresponding to the presence of a variation in the microenvironments around the phosphonate groups at this composition. The 31P spectrum of HP1–7 exhibited a narrower peak, shifted upfield to 17.8 ppm, corresponding to a more homogeneous microenvironment surrounding the ALN molecules. Moreover, the 31P of HP2–7 displayed two intense narrow peaks, which suggest the presence of well-ordered domains at this composition. These sharp peaks resembled the sharp peaks present in the 31P spectrum of pure ALN, but displayed a shift which might be caused by complexation with Ca2+.35,38 Importantly, this spectrum also revealed the existence of a signal from residual BG, which is in agreement with our previous SEM observations. Nevertheless, the BG signal showed a slight upfield shift which indicates that the microenvironment surrounding BG residues in the HP2–7 sample was different from the one in pure BG sample (Fig. S4, ESI†). Integration of phosphonate (10 to 27 ppm) and phosphate (−2 to 9 ppm) regions of 31P DP spectra of HP2–7 indicated that ∼96% of all phosphorous in this sample was derived from ALN molecules, while the remaining phosphorus (4%) was attributed to BG residues.
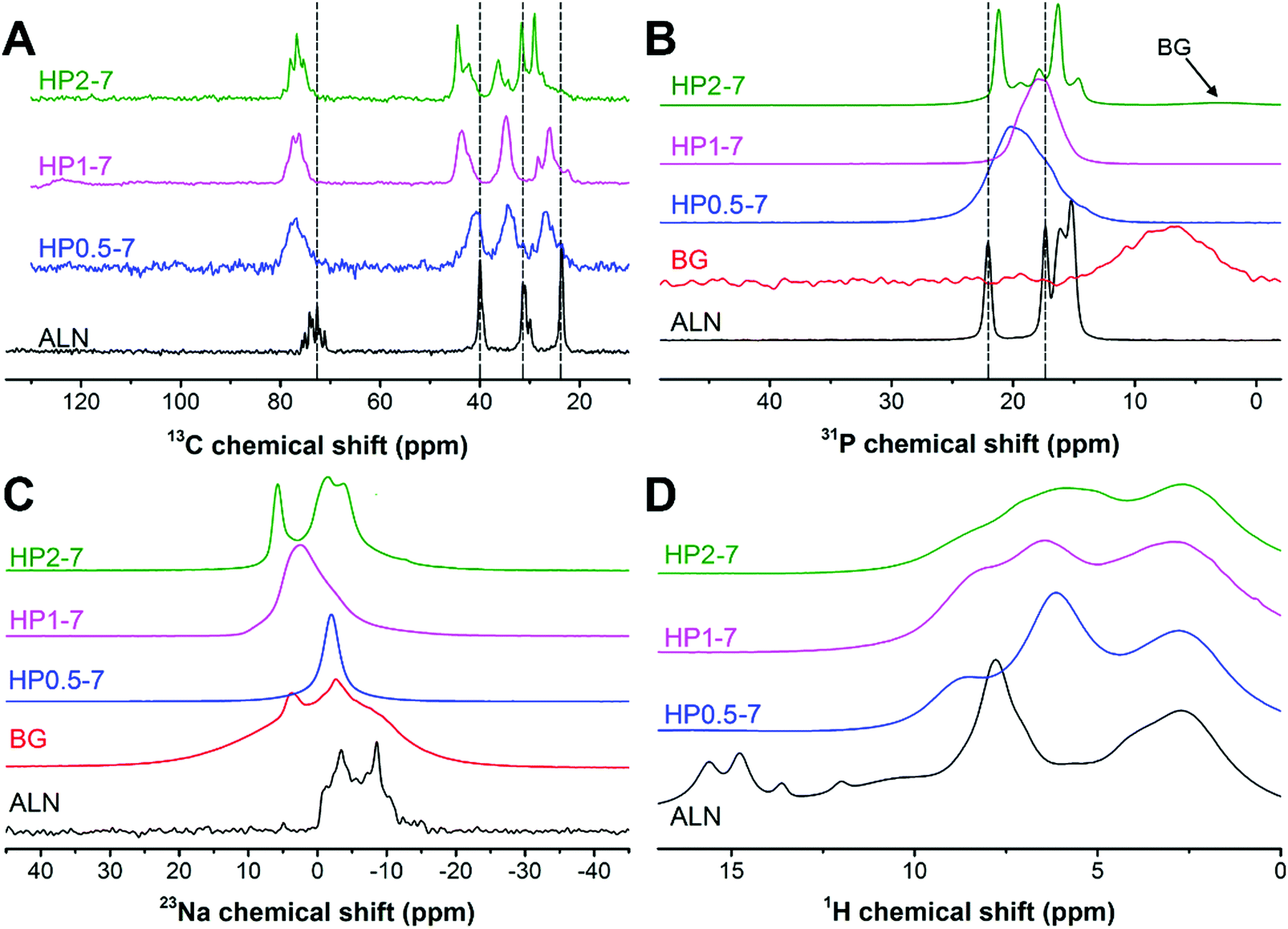 | ||
| Fig. 4 Solid-state CP-MAS NMR spectra of different types of hybrid particles. | ||
As shown in Fig. 4C, the 23Na spectrum of ALN displays a clear second-order quadrupolar pattern from a single Na site in a crystalline material (δiso = 0.64 ppm, e2qQ/h = 3.62 MHz, η = 0.33). The HP0.5–7 sample displays a fairly narrow 23Na resonance at around 0 ppm which is likely caused by Na in an octahedral oxygen coordination (e.g. coordinated by residual water molecules). The HP1–7 particles contained a larger variety of Na microenvironments as compared to HP0.5–7 and pure ALN. Finally, the HP2–7 sample displayed two 23Na resonances in relatively well-defined environments, a narrow peak at ∼6 ppm and a line displaying a fairly well-defined second-order quadrupolar interaction (δiso = 1.7 ppm, e2qQ/h = 3.13 MHz, η = 0.27). In analogy to the 31P spectra this is somewhat similar but certainly not identical to the ALN resonance.
Finally, the 1H spectrum of pure ALN displayed several low field signals (δH > 10 ppm) corresponding to the phosphonate groups (Fig. 4D). These signals were absent in the spectra of all hybrid particles, corresponding to deprotonation of phosphonate groups and a high degree of coordination complex formation in these samples.35,39
Overall, the presented results confirm that hybrid particles were formed with tunable morphology, composition and structure through a simple immersion strategy. Importantly, these results indicate: (i) formation of ALN-cation coordination complexes within the hybrid particles and (ii) a strong dependence of the chemical structure of the hybrid particles on the preparation conditions.
3.2 Precipitation kinetics
To elucidate the formation mechanism of hybrid particles and the interactions between BG and ALN, we selected HP1–7 particles to study the precipitation kinetics of these hybrid particles at different time points during the immersion process by analyzing the pH and elemental composition of supernatants as well as and the weight, morphology and chemical structure of the precipitated particles using weight measurements, SEM and NMR spectroscopy (Fig. 5). As shown in Fig. 5A, BG particles rapidly dissolved immediately after their immersion in the ALN solution; within the first minute of immersion, ∼20 wt% of BG particles dissolved causing a pH rise from 7 to 10. Until 200 min of immersion, dissolution of BG particles caused a continuous release of Ca, Na and Si ions into the supernatant, while the P content of the supernatant remained almost constant (Fig. 5B). After 200 min of immersion, a sharp reduction of the concentrations of Ca, Na and P in the supernatant coincided with a considerable mass gain of the particles (Fig. 5A), corresponding to the formation of precipitates containing these three specific elements. Nevertheless, the Si concentration in the supernatant continued to rise until the major part of the silica of the original BG particles was released (∼1000 min), revealing that the released Si ions did not play a dominant role in the formation of the precipitates. On the contrary, the maximum Ca content measured in the supernatant (∼200 min) corresponded to only half of the Ca content of the original BG particles, confirming that Ca ions were actively involved in the precipitation process. Importantly, the mass of particles as well as the pH and ionic concentrations of the supernatant stabilized after 1000 min, indicating that the reaction reached an equilibrium state. These results were in agreement with the SEM images of the particles (Fig. 5C), which revealed the presence of needle-like microparticles at 200 min and the formation of precipitates with identical morphology to the final HP1–7 particles (Fig. 1B) after 1000 min of immersion.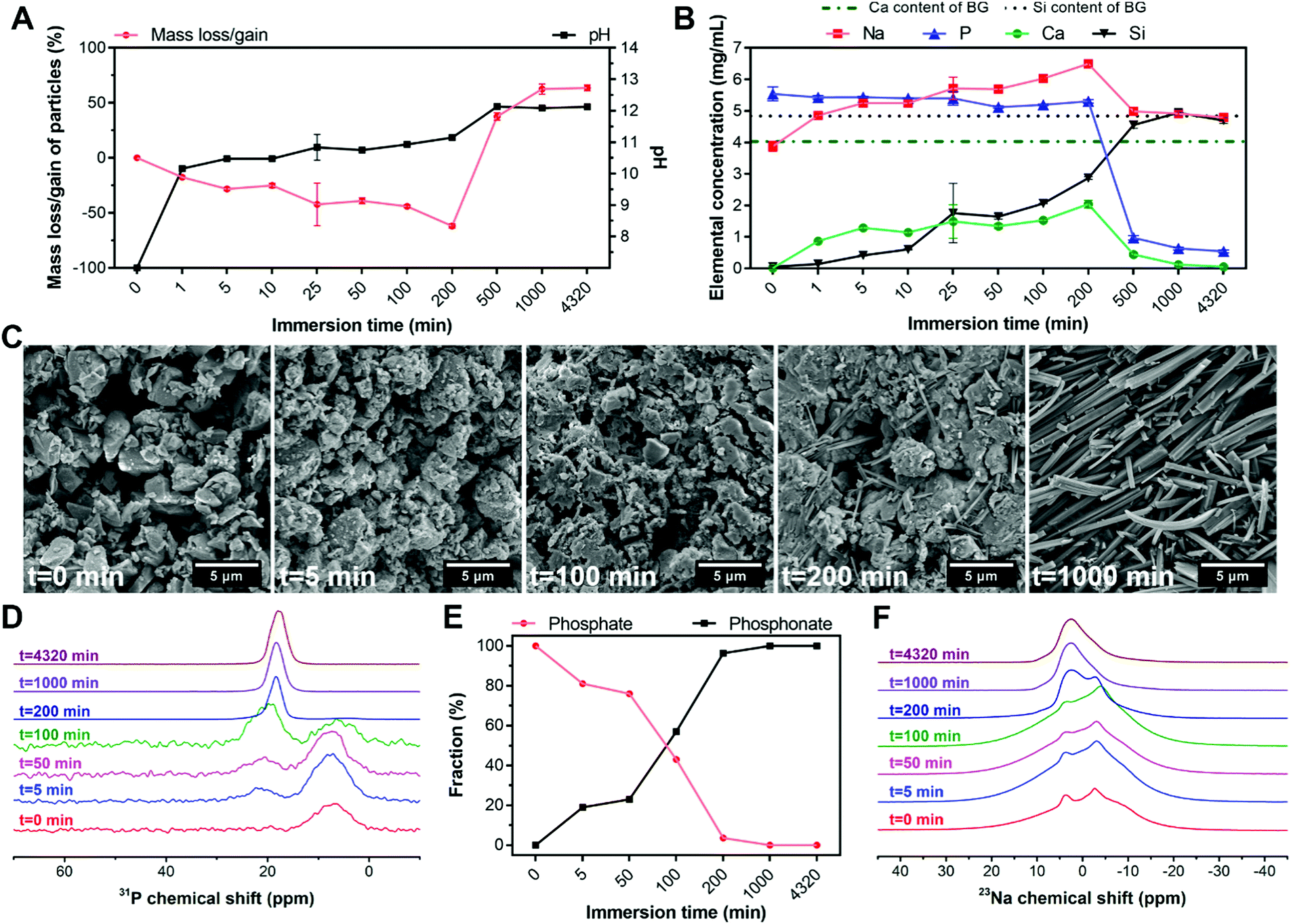 | ||
| Fig. 5 Precipitation kinetics of HP1–7 particles as a function of immersion time. (A) Mass loss/gain of particles and pH of supernatants. (B) Elemental composition of supernatants during the immersion period. (C) Scanning electron micrographs, (D) solid-state DP 31P NMR spectra, (E) phosphate and phosphonate weight fractions, and (F) solid-state MAS 23Na NMR spectra of particles at various time points. In (A and B), values are presented as average ± standard deviation for n = 3 per experimental condition. In (E), values represent the phosphate and phosphonate fractions calculated based on the NMR spectra shown in (D). | ||
Next, we studied 31P and 23Na NMR spectra of the particles as a function of immersion time. As shown in Fig. 5D, both phosphate and phosphonate signals were already evident at an early stage of the immersion process (5 min). Over time, the phosphonate fraction increased while the phosphate fraction gradually declined until its complete disappearance after 200 min (Fig. 5E). These results suggest that the formation of precipitates proceeded by substitution of phosphate groups of BG particles with phosphonate groups of ALN molecules, which is similar to previously reported mechanisms on the interaction of bisphosphonate drugs with calcium phosphate minerals.37,40 Considering the constant concentration of P in the supernatant during the first 200 min (Fig. 5B), we propose that this reaction occurs by release of ∼2 phosphate groups per ALN substitution. This substitution reaction did not affect the microenvironment of remaining phosphates in BG particles, since the phosphate peak did not shift substantially from its initial position until its disappearance (intensity <4%) from the spectrum at 200 min (Table S3, ESI†). Importantly, the phosphonate peak showed a significant change from 100 to 200 min, displaying an abrupt shift and narrowing of the signal. At this time point, significant changes were observed in the 23Na spectrum of the particles (Fig. 5F). Various Na sites were present in the samples up to 200 min, which reduced to a single Na resonance after 200 min. However, this single resonance showed some distribution in its quadrupolar interaction parameters, indicating that the Na microenvironment varied throughout the hybrid particles.
Overall, these results suggest that the precipitation of the hybrid particles proceeded through the complexation of ALN with Ca and Na cations as present in BG particles. This complexation process occurred by means of substitution of phosphate groups in BG particles by ALN molecules, leading to disruption of the BG silicate network and the release of Si, Ca and Na. The reaction between ALN and BG almost completely disrupted the BG structure after 200 min of immersion. This disruption was followed by a rapid precipitation and growth of hybrid particles, which stopped after 1000 min of immersion.
3.3 In vitro degradation and release behavior
The hybrid particles exhibited a pH- and composition-dependent degradation behavior in aqueous media (Fig. S5, ESI†). Therefore, to elucidate the degradability of these particles for applications in regenerative medicine, we studied the release of their degradation products in vitro at neutral pH (7.4) as well as acidic pH (5.5) to mimic acidosis.41 In addition, this acidic testing condition was selected since the interaction of ALN with BG was found to be strongly pH-dependent (see Section 3.1). Fig. 6 shows the cumulative release of various inorganic elements from hybrid particles, expressed as percentage of their original content in the hybrid particles, upon immersion in neutral or acidic solutions for up to 14 days. In general, the particles with a low ALN content (HP2–7) released more calcium than the two other particle types, but tended to retain their sodium content, whereas HP0.5–7 and HP1–7 showed an opposite trend by releasing less calcium but more sodium. At the neutral pH, HP0.5–7 and HP1–7 particles showed an initial rapid release of their P contents which stabilized at ∼20% upon 3 days of immersion, whereas the particles of low ALN content displayed a more gradual P release. Based on the composition of hybrid particles (Table S2, ESI†), the P released from HP0.5–7 and HP1–7 particles corresponds almost entirely to ALN, whereas the P released from the particles of low ALN content is related to a more even mixture of both ALN and BG constituents. Despite the low Si content of HP0.5–7 and HP1–7 particles (Table S2, ESI†), their Si release was more pronounced, suggesting that the minor trace of Si was not well integrated in the structure of these particle types. As hypothesized, the acidification of solutions strongly affected the release behavior of the hybrid particles. An enhancement of Ca release was observed for all particle types upon acidification. Na release from HP0.5–7 and HP1–7 particles was also increased at acid pH, but Na release from the particles of low ALN content was hardly affected by the acidification, suggesting that Na played a more dominant role in complex formation in HP0.5–7 and HP1–7. A strong enhancement of P release upon acidification was only observed for the particles of low ALN content, which is in agreement with our degradation results (Fig. S5, ESI†).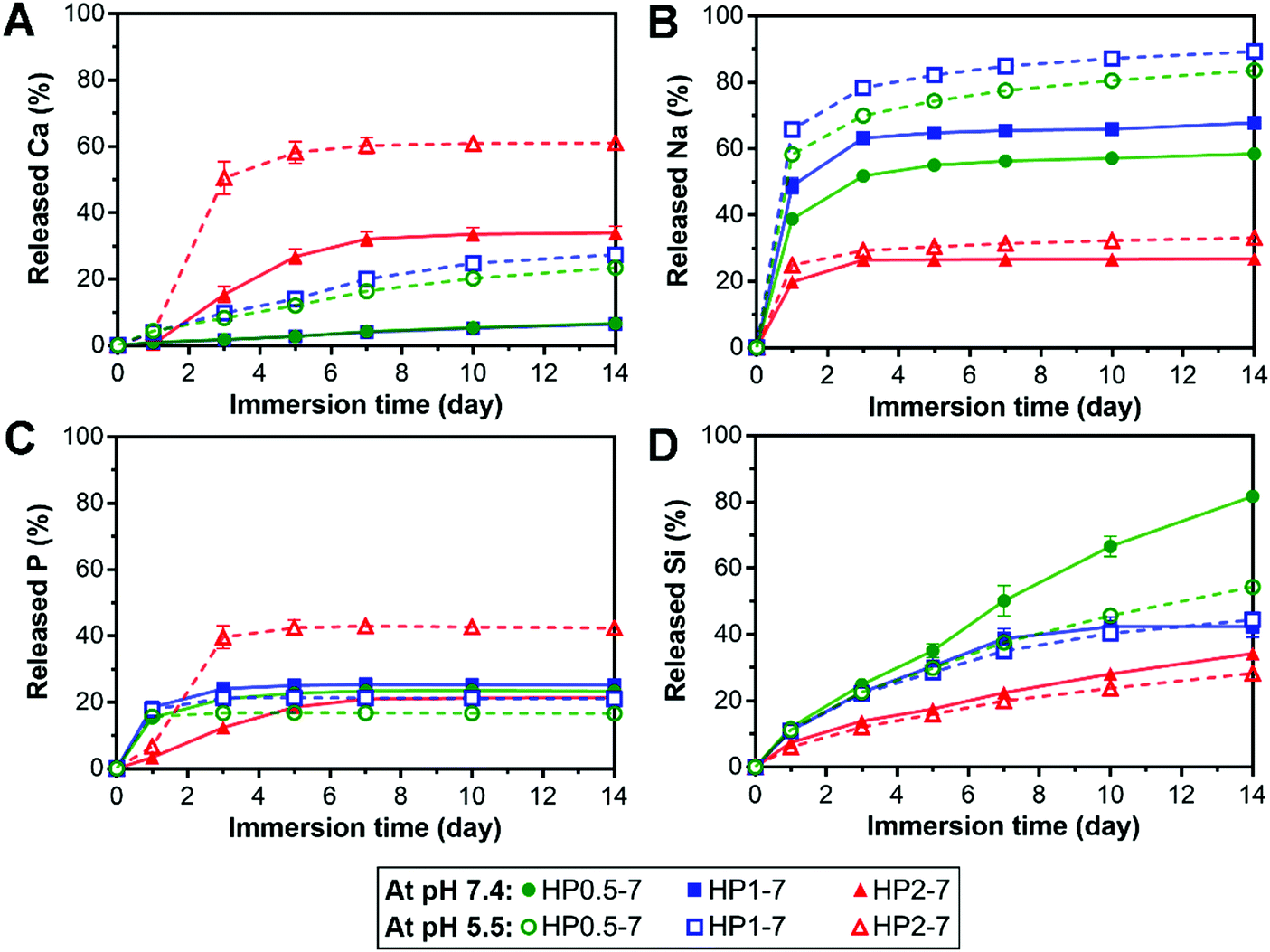 | ||
| Fig. 6 Cumulative release profiles of different elements from hybrid particles into buffered solutions at acidic (open symbols) or neutral pH (closed symbols). The release of each element was expressed relative to its original content in the hybrid particles. All values are presented as average ± standard deviation for n = 3 per experimental condition. | ||
Overall, these results indicate that the hybrid particles are degradable, which is an attractive feature of this novel class of biomaterials for application in bone tissue regeneration, since biomaterials should degrade to allow for replacement by newly formed bone tissue.42 In addition, anti-osteoporotic ALN molecules and BG-derived inorganic ions were released in a controllable manner. This controlled release is facilitated by the interactions between both components within the hybrids particles (e.g. through formation of coordination complexes). Inorganic ions are known to affect the regeneration of bone tissue in vivo.43 For example, specific concentrations of Ca ions were shown to stimulate proliferation and differentiation of osteoblastic cells and induce mineralization of the extracellular matrix.44 In addition, phosphate ions can promote the expression of matrix Gla protein (MGP) which is a key regulator for bone formation.45 Furthermore, Si is an important factor for bone formation which can affect the precipitation of hydroxyapatite.46,47 Nevertheless, it should be stressed that the biological effects of ALN and bioinorganic ions depend on their concentration in the extracellular matrix. Moreover, to determine the effectiveness of hybrid particles for the regeneration of osteoporotic bone defects, it is necessary to investigate their biological activity as compared to that of pure ALN and BG particles.
3.4 Influence of hybrid particles on osteoclastic cell function
In view of the excessive activity of bone-resorbing osteoclasts in osteoporotic bone, we studied in vitro the capability of the hybrid particles to reduce the activity of osteoclastic cells. To this end, we first selected the HP1–7 particles which contained an intermediate ALN content (Table S2, ESI†) to evaluate the influence of different concentrations of hybrid particles (0, 1, 10 and 100 μg mL−1) on the viability, proliferation, enzymatic activity and VEGF expression of osteoclast-like cells upon 72 h of in vitro incubation. We measured the viability and proliferation of osteoclastic cells by quantifying the formazan production of living cells and the LDH harvested upon cell lysis using WST-8 and LDH-activity assays, respectively (Fig. 7A). These tests revealed that osteoclast viability and proliferation decreased with increasing concentration of hybrid HP1–7 particles. At a concentration of 100 μg mL−1, HP1–7 particles showed the same effect in vitro as pure ALN, whereas BG particles did not affect osteoclast viability and proliferation. We then measured the TRAP-activity of the cells as a key marker for osteoclast activity48,49 to assess the effect of hybrid HP1–7 particles on osteoclastic cells (Fig. 7B). No TRAP enzyme activity was detected for osteoclasts cultured in the presence of 100 μg mL−1 of HP1–7 particles or pure ALN, while lower concentrations of hybrid particles did not reduce the TRAP activity. After identifying 100 μg mL−1 as the most effective anti-osteoclastic concentration for HP1–7 particles, we performed similar tests using HP0.5–7 or HP2–7 hybrid particles at the same particle concentration, which showed similar anti-osteoclastic effects (data not shown). These results were in agreement with our microscopic observations performed using actin/nucleus or TRAP staining of the cells (Fig. S6, ESI†). Importantly, bioactive glasses can release an excessive amount of alkaline ions into cell culture media, resulting in an undesirable pH increase of their environment, thus affecting cell viability during static culture conditions.50 Nevertheless, the BG control (100 μg mL−1) did not affect osteoclastic cells in our experiments, indicating that the anti-osteoclastic effect of the hybrid particles is based on the presence of functional ALN in their composition, and is not caused by a pH change that can occur due to the release of BG constituents.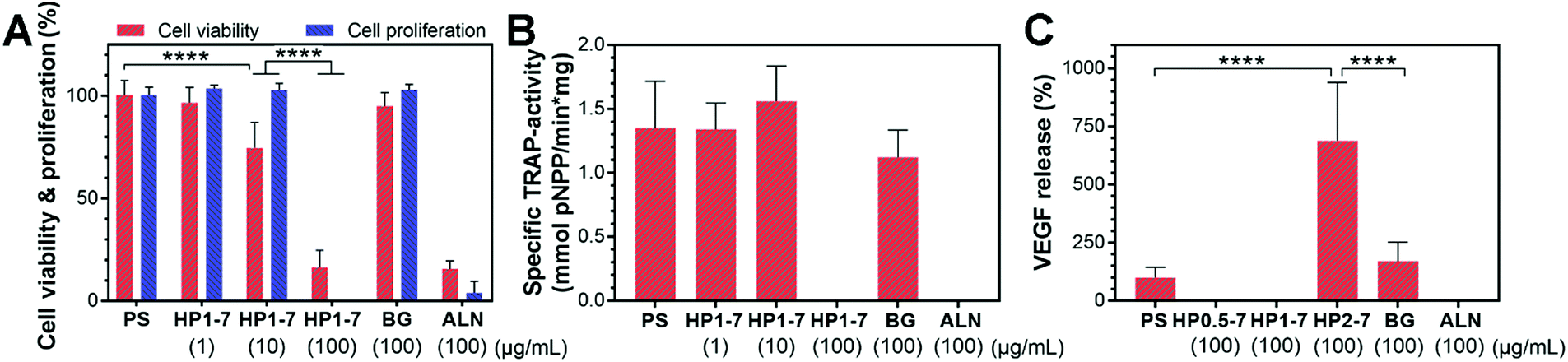 | ||
| Fig. 7 In vitro effects of hybrid particles on RAW 264.7-derived osteoclastic cells. (A) Viability and proliferation, (B) TRAP-activity, and (C) VEGF expression of cells upon incubation with different materials. Numbers in parentheses indicate the concentrations of particles in cell culture medium. PS, HP, BG, and ALN refer to particle-free polystyrene control, hybrid particles, bioactive glass, and alendronate, respectively. | ||
An important growth factor in the bone regeneration process that can be expressed by osteoclastic and osteoblastic progenitor cells is VEGF.25,26 VEGF expression can induce angiogenesis, which is an essential factor for the formation of healthy bone tissue.25 Accordingly, we measured the amount of VEGF expressed by the osteoclasts after exposure to hybrid particles of different composition prepared using CaBG/ALN ratios of 0.5, 1 or 2 (Fig. 7C). Interestingly, we observed a significant upregulation of VEGF expression when the cells were exposed to HP2–7 particles for 72 h. VEGF expression is usually mediated by hypoxia which activates hypoxia-inducible factor 1 (HIF-1) in cells.51,52 HIF-1 is a transcription factor that is composed of inducible α and constitutively expressed β subunits. Under hypoxia, the α subunit is stabilized enabling the expression of VEGF. Nevertheless, VEGF expression has also been found to occur under normoxic conditions. For instance, Trebec-Reynolds et al.26 reported that VEGF expression by RAW 264.7-derived osteoclasts can be induced though a non-hypoxic pathway. Consequently, further investigations are needed to determine the pathway through which HP2–7 particles induce VEGF expression in osteoclastic cells.
Lastly, the potential effect of ALN content of hybrid particles on bone-forming osteoblastic cells should also be considered. Our in vitro release experiments (Section 3.3) revealed that the HP1–7 particles release ∼20% of their ALN content during a 72 h incubation period, which translates to an ALN concentration of ∼50 μM for a particle concentration of 100 μg mL−1 in our cell culture experiments. Garcia-Moreno et al.53 studied the effect of different concentrations of ALN (10−1 to 10−12 M) on primary osteoblasts in vitro and found no effect on cell viability and proliferation at ALN concentrations of <100 μM. Considering the dynamic vs. static nature of in vivo vs. in vitro tests, respectively, we anticipate that ALN would not negatively affect the viability of osteoblasts in vivo. Nevertheless, it should be stressed that these claims can only be substantiated by performing in vivo implantation studies. Therefore, we performed an additional implantation study to investigate the osteocompatibility and bone regenerative capability of selected hybrid particles in osteoporotic rats.
3.5 Influence of hybrid particles on regeneration of osteoporotic bone
In view of the strong effects of hybrid HP1–7 and HP2–7 particles on osteoclastic cell function, we selected these two types of hybrid particles for an in vivo implantation study under osteoporotic conditions, while 45S5 BG particles were used as control in view of their well-known osteopromotive capacity.9 As shown in Fig. 8A and B, after 8 weeks of implantation of HP1–7 particles in osteoporotic rats, abundant bone formation was observed inside the original defect area without any visible inflammatory response, similar to the control BG group (Fig. 8E and F). However, the defects filled with HP2–7 particles showed low amounts of bone formation and contained material remnants after the implantation period (Fig. 8C and D). As shown in Fig. 8G, quantification of new bone formation inside the original defects indicated that the HP1–7 particles facilitated bone formation at levels similar to 45S5 BG particles, whereas the amount of newly formed bone was much lower upon implantation of HP2–7 particles. Such remarkable difference between the in vivo behavior of HP1–7 and HP2–7 particles can be attributed to the differences between their composition, structure and degradation behavior. For instance, upon implantation of biomaterials, the local pH of bone tissue drops from ∼7.4 to ∼5.5 due to an inflammatory response.54 As described in Section 3.3, such local acidification leads to the release of significantly higher amounts of Ca ions and ALN for HP2–7 particles as compared to HP1–7 particles. As shown in previous studies,44,55,56 such high concentrations of Ca and/or ALN might be cytotoxic for bone-forming osteoblast cells, thereby impeding in vivo bone regeneration. Interestingly, as shown in the inset of Fig. 8D, we observed macrophage-rich regions near HP2–7 remnants suggesting an inflammatory response caused by these particles. Although HP1–7 and 45S5 BG particles both stimulated the formation of bone tissue, the underlying stimulatory mechanism is expected to be different for both types of particles. While 45S5 BG particles can induce bone formation owing to their apatite-forming ability and osteogenic effects caused by their ionic dissolution products,9,43 HP1–7 particles can assist bone regeneration by inhibiting bone resorption through the release of ALN and promoting bone formation by releasing inorganic ions with osteogenic efficacy.44 Nevertheless, a more detailed in vivo investigation is required to unravel the exact mechanism by which HP1–7 particles are able to support bone formation.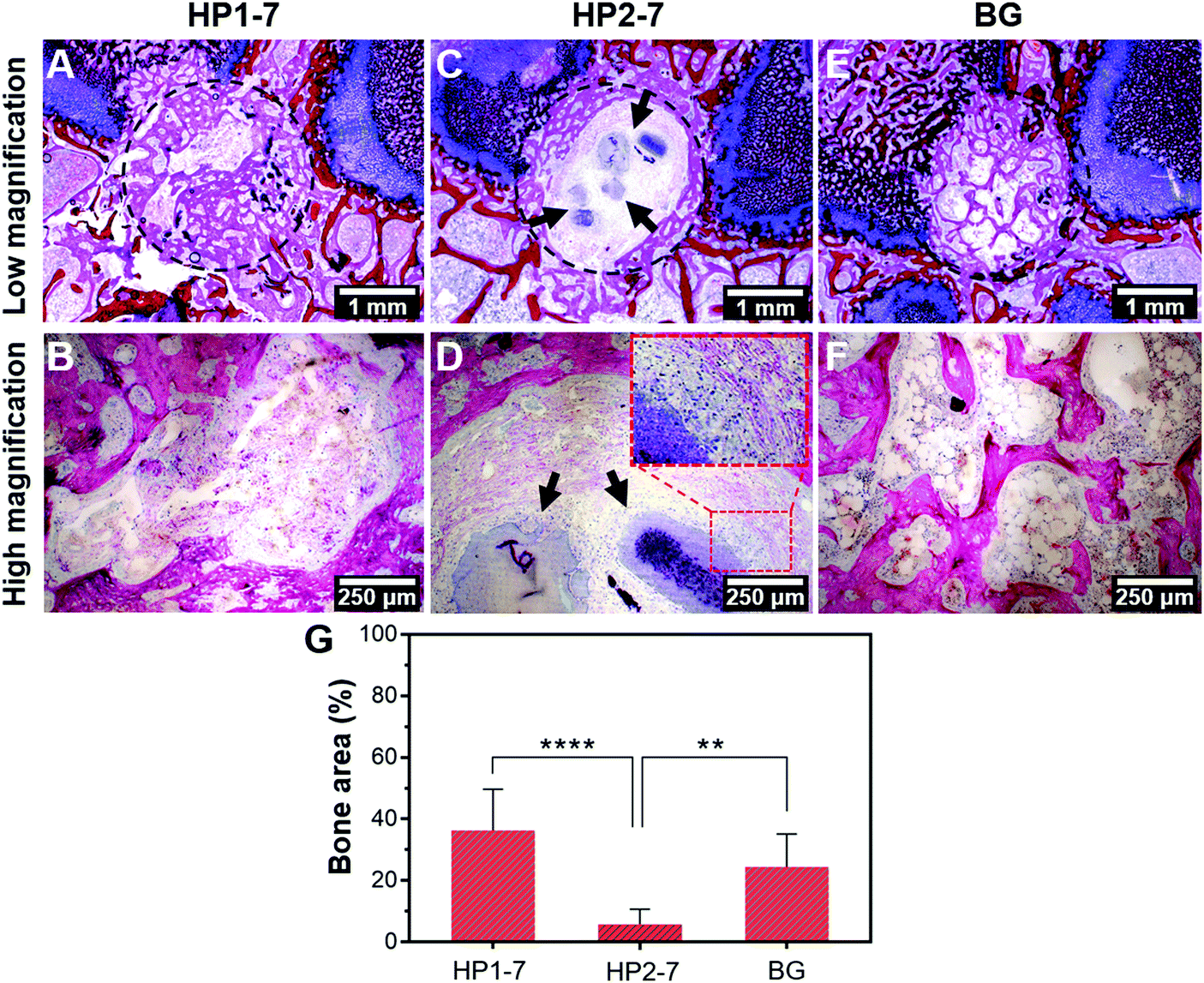 | ||
| Fig. 8 In vivo evaluation of hybrid particles. (A–F) Representative histological images and (G) quantified bone area (%) of different experimental groups after 8 weeks of implantation. Circles and arrows in images indicate the original defect area and material remnants, respectively. ** and **** indicate P < 0.005 and P < 0.0001, respectively. | ||
4. Conclusion
We have demonstrated the facile preparation of a novel class of hybrid particles of tunable morphology, chemical composition and structure. By immersing 45S5 BG particles in ALN solutions, phosphate ions of BG were substituted by ALN molecules, thereby forming ALN-cation coordination complexes. This synthesis procedure resulted in dissolution of BG particles and precipitation of novel hybrid particles. By varying immersion conditions, the morphology, chemical composition and structure of precipitated hybrid particles could be tuned. The hybrid particles facilitated controlled release of anti-osteoporotic ALN molecules and BG-derived inorganic ions, and displayed a strong anti-osteoclastic effect in vitro. In vivo evaluation of the hybrid particles revealed their strong capacity to facilitate regeneration of bone defects in an osteoporotic rat model. Our findings demonstrated that both in vitro and in vivo effects of the hybrid particles could be tuned by varying the chemical composition and structure of these novel hybrid particles. Consequently, considering this tunability of properties, this novel class of hybrid biomaterials opens up new avenues of research on the design of bone substitutes with specific activity to facilitate regeneration of bone in osteoporotic patients, for instance by applying the same synthetic procedures to BG compositions which contain osteogenic strontium ions to further boost the regeneration of osteoporotic bone defects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by Netherlands Enterprise Agency through Project No. SHM012014 in the theme IOP Self Healing Materials. Mani Diba would like to thank Prof. John A. Jansen and Dr. Matthew J. Harrington for valuable discussions and suggestions.References
- A. Klibanski, L. Adams-Campbell, T. Bassford, S. N. Blair, S. D. Boden, K. Dickersin, D. R. Gifford, L. Glasse, S. R. Goldring, K. Hruska, S. R. Johnson, L. K. McCauley and W. E. Russell, J. Am. Med. Assoc., 2001, 285, 785–795 CrossRef.
- O. Johnell and J. A. Kanis, Osteoporosis Int., 2006, 17, 1726–1733 CrossRef CAS PubMed.
- R. A. Perez, S.-J. Seo, J.-E. Won, E.-J. Lee, J.-H. Jang, J. C. Knowles and H.-W. Kim, Mater. Today, 2015, 18, 573–589 CrossRef CAS.
- D. Arcos, A. R. Boccaccini, M. Bohner, A. Díez-Pérez, M. Epple, E. Gómez-Barrena, A. Herrera, J. A. Planell, L. Rodríguez-Mañas and M. Vallet-Regí, Acta Biomater., 2014, 10, 1793–1805 CrossRef CAS PubMed.
- D. G. Little, M. Ramachandran and A. Schindeler, J. Bone Jt. Surg., Br. Vol., 2007, 89-B, 425–433 CrossRef PubMed.
- H. Fleisch, Endocr. Rev., 1998, 19, 80–100 CrossRef CAS PubMed.
- S. B. Woo, J. W. Hellstein and J. R. Kalmar, Ann. Intern. Med., 2006, 144, 753–761 CrossRef CAS PubMed.
- J. P. Cattalini, A. R. Boccaccini, S. Lucangioli and V. Mouriño, Tissue Eng., Part B, 2012, 18, 323–340 CrossRef CAS PubMed.
- J. R. Jones, Acta Biomater., 2013, 9, 4457–4486 CrossRef CAS PubMed.
- A. A. El-Rashidy, J. A. Roether, L. Harhaus, U. Kneser and A. R. Boccaccini, Acta Biomater., 2017, 62, 1–28 CrossRef CAS PubMed.
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res., 1971, 5, 117–141 CrossRef.
- A. K. Miri, N. Muja, N. O. Kamranpour, W. C. Lepry, A. R. Boccaccini, S. A. Clarke and S. N. Nazhat, Biomaterials, 2016, 85, 128–141 CrossRef CAS PubMed.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS PubMed.
- M. Bosetti and M. Cannas, Biomaterials, 2005, 26, 3873–3879 CrossRef CAS PubMed.
- S. Srisubut, A. Teerakapong, T. Vattraphodes and S. Taweechaisupapong, Oral Surg., Oral Med., Oral Pathol., 2007, 104, e11–e16 CrossRef PubMed.
- X. V. Bui, H. Oudadesse, Y. Le Gal, A. Mostafa, P. Pellen and G. Cathelineau, J. Aust. Ceram. Soc., 2010, 46, 24–28 CAS.
- J. H. Lee, K. M. Lee, S. J. Jang, H. S. Lee and H. R. Baek, Tissue Eng. Regener. Med., 2011, 8, 69–77 Search PubMed.
- K. Rosenqvist, S. Airaksinen, S. J. Fraser, K. C. Gordon and A. M. Juppo, Int. J. Pharm., 2013, 452, 102–107 CrossRef CAS PubMed.
- E. Boanini, S. Panseri, F. Arroyo, M. Montesi, K. Rubini, A. Tampieri, C. Covarrubias and A. Bigi, Materials, 2016, 9, 135 CrossRef PubMed.
- S. Ray, R. Acharya, S. Saha, A. Islam, S. Dey, S. K. Nandi, T. K. Mandal, G. Banerjee and J. Chakraborty, RSC Adv., 2016, 6, 89467–89483 RSC.
- M. S. Ironside, M. J. Duer, D. G. Reid and S. Byard, Eur. J. Pharm. Biopharm., 2010, 76, 120–126 CrossRef CAS PubMed.
- R. Detsch, M. Rübner, P. L. Strissel, D. Mohn, E. Strasser, W. J. Stark, R. Strick and A. R. Boccaccini, Nanomedicine, 2016, 11, 1093–1105 CrossRef CAS PubMed.
- R. Detsch, P. Stoor, A. Grünewald, J. A. Roether, N. C. Lindfors and A. R. Boccaccini, J. Biomed. Mater. Res., Part A, 2014, 102, 4055–4061 CrossRef PubMed.
- V. Breuil, F. Cosman, L. Stein, W. Horbert, J. Nieves, V. Shen, R. Lindsay and D. W. Dempster, J. Bone Miner. Res., 1998, 13, 1721–1729 CrossRef CAS PubMed.
- M. Grellier, L. Bordenave and J. Amédée, Trends Biotechnol., 2009, 27, 562–571 CrossRef CAS PubMed.
- D. P. Trebec-Reynolds, I. Voronov, J. N. M. Heersche and M. F. Manolson, J. Cell. Biochem., 2010, 110, 343–351 CAS.
- H. S. Alghamdi, J. J. J. P. van den Beucken and J. A. Jansen, Tissue Eng., Part C, 2014, 20, 493–505 CrossRef PubMed.
- H. B. M. van der Lubbe, C. P. A. T. Klein and K. de Groot, Stain Technol., 1988, 63, 171–176 CrossRef CAS PubMed.
- M. Diba, J. An, S. Schmidt, M. Hembury, D. Ossipov, A. R. Boccaccini and S. C. G. Leeuwenburgh, Macromol. Rapid Commun., 2016, 37, 1952–1959 CrossRef CAS PubMed.
- M. Diba, W. A. Camargo, M. Brindisi, K. Farbod, A. Klymov, S. Schmidt, M. J. Harrington, L. Draghi, A. R. Boccaccini, J. A. Jansen, J. J. J. P. van den Beucken and S. C. G. Leeuwenburgh, Adv. Funct. Mater., 2017, 27, 1703438 CrossRef.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689–702 RSC.
- W. L. Noorduin, A. Grinthal, L. Mahadevan and J. Aizenberg, Science, 2013, 340, 832–837 CrossRef CAS PubMed.
- C. N. Kaplan, W. L. Noorduin, L. Li, R. Sadza, L. Folkertsma, J. Aizenberg and L. Mahadevan, Science, 2017, 355, 1395–1399 CrossRef CAS PubMed.
- F. Gandara and T. D. Bennett, IUCrJ, 2014, 1, 563–570 CrossRef CAS PubMed.
- E. Alvarez, A. G. Marquez, T. Devic, N. Steunou, C. Serre, C. Bonhomme, C. Gervais, I. Izquierdo-Barba, M. Vallet-Regi, D. Laurencin, F. Mauri and P. Horcajada, CrystEngComm, 2013, 15, 9899–9905 RSC.
- G. Grossmann, A. Grossmann, G. Ohms, E. Breuer, R. Chen, G. Golomb, H. Cohen, G. Hägele and R. Classen, Magn. Reson. Chem., 2000, 38, 11–16 CrossRef CAS.
- S. Mukherjee, Y. Song and E. Oldfield, J. Am. Chem. Soc., 2008, 130, 1264–1273 CrossRef CAS PubMed.
- M. W. G. Lockyer, D. Holland and R. Dupree, J. Non-Cryst. Solids, 1995, 188, 207–219 CrossRef CAS.
- E. Boanini, P. Torricelli, M. Gazzano, R. Giardino and A. Bigi, Biomaterials, 2008, 29, 790–796 CrossRef CAS PubMed.
- F. Errassifi, S. Sarda, A. Barroug, A. Legrouri, H. Sfihi and C. Rey, J. Colloid Interface Sci., 2014, 420, 101–111 CrossRef CAS PubMed.
- T. R. Arnett, Arch. Biochem. Biophys., 2010, 503, 103–109 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757–2774 CrossRef CAS PubMed.
- S. Maeno, Y. Niki, H. Matsumoto, H. Morioka, T. Yatabe, A. Funayama, Y. Toyama, T. Taguchi and J. Tanaka, Biomaterials, 2005, 26, 4847–4855 CrossRef CAS PubMed.
- M. Julien, S. Khoshniat, A. Lacreusette, M. Gatius, A. Bozec, E. F. Wagner, Y. Wittrant, M. Masson, P. Weiss, L. Beck, D. Magne and J. Guicheux, J. Bone Miner. Res., 2009, 24, 1856–1868 CrossRef CAS PubMed.
- E. M. Carlisle, Calcif. Tissue Int., 1981, 33, 27–34 CrossRef CAS PubMed.
- J. J. M. Damen and J. M. Ten Cate, J. Dent. Res., 1992, 71, 453–457 CrossRef CAS PubMed.
- J. M. Halleen, S. L. Alatalo, H. Suominen, S. Cheng, A. J. Janckila and H. K. Väänänen, J. Bone Miner. Res., 2000, 15, 1337–1345 CrossRef CAS PubMed.
- R. Detsch and A. R. Boccaccini, J. Tissue Eng. Regener. Med., 2015, 9, 1133–1149 CrossRef CAS PubMed.
- F. E. Ciraldo, E. Boccardi, V. Melli, F. Westhauser and A. R. Boccaccini, Acta Biomater., 2018, 75, 3–10 CrossRef CAS PubMed.
- D. Shweiki, A. Itin, D. Soffer and E. Keshet, Nature, 1992, 359, 843–845 CrossRef CAS PubMed.
- M. C. Brahimi-Horn and J. Pouysségur, Biochem. Pharmacol., 2007, 73, 450–457 CrossRef CAS PubMed.
- C. García-Moreno, S. Serrano, M. Nacher, M. Farré, A. Díez, M. L. Mariñoso, J. Carbonell, L. Mellibovsky, X. Nogués, J. Ballester and J. Aubía, Bone, 1998, 22, 233–239 CrossRef.
- J. B. Park, Biomaterials: An introduction, Plenum Publishing Corporation, 1979 Search PubMed.
- J. Sun, F. Y. Song, W. P. Zhang, B. E. Sexton and L. J. Windsor, Arch. Oral Biol., 2012, 57, 728–736 CrossRef CAS PubMed.
- E. W. H. Bodde, R. S. Z. Kowalski, P. H. M. Spauwen and J. A. Jansen, Tissue Eng., Part A, 2008, 14, 29–39 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb03062f |
| ‡ Current address: Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |