
Probing the origins of photodegradation in organic–inorganic metal halide perovskites with time-resolved mass spectrometry†
Zhaoning
Song
 ,
Changlei
Wang
,
Adam B.
Phillips
,
Corey R.
Grice
,
Dewei
Zhao
,
Yue
Yu
,
Cong
Chen
,
Chongwen
Li
,
Xinxing
Yin
,
Randy J.
Ellingson
,
Michael J.
Heben
and
Yanfa
Yan
*
,
Changlei
Wang
,
Adam B.
Phillips
,
Corey R.
Grice
,
Dewei
Zhao
,
Yue
Yu
,
Cong
Chen
,
Chongwen
Li
,
Xinxing
Yin
,
Randy J.
Ellingson
,
Michael J.
Heben
and
Yanfa
Yan
*
University of Toledo, Wright Center for Photovoltaics Innovation and Commercialization, Department of Physics and Astronomy, 2801 W. Bancroft St., Toledo, OH, 43606 USA. E-mail: yanfa.yan@utoledo.edu
First published on 20th August 2018
Abstract
Operational and long-term stability of perovskite solar cells are critical for their commercialization on a large scale. To mitigate stability issues, a fundamental understanding of the physicochemical processes associated with the degradation of perovskite materials is needed. Here, we perform time resolved mass spectrometry of the gas species evolved during the photoinduced degradation of organic–inorganic lead trihalide perovskites made with commonly used monovalent cations, including methylammonium (MA), formamidinium (FA), and Cs. Our results indicate that the hot-carrier-induced deprotonation of MA+ cations is the fundamental origin of the photodegradation, which inevitably leads to the release of volatile species such as ammonia (NH3), aminocarbyne fragments (CNH2), hydrogen (H2), and iodine/hydrogen iodide (I/HI) from methylammonium lead iodide (MAPbI3) at different rates under simulated one sun solar illumination. Photodegradation processes can be mitigated by applying ultra-violet (UV) filters with suitable cutoff wavelengths to the light source. Additionally, we demonstrate that the incorporation of FA reduces the release of organic species but does not prevent the formation of I/HI. However, the addition of Cs effectively suppresses the release of all volatile gases. The best photostability is obtained with the FA/Cs mixed perovskites, showing that the complete removal of MA from mixed-cation perovskites is preferred for more photostable perovskites.
Introduction
In the past few years, organic–inorganic metal halide perovskites have emerged as an exciting class of photovoltaic (PV) materials.1,2 With high certified power conversion efficiencies that exceed 23.3% (ref. 3) and low-cost solution-based fabrication processes, perovskite solar cells (PVSCs) are projected to rival other more established PV technologies on the market, especially in the aspects of levelized cost of energy (LCOE),4 life-cycle environmental impact,5,6 and energy payback time (EPBT).7,8 Recent progress in large-area deposition techniques,9–11 combined with advances in materials engineering to improve device performance and durability,12–17 makes PVSCs attractive for more practical, commercial applications. However, major obstacles remain before high volume deployment of perovskite PV modules becomes feasible.18 The most critical one is the lack of operational and long-term stability. To be commercially viable, PVSCs need to retain robust stability throughout their lifetimes. The fact that most established PV companies provide a 25-year warranty for their products sets a high bar for perovskite PV technology to enter the energy production market.19Over the last decade, lifetimes of PVSCs reported in the literature have extended from a few hours to over a year.20 However, most high efficiency perovskite solar cells based on triple-cation (methylammonium, MA; formamidinium, FA; Cs) lead trihalide perovskites exhibit a certain degree of degradation under continuous operating conditions.21 In an attempt to understand and eliminate degradation, the origins and processes of device degradation are being intensively investigated.22 Degradation mechanisms of metal halide perovskite materials and devices under different environmental conditions have been extensively explored.23–35 In general, a variety of extrinsic factors, including light,36–42 heat,43–45 atmosphere (e.g., oxygen and moisture),42,46–48 and electric field,49,50 are considered to contribute to the degradation of perovskite materials and devices. Among them, heat, moisture, and electric field related degradation can be mitigated by employing appropriate encapsulation and interface protection,16,51–53 while solar radiation that contains a substantial quantity of energetic photons is an unavoidable stress during the operation of a solar cell.
A recent literature review that surveyed 50 publications shows that the stability of PVSCs is strongly reduced under illumination,36 which hinders practical application of PVSCs. To mitigate this photoinstability issue, a fundamental understanding of the physicochemical processes associated with the photodegradation of perovskite materials is needed. Thus far, only a few studies have investigated the degradation processes of perovskite films under illumination via X-ray photoelectron, diffraction, and absorption spectroscopies,40–42 with an emphasis on monitoring the chemical and structural changes in the perovskite films. Despite these efforts, little is known about the formation and escape rates of the volatile decomposition products during the photodegradation processes. Most recently, Nickel et al. reported an investigation on the photodegradation of MAPbI3 by analyzing the residual gas composition above the sample,39 providing insights for understanding the photodegradation mechanisms. However, the measurements of gas effusion were limited to a few selected low atomic weight (<32) gas molecules, and volatile species across the full spectrum of the photodegradation products, especially iodine or iodide vapors, have not yet been monitored. The missing information may hinder accurate understanding of the degradation mechanism and underestimate the impact of MA on the stability of state-of-the-art triple-cation (FA/MA/Cs) perovskites, which have demonstrated much better photostability than mixed FA/MA perovskites.13,14,54 Therefore, more detailed work is needed to develop a comprehensive understanding of the underlying degradation mechanisms and pathways.55
Herein, we address this need by employing high-resolution time resolved mass spectrometry (MS) to monitor the gas emission during the photodegradation of organic–inorganic halide perovskites in vacuum and under different illumination conditions. Starting with the archetypal perovskite, methylammonium lead iodide (CH3NH3PbI3 or MAPbI3), we measure the escape rates of volatile photodecomposition products and identify the photon energy threshold for the photodegradation of MAPbI3 using a filtered light source. For the first time, we observed that MA gas is released faster than HI under illumination, indicating that photodegradation likely originates from the hot carrier-induced deprotonation of MA+ cations. Moreover, we study the photostability of a series of FA/MA/Cs mixed-cation perovskites, demonstrating that the incorporation of FA does not fully stop the photodegradation, but the incorporation of nonvolatile Cs+ into perovskite films leads to improved photostability. The results show that the complete removal of MA from mixed-cation perovskites is preferred for more photostable perovskites.
Results and discussion
We start our investigation with the archetypal perovskite material, i.e., MAPbI3. An in-house-built temperature programmed MS spectroscopy system56 (Fig. S1†) was used to determine the chemical compositions of the gases released from MAPbI3 under simulated solar irradiation. Fig. 1a shows the evolution of the mass-to-charge ratio (m/z) spectrum recorded as a function of light exposure time (see the Experimental section for details). A variety of gas species including H2 (m/z = 2), NH3 (m/z = 17), H2O (m/z = 18), CH3NH2 (m/z = 28–31), HI (m/z = 64 or 128), and I2 (m/z = 127) were detected. By measuring these partial pressures as a function of time, the photodegradation dynamics of MAPbI3 were determined (vide infra). A comparison of the relevant peaks at the initial stage and at a time during the photodegradation is shown in Fig. 1b–f. The significant increase in the partial pressures of the gas signals clearly indicates the presence of the volatile decomposition products through photodecomposition of MAPbI3. It is worth noting that MS signals are comprised of the fragments of molecules due to the electron bombardment during the ionization process in the quadrupole ion analyzer. Multiple fragments from a gas can be detected, and one fragment species may originate from different parent molecules. Because of this, the chemical compositions revealed by MS are manifold, and chemical complexity needs to be considered while analyzing the data.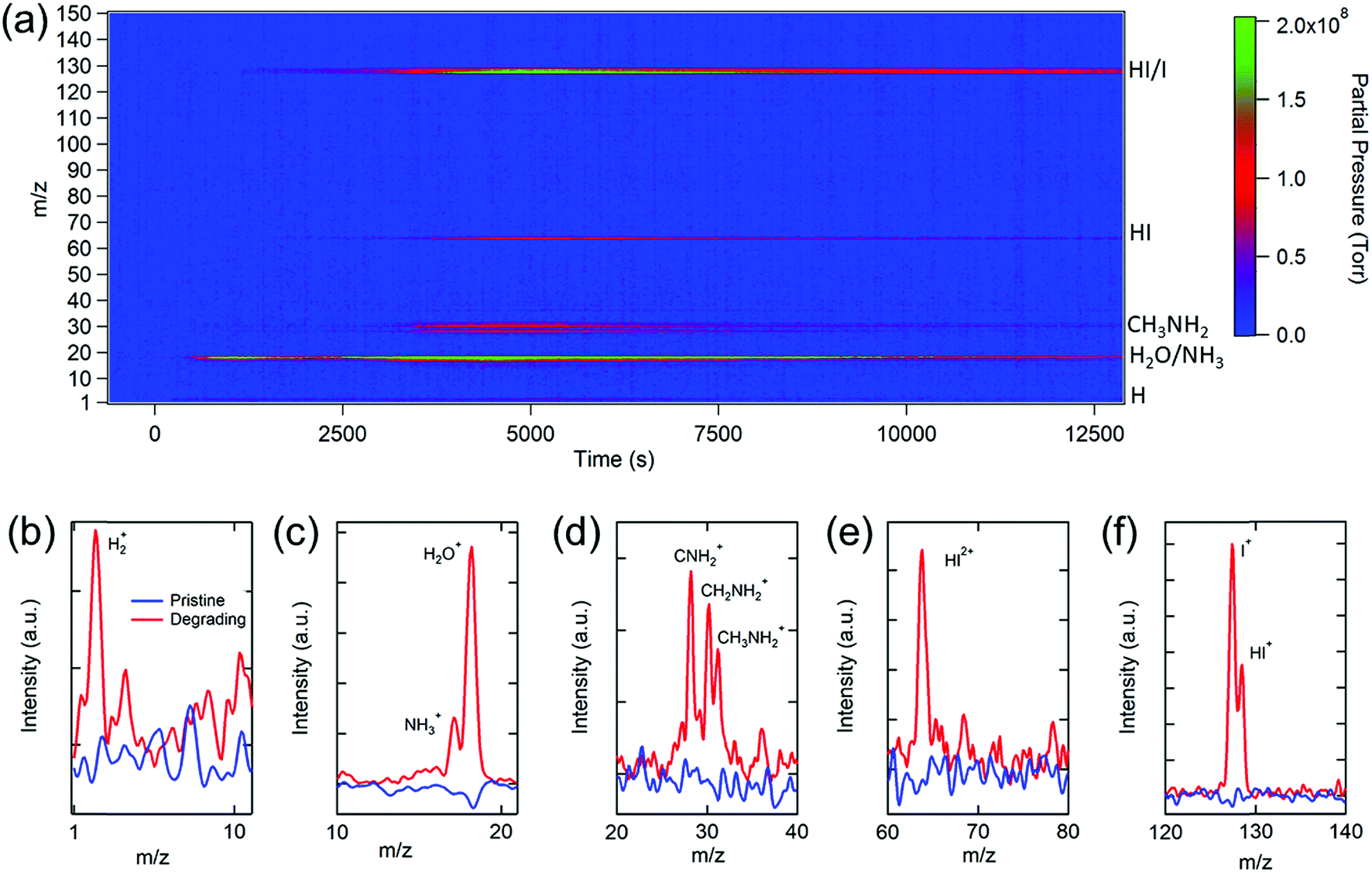 | ||
| Fig. 1 (a) Evolution of the mass spectrum (m/z) of the gases released during the photoinduced decomposition of CH3NH3PbI3. Comparison of the characteristic m/z peaks of the pristine and degraded samples, including (b) H+, (c) NH3+/H2O+, (d) CNH2+/CH2NH2+/CH3NH2+, (e) HI2+, and (f) I+/HI+. | ||
Turning to a detailed analysis of the photodegradation process, the release of H2O was initiated after MAPbI3 was exposed to light for ∼5 min (300 s). Due to the hygroscopic nature of perovskites,46 water molecules could either be adsorbed on the surface of the perovskite57 or infiltrate into grain boundaries to form the hydrated perovskite phases.58 The observation of photo-induced desorption of water indicates the high physisorption affinity of water molecules to perovskites, which leads to substantial water adsorption even for a short duration of exposure (2 to 3 min) to the air during the sample transfer. However, the amount of water desorbed from the perovskite is challenging to quantify because water contributes significantly to the base pressure of a typical high vacuum (∼10−7 torr) chamber (Fig. S2†), and its partial pressure is sensitive to subtle chamber condition changes and degassing of other gas species. Therefore, water is not used as the primary indicator for the photodegradation analysis in this study.
The release of methylamine (CH3NH2 or MA) and HI signals commenced at ∼20 min (1200 s), signifying the beginning of photodegradation of MAPbI3. The peaks observed at m/z = 2 (Fig. 1b) and m/z = 17 and 18 (Fig. 1c), corresponding to H2 and NH3/H2O, respectively, could be directly produced by the photodecomposition of MAPbI3 or from the ionization byproducts of MA. The released MA was characterized by three aminocarbyne fragments at m/z ratios of 28 (CNH2), 30 (CH2NH2), and 31(CH3NH2), as shown in Fig. 1d, which is consistent with the reference MS of MA (Fig. S3†). The release of NH3 and MA gas molecules can break the phase equilibrium homogeneity in MAPbI3 and worsen the decomposition of the pristine perovskite phase by triggering a phase transfer from the pure corner-sharing Pb–I octahedral perovskite network into an inhomogeneous combination of MAPbI3 perovskite, layered PbI2, and low-dimensional perovskites.59,60 This phase transition creates more vulnerable defect sites on the grain boundaries with loose bonding or dangling bonds that are prone to decomposition.
The release of HI gas was evident in the single and double charged HI with m/z ratios of 128 and 64, respectively (Fig. 1e and f), which was not found in the previous photodegradation study.39 The signal of m/z = 127 could be attributed to HI as well as the single charged I or double charged I2, indicating that an equilibrium between I2 and I− exists in the perovskite films during photodegradation. The condensation of I2 residues on the inner wall of the sample conditioning tube (Fig. S4†) reveals the simultaneous formation of I2 gas with HI, consistent with a recent report on measuring the optical absorption spectrum of I2 release from MAPbI3 in toluene under illumination.61 Both I2 and HI are detrimental to the stability of perovskite films. Iodine vapor could induce severe degradation of MAPbI3 due to chemical chain reactions.62 HI is a highly corrosive acid which could cause severe damage to the perovskite crystals upon contact and accelerate the degradation of perovskites.
The aforementioned results reveal that the MA derivatives and I2/HI gases are the primary volatile products during the photodegradation of MAPbI3. Interestingly, the photodegradation products of MAPbI3 differ from the thermal degradation products in which CH3I and NH3 were detected at above 300 °C by MS measurements.44 Although photo- and thermal degradation result in different products, there should be a chemical equilibrium between these gasses, which favors the formation of MA and HI at low temperatures (∼60 °C under illumination) but leads to the formation of CH3I and NH3 at high temperatures (>300 °C). Nonetheless, for both degradation pathways, the decomposition of perovskites is associated with the release of volatile gases, especially MA and HI. It is noteworthy that while some studies have demonstrated that photo-induced superoxide formed in the presence of oxygen or an oxide interface leads to the photodecomposition of perovskites,37,63–65 the results here show that oxygen is not necessary for the photodecomposition since no oxygen or superoxide signals were detected.
To further elucidate the degradation mechanisms, we measured the photon energy dependences of the photodegradation of MAPbI3. Long pass filters with various cutoff wavelengths were used to remove the high energy photons that are known to be particularly detrimental to PVSCs.39,64Fig. 2 shows the evolution of the MS traces of MA and HI from MAPbI3 films exposed to illumination with the spectra filtered with short wavelength photons being cut off at 280, 320, 370, 395, 420, and 455 nm. The transmittance spectra of the filters and full MS spectral data are shown in Fig. S5 and S6,† respectively.
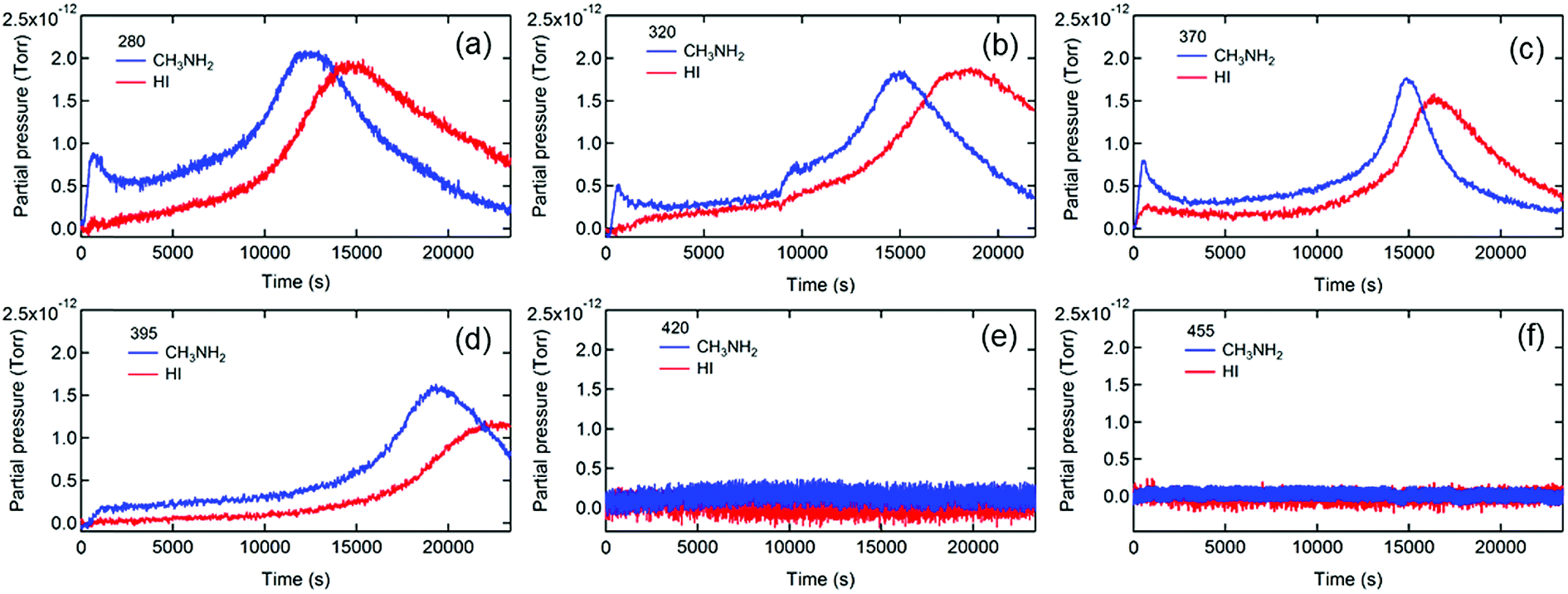 | ||
| Fig. 2 Mass spectroscopy traces of CH3NH2 (28–31) and HI (126–127) during the photodecomposition of MAPbI3 films under illumination from a xenon lamp equipped with a long pass optical filter with a cutoff wavelength of (a) 280, (b) 320, (c) 370, (d) 395, (e) 420, and (f) 455 nm. | ||
Insights into the dynamics and kinetics of photodegradation of MAPbI3 can be obtained from analyzing the gas emission processes. First, the MS traces of MA and HI (Fig. 2a–d) show a general trend of an increase in MA and HI partial pressures to the maxima followed by a decline to a residual level, indicating that the photodegradation of MAPbI3 is a self-limiting process that likely initiates on the surface of the film or at grain boundaries, reaches the pressure maxima when most surface bonds are cleaved, and then slows down due to the formation of more photostable PbI2 on the surface. These findings are consistent with the report that photodegradation of MAPbI3 became saturated after the decomposition of the surface layer.38 Beyond that, the escape rates of the residual gas determined by the time derivative of the partial pressure decreased with increasing the cutoff wavelength (Fig. S7†), likely due to the retarded photodegradation caused by removing highly energetic photons. The residual gas signals were in the minimum detectable range and were negligible when the 420 (Fig. 2e) and 455 nm (Fig. 2f) filters were applied, respectively, indicating that a minimum photon energy of 2.95 eV (∼420 nm) is needed for triggering light-induced degradation of MAPbI3. Considering the bandgap of 1.55 eV for MAPbI3,45 the excess kinetic energy of photoexcited charge carriers (∼1.4 eV) is close to the energy associated with the deprotonation of MA+ cations (1.49 eV) reported in the literature.66 It is worth noting that our result is more accurate than the minimum energy of 2.72 eV required for the light-induced dissociation of MAPbI3 perovskite reported by Nickel et al.39 To verify that the mitigation of photodecomposition is merely due to removal of high energy photons rather than reduction of light intensity, we performed the MS measurements under reduced illumination intensities (25%) achieved by using a neutral density filter with an optical density (OD) of 0.6. Fig. S8† shows evidence of photodecomposition even under this weak illumination condition (∼25 mW cm−2), confirming that the high energy photons are responsible for the photodegradation of MAPbI3.
The detailed analysis of in situ MS measurement data provides insights into the kinetics of the photodecomposition process. Interestingly, MA exhibits higher partial pressure than HI at the early stage of the photodecomposition process and reaches the peak prior to HI in most cases (Fig. 2a–d), indicating that MA is released faster than HI when photodegradation occurs. The result reveals that MA+, i.e., CH3NH3+ (resulting from the protonation of MA molecules), is more vulnerable to high energy photons than I− and is more likely to be the origin of the photodegradation of the perovskite. It is worth noting that a clear initial spike in the MA signal was observed in some MAPbI3 films (Fig. 2a–c), likely due to the existence of MA+ cations on film surfaces and in grain boundaries.67 It is known that MAPbI3 surfaces contain under-coordinated MA+ cations.68 Photoexcitation could release those loosely bounded MA gas molecules by a deprotonation process, leading to the initial spike in the MA signal.
Applying a filter to eliminate UV photons is an effective strategy to improve the photostability of perovskite materials and devices. Additionally, when the perovskite films are fully encapsulated, the volatile photodecomposition products cannot escape and thus persist in the films, retaining the perovskite phase. However, these approaches do not solve the intrinsic instability issue of MAPbI3. Alternatively, many studies suggest that incorporation of less volatile large organic (e.g., FA) and monovalent metal cations (e.g., Cs+) can significantly enhance photostability.1,14,69 To explore the effect of the A-site cation on the photostability of perovskite materials, we performed MS measurements on a variety of single- and mixed-cation perovskites. Fig. 3a shows the accumulated MS signals of organic species (i.e. MA and FA derivatives) and HI for perovskite films with various (CsxFAyMA1−x−y)PbI3 compositions, including MAPbI3, FAPbI3, MA0.7FA0.3PbI3, FA0.95Cs0.05PbI3, FA0.9Cs0.1PbI3, FA0.8Cs0.2PbI3, and (MA0.7FA0.3)0.8Cs0.2PbI3, after 7 h of light exposure. Detailed traces of the corresponding curves are shown in Fig. S9.† As expected, we found that MAPbI3 had the most severe photodecomposition. Interestingly, gravimetric analysis shows that MAPbI3 lost about ∼30% of its initial weight, more than the weight ratio (25.6%) of MAI (159 g mol−1) to MAPbI3 (620 g mol−1), indicating the further decomposition of PbI2 into metallic lead (Pb0) and I2 gas.41,70 FAPbI3 shows significantly lower emission of organic species (mainly in terms of CNH and CNH2) but comparable HI emission compared with MAPbI3 (Fig. 3b). Although improved photostability of FAPbI3 was reported,69 the significant release of HI could still fundamentally limit the durability of FAPbI3. In contrast, incorporating Cs into FAPbI3 significantly suppressed the photodecomposition of perovskite materials. The photostability of the perovskite increases with increasing Cs proportion, with FA0.8Cs0.2PbI3 showing two orders of magnitude less loss of materials during continuous illumination (Fig. 3c). Interestingly, the addition of MA to the mixed-cation perovskites (FA/MA and FA/MA/Cs) shows worse photostability than that of their pure FA and FA/Cs counterparts, indicating that the UV-photon induced degradation of the perovskites is mainly attributed to MA cations.
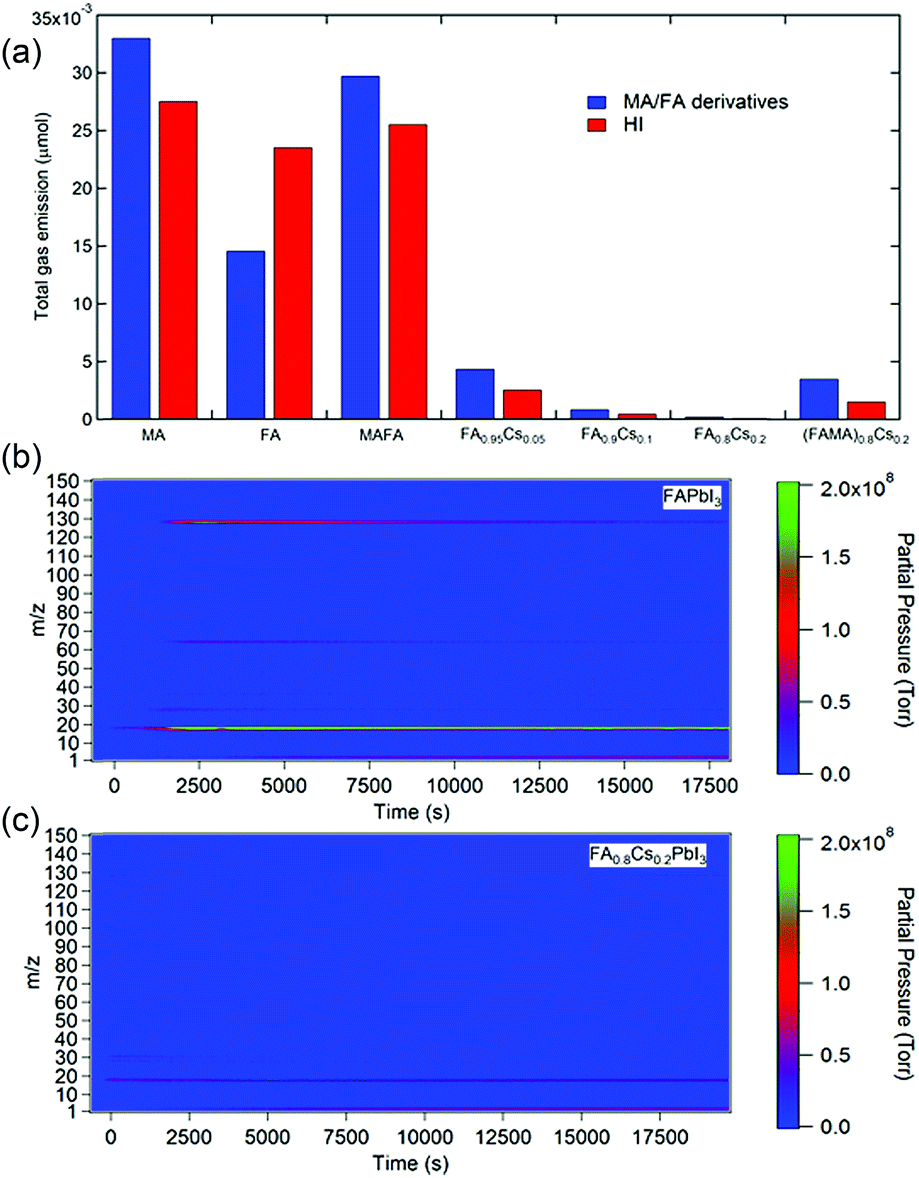 | ||
| Fig. 3 (a) Total amounts of organic species and HI gases released from various perovskite films, including MAPbI3, FAPbI3, MA0.7FA0.3PbI3, FA0.95Cs0.05PbI3, FA0.9Cs0.1PbI3, FA0.8Cs0.2PbI3, and (MA0.7FA0.3)0.8Cs0.2PbI3 under a simulated irradiance of 100 mW cm−2 for 7 h. Time-resolved mass spectra of (b) FAPbI3 and (c) FA0.8Cs0.2PbI3 under a simulated irradiance of 100 mW cm−2. | ||
The above observation leads us to hypothesize that the photodegradation mechanism of MA-containing perovskites proceeds through a sequence of steps involving (1) photoexcitation of hot carriers; (2) deprotonation of MA cations and formation of MA gas near surface or grain boundary regions; and (3) formation of HI gas and iodine vacancies (VI) (deterioration of the PbI6 octahedron), as described by eqn (1)–(3) and shown in Fig. 4.
| hν(>3 eV) → e−(hot) + h+(hot) | (1) |
| CH3NH3+ + e−(hot) → CH3NH2(g) + H+ + e− | (2) |
| H+ + I− → HI(g) + VI | (3) |
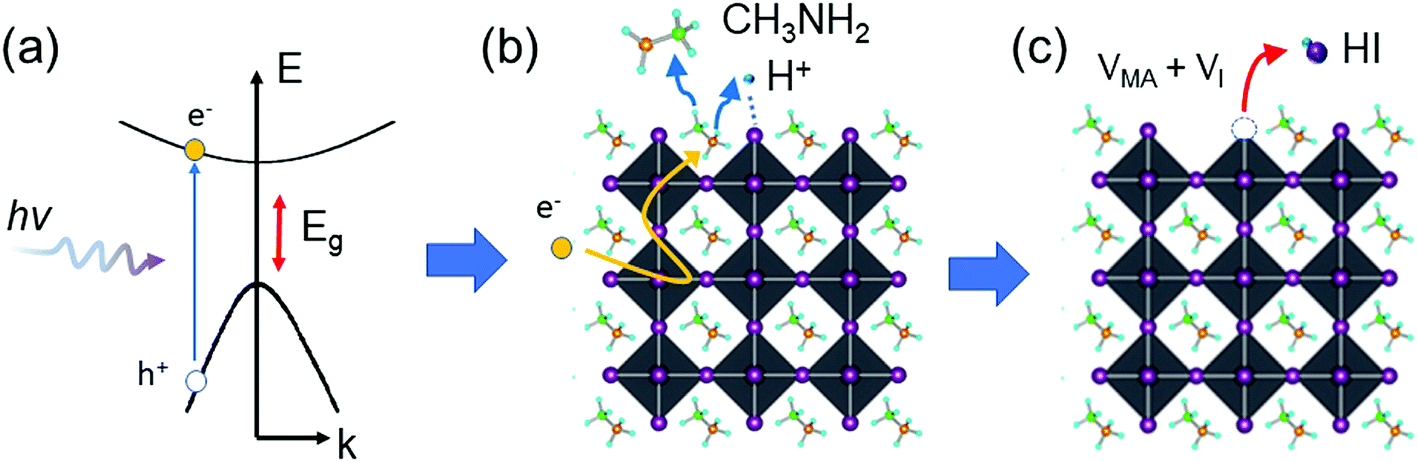 | ||
| Fig. 4 Schematic representation of the photodegradation mechanism. (a) Hot carrier generation, (b) deprotonation and release of MA gas, and (c) formation of HI gas and VI. | ||
The generation of long-lived hot electrons is a requisite for the observed photodegradation of MAPbI3 (eqn (1) and Fig. 4a). Recent studies reported that hot carriers in perovskites have a relatively long lifetime (∼100 ps) and can migrate a long distance (up to 600 nm) in perovskite crystals.71–73 A hot electron may interact with a MA cation via coulombic coupling. The hot electron may transfer its excess energy to the MA cation and deprotonate it, releasing free protons and volatile MA gas (eqn (2) and Fig. 4b). The free protons then pair with under-coordinated I atoms on the surface, forming HI gas and iodine vacancies (i.e., deteriorated PbI6 octahedra) (eqn (3) and Fig. 4c). We speculate that HI is the most detrimental species to the stability of the perovskite because the continuous loss of HI can cause the breakdown of the Pb–I framework via an equilibrium phase transfer from the corner-shared to the face-shared Pb–I octahedra, leading to the catastrophic decomposition of MAPbI3 into PbI2. The loss of HI completely changes the morphology of MAPbI3 films, turning the compact and smooth perovskite films into discrete PbI2 crystals (Fig. S10†). Although the primary gas products of the photodegradation are MA and HI, other gas species, such as H2, NH3, and I2, may coexist via a photochemical equilibrium in the atmosphere. Nonetheless, preventing the formation of volatile gases could be an effective strategy to mitigate the photoinstability issue of perovskites.
The mechanism of improved stability of Cs containing perovskites is attributed to the low volatility of Cs. Due to its small ionic size (167 pm) compared with MA+ (217 pm) and FA+ (253 pm),74 it is reasonable to expect Cs+ ions to segregate into surface and grain boundaries, a process that reduces strain and, therefore, is energetically favorable. As discussed earlier, organic MA and FA cations tend to deprotonate and vaporize after receiving energy from hot carriers generated by high energetic photons. In contrast, Cs has much lower vapor pressures and has no proton. Preserving monovalent cations limits the formation and release of HI gas, locking iodine atoms in the perovskite framework. Therefore, the intrinsic decomposition of iodine-based perovskites can be eliminated, showing potential for long-term stability. However, the triple cation (MA0.7FA0.3)0.8Cs0.2PbI3, unlike its FA/Cs counterpart, still released substantial amounts of organic species and HI gases under continuous illumination (Fig. 3), indicating the intrinsic instability of perovskite materials incorporating MA cations. To overcome the instability issue, the MA content in the perovskites should be reduced or eliminated.
Conclusion
In summary, we have investigated the photodecomposition of MAPbI3 under different illumination conditions using MS. We demonstrated that photodecomposition is a self-limited process, identified the photon energy threshold of ∼3 eV to trigger the decomposition of MAPbI3, and showed that the release of MA is prior to HI. Lastly, we found that incorporating Cs and removing MA can improve the photostability of perovskite materials. Our findings suggest that compositional engineering of perovskite materials and UV filtering can prevent the photodegradation of the organic components and thus increase the operational and long-term stability of organic–inorganic halide perovskite solar cells.Experimental
Synthesis of perovskite thin films
The perovskite films were synthesized following a previously published method. Details are included in the ESI.†Time-resolved mass spectroscopy measurements
The mass spectra of perovskite films were obtained using a custom dynamically pumped temperature programmed desorption system. For each degradation measurement, a perovskite film (1 inch by 0.25 inch) was placed in a quartz tube that was connected to the primary chamber of the system. The system was pumped down for ∼20 h to allow degassing of the specimen prior to the photon induced degradation measurement. The chamber was evacuated to <10−7 torr before each measurement. The degradation process was monitored using a quadrupole mass spectrometer (Stanford Research System, RGA 300). The mass spectra with a range of 1 to 150 atomic mass units (AMU) were recorded at 5 s intervals. Prior to the illumination, background signals were first collected for 10 min in the dark and the averages were subtracted from the main signals. After that, the sample was exposed to a simulated solar irradiance of ∼100 mW cm−2 generated by a xenon arc lamp. For the spectroscopy-dependent measurements, long wavelength filters with the cutoff edge at 280, 320, 370, 395, 420, or 455 nm were used to eliminate the influence of high energy photons.Author contribution
Z. S. and Y. F. Y. conceived the project. Z. S. carried out the MS measurements. C. W., Y. Y., C. C., X. Y. and C. L. synthesized the perovskite thin films and helped with characterization. C. R. G., A. B. P. and M. J. H. assisted with the MS measurements. Z. S. and Y. F. Y. analyzed the data and wrote the manuscript. D. Z., A. B. P., R. J. E. and M. J. H. helped with the manuscript preparation. All the authors discussed the results and commented on the manuscript. Y. F. Y. supervised the project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is financially supported by the Office of Naval Research under contract no. N00014-17-1-2223, the Air Force Research Laboratory, Space Vehicles Directorate (contract # FA9453-11-C-0253), the National Science Foundation under contract no. CHE-1230246 and DMR-1534686, the U.S. Department of Energy (DOE) SunShot Initiative under the Next Generation Photovoltaics 3 program (DE-FOA-0000990), and the Ohio Research Scholar Program.References
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- J. J. Berry, J. van de Lagemaat, M. M. Al-Jassim, S. Kurtz, Y. Yan and K. Zhu, ACS Energy Lett., 2017, 2, 2540–2544 CrossRef CAS.
- NREL, Solar Cell Efficiency Chart, https://www.nrel.gov/pv/assets/images/efficiency-chart-20180716.jpg, accessed 6 August 2018.
- Z. Song, C. L. McElvany, A. B. Phillips, I. Celik, P. W. Krantz, S. C. Watthage, G. K. Liyanage, D. Apul and M. J. Heben, Energy Environ. Sci., 2017, 10, 1297–1305 RSC.
- J. Gong, S. B. Darling and F. You, Energy Environ. Sci., 2015, 8, 1953–1968 RSC.
- I. Celik, Z. Song, A. J. Cimaroli, Y. Yan, M. J. Heben and D. Apul, Sol. Energy Mater. Sol. Cells, 2016, 156, 157–169 CrossRef CAS.
- I. Celik, A. B. Phillips, Z. Song, Y. Yan, R. J. Ellingson, M. J. Heben and D. Apul, Energy Environ. Sci., 2017, 10, 1874–1884 RSC.
- I. Celik, A. B. Phillips, Z. Song, Y. Yan, R. J. Ellingson, M. J. Heben and D. Apul, IEEE J. Photovolt., 2017, 8, 305–309 Search PubMed.
- Z. Li, T. R. Klein, D. H. Kim, M. Yang, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Rev. Mater., 2018, 3, 18017 CrossRef CAS.
- H. Chen, F. Ye, W. Tang, J. He, M. Yin, Y. Wang, F. Xie, E. Bi, X. Yang, M. Grätzel and L. Han, Nature, 2017, 550, 92 CAS.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Energy, 2017, 2, 17038 CrossRef CAS.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- J. A. Christians, P. Schulz, J. S. Tinkham, T. H. Schloemer, S. P. Harvey, B. J. Tremolet de Villers, A. Sellinger, J. J. Berry and J. M. Luther, Nat. Energy, 2018, 3, 68–74 CrossRef CAS.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497 CrossRef CAS PubMed.
- A. Abate, J. P. Correa-Baena, M. Saliba, M. S. Su'ait and F. Bella, Chem.–Eur. J., 2018, 24, 3083–3100 CrossRef CAS PubMed.
- M. A. Green, Nat. Energy, 2016, 1, 15015 CrossRef.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- K. Domanski, E. A. Alharbi, A. Hagfeldt, M. Grätzel and W. Tress, Nat. Energy, 2018, 3, 61–67 CrossRef CAS.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- D. Wang, M. Wright, N. K. Elumalai and A. Uddin, Sol. Energy Mater. Sol. Cells, 2016, 147, 255–275 CrossRef CAS.
- M. I. Asghar, J. Zhang, H. Wang and P. D. Lund, Renewable Sustainable Energy Rev., 2017, 77, 131–146 CrossRef CAS.
- X. Guo, C. McCleese, C. Kolodziej, A. C. S. Samia, Y. Zhao and C. Burda, Dalton Trans., 2016, 45, 3806–3813 RSC.
- Y. Yao, G. Wang, F. Wu, D. Liu, C. Lin, X. Rao, R. Wu, G. Zhou and Q. Song, RSC Adv., 2017, 7, 42973–42978 RSC.
- Z. Li, C. Xiao, Y. Yang, S. P. Harvey, D. H. Kim, J. A. Christians, M. Yang, P. Schulz, S. U. Nanayakkara, C.-S. Jiang, J. M. Luther, J. J. Berry, M. C. Beard, M. M. Al-Jassim and K. Zhu, Energy Environ. Sci., 2017, 10, 1234–1242 RSC.
- K. Domanski, B. Roose, T. Matsui, M. Saliba, S.-H. Turren-Cruz, J.-P. Correa-Baena, C. R. Carmona, G. Richardson, J. M. Foster, F. De Angelis, J. M. Ball, A. Petrozza, N. Mine, M. K. Nazeeruddin, W. Tress, M. Gratzel, U. Steiner, A. Hagfeldt and A. Abate, Energy Environ. Sci., 2017, 10, 604–613 RSC.
- Z. Song, J. Werner, S. C. Watthage, F. Sahli, N. Shrestha, S. D. Wolf, B. Niesen, A. B. Phillips, C. Ballif, R. J. Ellingson and M. J. Heben, IEEE J. Photovolt., 2017, 7, 1563–1568 Search PubMed.
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef.
- E. Smecca, Y. Numata, I. Deretzis, G. Pellegrino, S. Boninelli, T. Miyasaka, A. La Magna and A. Alberti, Phys. Chem. Chem. Phys., 2016, 18, 13413–13422 RSC.
- A. Gomez, S. Sanchez, M. Campoy-Quiles and A. Abate, Nano Energy, 2018, 45, 94–100 CrossRef CAS.
- N. N. Shlenskaya, N. A. Belich, M. Grätzel, E. A. Goodilin and A. B. Tarasov, J. Mater. Chem. A, 2018, 6, 1780–1786 RSC.
- F. U. Kosasih and C. Ducati, Nano Energy, 2018, 47, 243–256 CrossRef CAS.
- Y.-H. Kye, C.-J. Yu, U.-G. Jong, Y. Chen and A. Walsh, J. Phys. Chem. Lett., 2018, 9, 2196–2201 CrossRef CAS PubMed.
- F. Lang, O. Shargaieva, V. V. Brus, H. C. Neitzert, J. Rappich and N. H. Nickel, Adv. Mater., 2018, 30, 1702905 CrossRef PubMed.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- Y. Li, X. Xu, C. Wang, B. Ecker, J. Yang, J. Huang and Y. Gao, J. Mater. Chem. C, 2017, 121, 3904–3910 CAS.
- N. H. Nickel, F. Lang, V. V. Brus, O. Shargaieva and J. Rappich, Adv. Electron. Mater., 2017, 3, 1700158 CrossRef.
- R.-P. Xu, Y.-Q. Li, T.-Y. Jin, Y.-Q. Liu, Q.-Y. Bao, C. O'Carroll and J.-X. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 6737–6746 CrossRef CAS PubMed.
- X. Tang, M. Brandl, B. May, I. Levchuk, Y. Hou, M. Richter, H. Chen, S. Chen, S. Kahmann, A. Osvet, F. Maier, H.-P. Steinruck, R. Hock, G. J. Matt and C. J. Brabec, J. Mater. Chem. A, 2016, 4, 15896–15903 RSC.
- B.-A. Chen, J.-T. Lin, N.-T. Suen, C.-W. Tsao, T.-C. Chu, Y.-Y. Hsu, T.-S. Chan, Y.-T. Chan, J.-S. Yang, C.-W. Chiu and H. M. Chen, ACS Energy Lett., 2017, 2, 342–348 CrossRef CAS.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- Z. Song, S. C. Watthage, A. B. Phillips, B. L. Tompkins, R. J. Ellingson and M. J. Heben, Chem. Mater., 2015, 27, 4612–4619 CrossRef CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- A. J. Pearson, G. E. Eperon, P. E. Hopkinson, S. N. Habisreutinger, J. T.-W. Wang, H. J. Snaith and N. C. Greenham, Adv. Energy Mater., 2016, 6, 1600014 CrossRef.
- Z. Song, A. Abate, S. C. Watthage, G. K. Liyanage, A. B. Phillips, U. Steiner, M. Graetzel and M. J. Heben, Adv. Energy Mater., 2016, 6, 1600846 CrossRef.
- T. Leijtens, E. T. Hoke, G. Grancini, D. J. Slotcavage, G. E. Eperon, J. M. Ball, M. De Bastiani, A. R. Bowring, N. Martino, K. Wojciechowski, M. D. McGehee, H. J. Snaith and A. Petrozza, Adv. Energy Mater., 2015, 5, 1500962 CrossRef.
- Y. Yuan, Q. Wang, Y. Shao, H. Lu, T. Li, A. Gruverman and J. Huang, Adv. Energy Mater., 2016, 6, 1501803 CrossRef.
- F. Bella, G. Griffini, J.-P. Correa-Baena, G. Saracco, M. Grätzel, A. Hagfeldt, S. Turri and C. Gerbaldi, Science, 2016, 354, 203–206 CrossRef CAS PubMed.
- F. Matteocci, L. Cinà, E. Lamanna, S. Cacovich, G. Divitini, P. A. Midgley, C. Ducati and A. Di Carlo, Nano Energy, 2016, 30, 162–172 CrossRef CAS.
- R. Cheacharoen, N. Rolston, D. Harwood, K. A. Bush, R. H. Dauskardt and M. D. McGehee, Energy Environ. Sci., 2018, 11, 144–150 RSC.
- C. Wang, Z. Song, Y. Yu, D. Zhao, R. A. Awni, C. R. Grice, N. Shrestha, R. J. Ellingson, X. Zhao and Y. Yan, Sustainable Energy Fuels, 2018 10.1039/c8se00200b.
- J. A. McLeod and L. Liu, J. Phys. Chem. Lett., 2018, 9, 2411–2417 CrossRef CAS PubMed.
- K. E. Hurst, M. J. Heben, J. L. Blackburn, T. Gennett, A. C. Dillon and P. A. Parilla, Rev. Sci. Instrum., 2013, 84, 025103 CrossRef CAS PubMed.
- R. Long, W. Fang and O. V. Prezhdo, J. Phys. Chem. Lett., 2016, 7, 3215–3222 CrossRef CAS PubMed.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- Y. Zhao and K. Zhu, Chem. Commun., 2014, 50, 1605–1607 RSC.
- C. Li, S. Pang, H. Xu and G. Cui, Sol. RRL, 2017, 1, 1700076 CrossRef.
- G. Y. Kim, A. Senocrate, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Nat. Mater., 2018, 17, 445–449 CrossRef CAS PubMed.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Nat. Energy, 2016, 2, 16195 CrossRef.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 3885 Search PubMed.
- Q. Sun, P. Fassl, D. Becker-Koch, A. Bausch, B. Rivkin, S. Bai, P. E. Hopkinson, H. J. Snaith and Y. Vaynzof, Adv. Energy Mater., 2017, 7, 1700977 CrossRef.
- E. L. Øiestad, Å. M. L. Øiestad, H. Skaane, K. Ruud, T. Helgaker, E. Uggerud and T. Vulpius, Eur. Mass Spectrom., 1995, 1, 121–129 CrossRef.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulović, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, J. Am. Chem. Soc., 2015, 137, 10048–10051 CrossRef CAS PubMed.
- J.-W. Lee, D.-J. Seol, A.-N. Cho and N.-G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- J. Schoonman, Chem. Phys. Lett., 2015, 619, 193–195 CrossRef CAS.
- Z. Guo, Y. Wan, M. Yang, J. Snaider, K. Zhu and L. Huang, Science, 2017, 356, 59–62 CrossRef CAS PubMed.
- J. Huang, Y. Yuan, Y. Shao and Y. Yan, Nat. Rev. Mater., 2017, 2, 17042 CrossRef CAS.
- S. A. Bretschneider, F. Laquai and M. Bonn, J. Mater. Chem. C, 2017, 121, 11201–11206 CAS.
- G. Han, H. D. Hadi, A. Bruno, S. A. Kulkarni, T. M. Koh, L. H. Wong, C. Soci, N. Mathews, S. Zhang and S. G. Mhaisalkar, J. Mater. Chem. C, 2018, 122, 13884–13893 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00358k |
| This journal is © The Royal Society of Chemistry 2018 |