
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface enhanced dynamic nuclear polarization solid-state NMR spectroscopy sheds light on Brønsted–Lewis acid synergy during the zeolite catalyzed methanol-to-hydrocarbon process†
Abhishek
Dutta Chowdhury‡
 *a,
Irina
Yarulina‡
a,
Edy
Abou-Hamad
*b,
Andrei
Gurinov
b and
Jorge
Gascon
*a
*a,
Irina
Yarulina‡
a,
Edy
Abou-Hamad
*b,
Andrei
Gurinov
b and
Jorge
Gascon
*a
aKing Abdullah University of Science and Technology, KAUST Catalysis Center, Advanced Catalytic Materials, Thuwal 23955, Saudi Arabia. E-mail: abhishek.duttachowdhury@kaust.edu.sa; jorge.gascon@kaust.edu.sa
bKing Abdullah University of Science and Technology, KAUST Core Labs, Thuwal 23955, Saudi Arabia. E-mail: edy.abouhamad@kaust.edu.sa
First published on 21st August 2019
Abstract
After a prolonged effort over two decades, the reaction mechanism of the zeolite-catalyzed methanol-to-hydrocarbon (MTH) process is now well-understood: the so-called ‘direct mechanism’ (via direct coupling of two methanol molecules) is responsible for the formation of the initial carbon–carbon bonds, while the hydrocarbon pool (HCP)-based dual cycle mechanism is responsible for the formation of reaction products. While most of the reaction events occur at zeolite Brønsted acid sites, the addition of Lewis acid sites (i.e., via the introduction of alkaline earth cations like calcium) has been shown to inhibit the formation of deactivating coke species and hence increase the catalyst lifetime. With the aim to have an in-depth mechanistic understanding, herein, we employ magic angle spinning surface-enhanced dynamic nuclear polarization solid-state NMR spectroscopy to illustrate that the inclusion of Lewis acidity prevents the formation of carbene/ylide species on the zeolite, directly affecting the equilibrium between arene and olefin cycles of the HCP mechanism and hence regulating the ultimate product selectivity and catalyst lifetime.
Introduction
Understanding the cause of preferential formation of one product over others (i.e., product control) has always been a challenging task in heterogeneous catalysis.1–3 Although the zeolite H-ZSM-5 catalyzed methanol-to-hydrocarbon (MTH) process was discovered by Mobil Corporation more than four decades ago, it is still a fascinating research topic with numerous mechanistic aspects/complexity yet to be unveiled.2–9 Mechanistically, this reaction is typically subdivided into two segments: (i) the direct mechanism, followed by (ii) the hydrocarbon pool (HCP)/dual-cycle mechanism, during the induction and autocatalytic periods of the reaction, respectively.2–9 During a very short induction period, the direct mechanism is responsible for the formation of initial carbon–carbon bonds containing acetate/ether-type species.2 Next, these species initiate the formation of hydrocarbons through an auto-catalytic dual cycle/HCP mechanism during the steady-state of the MTH reaction. This dual cycle mechanism consists of two intra-dependent cycles based on alkylated aromatic and olefinic species, i.e., arene and alkene cycles, respectively.2–9 Herein, based on 12C/13C transient switching experiments, Svelle et al. earlier demonstrated that all olefins, except ethylene, are produced from the alkene cycle, whereas ethylene typically originates from the arene cycle.2,10,11 Similarly, after thorough kinetic investigations, Sun et al. later concluded that the arene cycle gives rise to similar selectivity for both olefins, while the alkene cycle preferentially produces propylene over ethylene.2,12,13 Thus, hypothetically, if both arene and alkene cycles contribute equally to the MTH reaction, ethylene will be predominantly formed from the arene cycle.2In this aspect, we recently reported that Lewis acidity in zeolites is an active participant in the MTH reaction mechanism rather than a spectator.1,14 Using calcium (Ca) as a showcase, we demonstrated that the incorporation of alkaline-earth metals results in a significant decrease and isolation of Brønsted acid sites (BAS) together with the generation of Lewis acid sites (LAS).1,14 Meticulous analysis of catalysts with different amounts of Lewis and Brønsted acid sites (achieved through different loading of alkaline-earth metals) allowed us to decouple the effects/implications of these two types of acidic functionalities from each other. Brønsted acidity was found to be linearly correlated with propylene and ethylene selectivity proving itself to be the principal product distribution descriptor. On the other hand, we also showed that the presence of Lewis acidity is beneficial for a prolonged catalyst lifetime, which is tuned by the LAS/BAS ratio.1,14 Having a destabilizing effect on important carbenium ions, Lewis acidity decreases the reactivity of intermediates belonging to the arene cycle, thus altogether suppressing the formation of aromatic species. In order to gain a complete mechanistic overview of this very relevant observation, it is necessary to identify the nature of formed/trapped co-catalytic organic HCP scaffolds within the inorganic zeolites (both H-ZSM-5 and Ca-ZSM-5)10,15 governing the autocatalytic part of the MTH process. Herein, we have employed solid-state NMR spectroscopy, which recently proved itself to be a compelling analytical technique for the characterization of biomolecules and catalytic materials.1,15–20
Solid-state NMR spectroscopy is a quantitative and non-destructive/invasive approach to elucidate molecular structures along with their physiognomies accurately.15,16,21–23 However, this technique has not (yet) entirely overcome all its challenges. The natural abundance of the element of interest is one of the primary criteria (or concerns) behind its success. For instance, 13C has only 1.1% natural abundance and thus absolute 13C-enrichment within a molecule results in a ≥90 times enhancement of signals compared to usual. Therefore, the utilization of 13C-labelled species/reactant(s) is a popular strategy in multiple research fields, including heterogeneous catalysis, to deal with the low sensitivity issue.2,3,15,16,21–28 However, absolute isotope-enrichment of an element with low natural abundance is not always a practical solution, particularly in the field of heterogeneous catalysis and surface/materials science. To overcome the low-sensitivity concerns, dynamic nuclear polarization surface-enhanced NMR spectroscopy (DNP SENS) has recently gained a lot of interest from the scientific community.29–44 Typically, a sample is initially impregnated with a DNP agent (i.e., a nitroxide biradical) containing solution to bring it into the vicinity of the surface.29,30,37–43,45,46 The DNP agent enhances the polarization of protons of the solvent and neighbouring surface regions, which are then transferred to the surface's hetero-nuclei through cross-polarization (CP) or similar coherence magnetization transfer schemes (as illustrated in Fig. 1). As a result, signal enhancement of the hetero-nuclei is observed without any isotope-enrichment.29,30,37,40,45,46
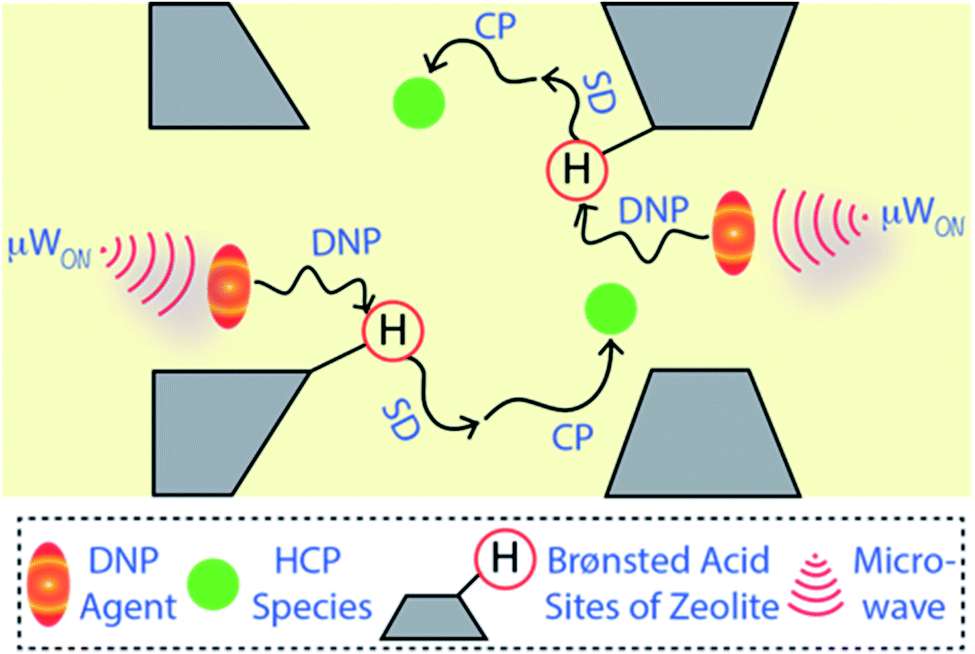 | ||
| Fig. 1 Simplified illustration of the hyperpolarization of carbon atoms of the hydrocarbon pool (HCP) species (in green spheres) using DNP SENS NMR spectroscopy. The yellow background represents the solvent matrix of the DNP agent (orange-yellow ellipsoids) within the post-impregnated zeolite. As a result of the microwave (μW) irradiation, electron polarization is transferred from the exogenous biradical (TEKPol) to the zeolitic Brønsted acid sites and protons from the solvent matrix, through electron–nuclear dipolar coupling. Next, 1H–1H spin-diffusion (SD) was allowed to occur at the cryogenic temperature, and then cross-polarization (CP) or similar coherence magnetization transfer schemes was used to transfer the enhanced polarization to the dilute spins of the medium (i.e., zeolite-trapped carbonaceous HCP species in the present case). | ||
In heterogeneous catalysis and materials science, until now, the concept of DNP SENS has been primarily utilized to characterize surface organometallic fragments/catalysts, metal–organic frameworks (MOFs), and amorphous aluminosilicates.40,45–48 To understand the reaction mechanism involving micro-porous zeolite catalysts, fully 13C-enriched reactants have typically been used to deal with the natural abundance issue of the hetero-nuclei, including in MTH catalysis.2,3,15,21–24,26–28,49–53 Although very informative, this approach primarily involves the use of non-ideal reactors (e.g., mostly in situ reaction cells with a very high ‘dead volume’), as well as shorter reaction times (cf. <60 min), which do not necessarily reflect the real reaction conditions.15,21,24,27,49–51 In contrast, the preparation of a fully deactivated catalyst (often requiring >10–20 hours of reaction time) using 13C-enriched reactant(s) in a conventional fixed-bed reactor is an unrealistically expensive process. Not only economic feasibility, but also the commercial availability of the 13C-enriched reactant, that too in a reasonable quantity, is another genuine concern. It is worth mentioning that the DNP SENS technique has recently been applied to MTH-reacted zeolite materials too.24,28 However, these post-reacted materials were prepared using fully 13C-enriched methanol, that too in a very short reaction time (20 min). Hence, through this current work, our DNP SENS approach offers a unique opportunity to perform mechanistic investigations using naturally abundant (without any 13C-enrichment) methanol on the more mechanistically relevant deactivated sample, which is indeed the primary aim/scope of the current work.
Results and discussion
In this work, magic angle spinning DNP SENS NMR spectroscopy was performed on deactivated un- and Ca-modified ZSM-5 materials after the MTH reaction (T = 500 °C, weight hourly space velocity (WHSV) = 8 gMeOH gcatalyst−1 h−1) using naturally abundant methanol (Fig. 2–6 and S1–S10 in the ESI; see also Section S1 and S2 in the ESI†) as a reactant, to identify the zeolite-trapped organic HCP species. Herein, both catalysts are synthesized aiming for the same amount of Brønsted acid sites but with strikingly different Lewis acid site-content to elucidate the effect of the latter on catalytic performance (Table S1 in the ESI†).1 Both catalysts show a rather similar selectivity to ethylene and propylene (Fig. 2). As was shown in our previous work,1,14 both propylene and ethylene selectivity were linearly correlated with the density of Brønsted acid sites. Being the principal descriptor of propylene selectivity, similar Brønsted acid sites yielded similar propylene selectivity (47.2% and 51% for zeolites ZSM-5 and Ca-ZSM-5, respectively, Fig. 2).1 Therefore, catalysts with the same amount of Brønsted acid sites show rather similar product distribution. Despite obvious similarities in product distribution, both catalysts show different resistance towards deactivation. While ZSM-5 shows no deactivation for ∼28 hours (corresponding to 224 gMeOH converted per gram of the catalyst), its Ca-modified counterpart is able to convert a twice higher amount of methanol (MeOH) before deactivation starts (up to 504 gMeOH converted per gram of the catalyst before deactivation compared to the Ca-free sample). Such lifetime improvement or stability towards deactivation was attributed to the Lewis acid site-promoted (as a result of Ca-incorporation) suppression of the aromatic cycle via destabilization of the key carbenium HCP species.1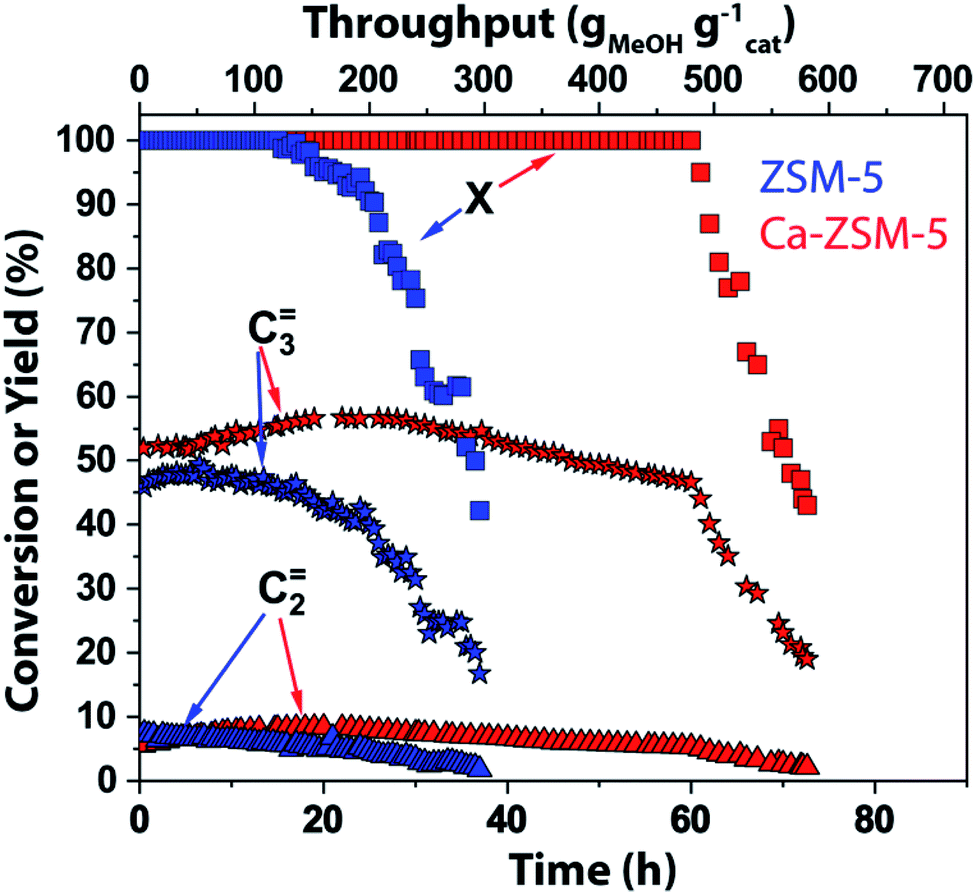 | ||
| Fig. 2 Catalytic testing results of unmodified ZSM-5 (Si/Al = 1280, in blue) and Ca-modified ZSM-5 (Si/Al = 40, in red) zeolites in the methanol-to-hydrocarbon reaction. Reaction conditions: T = 500 °C, weight hourly space velocity (WHSV) = 8 gMeOH gcatalyst−1 h−1 (methanol = MeOH, X represents conversion of methanol). The rest of the reaction products consist of paraffins, higher olefins, and aromatics (not shown). Herein, we refer to the previous publication from our group for detailed catalytic results.1 | ||
For DNP SENS measurements, samples were prepared using incipient wetness impregnation with the solution of 16 mM TEKPol in 1,1,2,2-tetrachloroethane (TCE) (see Section S3 in the ESI† for the detailed DNP sample preparation methods).39,40,42 Upon microwave irradiation, up to 75 times enhancement of the signal was observed on the DNP agent-impregnated post-reacted zeolite materials (Fig. S1 and S2 in the ESI†).39,40 There is a significant dissimilarity in the type of identified HCP species on both zeolites. The introduction of Ca led to the isolation of zeolitic Brønsted acid sites, which inhibit the formation of carbene/ylide species during the MTH reaction.21,26 Due to the absolute non-interference from Lewis acid sites (i.e., Ca), carbene/ylide-species were formed exclusively in the zeolite H-ZSM-5. Such subtle tweaking of the catalyst properties via the introduction of Lewis acid sites is ultimately proved to be the most decisive factor in catalysis.54
In the 1D 1H–13C cross-polarization magic-angle spinning DNP SENS of both post-reacted H-ZSM-5 and Ca-ZSM-5 zeolites (Fig. 3), the following features were primarily observed: (i) 15–38 ppm aliphatic/paraffinic species (i.e. saturated hydrocarbon moieties), (ii) ∼100 ppm acetal (–O–CH2–O) groups, and (iii) 118–142 ppm unsaturated hydrocarbon moieties (i.e., olefinics/aromatics, see Fig. 3a and b).2,21 The post-reacted zeolite ZSM-5 sample also showcases two additional peaks in both upfield (∼0.5 ppm, see Fig. 3a) and downfield (∼368 ppm, see Fig. 3c and d) regions, which were attributed to the zeolite-trapped methane/Si-methyl species and zeolite-surface bound carbene/ylide species, respectively.40 The upfield peak at 0.5 ppm is usually observed as a result of adsorption of methane in the zeolite framework.55 Methane is a common by-product in MTH catalysis as a result of disproportionation of methanol (2CH3OH → CH4 + HCHO).56 However, at least in principle, such shielded carbon could also originate from Si-methyl species (possibly at external surface/lattice defect sites in zeolites), since tetramethylsilane (or similar methylated silanes) usually appears at around 0 ppm in the 13C NMR spectrum.57
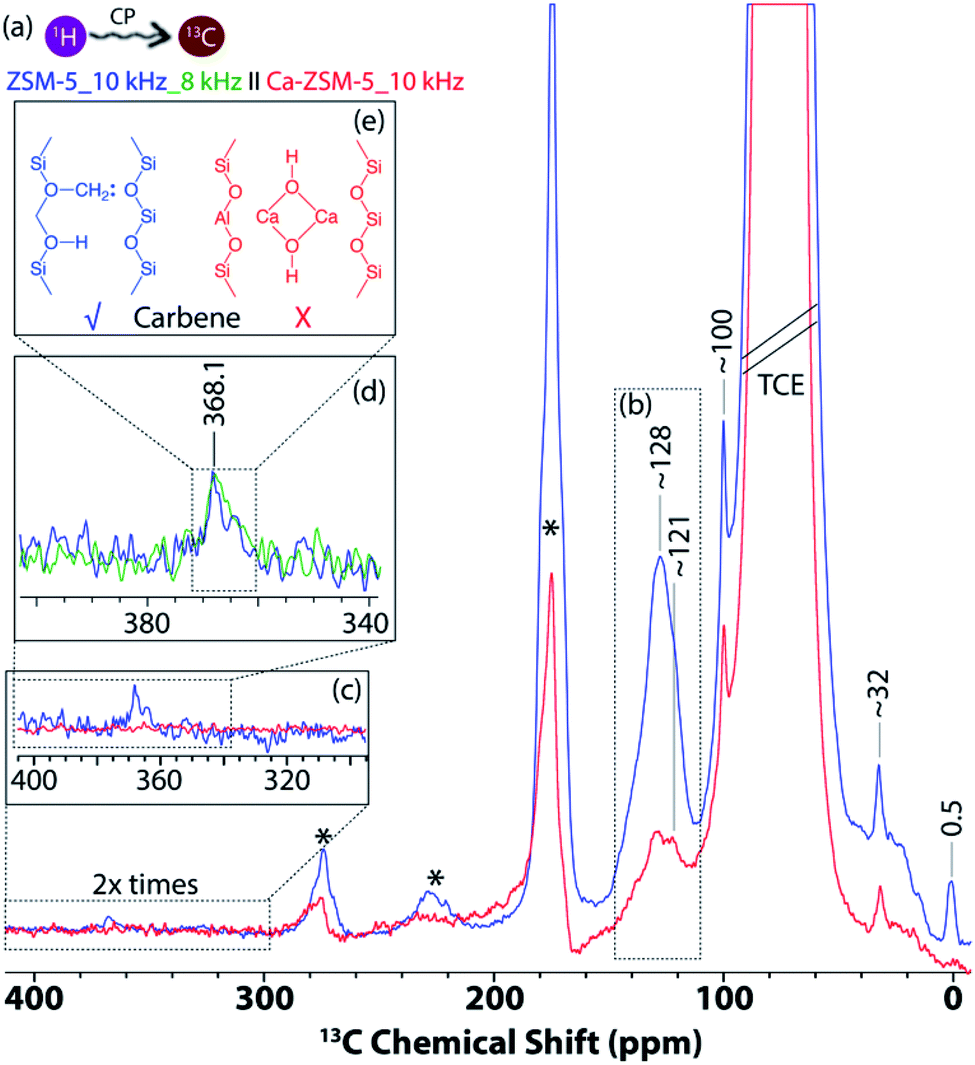 | ||
| Fig. 3 1D 1H–13C cross-polarization magic-angle spinning (MAS) DNP SENS results of post-reacted (a–c) H-ZSM-5 (blue) and Ca-ZSM-5 (red) zeolites in a 16 mM TEKPol solution in 1,1,2,2-tetrachloroethane (TCE) (* = spinning side-bands, 10 kHz MAS, intensity was multiplied by two in c). (b) Unsaturated hydrocarbon (aromatic/olefinic) region. Enlarged sections of (c and d) the carbene region. The (e) carbene peak at (c) 368.1 ppm (blue) was further verified under (d) 8 kHz MAS (green) to confirm its identity. | ||
The existence of a carbene insertion mechanism during the zeolite-catalyzed MTH process has been present in the literature for a while now, and it was primarily based on indirect evidence.2 For instance, the ability to form carbene with zeolite-surface methoxy species (SMS, i.e., formed upon adsorption of methanol onto zeolitic Brønsted acid sites) was first postulated by Chang and Silvestri58 as well as spectroscopically detected by Hunger et al.67 The Hunger group demonstrated that methylcyclohexane was formed through an insertion reaction of a carbene/ylide into the Csp3–H bond of cyclohexane.2,26,59,60 The polarization of a methyl C–H of zeolite-surface methoxy species by neighboring oxygen induces a carbene/ylide character on SMS and promotes direct coupling reactions with methanol, which was also verified by Kondo et al. using IR spectroscopy.2,61,62 Recently, this has been further confirmed by Chowdhury et al. using 2D solid-state NMR correlation (both 1H–13C and 13C–13C) spectroscopy to trap the precursor of the zeolite-surface carbene species (i.e., the adduct between zeolite-bound SMS and an incoming methanol).2,21 However, direct spectroscopic identification of the zeolite-bound carbene (or carbyne) species itself was still elusive. Indeed, this has always been an extremely challenging task due to its inherent lower signal-to-noise ratio and longer acquisition times (from weeks to months).40 Herein, DNP SENS is successfully implemented to overcome the sensitivity concern, providing direct spectroscopic identification of carbene-character containing carbons on zeolite, possibly for the first time in zeolite chemistry and catalysis (Fig. 3e).
Next, 1D 1H–13C cross-polarization magic-angle spinning DNP SENS was performed at different cross-polarization contact times (p15) on both post-reacted H-ZSM-5 and Ca-ZSM-5 zeolite samples to study diffusion characteristics (Fig. 4, also see Fig. S3 and S4 in the ESI† for a detailed study). In both cases, the intensity of the acetal peak (∼100 ppm region in Fig. 4, also see the blue-highlighted region in Fig. S3 and S4 in the ESI†) increases with the increasing contact time, which means a more efficient cross-polarization transfer was evidenced at longer contact times. This suggests that the acetal molecules are preferentially located within the zeolitic framework. On a closer inspection, it is also evident that the surface of the post-reacted H-ZSM-5 comparatively contains a much higher amount of unsaturated HCP species (like aromatics/polyaromatics).1 Although unsaturated aromatic-based HCP peaks were centred around 126–128 ppm at the longest cross-polarization contact times on both samples, an additional aromatic peak around 133 ppm in shorter contact time (p15 = 0.2–0.5 ms) experiments in H-ZSM-5 (Fig. 4a, also see the green highlighted region in Fig. S3 in the ESI†) implies the presence of polyaromatic products exclusively residing on the surface of the zeolite. However, this peak is completely absent in the post-reacted Ca-modified ZSM-5 zeolite, which clearly indicates the absence of any aromatic species in this case (Fig. 4b, also see Fig. S3 vs. Fig. S4 in the ESI† for a more detailed illustration), especially on the surface of the zeolite material.
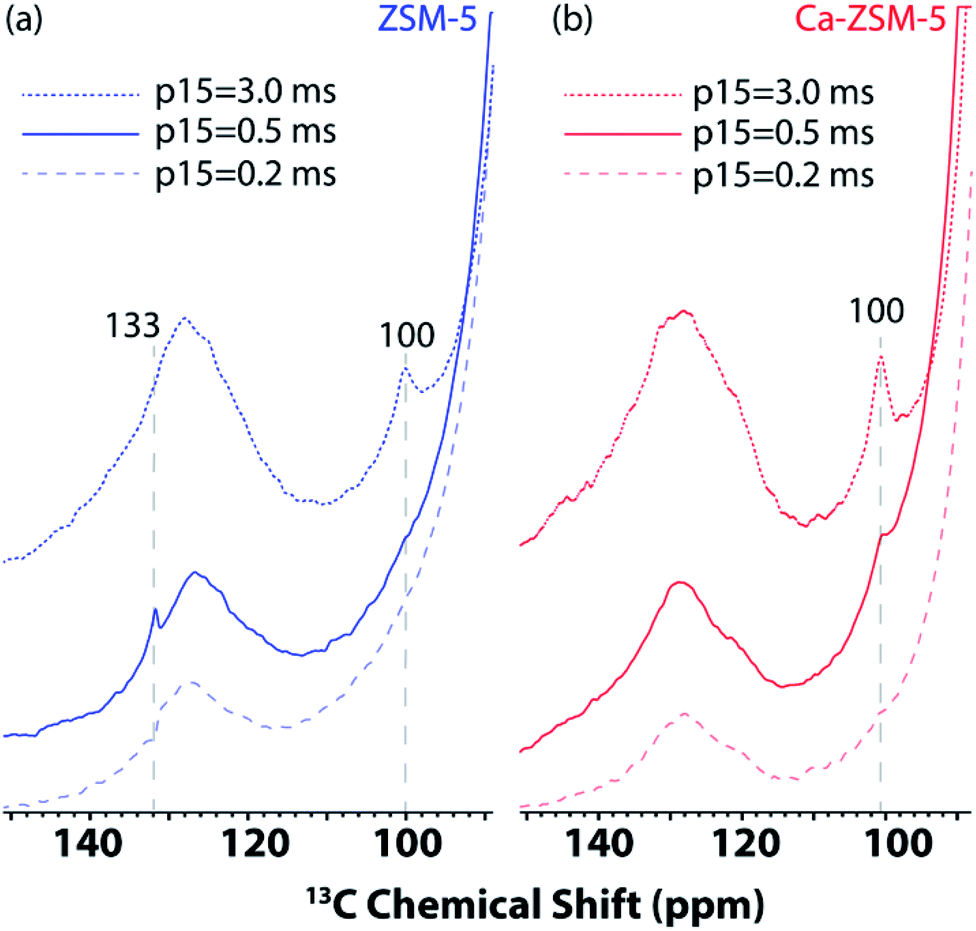 | ||
| Fig. 4 Enlarged section of 1D 1H–13C cross-polarization magic angle spinning DNP SENS spectra of post-MTH reacted zeolites (a) H-ZSM-5 (in blue, left) and (b) Ca-ZSM-5 (in red, right) in a 16 mM TEKPol solution in 1,1,2,2-tetrachloroethane (TCE) at different CP contact times (p15). See Fig. S3 and S4 in the ESI† for more detailed representation. The recycle delay was 3 s, and the magic angle spinning frequency was 8 kHz. | ||
Next, in order to achieve more in-depth information about the mechanism of DNP transfer inside both post-reacted materials, we performed several 1D 1H–13C cross-polarization magic-angle spinning DNP measurements, including saturation recovery experiments using different impregnation times (timp) (Fig. S5–S8 in the ESI†).40,47,48 At timp = 0 (immediately after sample preparation) in H-ZSM-5, fast-solvent build-up times (TB) and simultaneous high enhancement of both solvents and aromatics were concomitantly spotted (Fig. S5 and S6 in the ESI†). Both aliphatic and acetal resonances were only enhanced at longer impregnation times (timp = up to 90 days), which means both were primarily located within the micropores. Interestingly, both of them have slower TB at timp = 0 compared to aromatics, while all resonances eventually have faster/identical TB at longer impregnation times (timp = 7 days). Such time-dependent DNP-behaviour implies that solvent-spin diffusion into the zeolite-pores requires more time, which is a signature of the polarization process being relayed by spin diffusion inside the pores.40,47 This could be due to the fact that the pores are not fully impregnated immediately, and thus, diffusion of the solvent into the materials can continue over a period of days. Such pronounced characteristics of the polarization build-up are a typical signature of DNP relayed by spin diffusion, which is comparable with and analogous to the previously reported observations based on micro-crystalline solids, metal–organic frameworks, and zeolites.40,47,48,63,64 Therefore, it is safe to conclude that the post-reacted unmodified zeolite ZSM-5 sample is overwhelmed by surface (poly)aromatics species and it takes a while to diffuse into the micropores of the zeolite to enhance the signal originating from all species (i.e., aromatics, acetals and aliphatic moieties) within the micropore/framework.
In contrast, in the post-reacted Ca-ZSM-5, all resonances comparatively experienced similar signal enhancement over impregnation times (Fig. S7 and S8 in the ESI†). Herein, fast-solvent build-up times and a simultaneous high solvent enhancement are accompanied by the enhancement of all peaks (including aliphatic and acetals) at timp = 0. Later, such enhancement of all signals was observed at longer impregnation times (until a week) as well. Thus, the hydrocarbon pool species were uniformly distributed across the Ca-modified zeolite sample, which was yet to be deactivated and showed resistive characteristics towards coking/deactivation. Next, the instantaneous aromatic signal-enhancement in spent H-ZSM-5 indicates a higher concentration of deactivating coke-species, especially on the surface of the zeolite (Fig. S5 and S6 in the ESI†). This observation is also consistent with our earlier experiments at different cross-polarization contact times (Fig. 4 and Fig. S3 and S4 in the ESI†).
Next, 2D correlation solid-state NMR spectroscopy (i.e., both 1H–13C and 13C–13C) was performed to identify the nature of trapped organic species within both zeolites (Fig. 5 and 6 and Fig. S9 and S10 in the ESI†).15,21,22,40 In the 2D 1H–13C cross-polarization HETCOR (HETCOR: HETero nuclear CORrelation spectroscopy) spectra of post-MTH reacted unmodified ZSM-5 under both non-DNP (Fig. 5a) and DNP (Fig. 5c) conditions, saturated aliphatic groups (20–37 ppm (13C) and 1.1–3.1 (1H) ppm) along with unsaturated aromatic/olefinic moieties (130–145 ppm (13C) and 7.6–9 (1H) ppm) could be identified.
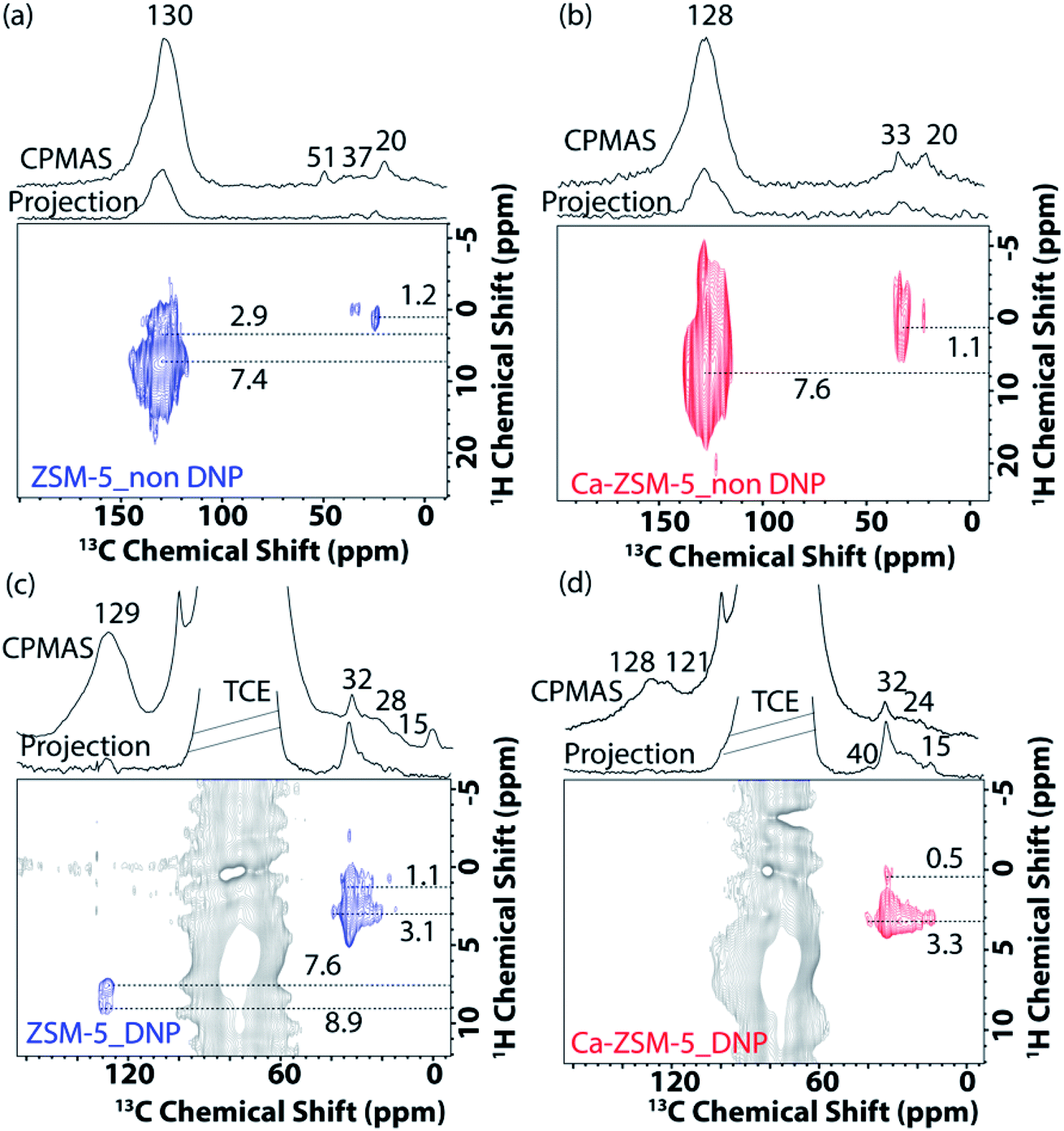 | ||
| Fig. 5 2D 1H–13C cross-polarization (CP) magic angle spinning (MAS) HETCOR (a and b) solid-state NMR and (c and d) DNP SENS HETCOR spectra (in a 16 mM TEKPol solution in 1,1,2,2-tetrachloroethane (TCE, in grey)) of post-MTH reacted (a and c) H-ZSM-5 (in blue) and (b and d) Ca-ZSM-5 (in red) zeolite materials with 13C-projections, including corresponding 1D 1H–13C CPMAS NMR spectra. | ||
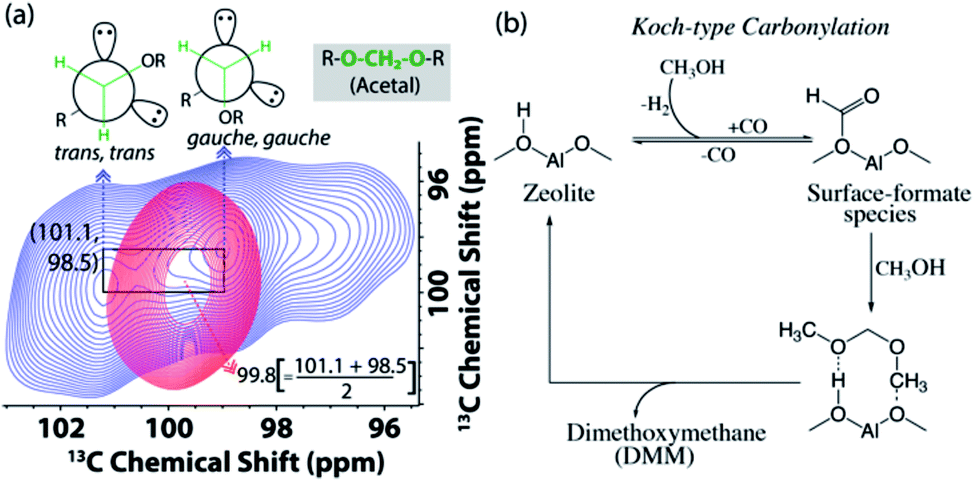 | ||
| Fig. 6 (a) Enlarged acetal region in the DNP enhanced 2D 13C–13C magic angle spinning spin-diffusion solid-state NMR spectra of the post-MTH reacted H-ZSM-5 (blue) and Ca-ZSM-5 (red) in a 16 mM TEKPol solution in 1,1,2,2-tetrachloroethane (TCE). See Fig. S9 and S10† for their full spectra. (b) The plausible mechanistic pathway to the formation of acetal species during the zeolite-catalyzed methanol-to-hydrocarbon process.21 | ||
In contrast, the presence of aliphatic/paraffinic species (15–32 ppm (13C) and 2.0–3.3 (1H) ppm) is relatively more pronounced in the 2D 1H–13C cross-polarization DNP SENS HETCOR spectra of post-MTH reacted Ca-ZSM-5 (Fig. 5bvs.Fig. 5d). Although Fig. 5b highlights the existence of unsaturated HCP moieties (cross-peak between 128 ppm (13C) and 7.6 (1H) ppm) in the Ca-modified zeolite, their non-existence under DNP conditions (Fig. 5d) could be attributed to the overwhelming majority of saturated moieties on the surface. This observation is fully consistent with our previously reported operando UV-visible diffuse reflectance study during the MTH reaction, where we observed that aromatic species were not accumulated at all in Ca-ZSM-5 (on the surface at least).1 However, it could also mean that DNP transfer in the post-reacted Ca-ZSM-5 sample is not as efficient as in non-modified ZSM-5. Overall, these results essentially justify that the incorporation/proximity of Lewis acid sites suppresses the formation of aromatics, preventing the dehydroaromatization route from forming coke from aliphatic/paraffinic species.15 The slight (∼2 ppm) up-field shifted peak maxima (∼128 ppm: more prone towards olefins, not aromatics) in Ca-ZSM-5 further confirms the destabilizing effect of Lewis acid sites on the arene cycle. This could be further observed in Fig. 3b, where the peak at around 121 ppm, due to the olefinic sp2-carbons, was clearly resolved in the post-reacted Ca-ZSM-5, indicating the dominance of the alkene cycle.
Since the availability/accessibility of zeolitic Brønsted acid sites within the zeolite micropores is a mandatory requirement for the formation of surface-carbene species, the close proximity of Lewis acid sites (presumably in the form of μ-hydroxo dimers, see Fig. 3e and Scheme 1) and/or highly isolated Brønsted acid sites eventually inhibits the formation of both carbene and coke species, contributing to a sharp increase in the catalyst lifetime (Fig. 2).1,14 Such synergy between Lewis and Brønsted acid sites could further be rationalized in the respective 1D 1H–13C cross-polarization magic angle spinning solid-state NMR spectrum (see top projections in Fig. 5a and c). The sole existence of a carbon signal at around 51 ppm in the post-MTH reacted zeolite H-ZSM-5 (Fig. 5avs.Fig. 3c), which typically originates from zeolite-adsorbed methanol, highlights the synergy between Lewis and Brønsted acid sites in the Ca-ZSM-5 material.21,26,54,59,60,65
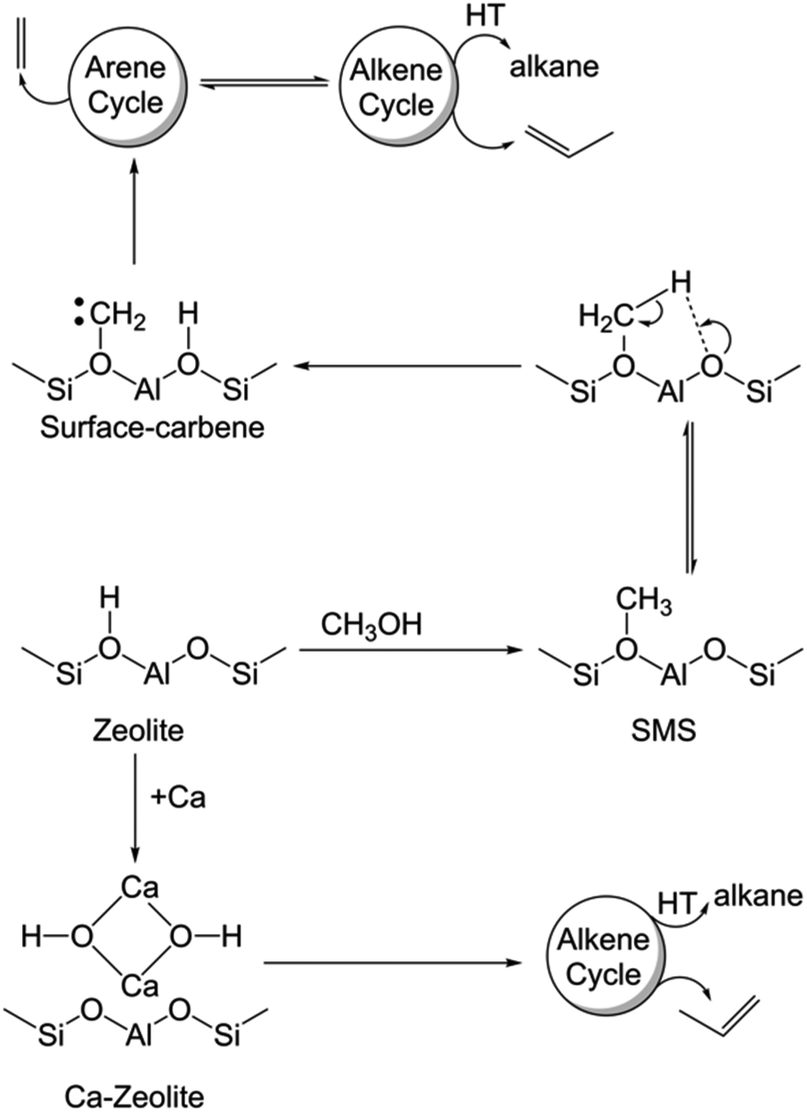 | ||
| Scheme 1 Mechanistic differences between the un- and Ca-modified zeolite ZSM-5 catalyzed methanol-to-hydrocarbon reaction (HT: hydrogen transfer, SMS: surface-methoxy species). | ||
Herein, we demonstrate that the carbene species is not merely a spectator in MTH chemistry.2,9,58,61,62,66,67 This concept in zeolite catalysis was first introduced by Chang and Silvestri more than four decades ago.58 However, this proposal did not get much attention from the community due to the lack of concrete experimental evidence. Recently, experimental evidence in support of the carbene species was provided by the groups of Hunger67 and Kondo,61,62 as already discussed previously. Notably, through in-depth spectroscopic investigations and isotope-labeling study, Kondo et al. clearly showed that both the carbene species and its precursor (i.e., SMS) were responsible for the direct formation of propylene from ethylene.61,62 In the present case, both SMS and carbene species promoted the homologation reaction during the MTH process, which ultimately led to the formation of aromatic species. In this regard, the effect of the homologation reaction and its exact mechanism for the arene cycle of the HCP mechanism during zeolite-catalyzed alcohol conversion have recently been established.68 Therefore, it is safe to conclude that the surface-carbene species on H-ZSM-5 is indeed primarily responsible for stimulating the arene cycle of the HCP-based dual cycle mechanism, which does not only promote the formation of coke species, but is also eventually accountable for the formation of ethylene (Scheme 1).1,10
Although the NMR samples were prepared with a naturally abundant reactant, 2D 13C–13C correlation DNP SENS experiments were still attempted to understand the local heterogeneity within the sample with respect to the post-reacted zeolite-trapped oxygenated species. For this purpose, the carbons were polarized through cross-polarization, and 13C–13C mixing (30 ms) was achieved through proton-driven spin-diffusion using phase-alternated recoupling irradiation schemes (PARIS, see Fig. S9 and S10 in the ESI†).15,22,69,70 Although the 2D 1H–13C cross-polarization magic angle spinning HETCOR spectrum (Fig. 5a) of unmodified post-reacted zeolite ZSM-5 already confirmed the presence of surface-adsorbed methanol, the 2D 13C–13C correlation DNP SENS spectrum further highlighted its heterogeneous nature as well as its multiple orientations. Herein, we identify two zeolite-surface bound oxygenated species in the methoxy region (Fig. S9b in the ESI†), which could be attributed to two different binding modes (∼49 and ∼51 ppm for end-on and side-on orientations, respectively) of methanol on zeolite.25,71,72 The relatively broad line-width of the ∼51 ppm peak (belonging to the side-on mode of zeolite-bound methanol) suggests heterogeneity in its molecular environment.23
In addition to the identification of acetal species (–O–CH2–O–) on both post-reacted zeolites (cf.Fig. 3), 2D 13C–13C correlation spectra also revealed its non-identical physical characteristics (Fig. 6). In MTH chemistry, an acetal typically symbolizes the presence of dimethoxymethane (DMM, CH3O–CH2–OCH3, an acetal of formaldehyde).56,58,73–78 Interestingly, methylene (–CH2–) in the post-MTH reacted unmodified zeolite ZSM-5 trapped DMM exhibits a twin signal (98.5 and 101.1 ppm), that shows an interchange of cross-peaks with each other.21 This is a typical signature of exchange between two anomeric DMM structures (trans,trans and gauche,gauche, see Fig. 6a) on a sub-second time-scale.21,79 In contrast in zeolite Ca-ZSM-5, a sole DMM-CH2 signal was observed at 99.8 ppm (i.e., exactly at an average of their anomeric resonances), implying that the exchange between the two anomeric DMM structures is too fast to be detected by NMR in this case. This is a distinctive signature of a mobile species, which typically displays much narrower features under cross-polarization-based dipolar/through-space magnetization transfer schemes.15,22,23
To produce acetal, methanol first undergoes dehydrogenation to form in situ carbon monoxide. Then, a surface-formate species was formed as a result of Koch-carbonylation of zeolite (Fig. 6b).15,21,75 Due to the highly electrophilic nature of the surface-formate species, it undergoes successive methanol nucleophilic attack to produce dimethoxymethane (DMM), an acetal of formaldehyde. Both formaldehyde and its acetal (i.e., DMM) are also reported to directly contribute to the formation of hydrocarbon pool species during the zeolite-catalyzed methanol-to-hydrocarbon process.21,80
In fact, the formation of DMM or its precursor, formaldehyde, further corroborates the occurrence of ongoing disproportionation of methanol (2CH3OH → CH4 + HCHO) during the zeolite-catalysed MTH process.56 This essentially further confirms that the ∼0.5 ppm peak in Fig. 3a indeed originated from the zeolite-trapped/adsorbed methane, which is nothing but a side-product of such a disproportionation reaction in MTH catalysis.
In terms of mechanistic aspects (cf.Scheme 1), this study unequivocally supports the founding postulation of Svelle et al. in which a hybrid inorganic–organic material (i.e., encompassing the inorganic zeolite and organic HCP reaction centers, presumably through host–guest interaction) is proposed as an active MTH catalyst.10,15,81,82 The same group also demonstrated that such a (in situ formed) hybrid material eventually controls the product selectivity: the formation of ethylene mechanistically originates from the arene cycle, while propylene is selectively produced within the alkene cycle (Scheme 1).1,10 Herein, the arene part of the HCP-based dual cycle is destabilized upon Ca-modification of zeolites and thus the hybrid ‘working catalyst material’ is primarily constituted by the inorganic zeolite and non-aromatic HCP-based organic reaction centers, which mechanistically prefer the selective formation of propylene during the MTH reaction.1,10,14 Alternatively, the accessibility of Brønsted acid sites in unmodified ZSM-5 promotes dehydroaromatization to form aromatics from olefins/paraffins/carbenes and therefore the arene cycle of the HCP mechanism would be equally operational along with the olefin cycle in the typical hydrocarbon pool mechanism of MTH chemistry. Not only the synergy between Brønsted and Lewis acid sites, but also their proximity plays a crucial role in catalysis. Herein, we demonstrated that the close-proximity of Ca nto conventional zeolite Brønsted acid sites was sufficient enough to prevent the formation of carbene species. Moreover, the carbene species on non-modified zeolite contributes to the formation of arene-based HCP species, which preferentially generates ethylene in MTH catalysis.2,83
Conclusions
In summary, the synergy and proximity between Lewis acid sites and Brønsted acid sites in the Ca modified zeolite material are indeed responsible for the inability to form surface-carbene species, hence strongly suppressing the propagation of the arene cycle. Such a shift in the equilibrium of the dual cycle mechanism towards the olefin cycle dramatically increases the catalyst lifetime due to the absence of ‘conventional’ deactivating (arene-based) coke species. The mobility-dependent host–guest chemistry between the inorganic zeolite and organic hydrocarbon pool reaction centers, i.e. the hybrid nature of the in situ formed active catalytic material, has also been explained. Such accurate elucidation of the zeolite-trapped organics along with their physiognomies highlighting Brønsted/Lewis acid site synergy/proximity by exploiting dynamic nuclear polarization enhanced NMR spectroscopy will not only be useful for the development of superior and/or upgraded catalyst materials, but will also contribute to the fundamental understanding of zeolite-catalyzed hydrocarbon conversion chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from the King Abdullah University of Science and Technology (KAUST).Notes and references
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martínez-Espín, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS
.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS
- H. Schulz, Catal. Lett., 2018, 148, 1263–1280 CrossRef CAS
- S. Xu, Y. Zhi, J. Han, W. Zhang, X. Wu, T. Sun, Y. Wei and Z. Liu, Adv. Catal., 2017, 61, 37–122 Search PubMed
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed
- S. Ilias and A. Bhan, ACS Catal., 2013, 3, 18–31 CrossRef CAS
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef
- X. Sun, S. Mueller, Y. Liu, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. van Veen and J. A. Lercher, J. Catal., 2014, 317, 185–197 CrossRef CAS
- X. Sun, S. Mueller, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. van Veen and J. A. Lercher, J. Catal., 2014, 314, 21–31 CrossRef CAS
- I. Yarulina, S. Bailleul, A. Pustovarenko, J. R. Martinez, K. De Wispelaere, J. Hajek, B. M. Weckhuysen, K. Houben, M. Baldus, V. Van Speybroeck, F. Kapteijn and J. Gascon, ChemCatChem, 2016, 8, 3057–3063 CrossRef CAS
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 8095–8099 CrossRef CAS PubMed
-
D. Apperley, R. Harris and P. Hodgkinson, Solid-State NMR: Basic Principles and Practice, Momentum Press, 2012 Search PubMed
-
D. Mance, M. Weingarth and M. Baldus, in Modern Magnetic Resonance, Springer International Publishing, Cham, 2016, pp. 1–17 Search PubMed
- T. Polenova, R. Gupta and A. Goldbourt, Anal. Chem., 2015, 87, 5458–5469 CrossRef CAS PubMed
- A. S. Andreev and V. Livadaris, J. Phys. Chem. C, 2017, 121, 14108–14119 CrossRef CAS
- H. Koller and M. Weiß, Solid State NMR of Porous Materials: Zeolites and Related Materials, Top. Curr. Chem., 2012, 306, 189–228 CrossRef CAS PubMed
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed
- A. D. Chowdhury, K. Houben, G. T. Whiting, S.-H. Chung, M. Baldus and B. M. Weckhuysen, Nat. Catal., 2018, 1, 23–31 CrossRef CAS
- Z. Ristanović, A. D. Chowdhury, R. Y. Brogaard, K. Houben, M. Baldus, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2018, 140, 14195–14205 CrossRef PubMed
- D. Xiao, S. Xu, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2017, 8, 8309–8314 RSC
- M. Zhang, S. Xu, J. Li, Y. Wei, Y. Gong, Y. Chu, A. Zheng, J. Wang, W. Zhang, X. Wu, F. Deng and Z. Liu, J. Catal., 2016, 335, 47–57 CrossRef CAS
- W. Wang and M. Hunger, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed
- D. Xiao, S. Xu, N. J. Brownbill, S. Paul, L.-H. Chen, S. Pawsey, F. Aussenac, B.-L. Su, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2018, 9, 8184–8193 RSC
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed
- B. Plainchont, P. Berruyer, J.-N. Dumez, S. Jannin and P. Giraudeau, Anal. Chem., 2018, 90, 3639–3650 CrossRef CAS
-
F. Blanc, in Nanotechnology in Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, pp. 1003–1028 Search PubMed
- N. J. Brownbill, R. S. Sprick, B. Bonillo, S. Pawsey, F. Aussenac, A. J. Fielding, A. I. Cooper and F. Blanc, Macromolecules, 2018, 51, 3088–3096 CrossRef CAS
- A. L. Paioni, M. A. M. Renault and M. Baldus, eMagRes, 2018, 7, 51–62 CAS
- A. Jantschke, E. Koers, D. Mance, M. Weingarth, E. Brunner and M. Baldus, Angew. Chem., Int. Ed., 2015, 54, 15069–15073 CrossRef CAS
- E. A. W. van der Cruijsen, E. J. Koers, C. Sauvée, R. E. Hulse, M. Weingarth, O. Ouari, E. Perozo, P. Tordo and M. Baldus, Chem.–Eur. J., 2015, 21, 12971–12977 CrossRef CAS PubMed
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed
- D. Mance, J. van der Zwan, M. E. Z. Velthoen, F. Meirer, B. M. Weckhuysen, M. Baldus and E. T. C. Vogt, Chem. Commun., 2017, 53, 3933–3936 RSC
- E. Pump, A. Bendjeriou-Sedjerari, J. Viger-Gravel, D. Gajan, B. Scotto, M. K. Samantaray, E. Abou-Hamad, A. Gurinov, W. Almaksoud, Z. Cao, A. Lesage, L. Cavallo, L. Emsley and J.-M. Basset, Chem. Sci., 2018, 9, 4866–4872 RSC
- E. Pump, J. Viger-Gravel, E. Abou-Hamad, M. K. Samantaray, B. Hamzaoui, A. Gurinov, D. H. Anjum, D. Gajan, A. Lesage, A. Bendjeriou-Sedjerari, L. Emsley and J.-M. Basset, Chem. Sci., 2017, 8, 284–290 RSC
- J. C. Mohandas, E. Abou-Hamad, E. Callens, M. K. Samantaray, D. Gajan, A. Gurinov, T. Ma, S. Ould-Chikh, A. S. Hoffman, B. C. Gates and J.-M. Basset, Chem. Sci., 2017, 8, 5650–5661 RSC
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed
- F. A. Perras, J. D. Padmos, R. L. Johnson, L. L. Wang, T. J. Schwartz, T. Kobayashi, J. H. Horton, J. A. Dumesic, B. H. Shanks, D. D. Johnson and M. Pruski, J. Am. Chem. Soc., 2017, 139, 2702–2709 CrossRef CAS PubMed
- D. Xiao, X. Han, X. Bao, G. Hou and F. Blanc, RSC Adv., 2019, 9, 12415–12418 RSC
- M. Valla, A. J. Rossini, M. Caillot, C. Chizallet, P. Raybaud, M. Digne, A. Chaumonnot, A. Lesage, L. Emsley, J. A. Van Bokhoven and C. Copéret, J. Am. Chem. Soc., 2015, 137, 10710–10719 CrossRef CAS PubMed
- W. R. Gunther, V. K. Michaelis, M. A. Caporini, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2014, 136, 6219–6222 CrossRef CAS PubMed
- A. J. Rossini, A. Zagdoun, M. Lelli, J. Canivet, S. Aguado, O. Ouari, P. Tordo, M. Rosay, W. E. Maas, C. Copéret, D. Farrusseng, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2012, 51, 123–127 CrossRef CAS PubMed
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed
- W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, ACS Catal., 2013, 3, 588–596 CrossRef CAS
- W. Dai, C. Wang, M. Dyballa, G. Wu, N. Guan, L. Li, Z. Xie and M. Hunger, ACS Catal., 2015, 5, 317–326 CrossRef CAS
- X. Wu, S. Xu, Y. Wei, W. Zhang, J. Huang, S. Xu, Y. He, S. Lin, T. Sun and Z. Liu, ACS Catal., 2018, 8, 7356–7361 CrossRef CAS
- X. Zhou, C. Wang, Y. Chu, J. Xu, Q. Wang, G. Qi, X. Zhao, N. Feng and F. Deng, Nat. Commun., 2019, 10, 1961 CrossRef PubMed
- C. Wang, Y. Chu, J. Xu, Q. Wang, G. Qi, P. Gao, X. Zhou and F. Deng, Angew. Chem., Int. Ed., 2018, 57, 10197–10201 CrossRef CAS PubMed
- P. Gao, Q. Wang, J. Xu, G. Qi, C. Wang, X. Zhou, X. Zhao, N. Feng, X. Liu and F. Deng, ACS Catal., 2018, 8, 69–74 CrossRef CAS
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev, I. G. Danilova, I. P. Prosvirin, V. V. Kriventsov, V. I. Zaikovskii, D. Freude and A. G. Stepanov, ACS Catal., 2017, 7, 1818–1830 CrossRef CAS
- A. Hwang, M. Kumar, J. D. Rimer and A. Bhan, J. Catal., 2017, 346, 154–160 CrossRef CAS
- B. K. Hunter and L. W. Reeves, Can. J. Chem., 1968, 46, 1399–1414 CrossRef CAS
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS PubMed
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260–15267 CrossRef CAS PubMed
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, J. Phys. Chem. C, 2012, 116, 24091–24097 CrossRef CAS
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Angew. Chem., Int. Ed., 2011, 50, 1853–1856 CrossRef CAS PubMed
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Núñez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Copéret and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS PubMed
- O. Lafon, A. S. L. Thankamony, T. Kobayashi, D. Carnevale, V. Vitzthum, I. I. Slowing, K. Kandel, H. Vezin, J.-P. Amoureux, G. Bodenhausen and M. Pruski, J. Phys. Chem. C, 2013, 117, 1375–1382 CrossRef CAS
- G. Qi, Q. Wang, J. Xu, J. Trébosc, O. Lafon, C. Wang, J.-P. Amoureux and F. Deng, Angew. Chem., Int. Ed., 2016, 55, 15826–15830 CrossRef CAS
- G. J. Hutchings and R. Hunter, Catal. Today, 1990, 6, 279–306 CrossRef CAS
- W. Wang and M. Hunger, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS PubMed
- A. D. Chowdhury, A. Lucini Paioni, G. T. Whiting, D. Fu, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2019, 58, 3908–3912 CrossRef CAS PubMed
- M. Weingarth, D. E. Demco, G. Bodenhausen and P. Tekely, Chem. Phys. Lett., 2009, 469, 342–348 CrossRef CAS
- J. Medeiros-Silva, S. Jekhmane, A. L. Paioni, K. Gawarecka, M. Baldus, E. Swiezewska, E. Breukink and M. Weingarth, Nat. Commun., 2018, 9, 3963 CrossRef
- I. I. Ivanova and A. Corma, J. Phys. Chem. B, 1997, 101, 547–551 CrossRef CAS
- W. Wang, M. Seiler and M. Hunger, J. Phys. Chem. B, 2001, 105, 12553–12558 CrossRef CAS
- J. Nováková, L. Kubelková and Z. Dolejšek, J. Catal., 1987, 108, 208–213 CrossRef
- Y. Liu, S. Müller, D. Berger, J. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 55, 5723–5726 CrossRef CAS PubMed
- P. N. Plessow and F. Studt, ACS Catal., 2017, 7, 7987–7994 CrossRef CAS
- P. N. Plessow and F. Studt, Catal. Lett., 2018, 148, 1246–1253 CrossRef CAS
- P. N. Plessow, A. Smith, S. Tischer and F. Studt, J. Am. Chem. Soc., 2019, 141, 5908–5915 CrossRef CAS PubMed
- Y. Liu, F. M. Kirchberger, S. Müller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef
- E. Juaristi and G. Cuevas, Tetrahedron, 1992, 48, 5019–5087 CrossRef CAS
- S. S. Arora, D. L. S. Nieskens, A. Malek and A. Bhan, Nat. Catal., 2018, 1, 666–672 CrossRef CAS
- C. Wang, J. Xu, Q. Wang, X. Zhou, G. Qi, N. Feng, X. Liu, X. Meng, F. Xiao and F. Deng, ACS Catal., 2017, 7, 6094–6103 CrossRef CAS
- C. Wang, Q. Wang, J. Xu, G. Qi, P. Gao, W. Wang, Y. Zou, N. Feng, X. Liu and F. Deng, Angew. Chem., Int. Ed., 2016, 55, 2507–2511 CrossRef CAS PubMed
- A. D. Chowdhury and J. Gascon, Angew. Chem., Int. Ed., 2018, 57, 14982–14985 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental procedure, supplementary NMR figures and analysis. See DOI: 10.1039/c9sc02215e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |