
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllosteric inhibition of α-thrombin enzymatic activity with ultrasmall gold nanoparticles†
André L.
Lira
a,
Rodrigo S.
Ferreira
a,
Ricardo J. S.
Torquato
a,
Maria Luiza V.
Oliva
a,
Peter
Schuck
 b and
Alioscka A.
Sousa
*a
b and
Alioscka A.
Sousa
*a
aDepartment of Biochemistry, Federal University of São Paulo, São Paulo, SP, Brazil. E-mail: alioscka.sousa@unifesp.br
bNational Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, MD, USA
First published on 24th September 2018
Abstract
The catalytic activity of enzymes can be regulated by interactions with synthetic nanoparticles (NPs) in a number of ways. To date, however, the potential use of NPs as allosteric effectors has not been investigated in detail. Importantly, targeting allosteric (distal) sites on the enzyme surface could afford unique ways to modulate the activity, allowing for either enzyme activation, partial or full inhibition. Using p-mercaptobenzoic acid-coated ultrasmall gold NPs (AuMBA) and human α-thrombin as a model system, here we experimentally tested the hypothesis that enzyme activity could be regulated through ultrasmall NP interactions at allosteric sites. We show that AuMBA interacted selectively and reversibly around two positively charged regions of the thrombin surface (exosites 1 and 2) and away from the active site. NP complexation at the exosites transmitted long-range structural changes over to the active site, altering both substrate binding affinity and catalysis. Significantly, thrombin activity was partially reduced – but not completely inhibited – by interactions with AuMBA. These findings indicate that interactions of proteins with ultrasmall NPs may mimic a typical biomolecular complexation event, and suggest the prospect of using ultrasmall particles as synthetic receptors to allosterically regulate protein function.
1. Introduction
Enzyme activity is tightly controlled in living systems by the action of endogenous regulators such as ions, small molecules and proteins, for example, to maintain homeostasis or to switch signaling pathways on or off. Conversely, failure to keep activity at the required level can lead to several diseases and disorders. Enzyme activity can be also controlled with suitable exogenous effectors, thus providing for a powerful means to modulate key biochemical reactions and their associated biological processes.1Most fundamentally, enzymes can be inhibited by blocking the active site with small-molecule drugs. This traditional mode of orthosteric inhibition is however limited in that, once bound to the active site, the drug effector completely abrogates enzyme activity. In contrast, targeting allosteric sites on the enzyme surface affords the unique opportunity to modulate the activity, allowing for either enzyme activation, partial or full inhibition.1–3
Catalytic and allosteric sites may differ significantly in amino acid composition and overall topology; e.g., the former consists typically of a deep cleft whereas the latter is generally shallow. Hence, it can be challenging to develop allosteric effector molecules according to the traditional drug discovery paradigm.4 This creates an opportunity to consider new allosteric modulators (other than the prototypical small-molecule drug) and study their novel modes of action.
Nanomaterials are an emerging class of enzyme regulators.5–24 They can be designed with tailored nanoscale sizes and be grafted with virtually any combination of organic surface moieties, thus being attractive as artificial receptors for protein surface recognition.25–28 As enzyme inhibitors, nanomaterials can bind around the active site of some enzymes to block access of substrate, a mechanism reminiscent of competitive inhibition by small-molecule probes.17 Full enzyme inactivation by nanomaterials can also originate from large changes in protein structure following adsorption.18,19 On the other hand, the use of nanomaterials as biomimetic allosteric modulators is a more challenging notion to realize: they must bind the enzyme reversibly at an allosteric site while eliciting the proper kind of conformational and/or dynamic changes at the ligand binding site. Recently, computer simulations have suggested that small gold particles (4 nm) may indeed be able to exert “subtle” allosteric long-range effects on protein structure or flexibility, while the overall protein would remain in a native folded structure.29
In this work we demonstrate the concept of monolayer-protected ultrasmall gold nanoparticles (NPs) as allosteric modulators of enzyme activity. As a model system, we employed α-thrombin as the enzyme and negatively charged p-mercaptobenzoic acid-coated gold NPs (AuMBA) of 2 nm in core diameter as the nanoscale effector. Unlike other types of nanostructures, ultrasmall particles decorated with suitable surface moieties can resemble protein receptors in both size and surface chemistry. Thrombin, a key serine protease of the blood coagulation cascade, contains two positively-charged domains (exosites 1 and 2) situated on opposite ends of the molecule.30,31 Both exosites bind several cofactors and substrates, including fibrinogen and thrombomodulin in exosite 1, and heparin and the platelet receptor GPIba in exosite 2.31 Thrombin is characterized as a highly plastic enzyme: it is known to exist as an ensemble of conformations in the fully unliganded state; and its structure and catalytic activity can be allosterically modulated by association with a diverse set of endogenous and exogenous ligands.32
Through a series of biophysical and biochemical measurements, we show that negatively charged ultrasmall AuMBA particles bind selectively and reversibly to both positively charged exosites of thrombin to allosterically disturb – without fully inhibiting – its enzymatic activity. To our knowledge, a sub-maximal inhibitory response has not been described before in the interactions of synthetic NPs with enzymes.
2. Materials and methods
2.1. Materials and reagents
Human α-thrombin, γ-thrombin and FITC-PPACK were from Molecular Innovations (Novi, MI). Biotinylated thrombin was from Haematologic Technologies (Essex Junction, VT). The substrate S-2238 was obtained from Diapharma (West Chester, OH). The aptamers HD1 and HD22 were synthesized by Exxtend (Campinas, SP) and hirudin was from Anaspec (Fremont, CA). Streptavidin-coupled magnetic beads were from Thermo Scientific (Waltham, MA). The peptide ECGK–biotin was synthesized by peptide 2.0 (Chantilly, VA). Sensor chip SA with pre-immobilized streptavidin was from GE Healthcare Life Sciences (Piscataway, NJ). The remaining reagents were from Sigma-Aldrich (Milwaukee, WI). Thrombin was labeled stoichiometrically with fluorescein (FITC) for subsequent use in fluorescence experiments. Site-selective labeling of thrombin was achieved through a FITC probe conjugated to the potent active-site inhibitor PPACK.33 Labeling was accomplished by mixing a 10-fold molar excess of FITC-PPACK with thrombin for 30 min. The conjugated protein was separated from excess FITC by gel filtration in a Sephadex-G75 column. The AuMBA nanoparticles were prepared and characterized as described in detail in several previous reports.34–372.2. Fluorescence and circular dichroism (CD) spectroscopy
The assays were performed in 20 mM phosphate buffer solution (pH 7.2) supplemented with 150 mM NaCl and 0.1% polyethylene glycol (PEG) 8000. Fluorescence measurements were performed on a Shimadzu spectrofluorimeter model RF-6000 at 25 °C. Native thrombin was loaded into a quartz cuvette at a concentration of 0.1 μM and titrated with AuMBA. Fluorescence spectra were acquired using an excitation wavelength of 280 nm and 5 nm bandpass. Fluorescence quenching was measured from the intensities of the emission spectra and plotted as a function of NP concentration. Fluorescence quenching of FITC-labeled thrombin was performed in a similar manner but with a protein concentration of 0.05 μM and excitation wavelength of 495 nm. The inner-filter effect from AuMBA was accounted for by titrating a solution of the amino acid tryptophan (or the FITC probe) with identical concentrations of NPs. Corrected quenching curves for native thrombin (or FITC-labeled thrombin) were generated by dividing the uncorrected data by the tryptophan (or FITC) reference curve.38 The effect of ionic strength on AuMBA–thrombin complexation was studied by recording the increase in fluorescence intensity of thrombin pre-mixed with AuMBA (0.1 μM for both) as the NaCl concentration was gradually increased from 0 to 1 M. For the evaluation of structural changes on thrombin due to NP complexation, Trp emission spectra were obtained in the absence and presence of AuMBA; the excitation wavelength was 295 nm. CD measurements were carried out on a Chirascan Plus instrument (Applied Photophysics, Leatherhead, UK); thrombin and NPs were used at concentrations of 3 and 0.5 μM, respectively.2.3. Fluorescence spectroscopy studies in the presence of competing exosite-directed ligands
The assays were performed in 20 mM phosphate buffer solution (pH 7.2) supplemented with 150 mM NaCl and 0.1% PEG 8000. 0.1 μM of native thrombin and 0.05 μM of FITC-labeled thrombin were equilibrated with HD1 (15 μM), hirudin (15 μM) and HD22 (5 μM) and titrated with AuMBA. The different sample combinations included thrombin + HD1, thrombin + HD22, thrombin + HD1 + HD22 and thrombin + hirudin + HD22. The remaining procedure was performed in a similar manner as described above (c.f. Section 2.2.). In another experiment, 0.1 μM of native thrombin was equilibrated with 0.1 μM AuMBA and titrated with HD1 followed by HD22 (also with HD22 followed by HD1). The Trp emission signal was recorded and plotted as a function of aptamer concentration. The inner-filter effect from the aptamers was accounted for by titrating thrombin with identical concentrations of aptamers and recording the intensities. Corrected quenching curves were generated by dividing the uncorrected data by the reference curves.2.4. Pull-down experiments with magnetic beads
AuMBA was biotinylated by ligand exchange with ECGK–biotin as previously described and immobilized onto streptavidin-coupled magnetic beads.39 The concentration of immobilized particles (∼2 μM) was calculated by measuring the concentration of free AuMBA in solution before and after addition of the beads. A control experiment using non-derivatized AuMBA showed the particles did not stick to the beads, as expected. In one experiment, 20 nM of thrombin was incubated with immobilized AuMBA in buffer solution (Tri–HCl, 20 mM to 1 M NaCl, 0.1% PEG 8000, pH 7.2). After separating the beads with a magnet, the presence of unbound thrombin at each NaCl concentration was evaluated by recording the initial velocity of substrate cleavage (v0). The values of v0 were normalized relative to thrombin in solution without beads. In another experiment, 20 nM of thrombin was incubated with immobilized AuMBA in buffer solution (Tris–HCl, 150 mM NaCl, 0.1% PEG 8000, pH 7.2) in the absence and presence of HD1 (20 μM), HD22 (6 μM) and HD1 + HD22. The remaining procedure was performed similarly as described above.2.5. Surface plasmon resonance (SPR)
Experiments were carried out in a Biacore T-200 SPR instrument (GE Healthcare Life Sciences). 170 response units (RU) of biotinylated thrombin were immobilized onto a commercial streptavidin surface. Phosphate buffer supplemented with 150 mM NaCl was used as running buffer. AuMBA was injected in the flow in the concentration range from 1 nM to 20 μM. The association and dissociation phases were recorded for 200 and 700 s, respectively, and the flow rate was 90 μL min−1. Regeneration of the sensor surface between injections was completed with 0.005% sodium dodecyl sulfate in water followed by a 2 M solution of NaCl in water; for both solutions the injection time was 30 s and the flow rate 30 μL min−1. Correction of bulk refractive index changes was performed by subtracting the responses from a reference surface from the raw SPR traces. Data analysis was carried out representing the surface sites as a continuous distribution of equilibrium and dissociation rate constants. This model was globally fit to the experimental data using the software EVILFIT.40,412.6. Analytical ultracentrifugation (AUC)
Sedimentation velocity experiments were carried out in an Optima XL-I analytical ultracentrifuge equipped with GUI version 5.7 and firmware version 5.06 (Beckman Coulter, Indianapolis). Samples of AuMBA in the presence of thrombin and aptamers were prepared in buffer solution and analyzed at 20 °C (20 mM phosphate, 150 mM NaCl, 0.1% PEG 8000, pH 7.2). AuMBA, thrombin, HD1 and HD22 were used at the concentrations of 1 μM, 5 μM, 75 μM and 20 μM, respectively. After acceleration to 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, absorbance scans at 520 nm were acquired in ∼6 min intervals for 4 h. Data were analyzed in the software SEDFIT with a c(s) sedimentation coefficient distribution.42
000 rpm, absorbance scans at 520 nm were acquired in ∼6 min intervals for 4 h. Data were analyzed in the software SEDFIT with a c(s) sedimentation coefficient distribution.42
2.7. Direct enzyme inhibition
The assays were performed in a Flexstation III from Molecular Devices (Sunnyvale, CA) at 25 °C. 2 μL from a series of AuMBA stocks were added to 183 μL of thrombin diluted in buffer solution (Tris–HCl, 100 mM NaCl, 0.1% PEG 8000, pH 7.2) in a 96-well plate. Next, 5 μL of the chromogenic substrate S-2238 was added to each well simultaneously to give a final volume of 190 μL and concentrations of 2 nM, 100 μM and 0–2 μM for thrombin, S-2238 and AuMBA, respectively. Control samples containing only AuMBA and substrate were prepared in a similar manner and used for background subtraction. Progress curves (absorbance readings over time) were recorded at 10 s intervals over 15–30 min, and enzyme activities (v0) were measured from the slope of the initial (linear) part of the curves.43 Fractional activities were calculated by dividing the v0 obtained at each AuMBA concentration by the v0 in the absence of particles. For the experiments in the presence of HD1 + HD22 and hirudin + HD22, the enzyme, NPs and competing ligands were pre-incubated in the wells before addition of substrate; HD1, hirudin and HD22 were used at the concentrations of 15, 15 and 5 μM, respectively.2.8. Michaelis–Menten kinetics analysis
2 μL from a series of AuMBA stocks were added to 183 μL of thrombin diluted in buffer solution (Tris–HCl, 100 mM NaCl, 0.1% PEG 8000, pH 7.2) in a 96-well plate. Next, 5 μL from a series of S-2238 stocks were added to each well simultaneously to give a final volume of 190 μL and concentrations of 2 nM, 5-100 μM and 0–0.1 μM for thrombin, S-2238 and AuMBA, respectively. The remaining procedure was carried out as described above. For each NP concentration, enzyme activities (v0) were measured from the slope of the initial part of the progress curves and plotted as a function of substrate concentration, thus yielding a series of Michaelis–Menten plots (v0vs. [S-2238]). Thrombin inhibition by AuMBA was evaluated within the framework of the general modifier mechanism (Fig. 9) and the corresponding velocity equation:44,45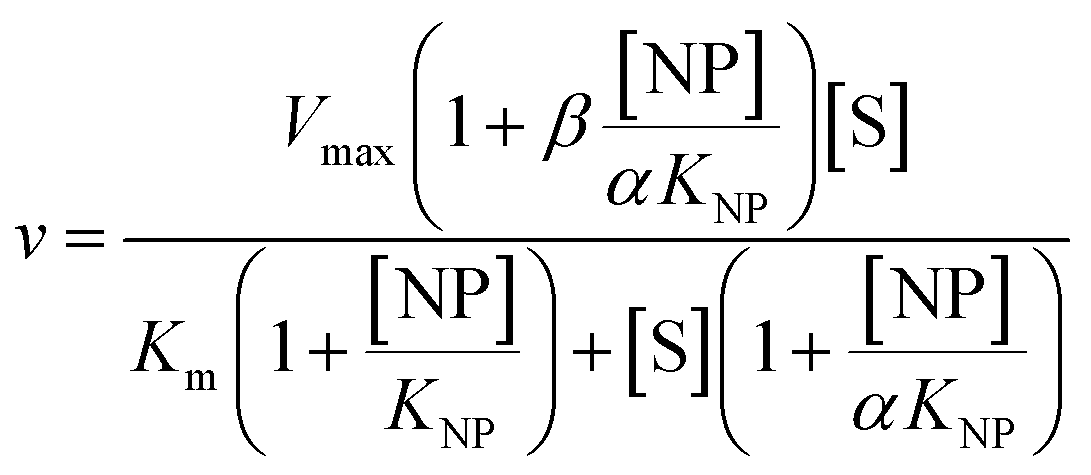 | (1) |
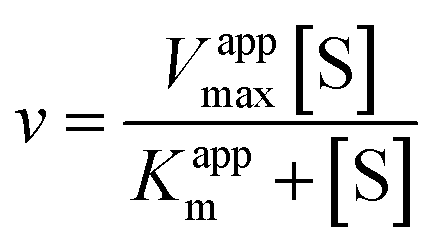 | (2) |
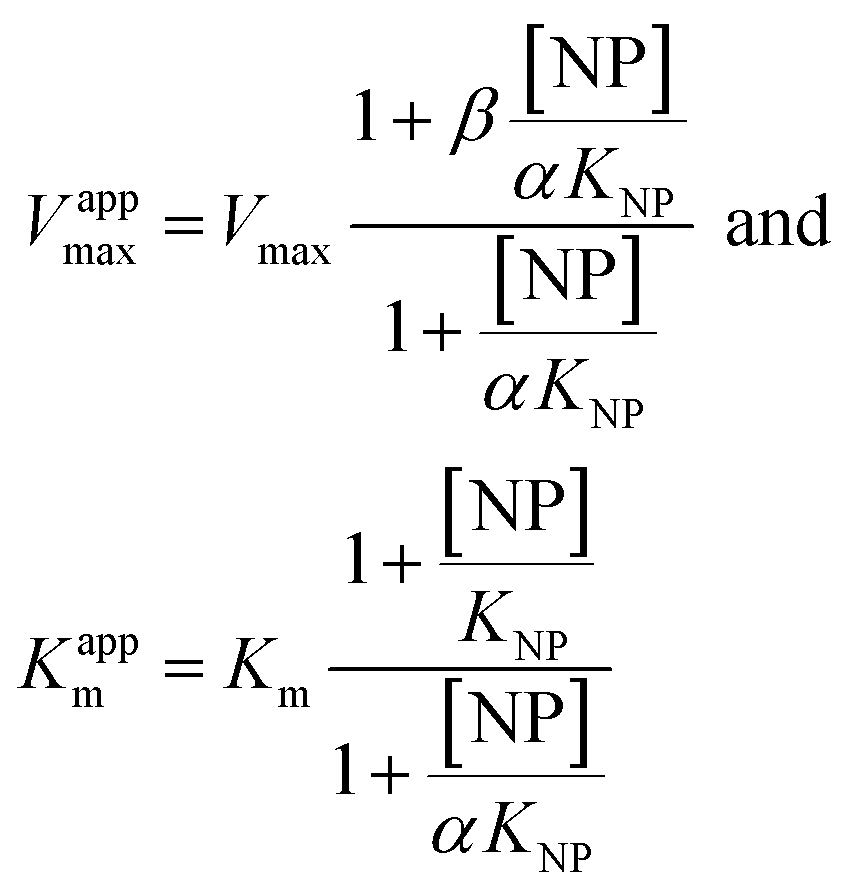 | (3) |
Eqn (2) was fitted to each of the Michaelis–Menten plots (v0vs. [S-2238]) to find the apparent Km and Vmax (Kappm and Vappmax) as a function of AuMBA concentration. For comparison, Kappmand Vappmax were also found by the direct linear plot method (see Fig. S10† for details).46 The dependence of Kappm and Vappmax on AuMBA concentration was then used to estimate the parameters α and β by fitting of eqn (3). Alternatively, the set of Michaelis–Menten plots was analyzed by global fitting of the velocity equation (eqn (1)).44 In eqn (1), [NP] was set to “number of independent binding sites” by multiplying the known particle concentration by 5 (the assumption of ∼5 binding sites/AuMBA was based on a previous estimate obtained by fluorescence quenching of ∼6 chymotrypsin molecules bound per AuMBA at saturation).38
3. Results and discussion
3.1. AuMBA and thrombin
The synthesis and physicochemical properties of ultrasmall AuMBA have been characterized in detail in previous publications.34–37 Briefly, the NPs are highly uniform with a core diameter around 2.0–2.5 nm (Fig. S1†). The NPs are negatively charged, with a zeta potential of −22 ± 0.5 mV at pH 7. Human α-thrombin is a roughly spherical protein (d ∼5 nm) with molecular mass of 37 kDa.47 Although thrombin has a nearly neutral isoelectric point, the surface charge distribution is highly anisotropic at pH 7.32,47 As shown by electrostatic surface potential calculations (performed with APBS48), the catalytic site is situated around an acidic surface patch, which is flanked by two highly positively charged domains denominated exosites 1 and 2 (Fig. 1). A schematic representation of AuMBA bound to thrombin's exosites is also depicted in Fig. 1.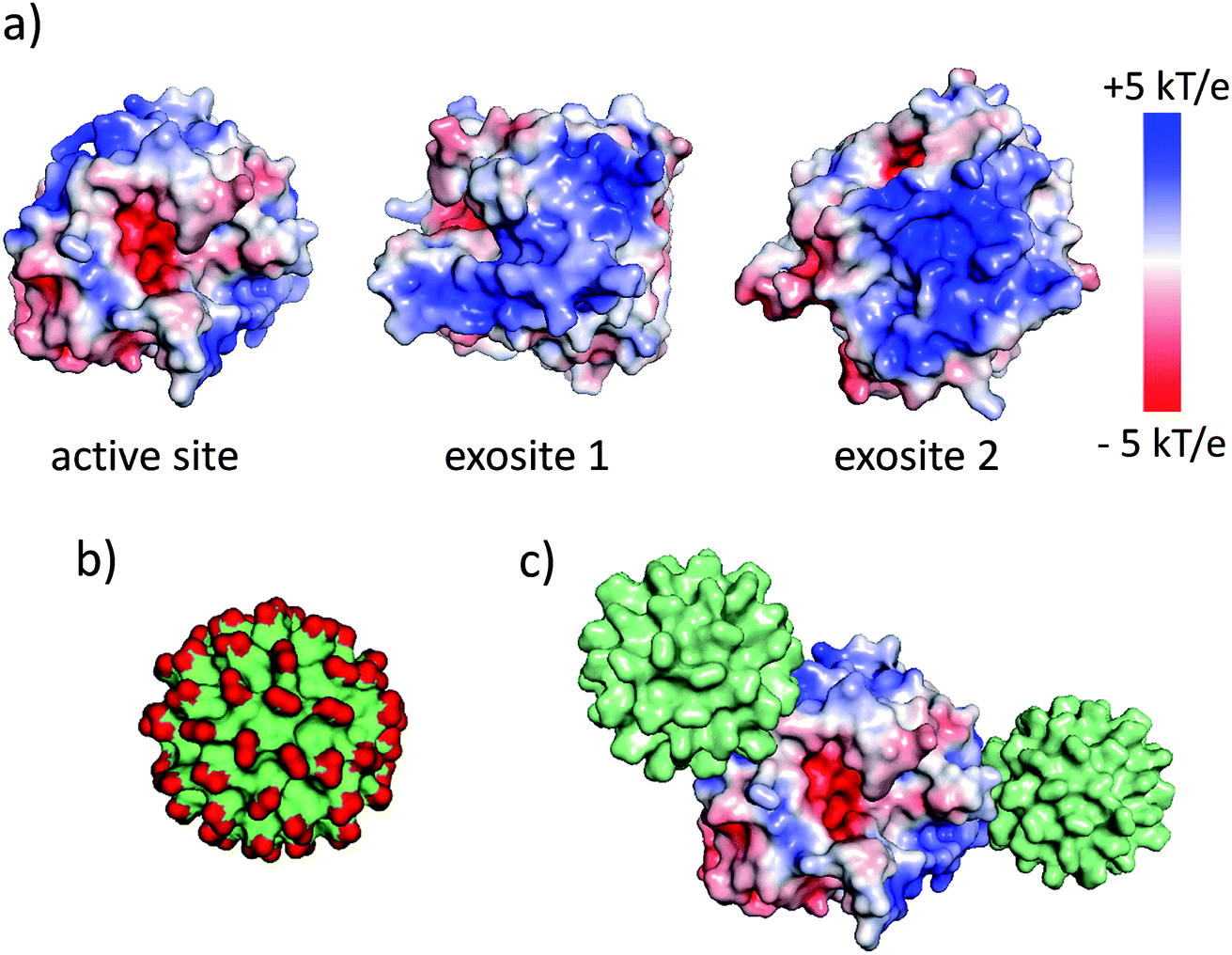 | ||
| Fig. 1 AuMBA and thrombin. (a) Electrostatic surface potential of α-thrombin (1PPB). (b) Molecular surface representation of AuMBA (red: –CO2− groups). (c) Schematic representation of AuMBA particles bound to thrombin's exosites. | ||
3.2. AuMBA–thrombin interactions
The AuMBA–thrombin interactions were first investigated by means of a separation experiment. Biotinylated AuMBA was immobilized onto streptavidin-coated magnetic beads and mixed with thrombin under different NaCl concentrations. After separating the beads from solution with a magnet, the amount of unbound thrombin left in the supernatant was measured by recording the initial velocity of substrate cleavage. Consistent with electrostatic-controlled complexation, the extent of binding depended strongly on solution ionic strength, going from complete to no binding as the NaCl concentration was increased from 20 mM to 1 M (Fig. S2†).In order to study this in more detail, we carried out binding experiments by fluorescence titration quenching.38 The native fluorescence of 0.1 μM thrombin was completely quenched with the addition of approximately 0.1 μM NPs (Fig. 2a). This clearly demonstrates strong binding of AuMBA to thrombin. Similar results were obtained by monitoring the decay of FITC fluorescence in active site-labeled FITC-thrombin (Fig. S3†). Next, we examined the salt dependence of this interaction. To this end, thrombin and AuMBA were first pre-incubated in buffer solution without NaCl, resulting in almost complete quenching of the native fluorescence signal. Increasing the NaCl concentrations led to progressively higher fluorescence readings, again consistent with electrostatic interactions between AuMBA and thrombin (Fig. 2b).
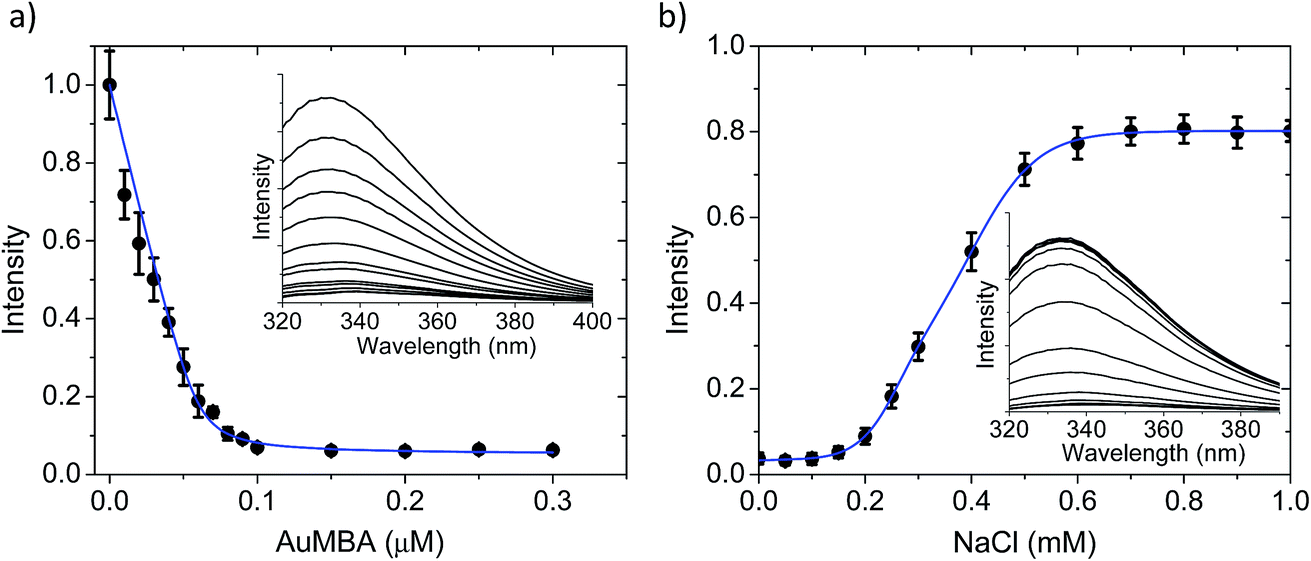 | ||
| Fig. 2 Fluorescence spectroscopy characterization of AuMBA–thrombin interactions. (a) Fluorescence titration quenching of native thrombin with AuMBA in phosphate buffer solution supplemented with 150 mM NaCl. Thrombin concentration, 0.1 μM. Inset: raw fluorescence spectra; the AuMBA concentration increases from top to bottom. (b) Dependence of fluorescence quenching on solution ionic strength. Intensities were normalized relative to the fluorescence signal recorded without NPs. Inset: raw fluorescence spectra; the NaCl concentration increases from bottom to top. Thrombin and AuMBA concentrations, 0.1 μM. Blue lines are a guide to the eye. | ||
A quantitative analysis of the fluorescence quench data in Fig. 2a is precluded by thrombin/particle aggregation (see below), in which case the observed fluorescence signal is not proportional to the degree of binding. AuMBA–thrombin binding affinity was therefore measured by surface plasmon resonance (SPR) by flowing AuMBA over a streptavidin sensor surface containing immobilized thrombin. Importantly, the spatial constraints of the surface immobilization in SPR hinder aggregation and thereby allow the measurement of binding affinity.49 Immobilization was accomplished through a biotin–PPACK label attached to thrombin's active site; therefore, the protein was uniformly oriented on the sensor surface with both exosites presumably exposed to allow interactions with AuMBA. The AuMBA–thrombin interactions were analyzed with a continuous surface-site distribution model,40,41,49,50 which can allow the distinction between mono- and multi-valently attached AuMBA populations based on their different dissociation kinetics. It can also naturally accommodate multiple binding sites on the immobilized thrombin. The calculated affinity and rate constant distributions showed a single major peak whose integrated value yielded a binding affinity in the low nM range (KD ∼40 nM), which is in the range expected from the fluorescence quench experiments (Fig. 3). The single value of KD found by SPR suggested that AuMBA binds with similar affinity to both exosites (c.f. Section 3.3.). The corresponding association and dissociation rate constants were 1.9 × 106 M−1 s−1 and 0.082 s−1, respectively. Additional details about the SPR characterization of AuMBA–thrombin interactions and the significance of the kinetic rate constants will be discussed in a subsequent publication.
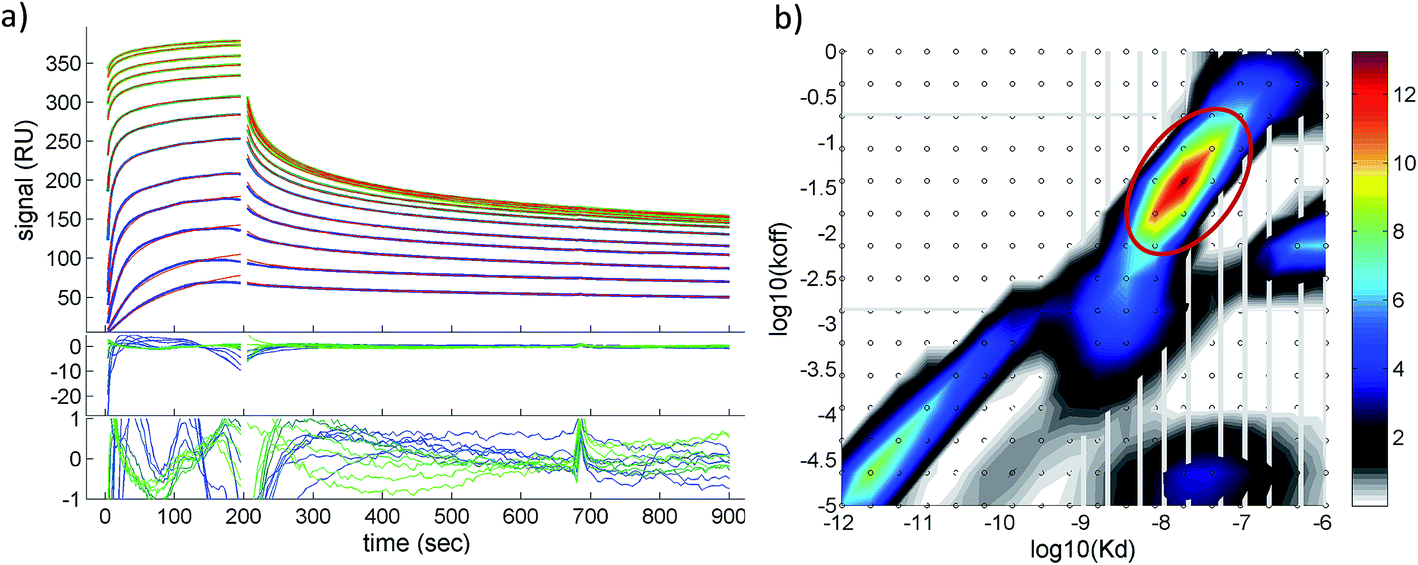 | ||
| Fig. 3 Surface plasmon resonance characterization of AuMBA–thrombin interactions. (a) Shown are the experimental binding traces (green and blue lines), best-fit curves (red lines), and fitting residuals. Assay performed in phosphate buffer solution supplemented with 150 mM NaCl. (b) Calculated affinity and rate constant distributions from corresponding traces shown in (a). Circled region indicates the major peak in the distribution. Integration of the peak provided the binding parameters KD, kon and koff. | ||
3.3. Binding sites on thrombin for AuMBA
It is usually challenging to determine precisely the orientation of bound proteins on NPs. For electrostatically driven interactions, it can be assumed that binding occurs predominantly through protein surface patches of highest electrostatic surface potential.51,52 In some cases, the approximate binding location might be inferred from measurements of hydrodynamic diameter, since these will be influenced by the orientation of the protein on the particle surface.51 Other advanced methods used to determine protein orientation on NPs include enzymatic digestion followed by mass spectrometry and nuclear magnetic resonance, among others.53–57Here, we attempted to establish the main interaction sites on thrombin for AuMBA, which we hypothesized would be situated around the two positively charged exosites. This can be tested by employing exosite-directed ligands in competition experiments with AuMBA. The exosite ligands included the aptamer HD1 and hirudin (54–65) for exosite 1 (KD ∼100 nM and 20 nM, respectively), and the aptamer HD22 for exosite 2 (KD ∼1 nM).58–61 In most experiments the ligands were used in high excess to minimize binding from AuMBA. For example, we calculated that <10% of exosite 1 (from a total thrombin concentration of 0.1 μM) would be occupied by AuMBA (0.1 μM) in the presence of 15 μM of HD1.
First, we used fluorescence titration quenching to characterize the interactions in the presence of the competing ligands. In one experiment, thrombin was pre-mixed with a large excess of aptamers and titrated with AuMBA (Fig. 4a). It can be seen that AuMBA still caused a significant fluorescence signal decrease when a single aptamer type was used as competing ligand, therefore suggesting the particles were still able to bind to the free exosite and quench the protein fluorescence (Fig. 4a, blue and red traces). Adding both aptamers together led to a much smaller signal decay, in agreement with reduced overall AuMBA binding to thrombin (Fig. 4a, green trace). Similar results were obtained using FITC-labeled thrombin (Fig. S4†). In another experiment, thrombin was first pre-mixed with AuMBA followed by titration with aptamers (Fig. 4b and c). Titration of the first aptamer (either HD1 or HD22) caused the fluorescence intensity to increase from zero to approximately 20% of the maximum, i.e., fluorescence emission from the ternary AuMBA–thrombin–aptamer complex remained highly quenched. Addition of the second aptamer increased the signal further up to ∼80%, now consistent with reduced AuMBA–thrombin complex formation. As a control, we also tested AuMBA pre-mixed with γ-thrombin, which does not bind HD1 due to a structurally compromised exosite 1.62 As expected, the fluorescence of γ-thrombin remained unaltered with HD1 titration; signal recovery was observed only with HD22 titration and with the addition of 1 M NaCl (Fig. S5†).
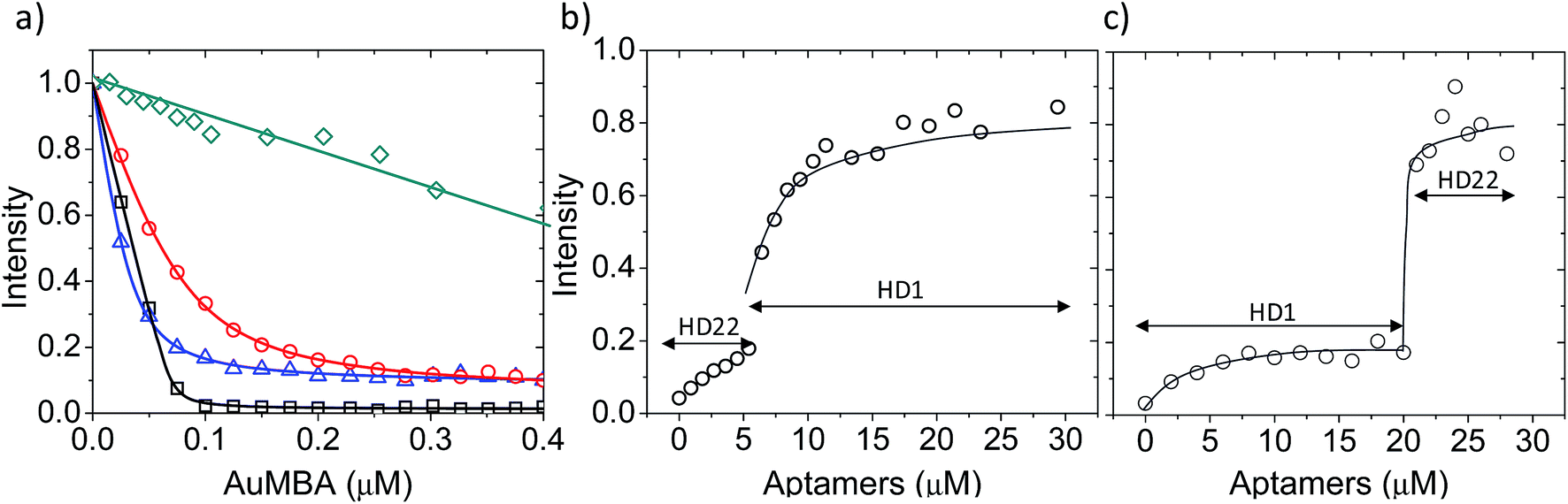 | ||
| Fig. 4 AuMBA–thrombin interactions in the presence of exosite-binding aptamers. (a) Fluorescence titration quenching of native thrombin with AuMBA in the absence (black squares) or presence of excess HD1 (blue triangles), HD22 (red circles), and HD1 + HD22 (green diamonds). Thrombin concentration, 0.1 μM; HD1, 15 μM; HD22, 5 μM. (b) Recovery of thrombin fluorescence by titration of AuMBA-bound thrombin with HD22 followed by HD1. (c) Recovery of thrombin fluorescence by titration of AuMBA-bound thrombin with HD1 followed by HD22. Thrombin and AuMBA concentrations, 0.1 μM. Assays performed in phosphate buffer solution supplemented with 150 mM NaCl. Lines are a guide to the eye. | ||
Next, we carried out quantitative analytical ultracentrifugation (AUC) experiments,35,40,63 monitoring absorbance profiles at 520 nm to exclusively record the sedimentation behavior of AuMBA when co-incubated with thrombin and aptamers. In the presence of thrombin, the AuMBA particles sedimented too rapidly and no absorbance signals were detected around the sedimentation coefficient range of AuMBA. This result indicated the formation of NP microaggregates, in agreement with the notion that a crosslinked network of AuMBA–thrombin complexes forms by mutual polyvalent interactions between thrombin and NPs. Interestingly, the sedimentation coefficient distributions (s) of AuMBA in the presence of thrombin plus either aptamer (Fig. 5, red and blue traces) appeared displaced towards lower values relative to the distributions of free AuMBA (black trace), consistent with an increased hydrodynamic friction experienced by the ternary AuMBA–thrombin–aptamer complex (the lower s-value of the complex is caused by the dissimilar density of the gold particles and the protein). Any crosslinking of two or more AuMBA would have led to a significantly higher s-value. In addition, the areas integrated under the distributions, which correspond to the loading concentration of species in this sedimentation coefficient range, were remarkably similar to one another, ruling out the loss of a significant fraction of material in very large, rapidly-sedimenting aggregates, and providing direct proof that AuMBA was colloidally stable in the samples. Hence, in the presence of a single aptamer type, each thrombin molecule could only bind to a single AuMBA particle through the aptamer-free exosite, and thrombin was unable to bridge NPs and induce their aggregation. The AUC analysis further revealed that the sedimentation velocity of AuMBA in the presence of thrombin plus both the aptamers (Fig. 5, green trace) was only partly lowered relative to pristine AuMBA (black trace), hence revealing incomplete competition but strongly reduced levels of AuMBA–thrombin complex formation.
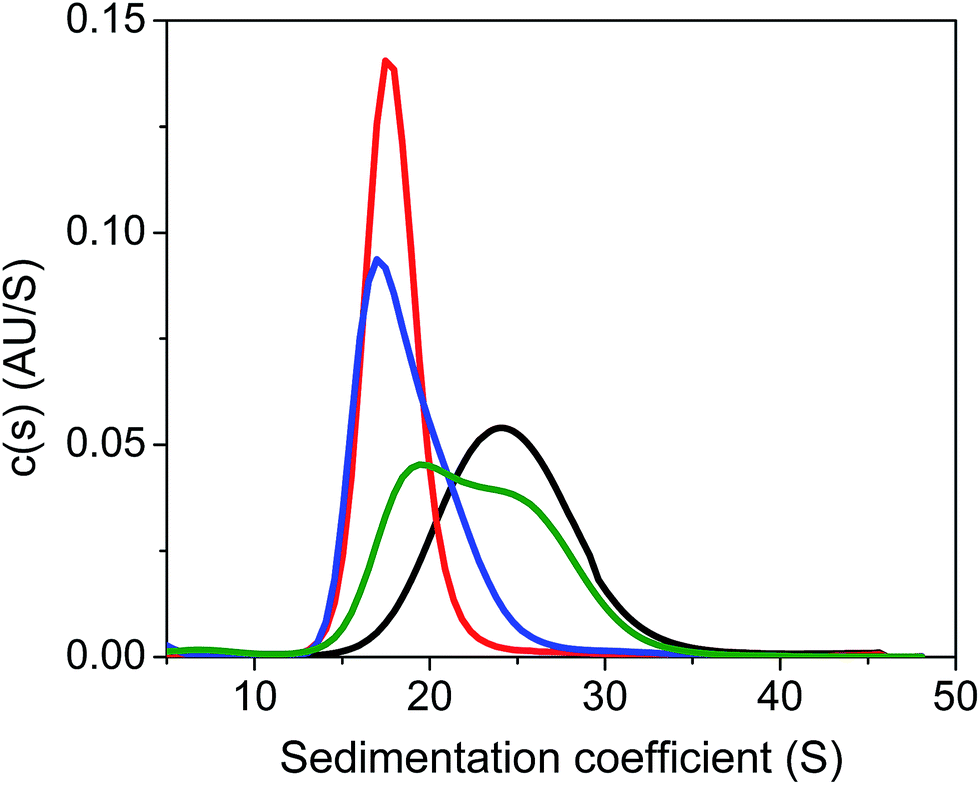 | ||
| Fig. 5 Analytical ultracentrifugation analysis of AuMBA in the presence of thrombin and aptamers. Shown are the sedimentation coefficient distributions for free AuMBA (black) and AuMBA in the presence of thrombin + HD1 (red), thrombin + HD22 (blue), and thrombin + HD1 + HD22 (green). The integrated areas under the distributions fall in the range from 0.54 to 0.58 AU. AuMBA concentration, 1 μM; thrombin, 5 μM; HD1, 75 μM; HD22, 20 μM. Assays performed in phosphate buffer solution supplemented with 150 mM NaCl. | ||
AuMBA–thrombin interactions were finally evaluated using the pull-down method with magnetic beads as described earlier. After allowing thrombin to bind onto immobilized AuMBA and separating the beads with a magnet, the presence of unbound thrombin in the supernatant was verified by measuring the initial velocity of substrate cleavage. In agreement with the above data, reduced levels of complexation were observed in the presence of excess HD1 + HD22 (Fig. S6†).
Collectively, the above results allowed the following conclusions to be drawn: (i) the ligands were able to effectively compete with AuMBA for thrombin, thus providing solid evidence that AuMBA interacts with both exosites 1 and 2. Moreover, it also follows that there are no major (“high affinity”) interaction regions on thrombin for AuMBA other than the two exosites themselves. (ii) Binding of AuMBA to one of the exosites still enabled ligand binding to take place at the free exosite to yield a ternary AuMBA–thrombin-ligand complex. This suggests that AuMBA complexation does not transmit structural changes from one exosite to another that prevent ligand binding. Previous investigations found no support for significant inter-exosite communication in thrombin, at least for some ligands. For instance, it was demonstrated that thrombin can bind HD1 and HD22 simultaneously through both exosites.64 Thrombin was also shown to bind hirudin and the Fbg γ′/GpIbα peptides simultaneously through exosites 1 and 2, respectively.62 On the other hand, several previous reports have described allosteric linkage between exosites. For example, it was reported that HD22 weakens thrombin interactions with γA/γA-fibrin, which takes place exclusively via exosite 1.65 (iii) Our data does not exclude the possibility that AuMBA may bind with “low affinity” (μM range) somewhere onto the protein surface, including onto the exosites themselves alongside the ligands.
3.4. Thrombin structural changes
Fluorescence and circular dichroism (CD) spectroscopy were first used to verify the occurrence of overall structural changes on thrombin upon NP complexation. Fluorescence measurements showed a small red-shift (∼2 nm) in the emission spectrum of Trp (Fig. S7a†). CD spectroscopy also revealed some changes to secondary structure (Fig. S7b†). Qualitatively, AuMBA binding did not induce substantial global structural changes on thrombin.Next, the occurrence of conformational changes near thrombin's active site was investigated by comparing the emission spectra of FITC-labeled thrombin in the absence and presence of AuMBA. The method is based on the principle that structural changes near the active site would affect the chemical environment around the FITC probe and therefore its emission spectral properties.66,67 It is important to note that the FITC molecule is positioned a few angstroms away from the catalytic triad, but still in sufficient proximity to sense changes in the dielectric environment caused by structural rearrangements at the active site.68 Interactions with AuMBA caused a large blue shift in the emission spectrum of FITC-labeled thrombin (reaching ∼13 nm at saturating NP concentrations) (Fig. 6a and c), therefore suggesting that the fluorophore experiences a distinct chemical environment near the orthosteric site after the NPs have bound. AuMBA did not produce significant spectral changes when HD1 + HD22 were introduced in excess as competing ligands, in agreement with lack of NP binding in the presence of both aptamers (Fig. 6b and c).
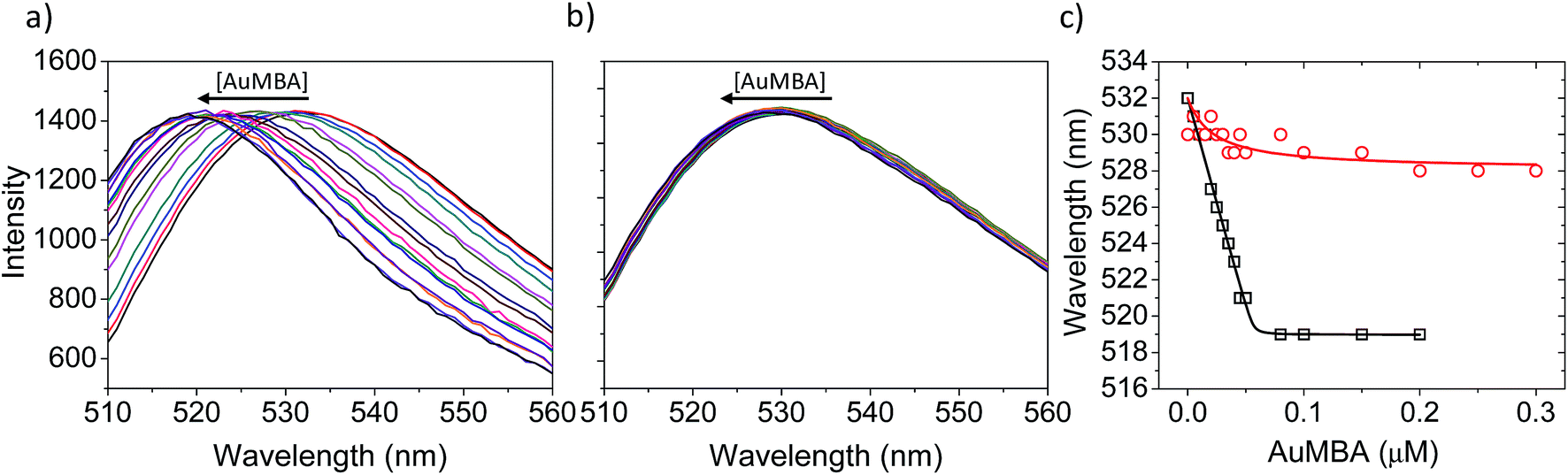 | ||
| Fig. 6 Characterization of structural changes near the active site of thrombin. (a) FITC emission spectra of FITC-labeled thrombin titrated with AuMBA. The spectra were normalized to the same maximum intensity. (b) The same as in (a) but in the presence of excess HD1 + HD22. Thrombin concentration, 0.05 μM; AuMBA, 0–0.3 μM; HD1, 15 μM; HD22, 5 μM. The AuMBA concentrations corresponding to each colored line in (a) and (b) can be read from the abscissa in (c). (c) Wavelengths of maximum emission obtained from the spectra in (a) and (b) (squares and circles, respectively). Assays performed in phosphate buffer solution supplemented with 150 mM NaCl. | ||
It is conceivable that the conformational changes described above could impair ligand binding into the active-site cleft of thrombin. To check this possibility, free and AuMBA-bound thrombin were titrated with the active-site probe PABA (KD ∼70 μM).69 PABA is weakly fluorescent when free in solution, but the fluorescence yield is enhanced significantly when associated with thrombin. The results showed that PABA fluorescence was higher in the presence of AuMBA–thrombin than in control solutions without thrombin (Fig. S8†). It can thus be concluded that, despite structural rearrangements near the active site upon interactions with NPs (Fig. 6), thrombin remained able to bind PABA.
3.5. Enzymatic inhibition by AuMBA
To assess the impact of AuMBA complexation on thrombin activity, initial rates of hydrolysis (v0) of the chromogenic substrate S-2238 were measured as a function of NP concentration. The dose–response profile revealed a rapid decrease in the residual activity of thrombin up to a NP concentration ∼0.1 μM, after which the activity remained constant at ∼40% (Fig. 7). The non-zero activity obtained at high NP concentrations suggested a partial, hyperbolic mechanism of inhibition. As expected, the inhibitory effect of AuMBA on thrombin activity was reduced significantly in the presence of excess HD1 + HD22 or hirudin + HD22 (Fig. 7). To rule out the possibility that the observed inhibition might be due to interactions of individual p-mercaptobenzoic acid (MBA) molecules with thrombin, we measured the kinetics of substrate cleavage in the presence of up to 100 μM of free MBA. The results confirmed that thrombin activity was unaffected by MBA alone (Fig. S9†).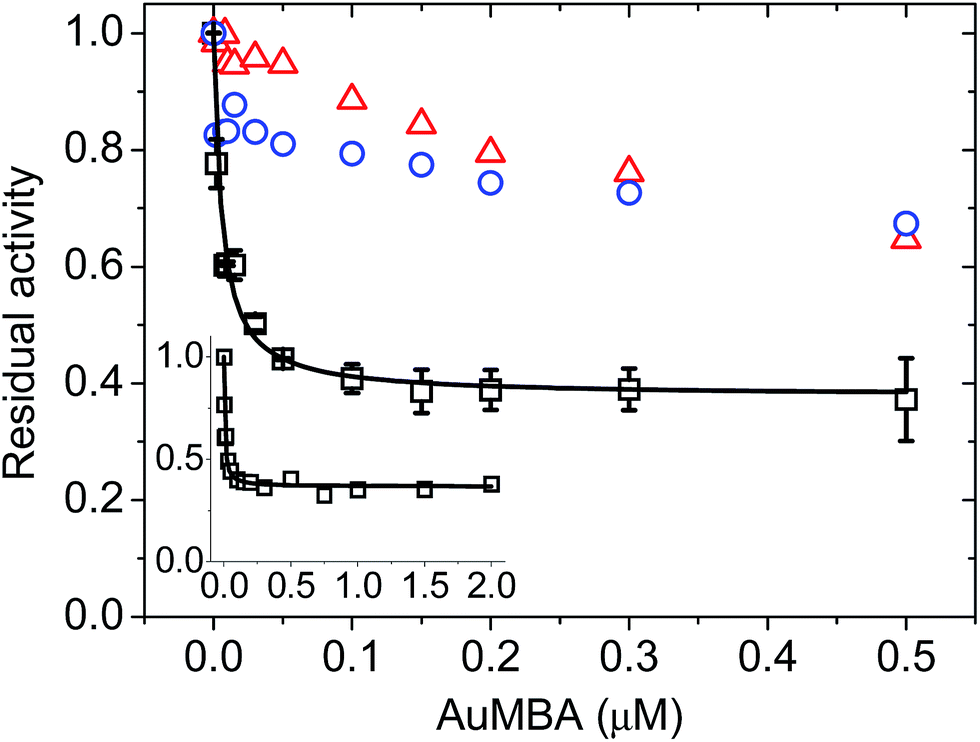 | ||
| Fig. 7 Concentration-dependent inhibition of thrombin by AuMBA. Data obtained in the absence of competing ligands (squares) and in the presence of excess HD1 + HD22 (circles) and hirudin + HD22 (triangles). The inset shows the dose–response profile up to an AuMBA concentration of 2 μM. Thrombin concentration, 2 nM; HD1 and hirudin, 15 μM; HD22, 5 μM; S-2238, 100 μM. Assay performed in Tris–HCl buffer solution supplemented with 100 mM NaCl. Lines are a guide to the eye. | ||
Additional details on the inhibition mechanism could be obtained by varying both the substrate and inhibitor concentrations ([S] and [NP], respectively), thus yielding a series of Michaelis–Menten plots (v0vs. [S]) as displayed in Fig. 8a. These sets of curves were described and analyzed in terms of the general modifier mechanism (Fig. 9).44,45 From Fig. 9, the inhibition mechanism can be described by the pair of values α and β. α determines the effect of AuMBA interactions on substrate-binding affinity, as such: for competitive inhibition, α → ∞; for uncompetitive inhibition, α → 0; for mixed inhibition the α values fall in between these extremes (the special case of mixed inhibition where α = 1 is termed non-competitive inhibition). β represents the factor by which the catalytic rate (k) is reduced in the complex E·S·NP relative to E·S; for complete inhibition, β = 0. It should be also noted that the α and β values must be strictly regarded here as apparent parameters describing the overall response of thrombin to NP binding with both exosites.
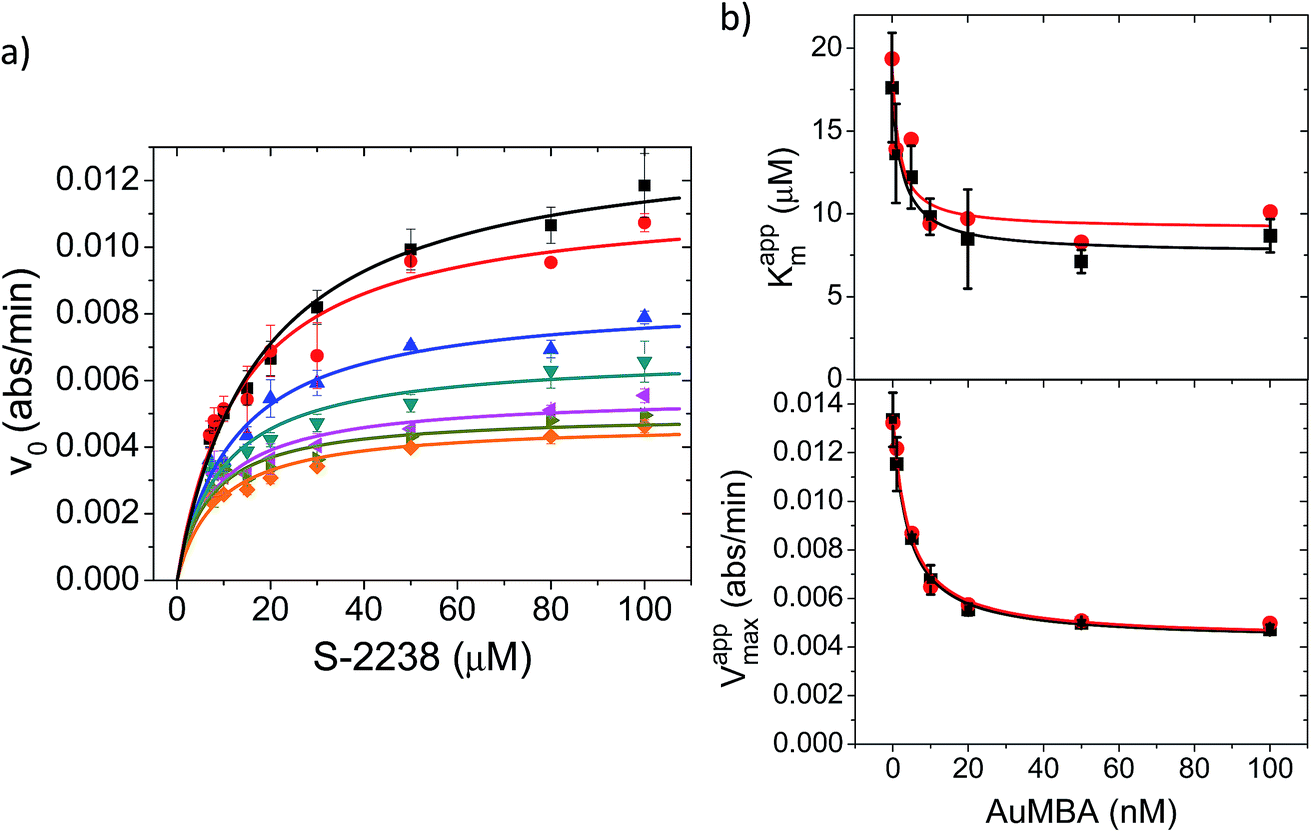 | ||
| Fig. 8 Michaelis–Menten kinetic analysis of thrombin inhibition by AuMBA. (a) Michaelis–Menten plots of thrombin activity as a function of AuMBA concentration (from top to bottom: 0, 1, 5, 10, 20, 50, 100 nM). Lines calculated by global fitting of eqn (1). Thrombin concentration, 2 nM; assay performed in Tris–HCl buffer solution supplemented with 100 mM NaCl. (b) Dependence of apparent Km and Vmax on AuMBA concentration. Squares: apparent Km and Vmax determined by fitting eqn (2) to the traces shown in (a). Circles: apparent Km and Vmax determined by the direct linear plot method from the data shown in (a) (see also Fig. S10†). Lines calculated by fitting of eqn (3). | ||
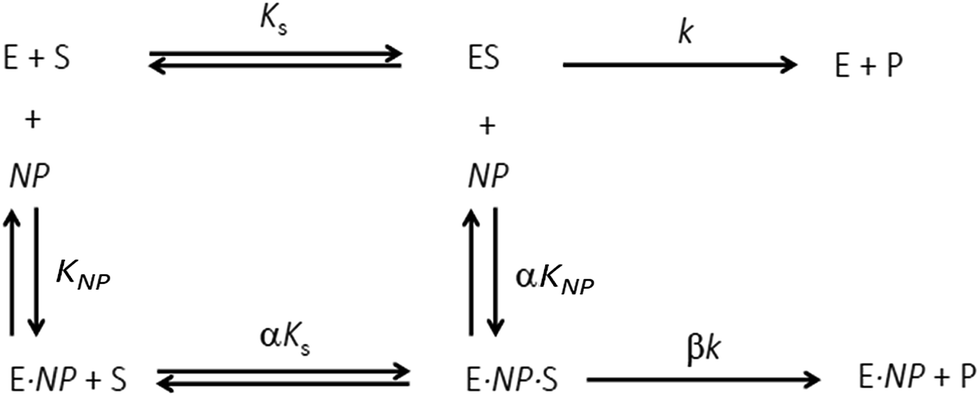 | ||
| Fig. 9 General modifier mechanism. E, S and NP refer to enzyme, substrate and nanoparticle, respectively. KS and KNP are equilibrium dissociation constants for the binding of S to E and of NP to E, respectively; αKS and αKNP are equilibrium dissociation constants for the binding of S to E·NP and of NP to E·S, respectively. The inhibition mechanism is described by the pair of values α and β. α determines the effect of the modifier (NP) on substrate-binding affinity: α → ∞, competitive inhibition; α → 0, uncompetitive inhibition; other values of α, mixed inhibition. β represents the factor by which the catalytic rate (k) is modified in the complex E·S·NP relative to E·S. | ||
The Michaelis–Menten kinetic data was analyzed quantitatively by fitting eqn (2) to each of the experimental traces in Fig. 8a, therefore yielding Kappm and Vappmax as a function of NP concentration (Fig. 8b). Kappm and Vappmax were also determined independently from Fig. 8a by the direct linear plot method (Fig. S10†), yielding similar results (Fig. 8b). Fig. 8b shows that the calculated Kappm and Vappmax first decreased but reached a plateau at higher NP concentrations. Fitting eqn (3) to the experimental points in Fig. 8b allowed α and β to be estimated as 0.44–0.48 and 0.32, respectively. Alternatively, the set of Michaelis–Menten plots (Fig. 8a) was analyzed by global fitting of the velocity equation (eqn (1)). The global fit yielded: α = 0.43, β = 0.33, KNP = 21 nM, Km = 17 μM and Vmax = 0.013 abs/min. The obtained KNP ∼21 nM was close to the KD ∼44 nM determined by SPR. β = 0.32, representing residual enzyme activity at saturating inhibitor concentrations, matched the β that could be estimated from the plateau in Fig. 7 (∼0.4). α = 0.43 indicated that substrate-binding affinity was enhanced in the enzyme–NP complex relative to the enzyme alone. Overall, these α and β values implied a hyperbolic mixed inhibition mechanism with predominant uncompetitive character.44
4. Conclusions
Enzyme activity can be modulated with NPs in several ways. For example, NPs that bind near the active site can block the access of substrate and thus inhibit the activity.17 NP interactions near the active site can also tune, rather than simply inhibit, the catalytic response by controlling substrate capture and product release at the enzyme–NP interface.70 Enzymes physically anchored onto the surface of NPs through a linker have been also shown to display increased activity.71–73To date, however, the characterization of NPs as allosteric modifiers has not been pursued in detail. Allostery requires that effectors bind to well-defined allosteric sites and induce specific changes in conformation and/or dynamics in the protein transmitted to the orthosteric site, by means of which substrate binding affinity and/or catalysis are modified. Allosteric regulation may be difficult to accomplish with conventionally large NPs. For example, large NPs may totally or partly occlude the active site given the extended contact area between particle and enzyme. In addition, large NPs may elicit broad, global changes in protein conformation, which may lead to partial denaturation and enzyme inactivation74–76 – incidentally, partial denaturation at the particle surface can support the formation of an irreversibly bound protein corona.77,78 On the other hand, interactions of proteins with ultrasmall NPs such as used in the present study may mimic a typical biomolecular complexation event, insofar as these interactions may take place through smaller binding interfaces and be fully reversible – i.e., a permanent protein corona may not form.49,79,80 Interestingly, computer simulations have suggested that small gold particles may indeed be able influence the structure and flexibility of protein regions distant from the binding site, while the overall protein would remain in a native folded structure.29
Here, as proof of concept, we showed that ultrasmall AuMBA particles can serve as synthetic protein receptors to allosterically modulate thrombin activity. We found that AuMBA interacted selectively with thrombin's exosites 1 and 2 with a KD ∼40 nM. The resulting complex was fully reversible, as the bound particles could be readily displaced from the enzyme surface by competing exosite-directed ligands. Interestingly, the NPs and ligands were able to form a ternary complex with thrombin by interacting simultaneously at opposite exosites, therefore revealing that NP complexation did not induce global, extensive structural changes across the protein to the point of hindering ligand binding to distant sites. We also found that interactions of AuMBA at the exosites transmitted long-range conformational changes over to the active site, modulating both substrate binding affinity and catalysis. An enzyme kinetics analysis performed within the framework of the general modifier mechanism implied a hyperbolic mixed inhibition mechanism with predominant uncompetitive character. To our knowledge, this type of allosteric response has not been described before in the interactions of synthetic NPs with enzymes.
Although ultrasmall AuMBA appears to share some of the attributes of traditional allosteric inhibitors, it must be noted that AuMBA interactions with thrombin are driven by electrostatic forces and thus are mostly nonspecific, whereas a typical inhibitor should ideally bind with high specificity to its enzyme target. Thus, in a complex biological fluid, AuMBA is expected to interact with other proteins that also exhibit clusters of positive charge on the surface. This problem may be partly overcome by rational design of the NP surface chemistry to increase the specificity of the interactions.81
Finally, we note that a number of exosite probes have been studied and shown to induce distinct effects on thrombin activity against synthetic substrates.82–85 Focusing on exosite 2-directed ligands, it was demonstrated that sulfated benzofuran scaffolds and the compound suramin could induce full inhibition (>80%) of thrombin.85,86 On the other hand, thrombin activity was not significantly affected by binding of heparin at exosite 2.83 Between these limiting cases, a set of sulfated coumarin molecules was recently introduced and demonstrated to induce partial inhibition of thrombin.84 Similarly to AuMBA, the coumarins were reported to bind to exosite 2, induce long-range structural changes near the active site, and produce a sub-maximal inhibitory response around 50%. Thus, it appears that allosteric partial inhibition of thrombin can be promoted through interactions with both synthetic molecules and particles. However, the underlying molecular mechanisms may be different, as the sulfated coumarins were proposed to bind primarily through hydrophobic interactions whereas AuMBA binding was mostly driven by electrostatics. Future work should be directed towards greater understanding of the allosteric mechanisms leading to thrombin inhibition by ultrasmall particles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Sergio A. Hassan for providing a computer model for AuMBA, Richard D. Leapman for images of AuMBA, and the Spectroscopy and Calorimetry facility at Brazilian Biosciences National Laboratory (LNBio), CNPEM, Campinas, Brazil, for their support with the analytical ultracentrifuge. This work was supported by the São Paulo Research Foundation (FAPESP: 2013/18481-5), and by the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, U.S.A.References
- N. V. Dokholyan, Chem. Rev., 2016, 116, 6463–6487 CrossRef CAS PubMed.
- R. Nussinov and C.-J. Tsai, Curr. Pharm. Des., 2012, 18, 1311–1316 CrossRef CAS PubMed.
- J. R. Wagner, C. T. Lee, J. D. Durrant, R. D. Malmstrom, V. A. Feher and R. E. Amaro, Chem. Rev., 2016, 116, 6370–6390 CrossRef CAS PubMed.
- R. Nussinov and C.-J. Tsai, Cell, 2013, 153, 293–305 CrossRef CAS PubMed.
- Y. Liu, D. A. Winkler, V. C. Epa, B. Zhang and B. Yan, Nano Res., 2015, 8, 1293–1308 CrossRef CAS.
- Y. Liu, B. Yan, D. A. Winkler, J. Fu and A. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 18626–18638 CrossRef CAS PubMed.
- S. Maiti, K. Das, S. Dutta and P. K. Das, Chem.–Eur. J., 2012, 18, 15021–15030 CrossRef CAS PubMed.
- T. Mizuhara, D. F. Moyano and V. M. Rotello, Nano Today, 2016, 11, 31–40 CrossRef CAS PubMed.
- E. Sanfins, C. Augustsson, B. r. Dahlbäck, S. Linse and T. Cedervall, Nano Lett., 2014, 14, 4736–4744 CrossRef CAS PubMed.
- S. Tomita and K. Shiraki, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3835–3841 CrossRef CAS.
- C.-S. Wu, C.-C. Lee, C.-T. Wu, Y.-S. Yang and F.-H. Ko, Chem. Commun., 2011, 47, 7446–7448 RSC.
- Z. Wu, B. Zhang and B. Yan, Int. J. Mol. Sci., 2009, 10, 4198–4209 CrossRef CAS PubMed.
- C.-C. You, M. De, G. Han and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 12873–12881 CrossRef CAS PubMed.
- S.-H. Cha, J. Hong, M. McGuffie, B. Yeom, J. S. VanEpps and N. A. Kotov, ACS Nano, 2015, 9, 9097–9105 CrossRef CAS PubMed.
- Y. C. Shiang, C. C. Huang, T. H. Wang, C. W. Chien and H. T. Chang, Adv. Funct. Mater., 2010, 20, 3175–3182 CrossRef CAS.
- A. Gole, C. Dash, V. Ramakrishnan, S. Sainkar, A. Mandale, M. Rao and M. Sastry, Langmuir, 2001, 17, 1674–1679 CrossRef CAS.
- Y. Liu, J. Fu, W. Pan, Q. Xue, X. Liu and A. Zhang, J. Environ. Sci., 2018, 63, 285–295 CrossRef PubMed.
- E. Tellechea, K. J. Wilson, E. Bravo and K. Hamad-Schifferli, Langmuir, 2012, 28, 5190–5200 CrossRef CAS PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- M. De, S. S. Chou and V. P. Dravid, J. Am. Chem. Soc., 2011, 133, 17524–17527 CrossRef CAS PubMed.
- J. Ge, J. Lei and R. N. Zare, Nat. Nanotechnol., 2012, 7, 428 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- L.-B. Wang, Y.-C. Wang, R. He, A. Zhuang, X. Wang, J. Zeng and J. Hou, J. Am. Chem. Soc., 2013, 135, 1272–1275 CrossRef CAS PubMed.
- Y. Zhang, J. Ge and Z. Liu, ACS Catal., 2015, 5, 4503–4513 CrossRef.
- M. De, C.-C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS PubMed.
- H. Koide, K. Yoshimatsu, Y. Hoshino, S.-H. Lee, A. Okajima, S. Ariizumi, Y. Narita, Y. Yonamine, A. C. Weisman and Y. Nishimura, Nat. Chem., 2017, 9, 715–722 CrossRef CAS PubMed.
- N. A. Kotov, Science, 2010, 330, 188–189 CrossRef CAS PubMed.
- A. Verma and V. M. Rotello, Chem. Commun., 2005, 303–312 RSC.
- Q. Shao and C. K. Hall, Nanoscale, 2017, 9, 380–390 RSC.
- E. Di Cera, Chest, 2003, 124, 11S–17S CrossRef CAS PubMed.
- J. Huntington, J. Thromb. Haemostasis, 2005, 3, 1861–1872 CrossRef CAS PubMed.
- J. A. Huntington, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 246–252 CrossRef CAS PubMed.
- P. E. Bock, Biochemistry, 1988, 27, 6633–6639 CrossRef CAS PubMed.
- C. L. Heinecke and C. J. Ackerson, Methods Mol. Biol., 2013, 950, 293–311 CAS.
- A. A. Sousa, S. A. Hassan, L. L. Knittel, A. Balbo, M. A. Aronova, P. H. Brown, P. Schuck and R. D. Leapman, Nanoscale, 2016, 8, 6577–6588 RSC.
- A. A. Sousa, J. T. Morgan, P. H. Brown, A. Adams, M. Jayasekara, G. Zhang, C. J. Ackerson, M. J. Kruhlak and R. D. Leapman, Small, 2012, 8, 2277–2286 CrossRef CAS PubMed.
- O. A. Wong, R. J. Hansen, T. W. Ni, C. L. Heinecke, W. S. Compel, D. L. Gustafson and C. J. Ackerson, Nanoscale, 2013, 5, 10525–10533 RSC.
- A. A. Sousa, J. Fluoresc., 2015, 25, 1567–1575 CrossRef CAS PubMed.
- L. L. Knittel, P. Schuck, C. J. Ackerson and A. A. Sousa, RSC Adv., 2016, 6, 46350–46355 RSC.
- J. Svitel, A. Balbo, R. A. Mariuzza, N. R. Gonzales and P. Schuck, Biophys. J., 2003, 84, 4062–4077 CrossRef CAS PubMed.
- J. Svitel, H. Boukari, D. Van Ryk, R. C. Willson and P. Schuck, Biophys. J., 2007, 92, 1742–1758 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS PubMed.
- H. Bisswanger, Perspect. Sci., 2014, 1, 41–55 CrossRef.
- A. Baici, Kinetics of Enzyme-Modifier Interactions, Springer, 2015 Search PubMed.
- A. Cornish-Bowden, Fundamentals of enzyme kinetics, Wiley-Blackwell, Weinheim, 2012 Search PubMed.
- R. Eisenthal and A. Cornish-Bowden, Biochem. J., 1974, 139, 715–720 CrossRef CAS PubMed.
- M. T. Stubbs and W. Bode, Trends Biochem. Sci., 1995, 20, 23–28 CrossRef CAS PubMed.
- E. Jurrus, E. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G.W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- A. Lira, R. Ferreira, R. Torquato, H. Zhao, M. L. Oliva, S. A. Hassan, P. Schuck and A. A. Sousa, Nanoscale, 2018, 10, 3235–3244 RSC.
- T. Vorup-Jensen, Methods Mol. Biol., 2012, 757, 55–71 CrossRef PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- A. Wang, Y. R. Perera, M. B. Davidson and N. C. Fitzkee, J. Phys. Chem. C, 2016, 120, 24231–24239 CrossRef CAS PubMed.
- J. A. Yang, B. J. Johnson, S. Wu, W. S. Woods, J. M. George and C. J. Murphy, Langmuir, 2013, 29, 4603–4615 CrossRef CAS PubMed.
- A. Wang, T. Vo, V. Le and N. C. Fitzkee, J. Phys. Chem. B, 2014, 118, 14148–14156 CrossRef CAS PubMed.
- W. Lin, T. Insley, M. D. Tuttle, L. Zhu, D. A. Berthold, P. Král, C. M. Rienstra and C. J. Murphy, J. Phys. Chem. C, 2015, 119, 21035–21043 CrossRef CAS PubMed.
- S. Zanzoni, M. Pedroni, M. D'Onofrio, A. Speghini and M. Assfalg, J. Am. Chem. Soc., 2015, 138, 72–75 CrossRef PubMed.
- S. Shrivastava, J. H. Nuffer, R. W. Siegel and J. S. Dordick, Nano Lett., 2012, 12, 1583–1587 CrossRef CAS PubMed.
- I. M. Verhamme, S. T. Olson, D. M. Tollefsen and P. E. Bock, J. Biol. Chem., 2002, 277, 6788–6798 CrossRef CAS PubMed.
- D. Breitsprecher, N. Schlinck, D. Witte, S. Duhr, P. Baaske and T. Schubert, Methods Mol. Biol., 2016, 1380, 99–111 CrossRef CAS PubMed.
- A. Pica, I. Russo Krauss, V. Parente, H. Tateishi-Karimata, S. Nagatoishi, K. Tsumoto, N. Sugimoto and F. Sica, Nucleic Acids Res., 2016, 45, 461–469 CrossRef PubMed.
- T. J. Rydel, K. Ravichandran, A. Tulinsky, W. Bode, R. Huber, C. Roitsch and J. W. Fenton, Science, 1990, 249, 277–280 CrossRef CAS PubMed.
- M. V. Malovichko, T. M. Sabo and M. C. Maurer, J. Biol. Chem., 2013, 288, 8667–8678 CrossRef CAS PubMed.
- A. Bekdemir and F. Stellacci, Nat. Commun., 2016, 7, 13121 CrossRef CAS PubMed.
- Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664–5669 CrossRef CAS PubMed.
- N. S. Petrera, A. R. Stafford, B. A. Leslie, C. A. Kretz, J. C. Fredenburgh and J. I. Weitz, J. Biol. Chem., 2009, 284, 25620–25629 CrossRef CAS PubMed.
- B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965–970 CrossRef CAS PubMed.
- N. Klonis, A. H. Clayton, E. W. Voss and W. H. Sawyer, Photochem. Photobiol., 1998, 67, 500–510 CAS.
- A. Y. Mehta and U. R. Desai, Biochem. Biophys. Res. Commun., 2014, 452, 813–816 CrossRef CAS PubMed.
- S. A. Evans, S. Olson and J. Shore, J. Biol. Chem., 1982, 257, 3014–3017 CAS.
- C.-C. You, S. S. Agasti, M. De, M. J. Knapp and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 14612–14618 CrossRef CAS PubMed.
- W. R. Algar, A. Malonoski, J. R. Deschamps, J. B. Blanco-Canosa, K. Susumu, M. H. Stewart, B. J. Johnson, P. E. Dawson and I. L. Medintz, Nano Lett., 2012, 12, 3793–3802 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- B. J. Johnson, W. R. Algar, A. P. Malanoski, M. G. Ancona and I. L. Medintz, Nano Today, 2014, 9, 102–131 CrossRef CAS.
- M. Karlsson, L.-G. Mårtensson, B.-H. Jonsson and U. Carlsson, Langmuir, 2000, 16, 8470–8479 CrossRef CAS.
- M. Lundqvist, I. Sethson and B.-H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- A. Assarsson, I. Pastoriza-Santos and C. Cabaleiro-Lago, Langmuir, 2014, 30, 9448–9456 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- J. Piella, N. G. Bastús and V. Puntes, Bioconjugate Chem., 2016, 28, 88–97 CrossRef PubMed.
- L. Boselli, E. Polo, V. Castagnola and K. A. Dawson, Angew. Chem., Int. Ed., 2017, 56, 4215–4218 CrossRef CAS PubMed.
- K. Zarschler, L. Rocks, N. Licciardello, L. Boselli, E. Polo, K. P. Garcia, L. De Cola, H. Stephan and K. A. Dawson, Nanomedicine, 2016, 12, 1663–1701 CrossRef CAS PubMed.
- H. Bayraktar, C.-C. You, V. M. Rotello and M. J. Knapp, J. Am. Chem. Soc., 2007, 129, 2732–2733 CrossRef CAS PubMed.
- B. L. Henry, B. H. Monien, P. E. Bock and U. R. Desai, J. Biol. Chem., 2007, 282, 31891–31899 CrossRef CAS PubMed.
- B. L. Henry, J. N. Thakkar, A. Liang and U. R. Desai, Biochem. Biophys. Res. Commun., 2012, 417, 382–386 CrossRef CAS PubMed.
- S. Verespy III, A. Y. Mehta, D. Afosah, R. A. Al-Horani and U. R. Desai, Sci. Rep., 2016, 6, 24043 CrossRef PubMed.
- R. Q. Monteiro, P. T. Campana, P. A. Melo and M. L. Bianconi, Int. J. Biochem. Cell Biol., 2004, 36, 2077–2085 CrossRef CAS PubMed.
- P. S. Sidhu, M. H. Abdel Aziz, A. Sarkar, A. Y. Mehta, Q. Zhou and U. R. Desai, J. Med. Chem., 2013, 56, 5059–5070 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00081f |
| This journal is © The Royal Society of Chemistry 2019 |