
Visible-light promoted arene C–H/C–X lactonization via carboxylic radical aromatic substitution†
Qi
Yang‡
ab,
Zongbin
Jia‡
ab,
Longji
Li
ab,
Long
Zhang
abc and
Sanzhong
Luo
 *abc
*abc
aKey Laboratory for Molecular Recognition and Function, Institute of Chemistry, Beijing, 100190, China. E-mail: luosz@iccas.ac.cn; Web: http://luosz.iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100490, China
cCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300071, China
First published on 9th October 2017
Abstract
We report herein a photocatalytic arene lactonization via carboxylic radical aromatic substitution. This procedure is characterized by its ability to cleave inert C–X bonds (X = H, F, Cl, Br, O, S) under mild and oxidant-free conditions, thus furnishing a general and operationally simple synthetic protocol for diverse substituted coumarins.
Introduction
Redox transformation of carboxylic acids is an ancient, yet fundamental transformation, as carboxylic acids are readily available from nature or the chemical industry. A prominent early example can be traced back to Kolbe electrolysis developed in 1840s,1 when organic synthesis was born. Upon oxidation by either a chemical or electrochemical approach, aliphatic carboxylic acids undergo facile decarboxylation via transient carboxylic radical species (Scheme 1A). This process underlies many of the synthetically useful radical reactions such as the Hunsdiecker reaction and the Barton reaction.2 Recently, visible light mediated photoredox catalysis has been developed as an enabling approach for radical decarboxylation, setting the basis for a number of intriguing C–X/C–C bond formations with the in situ generated alkyl radical species following decarboxylation.3,4 It is noted that the direct radical decarboxylation of unsaturated acids, such as aryl and alkenyl acids, is more sluggish than their aliphatic homologues,5,6 which usually requires high temperature or transition-metal assistance.7 Capitalizing on this feature, we herein uncovered a distinctive reactivity of unsaturated carboxylic acids, which underwent radical aromatic substitution instead of decarboxylation, resulting in atom-economical and straightforward synthesis of coumarins via C–H or C–X bond cleavage (Scheme 1B).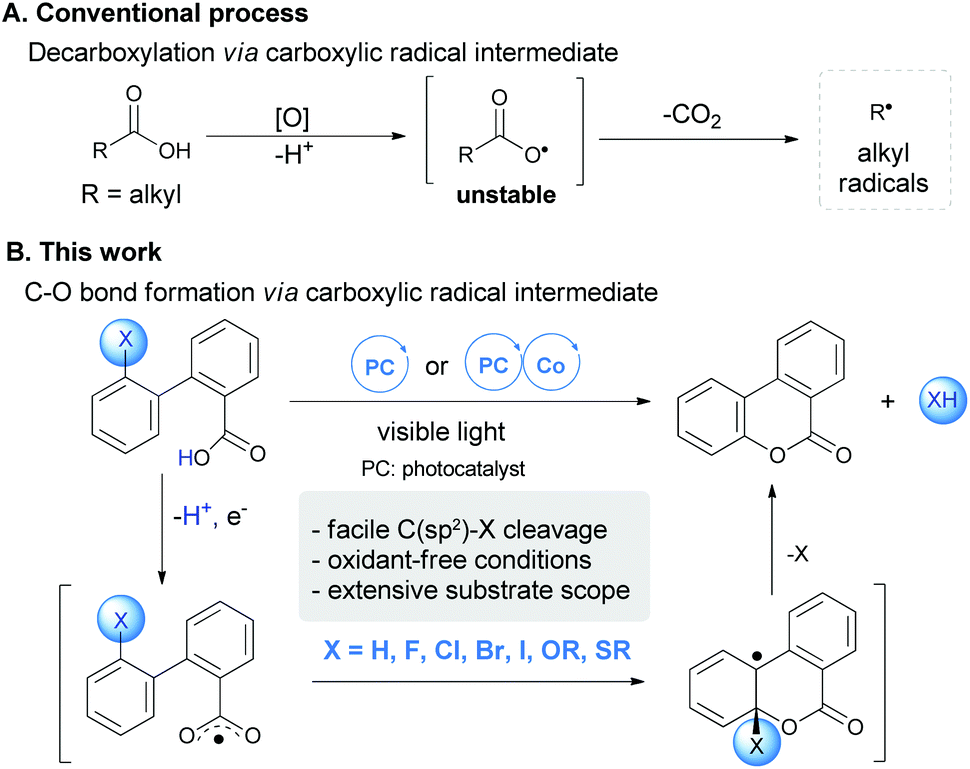 | ||
| Scheme 1 Hydrodecarboxylation and dehydrogenation of carboxylic acid via radical intermediates. | ||
Substitutional coumarin derivatives are widely found in natural and bioactive molecules, as well as in fluorescent materials, axially chiral products, and molecular rotary motors.8 Direct arene C–H oxidative lactonization of 2-alkenylbenzoic acids represents one of the most straightforward approaches for the synthesis of coumarins.9 However, most of the available methods require stoichiometric or excess amounts of oxidants under harsh conditions, thus limiting the scope as well as the applicability. Recently, Gonzalez-Gomez and coworkers reported a photo-catalytic dehydrogenative lactonization of 2-arylbenzoic acids by utilizing peroxysulphates as the oxidants with an organic photoredox catalyst.9h However, this method still required an excess of chemical oxidant and the protocol was not workable for alkenyl carboxylic acids. Recently, we reported an oxidant-free asymmetric cross-dehydrogenative coupling by merging a photocatalyst and a cobalt catalyst.10 The combination of the photocatalyst and the cobalt complex has been shown to mediate efficient hydrogen-evolution accompanying C–X/C–C bond formations.11 In order to expand this strategy to radical carboxylic acid transformations, an oxidant-free C–H lactonization process was developed, leading to the facile formation of coumarins under rather mild conditions. This method was not limited to C(sp2)–H bond cleavage, and the radical aromatic substitution could be well applied to other C(sp2)–X bonds, including the most challenging C(sp2)–F bond.12 Very recently, Weaver and Hashmi have independently achieved mild C–F bond functionalization of multifluoroarenes by photocatalysis and the reactions were proposed to proceed via an arene C–F radical anion in a formal SNAr-type pathway.13 We report herein a mechanistically distinctive radical substitution with an arene C–F bond.
Results and discussion
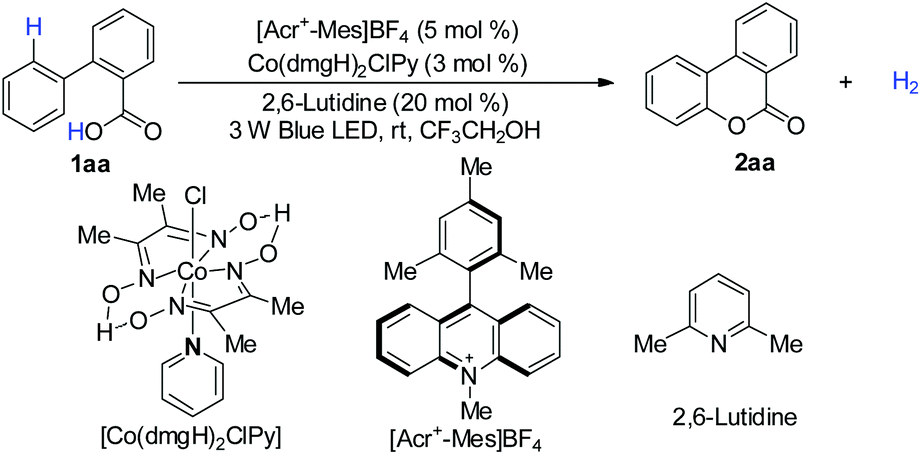
We first explored the proposed arene lactonization using 2-phenylbenzoic acid 1aa as the model substrate. We envisioned using [Acr+-Mes]BF4 in the reaction for its strong absorption band in visible light (λmax = 430 nm) and high excited state reduction potentials (>2.0 V vs. SCE).14a In further optimizations, the best results were obtained with a quantitative conversion (Table 1, entry 1). Variation of photocatalysts (Table 1, entries 2 and 3), bases (Table 1, entries 4 and 5) and solvents (Table S2†) led to a decrease in the yield of 2aa and H2 evolution. It is interesting that, under sunlight irradiation, 2aa was obtained with 88% yield and 95% H2 after 5 h (Table 1, entry 8). Control experiments revealed that photocatalyst, cobalt catalyst, and visible light were essential in the whole reaction (Table 1, entries 10–12), and no reaction or a poor yield was observed in their absence.|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) | H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (%) c (%) |
| a Reaction conditions: 1aa (0.1 mmol), [Acr+-Mes]BF4 (5 mol%), 2,6-lutidine (20 mol%) and Co(dmgH)2ClPy (3 mol%) were added to 1.0 mL CF3CH2OH, then deaerated and irradiated for 24 h by using a 3 W blue LED. b Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c Determined by gas chromatography using methane as an internal standard. d Isolated yield. e n.r. = no reaction. f n.d. = no detection. | |||
| 1 | None | 99 (95d) | >99 |
| 2 | Eosin Y instead of [Acr+-Mes]BF4 | Trace | Trace |
| 3 | Ru(bpy)3(PF6)2 instead of [Acr+-Mes]BF4 | Trace | Trace |
| 4 | DBU instead of lutidine | 90 | 95 |
| 5 | Cs2CO3 instead of lutidine | 76 | 83 |
| 6 | 2,6-Lutidine (100 mol%) | 99 | >99 |
| 7 | 2,6-Lutidine (10 mol%) | 86 | 90 |
| 8 | Under sunlight (5 h) | 88 | 95 |
| 9 | No 2,6-lutidine | 60 | 31 |
| 10 | No Co(dmgH)2ClPy | 22 | Trace |
| 11 | No [Acr+-Mes]BF4 | n.r.e | n.d.f |
| 12 | In the dark | n.r. | n.d. |
Under optimized conditions, the scope was then examined. Diverse substituted benzoic acids furnished the corresponding benzocoumarins in good to excellent yields (Scheme 2). 2-Pyrrolyl benzoic acid also furnished the desired product (Scheme 2, 2as). The reaction worked equally well on a gram scale (Scheme 2, 2aa). 4-Arylcoumarins (neoflavones), as the family members of flavonoids, are widely distributed in natural products and exhibit promising biological activities.8 A variety of 2-substituted cinnamic acids, including alkyl and unsymmetrical phenyl substituted aryl acids,8h gave the corresponding cyclization products without isomerization of the double bond (Scheme 3), which makes it possible to construct diverse unsymmetrical 4-substituted coumarins.
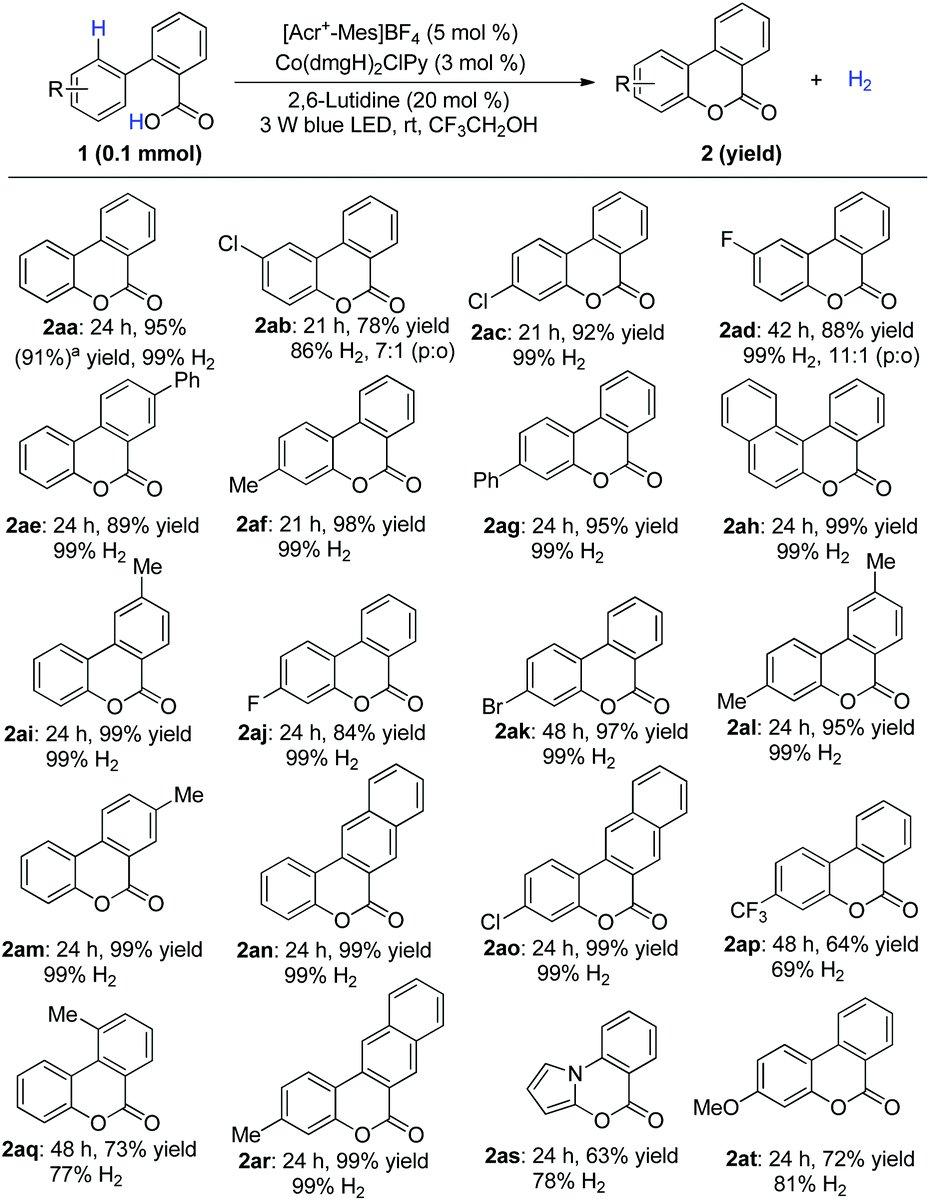 | ||
| Scheme 2 Scope of substituted 2-arylbenzoic acids. a8 mmol scale, [Acr+-Mes]BF4 (3 mol%), Co(dmgH)2ClPy (1.5 mol%) and 2,6-lutidine (20 mol%) under 30 W blue LED. | ||
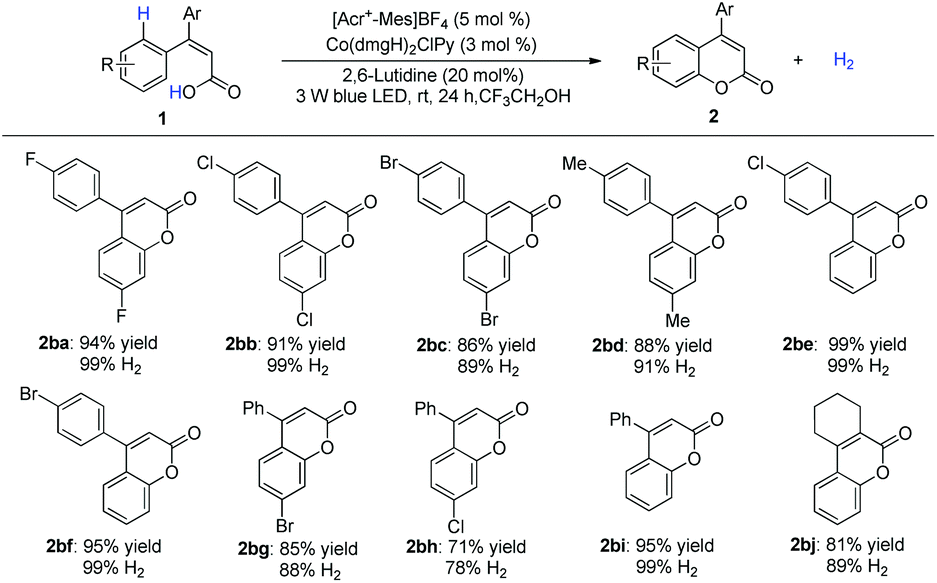 | ||
| Scheme 3 Scope of aryl cinnamic acids. | ||
To explore the synthetic potential of arene lactonization, a series of ortho-enyl benzoic acids were synthesized and tested under standard conditions (Scheme 4). 2ca, 2cb and 2cc could be obtained as five-exo adducts. It is interesting that the cyclization of 1cc resulted in double bond migration, and 1cd reacted to form a six-endo adduct 2cd without hydrogen evolution. In the latter case, the reaction likely proceeded via an anti-Markovnikov hydrocarboxylation process.14b
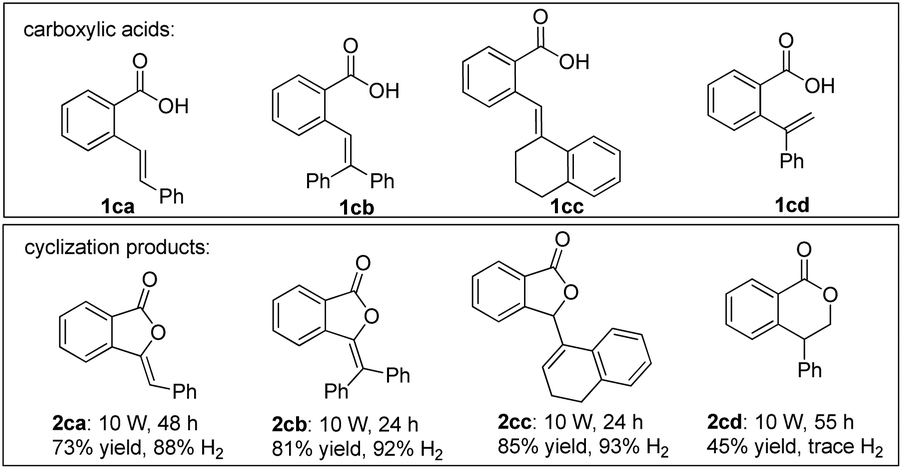 | ||
| Scheme 4 Scope of ortho-enyl benzoic acids. | ||
During the study, we found that this method could even cleave C–F in the reaction. In this context, a cobalt catalyst was not necessary. This result promoted us to explore arene C–X radical substitutions further. Substituted biphenyl acid and 2-aryl cinnamic acid reacted to give the desired products in good yield (Scheme 5, 2aa). Arene halides, including chloride and bromide, could be incorporated. Other C–X bonds such as phenol ether or thioether also reacted smoothly to give the lactonization adducts (Scheme 5). Control experiments revealed that no reaction was observed without a photocatalyst and visible light, which excluded an aromatic nucleophilic substitution process with the carboxylate as the attacking nucleophile.
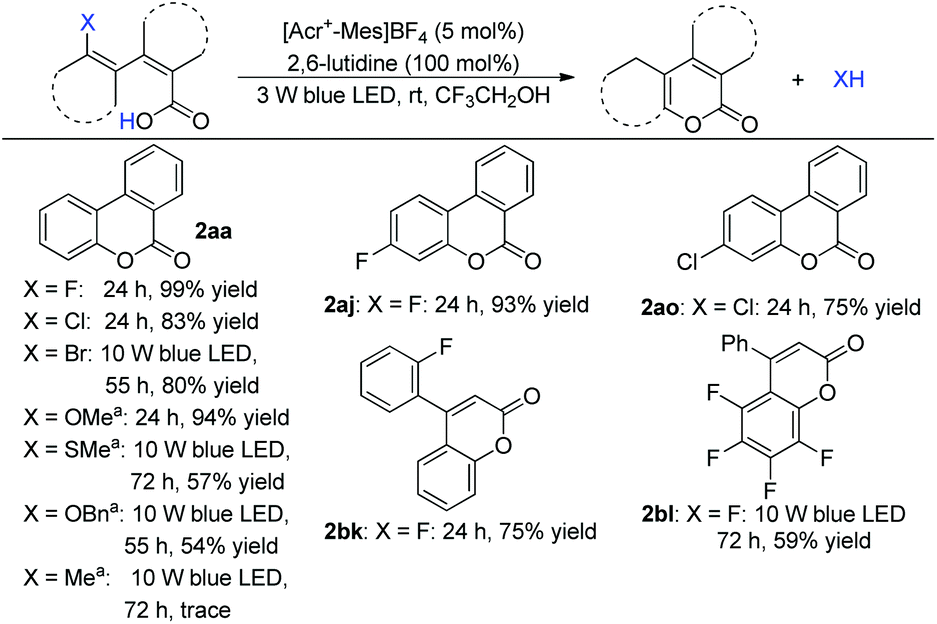 | ||
| Scheme 5 C(sp2)–X bond cleavage. a20 mol% 2,6-lutidine was added. | ||
To gain more insights into the mechanism, the kinetic isotope effect (KIE) experiments were examined by using in situ IR. A new peak at 1612 cm−1 belonging to the ester group of 2aa could be observed accompanied by the consumption of 1aa (Scheme 6b). The initial rate of oxidation of 1aaversus its deuterated analogue 1aa-D5 provided a KIE of 1.05. Similar kH/kD was obtained in mono-deuterated 1aa-D1 (determined by 1H NMR, Scheme 6a). Combined, these results suggested that C–H bond cleavage was not involved in the rate-determining step.
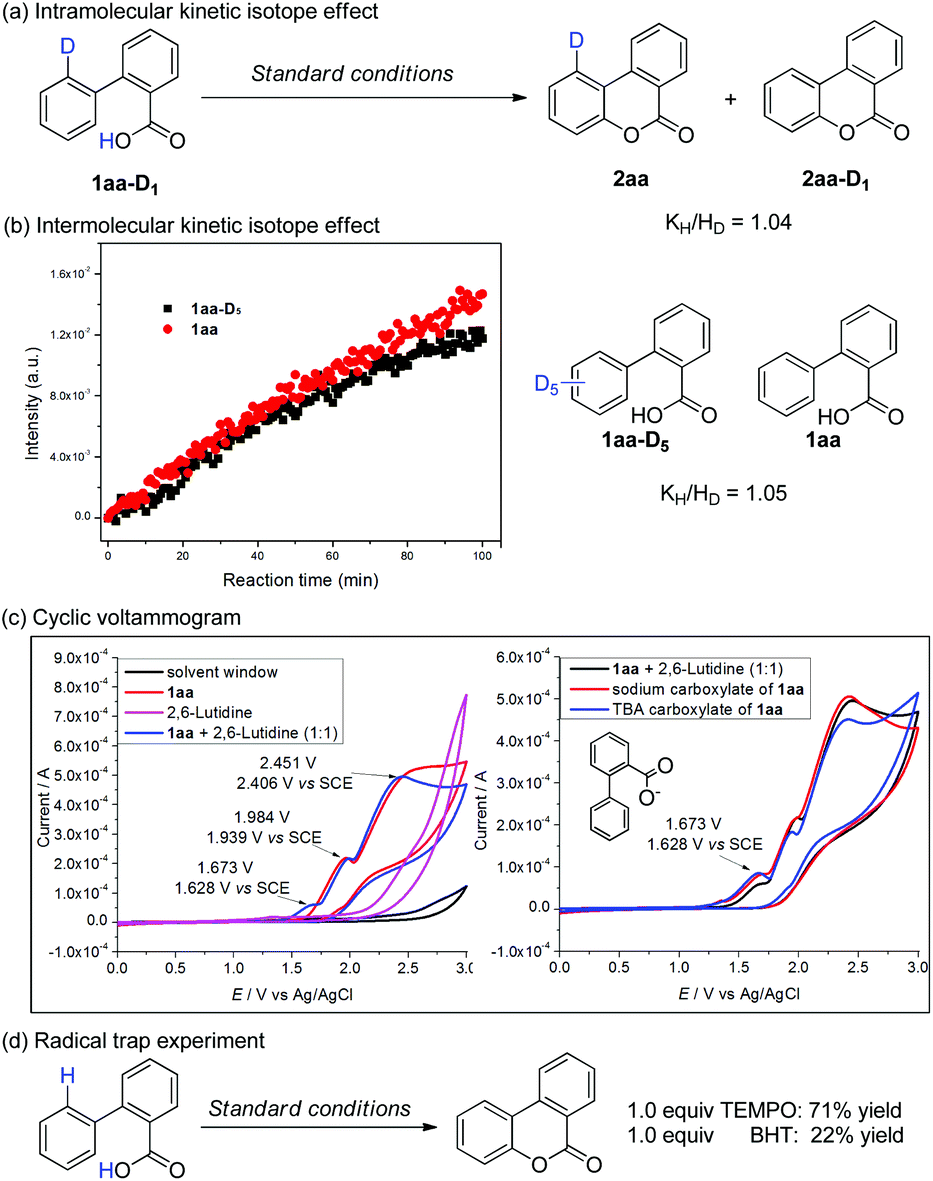 | ||
| Scheme 6 Mechanism studies. | ||
A cyclic voltammogram indicated that the oxidation potential of 1aa is 1.939 V vs. SCE. Once 2,6-lutidine was added, a lower oxidative peak appeared at 1.628 V vs. SCE, which could be ascribed as the oxidation of carboxylate (Scheme 6c). Direct CV studies of purified sodium and ammonium tetrabutyl salts (TBA) of 1aa showed nearly identical oxidative peaks (Scheme 6c).4d These results suggested that SET of carboxylate is more feasible than that of the arene ring under our catalytic conditions. In addition, control experiments indicated the inhibition effect of radical scavengers, such as TEMPO and BHT (Scheme 6d), which suggested that radical species were likely involved in this intramolecular radical process.
On the basis of the above observations, a plausible catalytic cycle is proposed in Scheme 7. Upon deprotonation, the carboxylic acid is oxidized via SET by *[Acr+-Mes], resulting in the formation of a carboxylic radical. The subsequent radical addition to an arene results in an aryl radical species and further oxidation by the cobalt catalyst leads to the desired product. The latter step may proceed via hydrogen atom abstraction (HAT) or e/H+ transfer sequence. The cobalt catalyst would serve as the proton and electron reservoir, leading to hydrogen release.15,16 An alternative mechanism scenario involving nucleophilic addition of carboxylate to the arene radical cation,11 though cannot be completely excluded, is not supported by the available experimental observations. In the C–X cleavage process in the absence of the cobalt catalyst, the generated aryl radical intermediate is reduced by [Acr˙-Mes], and elimination of X– gives the lactonization product (Scheme 7).
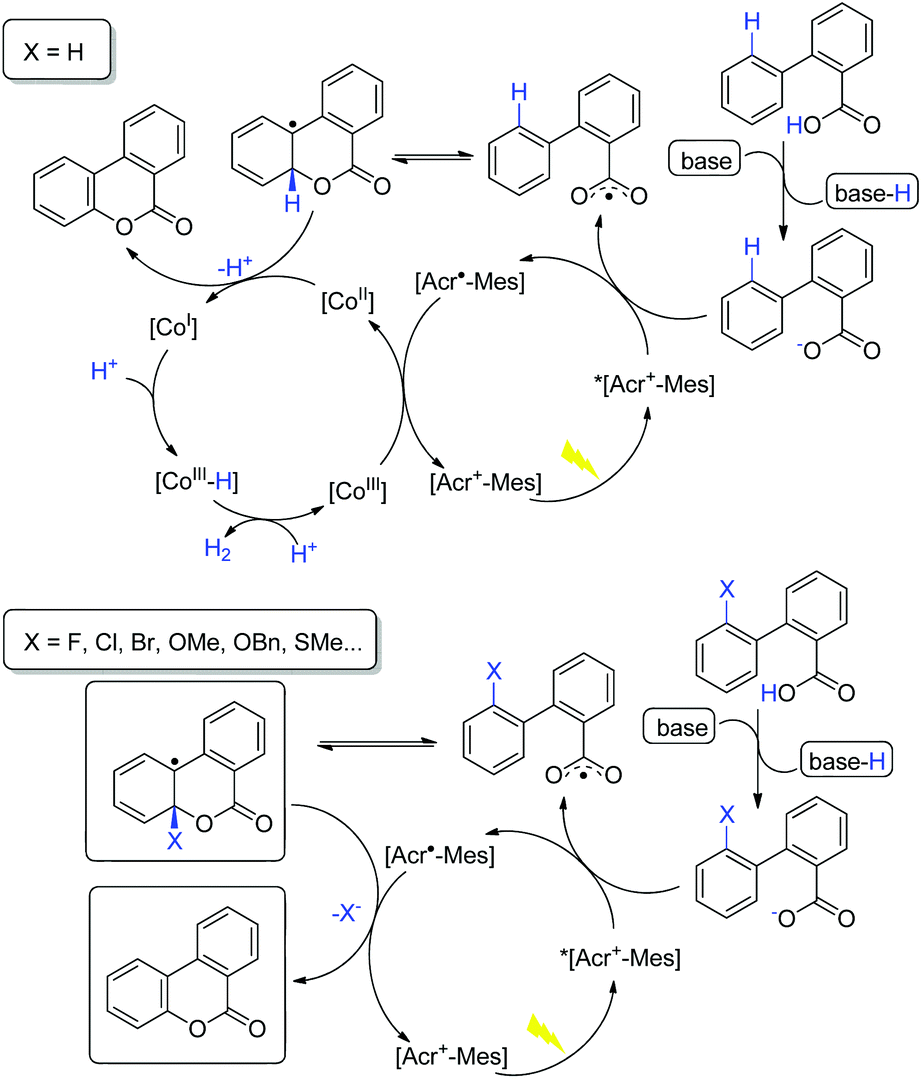 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In summary, we have developed a visible-light promoted arene C–H/C–X lactonization via a carboxylic radical intermediate. This protocol constitutes a facile, environmentally benign, and general method for preparing diverse coumarin derivatives under mild reaction conditions. This oxidant-free system, mediated by a photocatalyst and a cobalt catalyst, offers a new strategy beyond traditional catalysis using stoichiometric or excess amounts of oxidants. We believe that the current strategy via carboxylic radicals will find more application in intermolecular C–H and C–X bond functionalization processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21390400, 21572232, 21672217 and 21521002) for financial support. S. L. is supported by the National Program of Top-notch Young Professionals and CAS One-hundred Talents Program.Notes and references
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623 CrossRef CAS.
- (a) D. H. R. Barton, H. A. Dowlatshahi, W. B. Motherwell and D. Villemin, J. Chem. Soc., Chem. Commun., 1980, 732 RSC; (b) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939 RSC; (c) D. H. R. Barton, D. Crich and W. B. Motherwell, Tetrahedron Lett., 1985, 41, 3901 CrossRef CAS; (d) M. F. Saraiva, M. R. C. Couri, M. Le Hyaric and M. V. de Almeida, Tetrahedron, 2009, 65, 3563 CrossRef CAS.
- For selected reviews, see: (a) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (b) H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983 CrossRef CAS; (c) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- (a) J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854 RSC; (c) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed; (d) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340 CrossRef CAS PubMed; (e) L. Candish, L. Pitzer, A. Gómez-Suárez and F. Glorius, Chem. – Eur. J., 2016, 22, 4753 CrossRef CAS PubMed; (f) A. Millet, Q. Lefebvre and M. Rueping, Chem. – Eur. J., 2016, 22, 13464 CrossRef CAS PubMed.
- L. Candish, M. Freitag, T. Gensch and F. Glorius, Chem. Sci., 2017, 8, 3618 RSC.
- (a) J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683 CrossRef CAS; (b) A. Fraind, R. Turncliff, T. Fox, J. Sodano and L. R. Ryzhkov, J. Phys. Org. Chem., 2011, 24, 809 CAS.
- (a) N. Rodrígueza and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864 CrossRef CAS PubMed.
- (a) B. I. Alo, A. Kandil, P. A. Patil, M. J. Sharp, M. A. Siddiqui and V. Snieckus, J. Org. Chem., 1991, 56, 3763 CrossRef CAS; (b) N. Tibrewal, P. Pahari, G. Wang, M. K. Kharel, C. Morris, T. Downey, Y. Hou, T. S. Bugni and J. Rohr, J. Am. Chem. Soc., 2012, 134, 18181 CrossRef CAS PubMed; (c) R. Omar, L. Li, T. Yuan and N. P. Seeram, J. Nat. Prod., 2012, 75, 1505 CrossRef CAS PubMed; (d) M. Nakashima, R. Clapp and J. Sousa, Nature Phys. Sci., 1973, 245, 124 CrossRef CAS; (e) C.-W. Yang, T.-H. Hsia, C.-C. Chen, C.-K. Lai and R.-S. Liu, Org. Lett., 2008, 10, 4069 CrossRef CAS PubMed; (f) S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80 CrossRef CAS PubMed; (g) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615 CrossRef CAS PubMed; (h) J. Li, H. Chen, D. Zhang-Negrerie, Y. Du and K. Zhao, RSC Adv., 2013, 3, 4311 RSC.
- (a) G. W. Kenner, M. A. Murray and C. M. B. Tylor, Tetrahedron, 1957, 1, 259 CrossRef CAS; (b) D. Davies and C. Waring, Chem. Commun., 1965, 263 RSC; (c) Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574 CrossRef CAS PubMed; (d) Y. Wang, A. V. Gulevich and V. Gevorgyan, Chem. – Eur. J., 2013, 19, 15836 CrossRef CAS PubMed; (e) J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed; (f) X. Wang, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2014, 53, 11084 CrossRef CAS PubMed; (g) H. Togo, T. Muraki and M. Yokoyama, Tetrahedron Lett., 1995, 36, 7089 CrossRef CAS; (h) N. P. Ramirez, I. Bosque and J. C. Gonzalez-Gomez, Org. Lett., 2015, 17(18), 4550–4553 CrossRef CAS PubMed; (i) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef CAS PubMed.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694 CrossRef CAS PubMed.
- For selected examples, see: (a) J.-J. Zhong, Q.-Y. Meng, B. Liu, X.-B. Li, X.-W. Gao, T. Lei, C.-J. Wu, Z.-J. Li, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 1988 CrossRef CAS PubMed; (b) X.-W. Gao, Q.-Y. Meng, J.-X. Li, J.-J. Zhong, T. Lei, X.-B. Li, C.-H. Tung and L.-Z. Wu, ACS Catal., 2015, 5, 2391 CrossRef CAS; (c) G. Zhang, C. Liu, H. Yi, Q.-Y. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273 CrossRef CAS PubMed; (d) G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037 CrossRef CAS PubMed; (e) H. Yi, L. Niu, C. Song, Y. Li, B. Dou, A. K. Singh and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 1120 CrossRef CAS PubMed; (f) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X. L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080 CrossRef CAS PubMed; (g) Y.-W. Zheng, P. Ye, B. Chen, Q.-Y. Meng, K. F. W. Wang, L.-Z. Wu and C.-H. Tung, Org. Lett., 2017, 19, 2206 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413 CrossRef CAS; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed; (c) T. Ahrens, J. Kohlmann, M. Ahrens and T. Brau, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed; (d) M. F. Juehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed.
- (a) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002 CrossRef CAS PubMed; (b) A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206 RSC; (c) A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC; (d) S. Senaweera and J. D. Weaver, J. Am. Chem. Soc., 2016, 138, 2520 CrossRef CAS PubMed; (e) J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416 CrossRef CAS PubMed; (f) J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266 CrossRef CAS PubMed; (g) S.-F. Wang, X.-P. Cao and Y. Li, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201706597.
- (a) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS PubMed; (b) A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334 CrossRef CAS PubMed.
- M. Xiang, Q.-Y. Meng, J.-X. Li, Y.-W. Zheng, C. Ye, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2015, 21, 18080 CrossRef CAS PubMed.
- The cobalt catalyst showed rather lower oxidation potential Co(dmgH)2Cl Py (E°(CoIII/II) = −0.68 V vs. SCE and E°(CoII/I) = −1.13 V vs. SCE, J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed ), excluding the possibility of direct participation of Co catalyst in substrate oxidation (oxidation potential of 1aa is 1.939 V vs. SCE).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00826k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |