DOI:
10.1039/C9SC01739A
(Edge Article)
Chem. Sci., 2019,
10, 8939-8945
Extreme multi-point van der Waals interactions: isolable dimers of phthalocyanines substituted with pillar-like azaacenes†
Received
9th April 2019
, Accepted 29th August 2019
First published on 29th August 2019
Abstract
How strong are van der Waals interactions in determining the final outcome of self-assembled structures of small molecular systems? Herein we report isolable phthalocyanine (Pc) dimers bound by π–π interactions between monomeric Pcs which can be handled as single entities. Pc dimers have been continuously investigated as one of the simplest models of Pc aggregates. Pcs were substituted with eight dihydrodiazapentacene (DHDAP) moieties on the periphery, which act as pillar-like π-planes and these molecules form H-type dimers with the help of synergetic π–π interactions between two co-facial Pc rings and among the pillar-like DHDAP moieties. The dimer structures were fully confirmed by 1D and 2D NMR, ESR, and electronic absorption measurements. The dissociation of these dimers was observed in particular solvents such as o-dichlorobenzene, due to the good solubility of the larger π-conjugated molecules. On the other hand, in ethyl acetate the monomers were metastable species and underwent selective dimerization. Interestingly in THF, neither the dimerization of the monomers nor the dissociation of dimers was observed, suggesting that both the dimers and the monomers were kinetically well stabilized. For hours to days, we can handle these dimers as “a molecule” not only in solution but also even in mass spectrometry under ionization conditions without significant dissociation.
Introduction
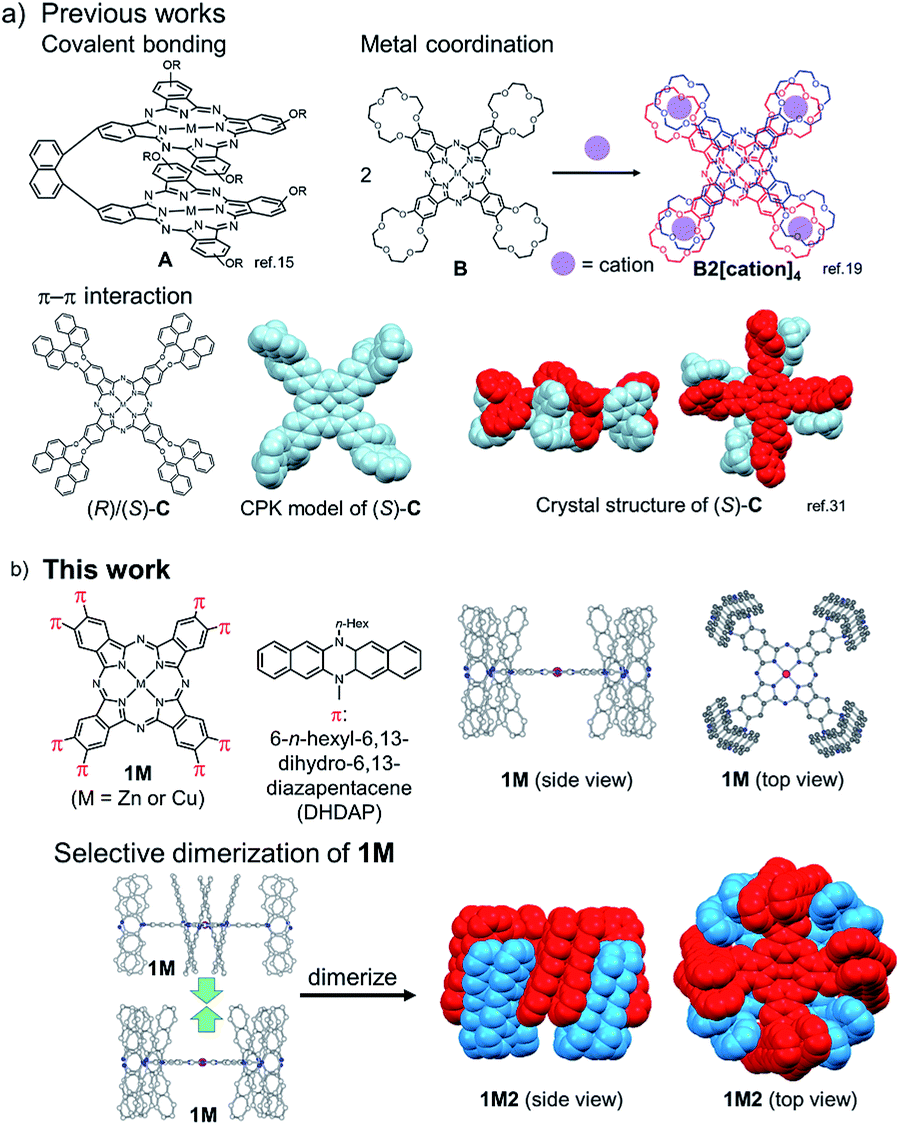
Phthalocyanines (Pcs) are one of the special classes of functional dyes and pigments due to their high photostability and wide range of applications.1,2 The photophysical properties of dye aggregates differ considerably due to the relative arrangement of dye molecules,3 leading to different types of exciton coupling.4 Hence, it is important and challenging to control the mode of aggregation of dye molecules. Until now, various kinds of Pc derivatives have been synthesized and their aggregates were investigated to understand the relationship between the relative arrangement of the Pcs and the electronic properties.5–8 In particular, as the simplest model of Pc aggregates, various Pc dimers have been intensively studied.9 In the pioneering studies on covalently bound Pc dimers, Leznoff, Lever and coworkers reported tethered Pc dimers, which can stack in a “clamshell” fashion.10–13 After these studies, various covalently linked Pc dimers were reported, where Pcs were differently arranged such as in a coplanar,14–16 cofacial (A in Fig. 1a),17,18 and slipped-stack manner.19 In 1986, three independent research groups consecutively reported the selective dimerization of Pcs by means of metal complexation using Pcs (B in Fig. 1a) substituted by four crown ether moieties (15-crown-5), which form co-facial dimer (H-type) triggered by the addition of cations such as alkaline or alkaline earth metal cations (Fig. 1a).20–22 The dimerization of these “crowned” Pcs proceeds in a two-step manner upon gradual addition of specific cations; at the first stage, linear dimers are formed which share one guest cation and are then converted to co-facial dimers by capturing additional cations.23 Due to high directionality and binding strength of coordination bonds, various other Pc dimers based on metal–ligand interactions have been designed.24–27 Other intermolecular interactions such as hydrogen bonds28,29 or hydrophobic forces30 have been also adopted as the driving force for the selective dimerization of Pcs. In this context, π–π interactions31 are also considered as an important intermolecular force to control molecular assembly of Pcs because of their large π plane.32 A few examples are reported for the selective dimerization of Pcs using π–π interactions; however it is difficult to avoid the formation of higher aggregates. In 1993, Kobayashi et al. reported that a Pc substituted with two enantiopure 2,2′-dihydroxy-1,1′-binaphthyl at the periphery of the Pc ring is in equilibrium between the monomer and the co-facial dimer in solution.33 Jiang et al. also reported the co-facial dimerization of the Pc with four 2,2′-dihydroxy-1,1′-binaphthyl groups, and they revealed the dimer structures by X-ray single crystal analysis, indicating the presence of multiple π–π interactions between the two Pc planes and among the peripheral binaphthyl substituents (C in Fig. 1a).34 However, in these cases, the separation of the monomer and the dimer was not achieved due to the fast interconversion between the monomeric and dimeric forms in solution. Other than Pc derivatives, few examples are known with supramolecular dimers bound by π–π interactions with high kinetic stability, such as the hexa-peri-hexabenzocoronenes (HBCs) with oligophenylene dendrons reported by Müllen et al.35,36 or a tweezer-type perylene bisimide (PDI) reported by Würthner et al.37 However, to the best of our knowledge, there are no examples of π-stacking molecules whose monomeric and dimeric forms are kinetically well-stabilized to be separated from each other. Hence, in this manuscript, we aimed to provide the first example of a Pc dimer bound by π–π interactions, which can be isolated from its monomer and handled as a single chemical entity. To obtain enough stabilization energy by π–π interactions, we conceived the idea to introduce wide π-systems perpendicularly arranged at the Pc periphery to use π–π interactions among the perpendicular π-systems in the dimer form (Fig. 1b). Recently, we have reported the synthesis of a phthalonitrile substituted with two 6,13-dihydro-6,13-diazapentacene (DHDAP) units (2), whose DHDAP moieties are fixed in a face-to-face manner and perpendicular to the benzene ring of phthalonitrile.38 This molecule could be converted to Pcs with eight pillar-like DHDAP units (1M), which can potentially form a tightly bound dimer (1M2) as shown in Fig. 1b. DFT calculations with the model compounds suggested that such dimers can be reasonably formed (see the ESI†). The stabilization energy for the dimerization was calculated to be −399.2 kJ mol−1, and this value is much larger than that of C calculated at the same theoretical level (−321.5 kJ mol−1).34 In this work, we describe the synthesis and unique dimerization behavior of the zinc and copper Pcs, 1Zn and 1Cu.
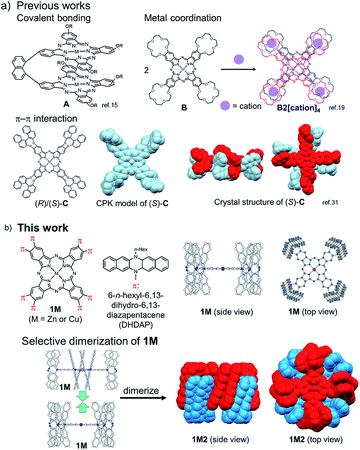 |
| | Fig. 1 (a) Previous examples of cofacially stacked Pc dimers bound by covalent bonding, metal coordination, and π–π interactions. (b) Molecular structure of Pcs with eight diazapentacene moieties (1M) and the schematic representation of the formation of the dimers (1M2). | |
Results and discussion
Synthesis
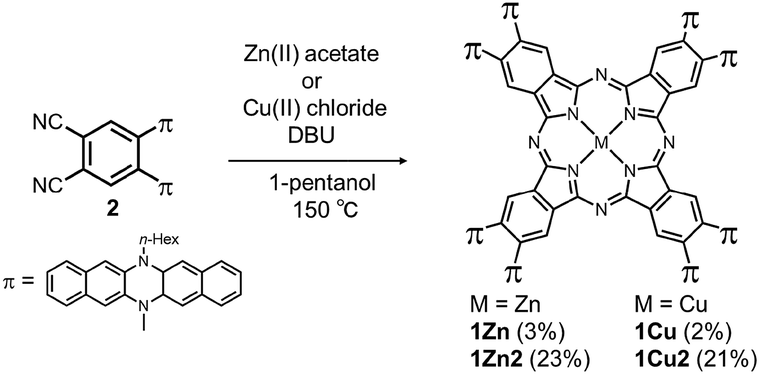
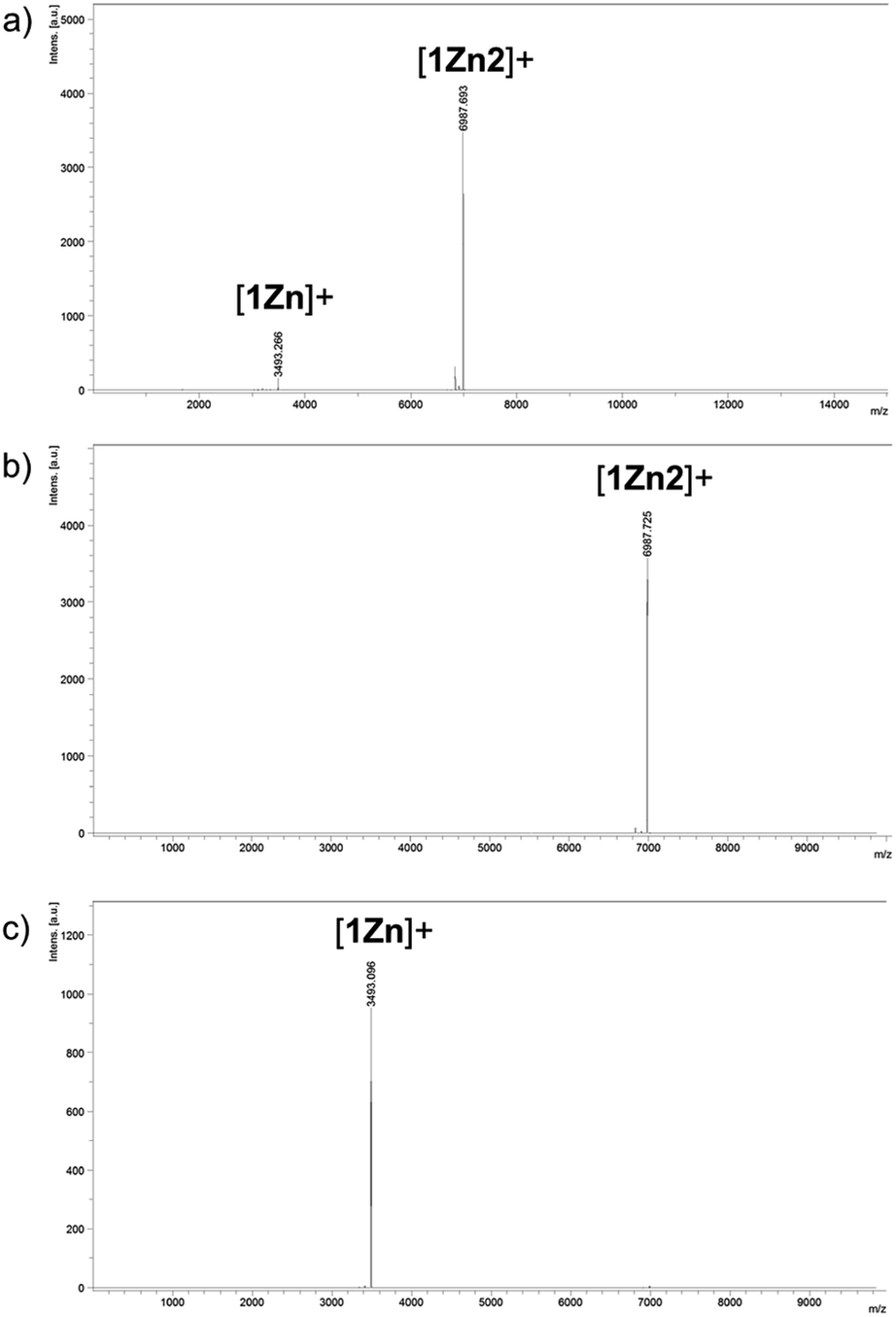
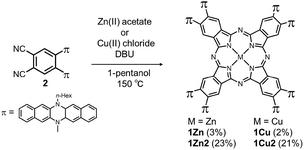
Two kinds of Pcs 1Zn and 1Cu were synthesized as examples of diamagnetic and paramagnetic (S = 1/2) Pcs according to Scheme 1. The precursor 2 was condensed in the presence of metal templates (Zn(OAc)2 for 1Zn and CuCl2 for 1Cu) and 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) by refluxing in 1-pentanol for 24 h. The reaction mixture was cooled down to room temperature and precipitated by adding methanol. The crude products were monitored by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS). Fig. 2a is the MALDI-TOF-MS spectrum of the crude product of the reaction with Zn(OAc)2 as the metal template. The peaks derived from 1Zn2 were clearly observed along with the peaks of 1Zn, and the peak intensity of 1Zn2 was much larger than that of 1Zn. No peaks corresponding to the trimer or higher oligomers were found. The lack of the trimer and higher oligomers was understood from the structure of 1Zn2, in which no spaces for the third monomer are left. The third monomer can have only minimal contact with 1Zn2 as understood from the molecular modeling of the trimer (Fig. S37†). We successfully isolated the dimers and monomers by recycling preparative gel permeation chromatography (GPC) (Fig. S1†). The MALDI-TOF-MS spectra of the first and the second eluting fractions unequivocally certified the isolation of 1Zn2 and 1Zn (Fig. 2b and c). It is noteworthy to mention that no peaks derived from 1Zn were observed from the measurements of isolated 1Zn2, showing no dissociation of 1Zn2 during the ionization process of MALDI (Fig. 2b). The separation of 1Cu and 1Cu2 was also accomplished in a similar way (Fig. S2†). We have done several trials and errors to obtain single crystals of these compounds, but unfortunately, only brittle crystals giving poor X-ray diffraction patterns were obtained in all cases. Therefore, the structures of the dimers were investigated by combining various spectroscopic measurements as described below.
 |
| | Scheme 1 Synthesis of target molecules. | |
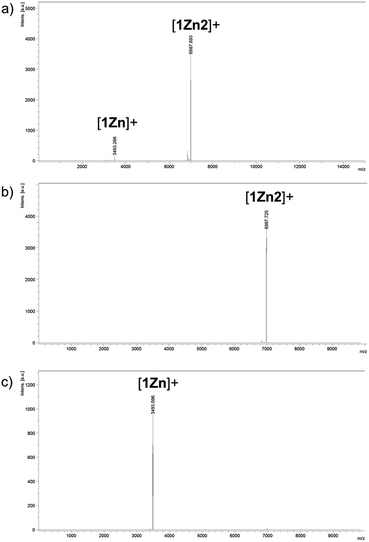 |
| | Fig. 2 MALDI-TOF-MS spectra (positive) of (a) the reaction crude containing 1Zn and 1Z2 and of (b) 1Zn2 and (c) 1Zn isolated by HPLC. | |
NMR spectroscopy
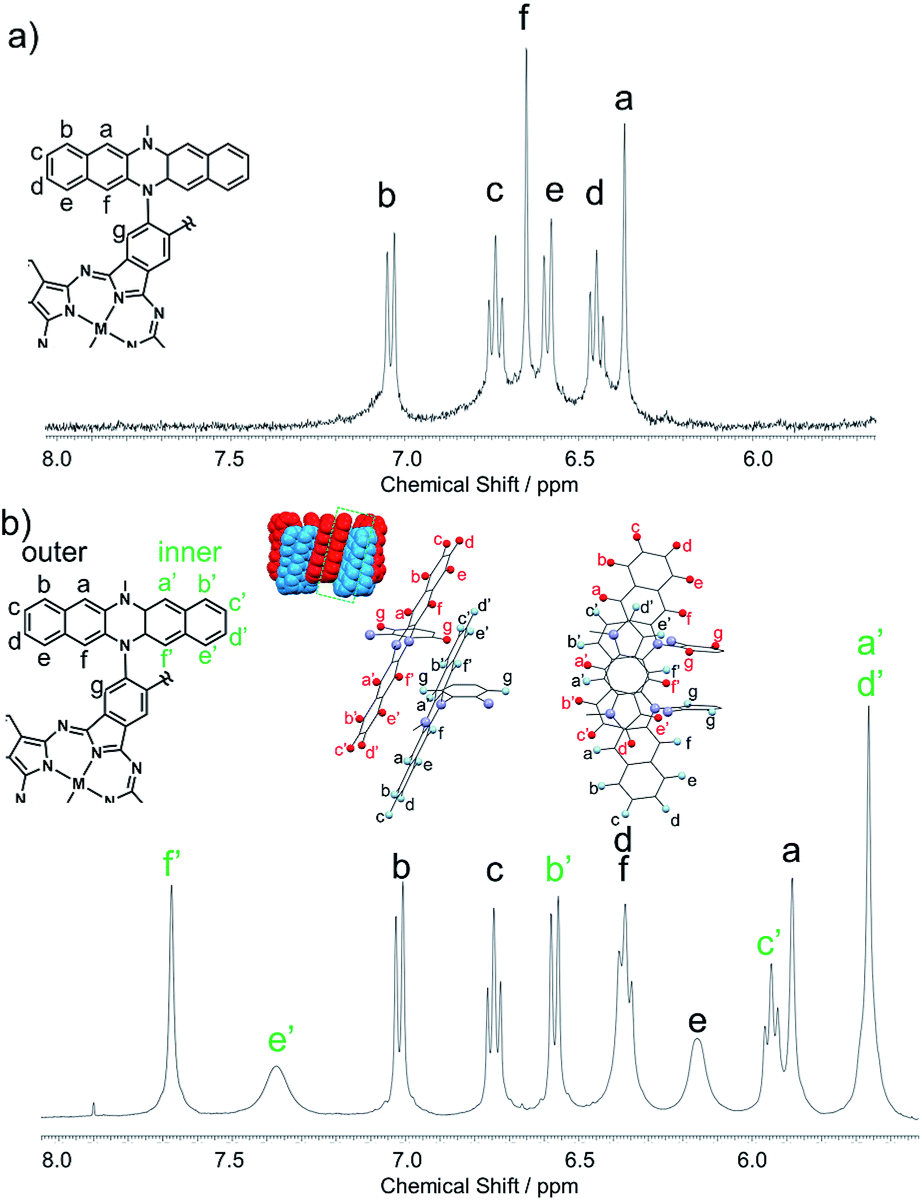
The structures of 1Zn and 1Zn2 were thoroughly investigated using various 1D and 2D NMR techniques. The 1H NMR spectrum of 1Zn in tetrahydrofuran-d8 shows seven peaks in the aromatic region (Fig. S4†). The protons of the benzo moieties of phthalocyanine were observed as a single peak at 10.27 ppm, and the other six peaks in the range of 6.3–7.1 ppm were attributed to the protons of the DHDAP units. The 13C NMR spectrum of 1Zn showed six aliphatic peaks and fourteen aromatic peaks (Fig. S5†). The results of 1H and 13C NMR spectra were fully consistent with the D4 symmetric structure of 1Zn. By combining the results of 1D and 2D NMR measurements, the peaks of DHDAP moieties of 1Zn were successfully assigned as shown in Fig. 4a (details are shown in Fig. S6–S10†). The 1H NMR spectrum of 1Zn2 in tetrahydrofuran-d8 showed a greater number of peaks in the aromatic region than that observed in 1Zn (Fig. S11†). The protons of the benzo moieties of 1Zn2 were also observed as a single peak at 10.27 ppm as in the case of 1Zn, indicating the existence of a four-fold axis in 1Zn2. The 13C NMR spectrum of 1Zn2 showed six peaks of aliphatic carbons and twenty four peaks of aromatic carbons (Fig. S12†). The HMQC measurements based on the result of the 1D 13C NMR measurement clearly showed the existence of six different aliphatic protons and thirteen different aromatic protons in 1Zn2, also being consistent with the interdigitated dimeric structure (Fig. S14†). Assuming the dimer structure shown in Fig. 1b, the outer (opposite side to the Pc of the complementary monomer) and inner (same side as the Pc of the complementary monomer) half of each DHDAP unit should be non-equivalent. Herein the protons of the inner naphthalene moiety of each DHDAP unit are labeled with apostrophes (a′ to f′), and all the protons of the DHDAP moieties were consistently assigned by combining the results of 1D and 2D NMR measurements as shown in Fig. 3b (details are shown in Fig. S13–S17†). The protons existing in the outermost part of the DHDAP units (b, c, and d) did not shift compared with those of 1Zn, suggesting the negligible interaction from the other 1Zn in the dimer. On the other hand, the protons of the inner half parts of DHDAP units (a′ to f′), which penetrate the pockets of the other monomer, showed significant shifts from those of 1Zn. In particular, the protons c′ and d′ showed significant high-field shifts of 0.80 and 0.79 ppm respectively compared with those of 1Zn. Judging from the optimized structure of the dimer, these protons exist above the π-plane of DHDAP of the other monomer, and the high-field shifts could be ascribed to the shielding effect from the DHDAP moiety. The protons f′ showed the largest down-field shift of 1.03 ppm compared with 1Zn due to the strong deshielding effect from nearby Pc ring of the other monomer. The 1H-DPFGSE-NOE spectra,39 which show weak NOE signals, also indicated the proposed dimer structure (Fig. S18†). We carried out DOSY measurements for a mixed solution of 1Zn and 1Zn2 in tetrahydrofuran-d8, and the diffusion constants of 1Zn and 1Zn2 were evaluated to be 3.0 × 10−10 m2 s−1 and 2.7 × 10−10 m2 s−1, respectively. The value for 1Zn2 was smaller than that for 1Zn, but the difference was not so large. This result implies the compactly interdigitated structure of 1Zn2.
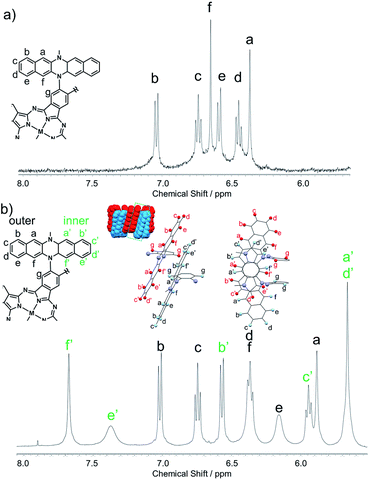 |
| | Fig. 3 Aromatic regions of 1H NMR spectra for (a) 1Zn and (b) 1Zn2 in tetrahydrofuran-d8. | |
ESR spectroscopy
Electron spin resonance (ESR) is a powerful tool to investigate the way of association of Pcs with a paramagnetic central metal such as Cu(II). To obtain further information about the dimer structure, we measured ESR spectra of the frozen solutions of 1Cu and 1Cu2 in CH2Cl2. Fig. 4a shows the ESR spectrum of the frozen solution of 1Cu in CH2Cl2 at 123 K. The spectral shape was a typical pattern of monomeric CuPcs23,40 and well reproduced by spectral simulation using the axial g-value (g‖ = 2.044, g⊥ = 2.164), the hyperfine interaction from four equivalent nitrogen nuclei (ANiso = 1.85 mT) and one Cu nucleus (ACu‖ = 3.06 mT, ACu⊥ = 21.6 mT). On the other hand, a frozen solution of 1Cu2 showed a completely different spectral pattern (Fig. 4b). The transition for ΔMs = ±1 showed a characteristic fine structure of a triplet species with axial symmetry (g‖ = 2.045, g⊥ = 2.081, D = 48.9 mT, E ≃ 0 mT) in addition to the hyperfine splitting from two equivalent Cu nuclei (ACu‖ = 1.40 mT, ACu⊥ = 10.1 mT). Furthermore, the forbidden transition signal (ΔMs = ±2) with a seven-line splitting arises due to the two equivalent Cu nuclei (I = 3/2) in the half-field region, clearly indicating the spin–spin interaction between two CuPc moieties. The ESR spectral features of 1Cu and 1Cu2 were completely in accordance with those of the monomers and H-type dimers of the crown ether substituted CuPcs,23 definitely verifying the proposed dimer structure of 1Cu2.
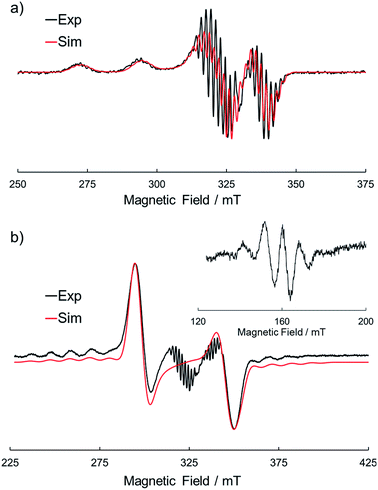 |
| | Fig. 4 ESR spectra of (a) 1Cu and (b) 1Cu2 in CH2Cl2 at 123 K. The black and red lines represent the experimental and simulated spectra, respectively. | |
Electronic absorption spectra
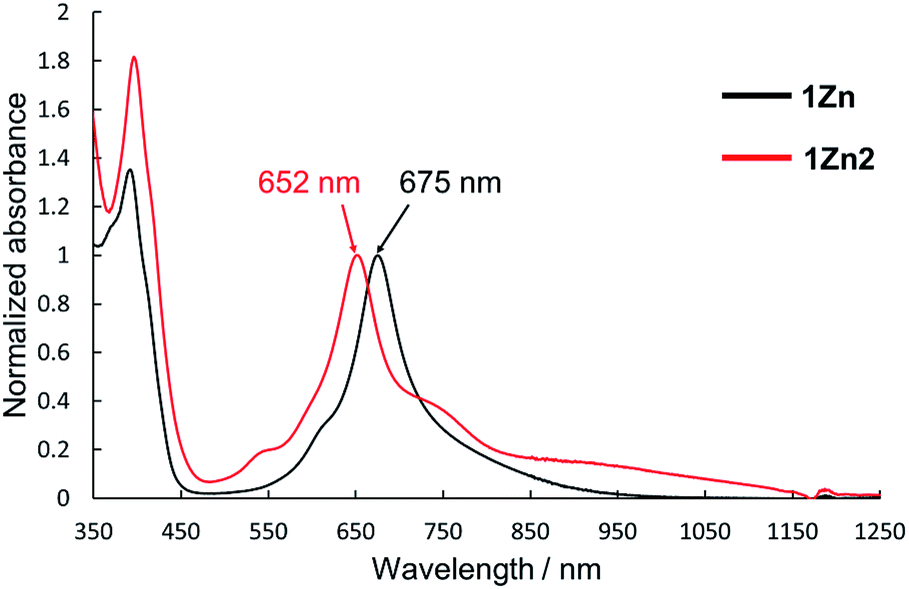
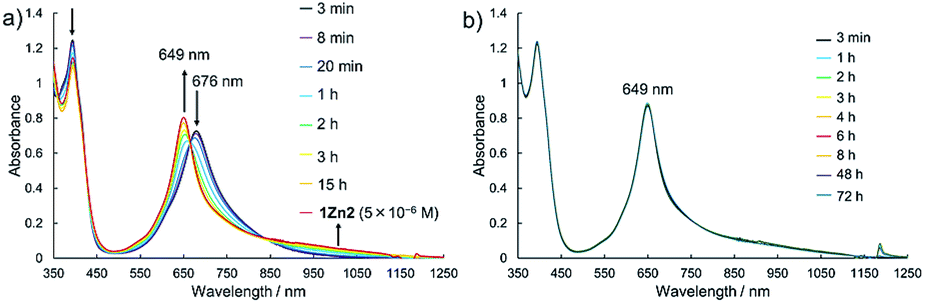
Electronic absorption measurement is one of the important methods to obtain information about the mode of aggregation of dye molecules in solution. We measured the UV-vis-NIR absorption spectra of the monomeric and the dimeric Pcs in various solvents, and they showed different time evolutions of the absorption spectra depending on solvents. The solution of 1Zn in tetrahydrofuran showed absorption bands corresponding to the Q-band of the Pc with a peak maximum at 675 nm and the bands ranging from 300 to 450 nm corresponding to the superposition of the absorption of DHDAP moieties and the Soret band of the Pc (Fig. 5). The solution of 1Zn2 in tetrahydrofuran also showed absorption bands ranging from 300 to 450 nm, similar to those of 1Zn but the Q-band of 1Zn2 showed a hypsochromic shift to 652 nm, suggesting an H-type association of two Pc moieties (Fig. 5). Furthermore, the Q-band of 1Zn2 had a broad low-energy tail up to 1200 nm. It is important to note that the spectral shapes of both 1Zn and 1Zn2 in tetrahydrofuran showed no temporal change over 72 h at room temperature, suggesting that both the dimerization of 1Zn and the dissociation of 1Zn2 were negligible in tetrahydrofuran (Fig. S20†). The hypsochromic shift of the Q-band and the broad low-energy tail of 1Zn2 were observed in all the solvents tested, but the temporal changes of the absorption spectra depended on the solvents. In CH2Cl2, the absorption spectra of 1Zn did not show any temporal change over 72 h at room temperature (Fig. 6a), whereas the absorption spectra of 1Zn2 slowly changed under the same conditions in the order of hours to days with isosbestic points at 659 nm and 890 nm (Fig. 6b). The original Q-band of 1Zn2 at 649 nm gradually decreased and a new peak at 677 nm emerged corresponding to the Q-band of 1Zn, indicating the dissociation of 1Zn2 to 1Zn. The spectral change of 1Zn2 in CH2Cl2 almost reached a steady state after 72 hours, and the spectral shape at this stage was completely in accordance with that of 1Zn in CH2Cl2. In toluene, the solution of 1Zn showed no temporal change (Fig. 6c) and 1Zn2 exhibited a similar dissociation behavior as in CH2Cl2, although the rate of dissociation was much slower (Fig. 6d). We found that the dissociation of 1Zn2 was remarkably accelerated in o-dichlorobenzene. The spectral shape of 1Zn2 in o-dichlorobenzene was almost completely changed to that of 1Zn within 2 h (Fig. 6f), whereas the spectral shape of 1Zn remains unchanged (Fig. 6e). The activation energy of the dissociation of 1Zn2 in o-dichlorobenzene was estimated to be about 72 kJ mol−1 from the Arrhenius plot by assuming a first-order reaction (Fig. S21 and S22†). The results in CH2Cl2, toluene, and o-dichlorobenzene indicate that the dimeric form is a metastable species and the monomeric state is thermodynamically favored, probably due to the strong solvation of the monomeric species. In particular, the faster dissociation in o-dichlorobenzene could be ascribed to the high affinity of o-dichlorobenzene for π-conjugated molecules, which can penetrate into the spaces between the interdigitated DHDAP moieties and break up the dimer bound by π–π interactions. On the other hand, different behaviors were observed in ethyl acetate. Interestingly, the solutions of both 1Zn and 1Zn2 in ethyl acetate showed almost no changes at room temperature over 72 h (Fig. S23†). However, when the solution of 1Zn in ethyl acetate was kept at 333 K, 1Zn gradually dimerized and the spectral shape became similar to that of 1Zn2 after 15 h (Fig. 7). This result indicates that 1Zn is metastable and 1Zn2 is thermodynamically favored in ethyl acetate. The activation energy of the dimerization of 1Zn in ethyl acetate was estimated to be about 134 kJ mol−1 from the Arrhenius plot by assuming that the dimerization obeys a second-order reaction (Fig. S24 and S25†). Furthermore, 1Zn also gradually dimerized in 1-pentanol, which was used as the reaction solvent in the final step of the synthesis, upon heating at 373 K (Fig. S26†). The fact that the yield of 1Zn2 was much higher than that of 1Zn could be understood by this dimerization of 1Zn during the reaction in the refluxing 1-pentanol. In tetrahydrofuran, the dimerization of 1Zn was not observed even at 333 K (Fig. S27†), suggesting that both 1Zn and 1Zn2 were kinetically well stabilized. The results of electronic absorption measurements of 1Cu and 1Cu2 were similar to those of 1Zn and 1Zn2 (Fig. S30–S35†).
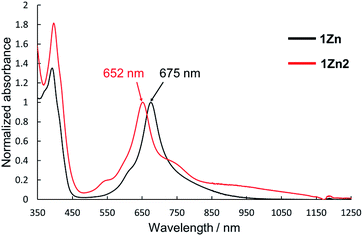 |
| | Fig. 5 UV-Vis-NIR absorption spectra of 1Zn (1 × 10−5 M, black) and 1Zn2 (5 × 10−6 M, red) in tetrahydrofuran. | |
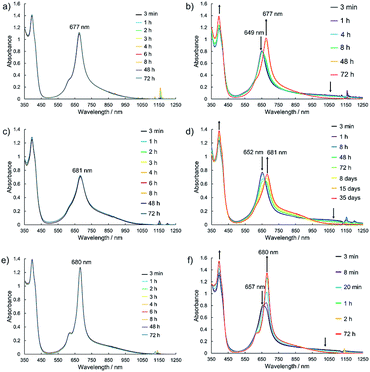 |
| | Fig. 6 Temporal changes of the UV-vis-NIR absorption spectra of 1Zn (1 × 10−5 M) in (a) CH2Cl2, (c) toluene, and (e) o-dichlorobenzene and of 1Zn2 (5 × 10−6 M) in (b) CH2Cl2, (d) toluene, and (f) o-dichlorobenzene at room temperature. | |
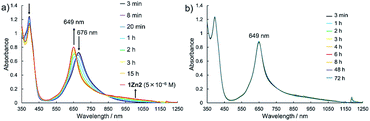 |
| | Fig. 7 Temporal changes of the UV-vis-NIR absorption spectra of (a) 1Zn (1 × 10−5 M) at 333 K and of (b) 1Zn2 (5 × 10−6 M) at room temperature in ethyl acetate. | |
Electrochemistry
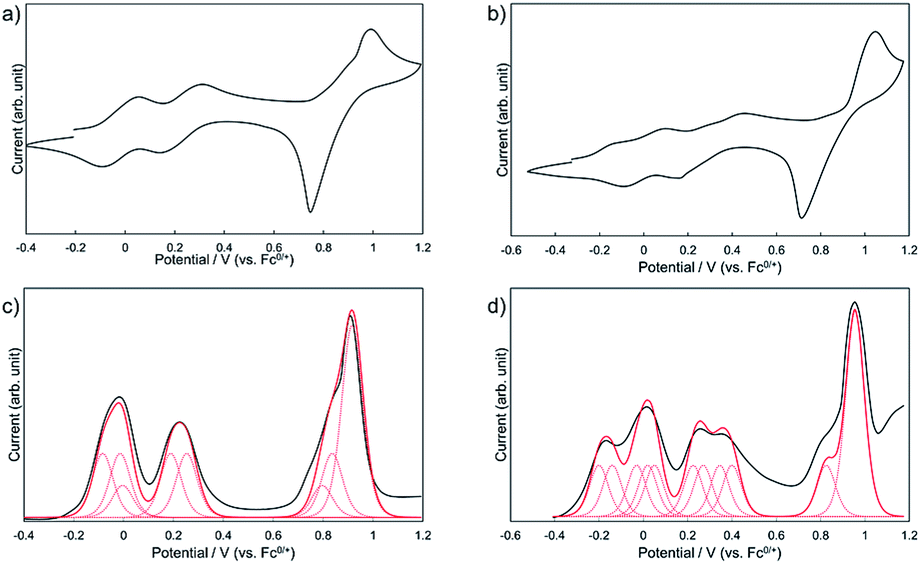
To investigate the effect of the dimerization on the redox properties, electrochemical measurements were performed for 1Zn and 1Zn2. Fig. 8a shows the cyclic voltammogram (CV) and the differential pulse voltammogram (DPV) of 1Zn in CH2Cl2 using [nBu4N][BF4] as the electrolyte. The oxidation processes were roughly grouped into three parts: ∼0 V, ∼0.25 V, and ∼0.9 V (vs. ferrocene/ferrocenium), and the shapes of the voltammograms are basically similar to those of 2.38 In the CV measurement, 2 shows three reversible oxidation waves. The first and the second oxidation waves of 2 were assigned to the consecutive one-electron oxidation from each DHDAP unit at 0.10 and 0.28 V, and the third was assigned to the quasi two-electron oxidation from each DHDAP (0.82 and 0.84 V), reflecting its twin donor structure. The similarity between the voltammograms of 1Zn and 2 strongly indicates that the redox properties of 1Zn could be attributed to the four independent co-facially stacked DHDAP pairs and one ZnPc, and actually the observed DPV of 1Zn was well simulated by overlapping the oxidation waves of the four co-facially stacked DHDAP pairs like in 2 and the two one-electron oxidations of ZnPc at ∼0 V and ∼0.8 V (Fig. 8c). This result indicates that the electronic interaction among each co-facially stacked DHDAP pair in 1Zn is small. The voltammograms of 1Zn2 in CH2Cl2, on the other hand, show more redox couples than 1Zn (Fig. 8b). The DPV of 1Zn2 clearly showed the splitting of the first and second groups of the redox couples (Fig. 8d), suggesting the existence of through-space electronic communication among DHDAP pairs belonging to different monomers. Furthermore, the first oxidation potential showed a cathodic shift of 0.12 V, indicating the increased electron donating ability of 1Zn2 by forming the interdigitated dimer (Table 1).
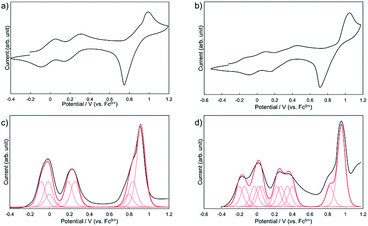 |
| | Fig. 8 Cyclic voltammograms of (a) 1Zn and (b) 1Zn2 and differential pulse voltammograms of (c) 1Zn and (d) 1Zn2 measured in CH2Cl2 containing 0.1 M [nBu4N][BF4] at 298 K (scan rate 100 m V s−1). | |
Table 1 Oxidation potentials (V vs. ferrocene/ferrocenium) of 1Zn, 1Zn2, and 2 determined by the digital simulations of DPV in CH2Cl2 (0.1 M [nBu4N][BF4])
|
|
E
1
|
E
2
|
E
3
|
E
4
|
E
5
|
E
6
|
E
7
|
E
8
|
E
9
|
E
10
|
E
11
|
|
1Zn
|
−0.08 (2e) |
−0.01 (2e) |
0.00 (1e) |
0.19 (2e) |
0.26 (2e) |
0.80 (1e) |
0.84 (2e) |
0.92 (6e) |
|
|
|
|
1Zn2
|
−0.20 (2e) |
−0.14 (2e) |
−0.03 (2e) |
0.02 (2e) |
0.05 (2e) |
0.23 (2e) |
0.27 (2e) |
0.35 (2e) |
0.40 (2e) |
0.83 (2e) |
0.96 (8e) |
|
2
|
0.10 (1e) |
0.28 (1e) |
0.82 (1e) |
0.84 (1e) |
|
|
|
|
|
|
|
Conclusions
We have prepared novel phthalocyanine (Pc) derivatives substituted with eight pillar-like DHDAP units on the periphery to maximize van der Waals interaction points and form selectively the H-type dimers of co-facial Pc rings. The dimers exhibited exceptional stability to be isolated from their monomeric forms in gel permeation chromatography protocols; even no dissociation was observed during the ionization process of MALDI-TOF-MS measurements. Both the monomers and dimers were well kinetically stabilized in solvents such as THF and ethyl acetate at room temperature and showed no temporal change over several days. In contrast, the dissociation of the dimers was observed in solvents with a high affinity for larger π-conjugated molecules such as o-dichlorobenzene. The metastable monomers were converted to the dimers by heating in solvents with less affinity for larger π-conjugated molecules such as ethyl acetate or 1-pentanol with a high activation energy of 134 kJ mol−1 in the former. The findings of this work will provide new molecular designs of supramolecular complexes and aggregates of π-conjugated molecules with an extremely strong bimolecular binding energy competitive to the chemical bonds by multi-point van der Waals interactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to Ms. Karin Nishimura for MS analyses and Ms. Eriko Kusaka for NMR analyses. This work was supported by a Grant-in-Aid for Young Scientists (A) (17H04874) and a Grant-in-Aid for Scientific Research (A) (18H03918) from the Japan Society for the Promotion of Science (JSPS) and a Grant-in-Aid for Scientific Research on Innovative Areas (“π-System Figuration” Area, 26102011). The theoretical calculations were performed at the Research Center for Computational Science, Okazaki, Japan.
Notes and references
-
Phthalocyanines: Properties and Applications, ed. C. C. Leznoff, and A. B. P. Lever, VCH, Weinheim, Germany, 1989, vol. 1–4 Search PubMed.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- A. R. Monahan, J. A. Brado and A. F. DeLuca, J. Phys. Chem., 1972, 76, 446–449 CrossRef CAS.
- A. W. Snow and N. L. Jarvis, J. Am. Chem. Soc., 1984, 106, 4706–4711 CrossRef CAS.
- W. J. Schutte, M. Sluyters-Rehbach and J. H. Sluyters, J. Phys. Chem., 1993, 97, 6069–6073 CrossRef CAS.
- X.-F. Zhang, Q. Xi and J. Zhao, J. Mater. Chem., 2010, 20, 6726–6733 RSC.
- N. Kobayashi, Coord. Chem. Rev., 2002, 227, 129–152 CrossRef CAS.
- C. C. Leznoff, S. Greenberg, S. M. Marcuccio, P. C. Minor, P. Seymour, A. B. P. Lever and K. B. Tomer, Inorg. Chim. Acta, 1984, 89, L35–L38 CrossRef CAS.
- C. C. Leznoff, S. M. Marcuccio, S. Greenberg, A. B. P. Lever and K. B. Tomer, Can. J. Chem., 1985, 63, 623–631 CrossRef CAS.
- S. M. Marcuccio, P. I. Svirskaya, S. Greenberg, A. B. P. Lever, C. C. Leznoff and K. B. Tomer, Can. J. Chem., 1985, 63, 3057–3069 CrossRef CAS.
- E. S. Dodsworth, A. B. P. Lever, P. Seymour and C. C. Leznoff, J. Phys. Chem., 1985, 89, 5698–5705 CrossRef CAS.
- C. C. Leznoff, H. Lam, S. M. Marcuccio, W. A. Nevin, P. Janda, N. Kobayashi and A. B. P. Lever, J. Chem. Soc., Chem. Commun., 1987, 699–701 RSC.
- N. Kobayashi, H. Lam, W. A. Nevin, P. Janda, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879–890 CrossRef CAS.
- N. Kobayashi, T. Fukuda and D. Leliévre, Inorg. Chem., 2000, 39, 3632–3637 CrossRef CAS.
- C. C. Leznoff, H. Lam, W. A. Nevin, N. Kobayashi, P. Janda and A. B. P. Lever, Angew. Chem., Int. Ed., 1987, 26, 1021–1023 CrossRef.
- Z. Odabaş, A. Altındal, A. R. Özkaya, M. Bulut, B. Salih and Ö. Bekaroğlu, Polyhedron, 2007, 26, 695–707 CrossRef.
- Y. Asano, A. Muranaka, A. Fukusawa, T. Hatano, M. Uchiyama and N. Kobayashi, J. Am. Chem. Soc., 2007, 129, 4516–4517 CrossRef CAS.
- A. R. Koray, V. Ahsen and Ö. Bekaroğlu, J. Chem. Soc., Chem. Commun., 1986, 932–933 RSC.
- N. Kobayashi and Y. Nishiyama, J. Chem. Soc., Chem. Commun., 1986, 1462–1463 RSC.
- R. Hendriks, O. E. Sielcken, L. A. Van de Kuil, W. Drenth and R. J. M. Nolte, J. Chem. Soc., Chem. Commun., 1986, 1464–1465 RSC.
- N. Kobayashi and A. B. P. Lever, J. Am. Chem. Soc., 1987, 109, 7433–7441 CrossRef CAS.
- T. F. Baumann, A. G. M. Barrett and B. M. Hoffman, Inorg. Chem., 1997, 36, 5661–5665 CrossRef CAS.
- S. J. Lange, H. Nie, C. L. Stern, A. G. M. Barrett and B. M. Hoffman, Inorg. Chem., 1998, 37, 6435–6443 CrossRef CAS.
- N. Kobayashi, A. Muranaka and V. N. Nemykin, Tetrahedron Lett., 2001, 42, 913–915 CrossRef CAS.
- K. Kameyama, M. Morisue, A. Satake and Y. Kobuke, Angew. Chem., Int. Ed., 2005, 44, 4763–4766 CrossRef CAS.
- M. V. Martínez-Díaz, M. S. Rodrígues-Morga, M. C. Feiters, P. J. van Kan, R. J. M. Nolte, J. F. Stoddart and T. Torres, Org. Lett., 2000, 2, 1057–1060 CrossRef.
- Y. Yamada, M. Okamoto, K. Furukawa, T. Kato and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 709–713 CrossRef CAS.
- M. Morisue, S. Ueda, M. Kurasawa, M. Naito and Y. Kurida, J. Phys. Chem. A, 2012, 116, 5139–5144 CrossRef CAS.
- C. A. Hunter and J. K. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- G. Bottari, O. Trukhina, A. Kahnt, M. Frunzi, Y. Murata, A. Rodríguez-Fortea, J. M. Poblet, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2016, 55, 11020–11025 CrossRef CAS.
- N. Kobayashi, Y. Kobayashi and T. Osa, J. Am. Chem. Soc., 1993, 115, 10994–10995 CrossRef CAS.
- K. Wang, D. Qi, H. Wang, W. Cao, W. Li and J. Jiang, Chem.–Eur. J., 2012, 18, 15948–15952 CrossRef CAS.
- J. Wu, M. D. Watson and K. Müllen, Angew. Chem., Int. Ed., 2003, 42, 5329–5333 CrossRef CAS.
- J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kastler, C. Fechtenkötter, M. Wagner and K. Müllen, J. Am. Chem. Soc., 2004, 126, 11311–11321 CrossRef CAS.
- C. Shao, M. Stolte and F. Würthner, Angew. Chem., Int. Ed., 2013, 52, 7482–7486 CrossRef CAS.
- D. Sakamaki, H. Saeki and S. Seki, Mater. Chem. Front., 2018, 2, 530–536 RSC.
- K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- C. M. Guzy, J. B. Raynor and M. C. R. Symons, J. Chem. Soc. A, 1969, 2299–2303 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01739a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Shu
Seki
b and
Shu
Seki