
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of a tetra[6,7]quinoxalinoporphyrazine-based near infrared photosensitizer†
Pin Shaoa,
Shaojuan Zhanga,
Shudong Hub,
Le Hanc,
Ningyang Jia*d and
Mingfeng Bai *aefg
*aefg
aDepartment of Radiology, University of Pittsburgh, 100 Technology Drive, Pittsburgh, PA 15219, USA. E-mail: mingfeng.bai@vanderbilt.edu
bDepartment of Radiology, The Affiliated Renmin Hospital, Jiangsu University, Zhenjiang, Jiangsu 212002, China
cVanderbilt University Institute of Imaging Sciences, Vanderbilt University Medical Center, Nashville, TN 37232, USA
dDepartment of Radiology, Shanghai Eastern Hepatobiliary Surgery Hospital, Shanghai 200438, China
eDepartment of Medicine, University of Pittsburgh, 3501 Fifth Ave, Pittsburgh, PA 15213, USA
fDepartment of Bioengineering, University of Pittsburgh, Pittsburgh, PA 15261, USA
gUniversity of Pittsburgh Cancer Institute, Pittsburgh, PA 15232, USA
First published on 30th October 2017
Abstract
Here we report a near infrared, water-soluble, functional and dendrimeric photosensitizer (PS) based on quinoxalinoporphyrazine structure. The photophysical properties and in vitro photodynamic therapy results suggest that this quinoxalinoporphyrazine-based dendrimer may serve as an efficient near infrared (NIR) PS platform.
Introduction
Photodynamic therapy (PDT) is a minimally invasive, effective and controllable cancer treatment technique and has received increasing attention recently, largely due to the development of new light sources and photosensitizers (PSs).1 PDT treatment relies on three elements: PS, oxygen and light irradiation. PS can react with oxygen and generate toxic reactive oxygen species (ROS) under light irradiation with specific wavelength to kill cancer cells. The treatment can occur mainly at the diseased area by using localized light irradiation and targeted PSs, and therapeutic efficacy can be improved by repeated light irradiation without increasing toxicity (e.g. one injection of PS and multiple light irradiations). Compared to surgery, radiation, and chemo therapy, PDT has less side effects and usually causes little or no physical damage after the site recovery.2 In addition, the intrinsic fluorescence of PS allows for fluorescence imaging-guided therapy, offering PDT a desired “see and treat” approach.The success of PDT largely depends on the wavelength at which PSs operate. A major limitation of any optical technology is related to tissue absorption and scattering of light. This problem can be partially resolved by employing near infrared (NIR) light (700–900 nm) under which tissue absorption and scattering is relatively low,3 allowing for deep tissue imaging and treatment. In addition, tissue autofluorescence is negligible in the NIR region, allowing for high-contrast optical imaging. Moreover, as compared to light in the visible region where most of the current PSs operate, NIR light causes significantly less damage to normal tissue in the irradiated region due to lower energy. As such, NIR PSs have great potential to treat tumors with high efficacy and low side effects, as well as providing fluorescence imaging guidance with high contrast.
Currently, most clinically approved PSs are based on porphyrin structure. However, most porphyrin derivatives possess deficient light absorption ability and usually require strong light irradiation source such as laser for the treatment, which may induce serious normal tissue burning. In addition, their maximum absorption is typically in the visible region (below 700 nm), which limits the light penetration and only superficial tissue can be treated.4,5 For example, Photofrin, the first FDA approved and the most widely used PS, has peak absorption at 632 nm with a low molar extinguish coefficient (ε = 3000 M−1 cm−1).5 These disadvantages limit the extensive clinic application of PDT. Therefore, significant effort has been devoted to developing more efficient PSs.6–10 Phthalocyanine (Pc) derivatives stand out as new generation PSs for their extraordinary light absorption ability (ε = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 M−1 cm−1) in the red to NIR region and large singlet oxygen generation yields.11–16 In particular, as part of the Pc family, several quinoxalinoporphyrazine derivatives with intense NIR absorption as well as efficient singlet oxygen generation ability in organic solvents have been reported.17,18 However, these Pc derivatives are not suitable for biomedical applications due to significant aggregation and solubility issues owing to the extended π systems.19,20 Further modification with four quaternized amine groups improved the water solubility; however, significant aggregation still exists in water.21
000–200000 M−1 cm−1) in the red to NIR region and large singlet oxygen generation yields.11–16 In particular, as part of the Pc family, several quinoxalinoporphyrazine derivatives with intense NIR absorption as well as efficient singlet oxygen generation ability in organic solvents have been reported.17,18 However, these Pc derivatives are not suitable for biomedical applications due to significant aggregation and solubility issues owing to the extended π systems.19,20 Further modification with four quaternized amine groups improved the water solubility; however, significant aggregation still exists in water.21
In this study, we aimed to synthesize a water-soluble, stable, functional and dendrimeric tetra[6,7]quinoxalinoporphyrazine PS for PDT treatment in the NIR region. The dendrimeric structure was adopted to address aggregation issue with surface carboxylic acid groups that provide water solubility and conjugation capability to targeting molecules.22 The photophysical properties were investigated and the in vitro PDT effect was evaluated.
Results and discussion
The synthetic route of our dendrimeric tetra[6,7]quinoxalinoporphyrazine PS is displayed in Scheme 1. The azide-functionalized polyamide 3 was synthesized through the conjugation of 2-azidoacetic acid to tri-ester amine 1 using DCC/HOBt. Compound 3 was obtained as a white solid with a moderate yield of 48%. Bromination of 2,2′-thenil 4 in chloroform using bromide led to compound 5 in a good yield of 78%. Condensing 5 with equal amount of 4,5-diaminophthalonitrile in acetic acid at room temperature produced dicyanoquinoxaline derivative 6 in a high yield (85%), and the next Sonogashira coupling reaction of 6 afforded dialkynylated compound 7 in a high yield as well (88%). The backbone of tetra[6,7]quinoxalinoporphyrazine was assembled by cyclotetramerisation of 7 at a high temperature with base. TQP1 was obtained as a green solid in a relatively low yield (19%), and it showed good solubility in organic solvents due to eight peripheral triisopropylsilane groups. After TQP1 was treated with tetrabutylammonium fluoride, the derivative with free terminal alkyne groups was obtained with poor solubility and used directly in the next “click” reaction with the azide-functionalized polyamide 3. The obtained green solid TQP2 is highly soluble in organic solvents due to the bulky dendronized polyamide arms. The next deprotection reaction in formic acid yielded TQP3. The disappearance of proton peak at 1.43 ppm in 1H NMR spectrum indicated that the t-butyl groups were completely removed. The molecular weight of TQP3 was analysed using a 15 Tesla Bruker matrix assisted laser desorption/ionization Fourier transform ion cyclotron resonance (MALDI-FTICR) mass spectrometer and the spectra was internally calibrated using multiple isotopes of insulin chain B (oxidized) and ubiquitin. The highest intensity signal (Fig. S1a† measured at m/z 4277.9524 was compared to the theoretically calculated value at m/z 4277.9565 in Fig. S1b†) (Compass Isotope Pattern; Bruker) which corresponds to <1 ppm error. With twenty-four carboxylic acid groups, the acid form of TQP3 is soluble in water. Its water solubility can be further improved by treating TQP3 with NaHCO3 to yield its sodium salt TQP3-Na.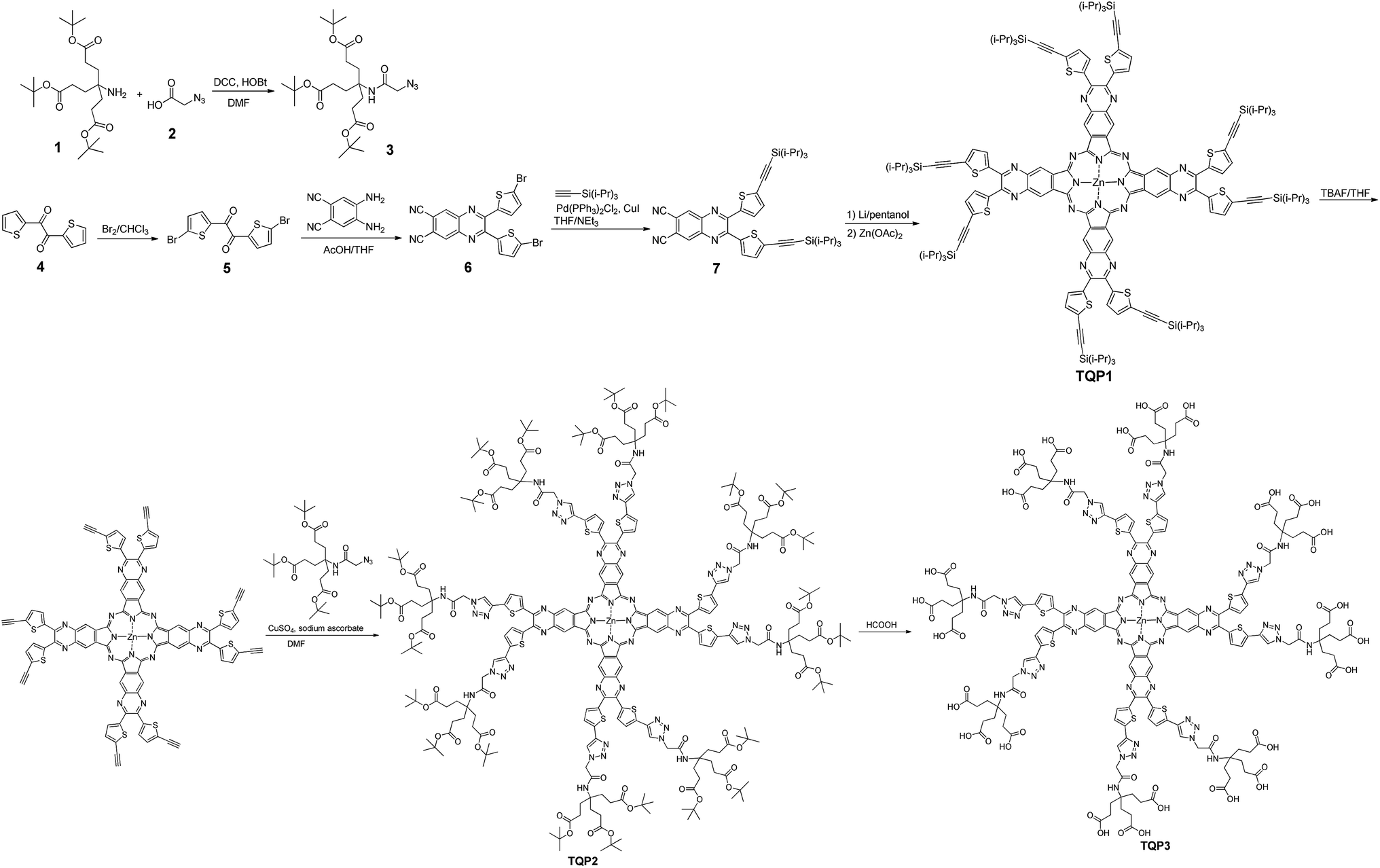 | ||
| Scheme 1 The synthesis of TQP3. | ||
The absorption and emission spectra of TQP3 and TQP3-Na are displayed in Fig. 1. As expected, both TQP3 and TQP3-Na are soluble in water. In DMSO, TQP3 showed a sharp and intense NIR absorption spectrum peaked at 781 nm (ε = 1.51 × 105 M−1 cm−1) and similar to the other Pc PSs, it possessed a small Stokes shift with the weak emission peak at 788 nm. As we expected, the excitation spectrum (Fig. S2†) peaked at around the absorption maximum without a shoulder peak (seen in the absorption spectrum at around 725 nm), suggesting that the aggregated TQP3 was not responsible for the fluorescence around 788 nm. In water, TQP3 showed significant aggregation, with or without treatment with surfactant (0.01% Kolliphor RH40), as evidenced by the broad absorption band with peak at around 725 nm (Fig. 1). Such aggregation in aqueous solutions led to negligible fluorescence, as shown in Fig. 1.
 | ||
| Fig. 1 The absorption (solid) and emission (dot) spectra of 1 μM dye solutions. (TQP3 in water: green; TQP3 in DMSO: blue; TQP3 in 0.01% Kolliphor RH40: red; TQP3-Na in water: black; TQP3-Na in 0.01% Kolliphor RH40: orange. λex = 730 nm). | ||
It has been previously reported that electrostatic repulsion is an efficient method to reduce the aggregation of aromatic molecules.23,24 We therefore deprotonated the peripheral carboxylic acid groups with sodium bicarbonate to reduce the aggregation and studied the photophysical properties. As shown in Fig. 1, the absorption of TQP3-Na in the NIR region is stronger than that of TQP3, although shoulder absorption around 725 nm that is associated with aggregation still dominates the absorption spectrum. This suggests that electrostatic repulsion only partially suppressed aggregation of this big π system. The aggregation may be further reduced though, if the alkynyl substitution was introduced non-peripherally, instead of peripherally. A recent study has shown that non-peripheral isomer showed less degree of aggregation than peripheral dendrimeric Pc PS, as evidenced by the minimal shoulder absorption peak even at higher concentrations.25 Because aggregation is associated with fluorescence and singlet oxygen quenching, accurate fluorescence and singlet oxygen quantum yields were not able to be measured and are therefore not reported.
We then added surfactant Kolliphor RH40 to further reduce the aggregation and the resulting micelles showed improved absorption at 781 nm as compared to TQP3-Na (Fig. 1). In addition, emission peak at 788 nm was observed whereas TQP3-Na without surfactant showed minimal fluorescence. In our previous studies, we have shown that surfactants were able to enhance fluorescence of dyes with big π systems.26,27 These results suggest that the dendrimeric structure offered water solubility of quinoxalinoporphyrazine, and introducing electrostatic repulsion and surfactant could further reduce aggregation.
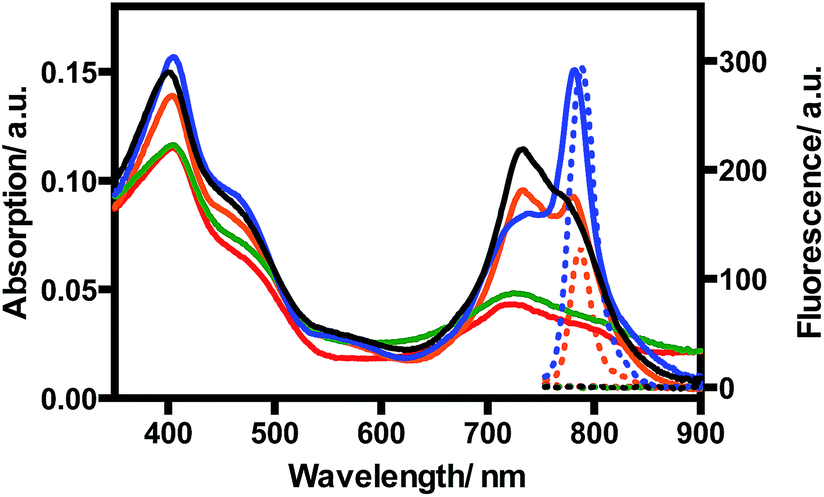
TQP3-Na incorporated in Kolliphor RH40 micelles (TQP3-Na-M), which has the most favorable photophysical properties, was selected as the PS in the in vitro PDT study. As a proof-of-principle, we chose widely used murine raw 264.7 macrophage cells to evaluate the in vitro PDT effect of TQP3-Na-M. We first evaluated the singlet oxygen generation ability of TQP3-Na-M in water by monitoring the fluorescence change of singlet oxygen sensor green (SOSG). As shown in Fig. 2, when TQP3-Na-M was irradiated by LED light at 780 nm, increasing green fluorescence from SOSG was observed, indicating that TQP3-Na-M can efficiently generate singlet oxygen in water. We then investigated the singlet oxygen generation of TQP3-Na-M in the cell culture medium RPMI. Similar to the results in Fig. 2, in RPMI (Fig. S3a†) or saline (Fig. S3c†), TQP3-Na-M showed gradually increasing green fluorescence during the NIR irradiation. Without NIR irradiation, no significant fluorescence intensity change was observed (Fig. S3b and d†). This result indicates that TQP3-Na-M was effective in singlet oxygen production in RPMI.
 | ||
| Fig. 2 The fluorescence spectra change of SOSG (5 μM) treated with TQP3-Na (1 μM) in 0.01% Kolliphor RH40 and irradiation by LED light at 780 nm. | ||
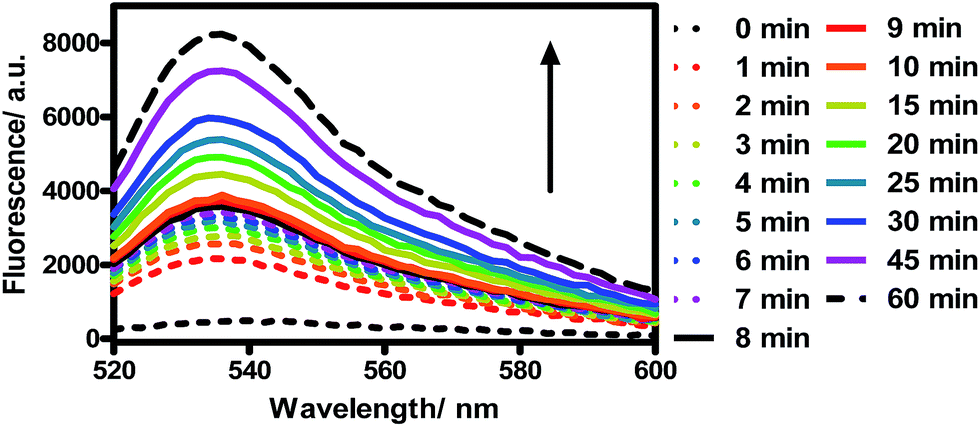
Next, we evaluated the cytotoxicity of TQP3-Na-M (5 μM TQP3-Na) and the Kolliphor RH40 surfactant (micelles without TQP3-Na) against raw 264.7 cells. As shown in Fig. 3, no significant cell death was observed when the cells were treated with 0.01% Kolliphor RH40 surfactant, with or without NIR light irradiation. Treatment with TQP3-Na-M without NIR light irradiation did not lead to cell death either, and even with 30 minutes NIR light irradiation, no immediate cell death was observed. However, after the cells were returned to incubator for 18 hours after light treatment, almost all cells were dead, suggesting programmed cell death (apoptosis). As a control group, cells incubated for 18 hours after treatment with 0.01% Kolliphor RH40 and NIR light irradiation did not lead to significant cell death (Fig. 3). These results suggest that TQP3-Na-M was phototoxic to cells but not toxic without light treatment.
 | ||
| Fig. 3 The cell viability of raw 264.7 macrophage when incubated with RPMI, 0.01% Kolliphor RH40 micelles (0 and 18 h incubation), or TQP3-Na-M (5 μM TQP3-Na, 0 and 18 h incubation) in the absence and presence of LED light irradiation at 780 nm for 30 minutes. | ||
The in vitro PDT effect of TQP3-Na-M was evaluated at various concentrations and light doses. As shown in Fig. 4, the dark toxicity of TQP3-Na-M was low at all concentrations ranged from 0.1 to 10 μM. After 780 nm light irradiation for 30 minutes, TQP3-Na-M showed concentration-dependent photo cytotoxicity. PDT with 5 μM TQP3-Na-M induced cell death as high as 96%. We then investigated the effects of light dose and post-PDT incubation time on the therapeutic outcome. Similar to the concentration dependency, TQP3-Na-M showed light dose-dependent and post-PDT incubation time-dependent PDT effects. As displayed in Fig. 5, 1 minute, 10 minute and 30 minute light irradiation induced negligible, 64% and 96% cell death, respectively. Post-PDT incubation time also has large effect on the therapeutic outcome. As shown in Fig. S4,† more cell death was observed with longer post-PDT incubation time. While roughly 50% cell death was observed after 4 hours of post-PDT incubation, almost all cells were dead at 18 hour post-PDT incubation time. This programmed cell death appears to correlate with apoptosis, as the duration of apoptosis is estimated to be from 12 to 24 hours, although morphologic changes can be visualized in less than two hours.28 The morphology of cells at 4 hours after PDT treatment also supports apoptotic cell death (Fig. 6), characterized by cell shrinkage, membrane blebbing and apoptotic bodies formation.29 The in vitro PDT results suggest that PDT with TQP3-Na-M can cause effective apoptotic cell death in a concentration- and light dose-dependent manner.
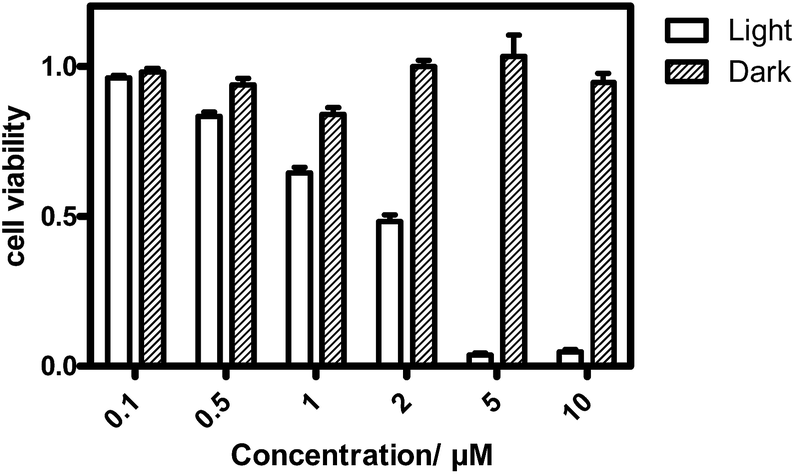 | ||
| Fig. 4 The concentration-dependent PDT effect of TQP3-Na-M in the absence and presence of LED light at 780 nm for 30 min. After PDT treatment, the cells were incubated for 18 hours before the cell viability test. | ||
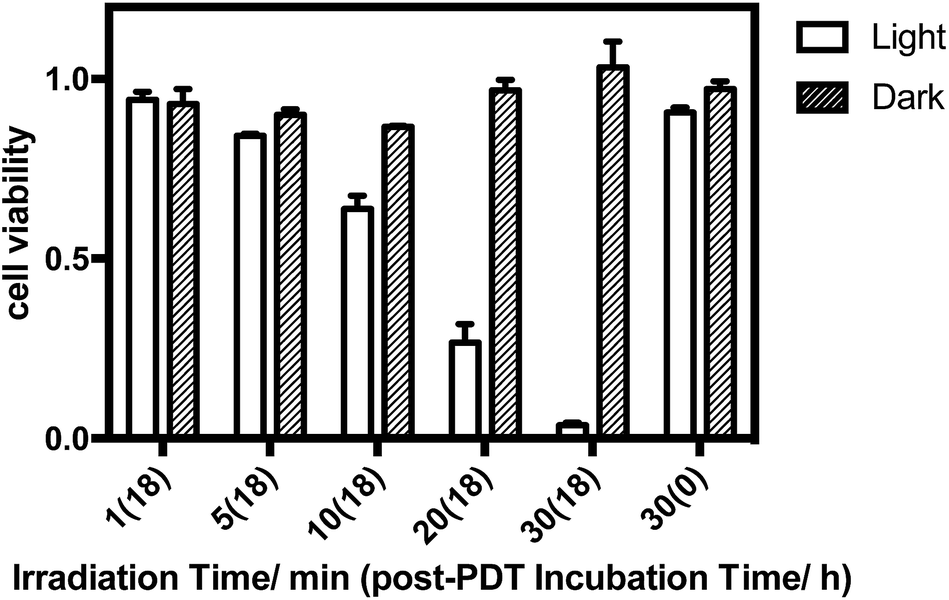 | ||
| Fig. 5 The light dose-dependent PDT effect of TQP3-Na-M (5 μM TQP3-Na). | ||
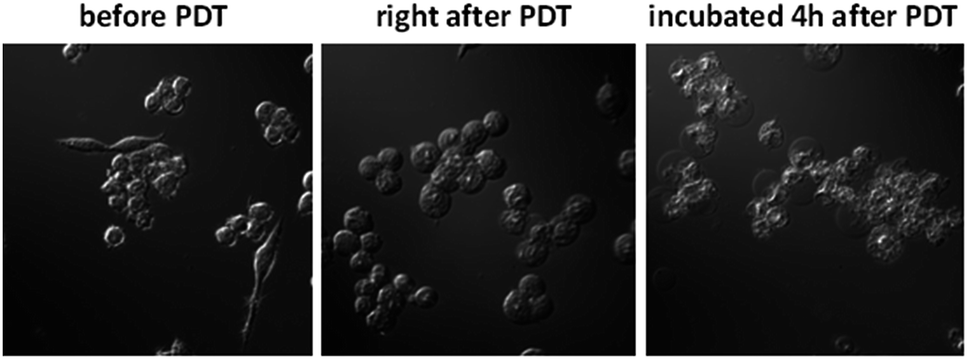 | ||
| Fig. 6 The cell morphologic change after PDT treatment. | ||
Experimental
General
The solvents used are of commercial grade. Column chromatography was performed on the silica gel (standard grade, 60A, Sorbtech). 1H and 13C NMR spectra were recorded on a Brucker Avance III 400 MHz instrument. MALDI-TOF mass spectra were recorded on a PerSeptive Voyager STR MS spectrometer. HRMS of TQP3 was performed using a 15 Tesla Bruker MALDI-FTICR mass spectrometer (Bruker Daltonics, Billerica, MA, USA). The instrument was equipped with an Apollo II dual MALDI/Electrospray (ESI) ion source, a Smartbeam II 2 kHz Nd:YAG (355 nm) laser and a Para ICR cell. UV/Vis spectra were recorded on a Cary 100 Bio UV-Vis spectrophotometer, and fluorescence spectra were recorded on a Cary Eclipse fluorescence spectrophotometer. Raw 264.7 cells were purchased from ATCC (Catalog #TIB-71, Manassas, VA, USA). The cell viability was evaluated by Synergy™ H4 Hybrid Multi-Mode Microplate Reader using CellTiter-Glo Luminescent Cell Viability Assay kit. The cell morphology change after PDT was monitored by a Zeiss Axio Observer fluorescent microscopy system.Di-tert-butyl-4-(2-azidoacetamido)-4-(3-(tert-butoxy)-3-oxoproyl)heptanedioate (3)
A mixture of triester amine 1 (1.30 g, 3.1 mmol), 2-azidoacetic acid 2 (0.30 g, 3 mmol), N,N′-dicyclohexylcarbodiimide (0.62 g, 3 mmol) and 1-hydroxybenzotriazole hydrate (0.41 g, 3 mmol) in anhydrous DMF (10 mL) was stirred at room temperature for 24 hours under argon. The white precipitate was removed by filtration and DMF was evaporated by rotary evaporator. The crude product was purified over silica gel column chromatography using hexane/ethyl acetate (7/3) as eluent resulting in 3 (0.71 g, 48%) as a white solid. 1H NMR (CDCl3, 273 K): δ = 6.52 (s, 1H), 3.86 (s, 2H), 2.17 (t, 6H, J = 3.2 Hz), 1.94 (t, 6H, J = 3.2 Hz), 1.39 (s, 27H). 13C NMR (CDCl3, 273 K): δ = 172.72, 166.43, 80.90, 57.98, 52.85, 29.88, 29.69, 28.08.1,2-Bis(5-bromothiophen-2-yl)ethane-1,2-dione (5)
A mixture of 2,2′-thenil 4 (1.6 g, 7.2 mmol) and sodium bicarbonate (2.68 g, 31.9 mmol) in chloroform (8 mL) was cool to 0 °C, and then bromine (5.12 g, 32 mmol) was added dropwise. The resulting mixture was heated to 80 °C for 4 hours and then cooled to room temperature. The excess bromine was quenched with sodium thiosulfate solution, and the crude product was purified over silica gel column chromatography using hexane/dichloromethane (6/1) as eluent. 5 (2.1 g, 78%) was obtained as yellow solid. 1H NMR (CDCl3, 273 K): δ = 7.90 (d, 2H, J = 4.4 Hz), 7.19 (d, 2H, J = 4.4 Hz).2,3-Bis(5-bromothiophen-2-yl)quinoxaline-6,7-dicarbonitrile (6)
4,5-Diaminophthalonitrile (0.3 g, 1.9 mmol) was dissolved in THF/AcOH (36 mL, 4/5) and then 5 (0.71 g, 1.9 mmol) was added. The resulting mixture was stirred at room temperature for 1 hour, and then solvents were removed by rotary evaporation. 6 was obtained as an orange solid (0.8 g, 85%) after being purified over silica gel column using gradient eluent from hexane/dichloromethane (4/1) to dichloromethane. 1H NMR (CDCl3, 273 K): δ = 8.43 (s, 2H), 7.39 (d, 2H, J = 4.0 Hz), 7.06 (d, 2H, J = 4.0 Hz). 13C NMR (CDCl3, 273 K): δ = 148.55, 141.52, 140.79, 136.08, 131.71, 131.21, 120.40, 114.99, 114.41.2,3-Bis(5-((triisopropylsilyl)ethynyl)thiophen-2-yl)quinoxaline-6,7-dicarbonitrile (7)
A mixture of 6 (0.50 g, 1 mmol), copper(I) iodide (0.023 g, 0.12 mmol), Pd(PPh3)2Cl2 (0.083 g, 0.12 mmol), ethynyltriisopropylsilane (0.66 mL, 2.9 mmol) and trimethylamine (1 mL) in anhydrous THF (15 mL) was refluxed under argon for 24 hours. To the cooled reaction mixture ethyl acetate (30 mL) and water (15 mL) were added. The organic phase was separated and dried over anhydrous Na2SO4. The crude product was purified over silica gel column using hexane/dichloromethane (4/1 to 1/1) as the eluent. 7 was obtained as an orange solid (0.62 g, 88%). 1H NMR (CDCl3, 273 K): δ = 8.42 (s, 2H), 7.44 (d, 2H, J = 4.0 Hz), 7.14 (d, 2H, J = 4.0 Hz), 1.15 (s, 42H). 13C NMR (CDCl3, 273 K): δ = 149.14, 140.91, 140.62, 136.10, 132.97, 131.18, 130.05, 115.07, 114.25, 100.70, 98.49, 18.77, 11.40.Zinc(II) tetra[6,7]quinoxalinoporphyrazine TQP1
Metal lithium (0.014 g, 4.7 mmol) was dissolved in anhydrous pentanol (5 mL) at 100 °C under argon. To the cooled lithium/pentanol solution, compound 7 (0.13 g, 0.18 mmol) was added, and the resulting mixture was heated to 150 °C under argon. After 1 hour, zinc acetate (0.13 g, 0.7 mmol) was added and continued heating for another 3 hours. The reaction mixture was cooled to room temperature and pentanol was removed under high vacuum. The resulting green solid was extracted with THF. TQP1 (0.025 g, 19%) was purified by flash chromatography twice. At the first time, toluene/THF (100% toluene to 50% toluene) was applied, and at the second time hexane/THF (100/1 to 5/1) was applied. 1H NMR (d8-THF, 273 K): δ = 9.31 (s, 8H), 7.44 (d, 8H, J = 3.6 Hz), 7.32 (d, 8H, J = 3.6 Hz), 1.35 (s, 168H). Due to the aggregation at high concentration, the 13C NMR spectrum could not be obtained. MS (MALDI-TOF): m/z (M)+ = 2881.95, calcd. for C160H192N16S8Si8Zn: 2882.08.Zinc(II) tetra[6,7]quinoxalinoporphyrazine TQP2
To a THF (2 mL) solution of TQP1 (0.019 g, 6.59 μmol), tetrabutylammonium fluoride solution 1.0 M in THF (10 μL) was added. The reaction was quenched by water (5 mL) after being stirred at room temperature overnight. The green solid was collected by centrifuge and washed using methanol. After being dried in vacuum overnight, the green solid was used directly for the next step. To a suspension solution of the green solid in THF/DMF (10 mL, 1/1), compound 3 (0.092 g, 185 μmol), copper(I) iodide (0.004 g, 21 μmol) and DIEA (64 μL, 0.37 mmol) were added under argon. The resulting reaction mixture was stirred at room temperature for 48 hours. After d the solvents were removed under vacuum, the crude product was purified by flash chromatography twice. At the first time, toluene/THF (100% toluene to 50% toluene) was applied, and at the second time hexane/THF (100% hexane to 100% THF) was applied. The green band was washed with methanol (2 mL × 3) and dried under vacuum. TQP2 was obtained as a green solid (0.008 g, 21%). 1H NMR (d6-DMSO/CDCl3, 273 K): δ = 9.31 (s, 8H), 8.53 (s, 8H), 7.90 (s, 8H), 7.52 (br.s, 8H), 7.46 (br.s, 8H), 5.25 (br.s, 16H), 2.23 (s, 48H), 1.93 (s, 48H), 1.43 (s, 216H). Due to the aggregation at high concentration, the 13C NMR spectrum could not be obtained.Zinc(II) tetra[6,7]quinoxalinoporphyrazine TQP3
A solution of TQP2 (0.005 g, 0.89 μmol) in formic acid (1 mL) was stirred at room temperature for 24 hours, and then the acid was removed by rotary evaporation. The residue was dissolved in 1 M NaHCO3 solution (200 μL) and then dialyzed (membrane cutoff = 1 kD) in water for 2 days. The green solution was acidified to pH ∼ 3 with HCl. The green precipitate was collected by centrifuge and washed with water (5 mL × 3). After being dried in vacuum overnight, 0.003 g TQP3 was obtained with a yield of 79%.1H NMR (d6-DMSO, 373 K): δ = 10.10 (br.s, 8H), 8.51 (s, 8H), 7.68 (s, 8H), 7.60 (s, 16H), 5.21 (s, 16H), 2.26–2.29 (m, 48H), 1.98–2.02 (m, 48H). Due to the aggregation at high concentration, the 13C NMR spectrum could not be obtained.MALDI-FTICR data were collected in positive-ion mode from m/z 1000–30000 with a resolving power of ∼30000 at m/z 4300. Special tuning of the funnel RF amplitude (230 Vpp), accumulation hexapole (1.4 MHz, 1900 Vpp), transfer optics (1 MHz, 410 Vpp), time of flight delay (2.5 ms), and ICR cell (sweep excitation power: 50%) were required for high m/z analysis. TQP3 was dissolved in dimethyl sulfoxide and mixed 1:10 with the MALDI matrix solution (40 mg mL−1 2,5-dihydroxybenzoic acid in 90:10 acetonitrile:water) that contained mass calibration standards (oxidized insulin chain B and ubiquitin). DataAnalysis 5.0 (Bruker Daltonics) was used to internally calibrate and process the spectra.
Cell culture
Raw 264.7 cells were cultured in Gibco™ RPMI 1640 medium containing 10% fetal bovine serum (FBS). Cells were incubated in a water jacketed incubator (37 °C, 5% CO2).In vitro PDT study
A typical 1 mL of TQP3-Na-M with 5 μM TQP3-Na was formulated by the following procedure: 5 μL TQP3 stock (1 mM in DMSO) was treated with 5 μL NaHCO3 solution (1 M), then 100 μL of 0.1% Kolliphor® RH40 was added, and the resulting mixture was diluted with 890 μL RPMI medium without FBS.A typical in vitro PDT treatment: raw 264.7 cells were seeded into 96-well optical black plates and incubated for 24 hours prior to treatment. The cell medium was removed from the well and 100 μL TQP3-Na-M was added. The cells were incubated with TQP3-Na-M at 37 °C. After 4 hours, cells were washed once to remove the free PDT reagent using RPMI medium without FBS. Then cells were irradiated with LED light (L780-66-60, Marubeni America Co., New York, NY) at a peak wavelength of 780 nm (half width = 30 nm) and a power density of 25 mW cm−2 for the time experiment required. After the treatment of light irradiation, cells were returned to incubator for an additional time.
For concentration-dependent PDT experiment, the cells were irradiated with LED light for 30 minutes and then incubated for an additional 18 hours. In light dose-dependent experiment, the cells were treated with TQP3-Na-M (5 μM TQP3-Na), and were incubated for an additional 18 hours after irradiation. To study the effect of post-PDT incubation time on the cell viability, cells were treated with TQP3-Na-M (5 μM TQP3-Na) and light irradiation time was 30 min.
Cell viability was determined by CellTiter-Glo assay per the manufacturer's instructions. The luminescent intensity was directly proportional to the amount of remaining viable cells.
Cell morphologic change after PDT
Raw 264.7 cells were seeded into 35 mm MatTek dishes. Cells were washed once with RPMI medium without PBS and then treated with TQP3-Na-M (5 μM TQP3-Na) for 4 hours. Cells were washed once before being exposed to 780 nm LED light for 1 hour. The morphologic changes in living cells were recorded by continuous videos using a Zeiss Axio Observer fluorescent microscopy system. Cells were kept at room temperature during the video recording.Conclusions
In conclusion, we have developed a new NIR dendrimeric PS based on tetra[6,7]quinoxalinoporphyrazine. This PS showed NIR absorption and emission close to 800 nm. Our preliminary in vitro PDT study showed that this PS formulated with surfactant could induce effective cell death under NIR light irradiation at 780 nm. The combined results suggest that this new dendrimeric tetra[6,7]quinoxalinoporphyrazine-polyamide structure can be used as an efficient NIR PS platform. Future studies will be focused on increasing the dendron size to overcome the aggregation issue.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Michelle L. Reyzer and Lisa M. Manier at Vanderbilt University for their assistance on MALDI-FTICR study. This work was supported by the NIH Grant # R21CA174541 (PI: Bai), the startup fund provided by the Department of Radiology, University of Pittsburgh, Science and Technology Commission of Shanghai Municipality #14430723200 & 14430723201 (PIs: Jia & Bai) and National Nature Science Foundation of China #81671739 (PI: Jia).Notes and references
- Z. Huang, Technol. Cancer Res. Treat., 2005, 4, 283–293 CrossRef CAS PubMed.
- M. Triesscheijn, P. Baas, J. H. M. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034–1044 CrossRef CAS PubMed.
- R. M. Strongin, J. O. Escobedo, O. Rusin and S. Lim, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef PubMed.
- R. R. Allison, G. H. Downie, R. Cuenca, X. H. Hu, C. J. Childs and C. H. Sibata, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS PubMed.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS PubMed.
- T. S. Hsieh, J. Y. Wu and C. C. Chang, Chem.–Eur. J., 2014, 20, 9709–9715 CrossRef CAS PubMed.
- O. Taratula, C. Schumann, T. Duong, K. L. Taylor and O. Taratula, Nanoscale, 2015, 7, 3888–3902 RSC.
- L. D. Costa, J. I. T. Costa and A. C. Tome, Molecules, 2016, 21, 320 CrossRef PubMed.
- S. K. Sharma, M. Krayer, F. F. Sperandio, L. Y. Huang, Y. Y. Huang, D. Holten, J. S. Lindsey and M. R. Hamblin, J. Porphyrins Phthalocyanines, 2013, 17, 73–85 CrossRef CAS PubMed.
- Y. C. Yang, Q. L. Guo, H. C. Chen, Z. K. Zhou, Z. J. Guo and Z. Shen, Chem. Commun., 2013, 49, 3940–3942 RSC.
- L. Lamch, J. Kulbacka, J. Pietkiewicz, J. Rossowska, M. Dubinska-Magiera, A. Choromanska and K. A. Wilk, J. Photochem. Photobiol., B, 2016, 160, 185–197 CrossRef CAS PubMed.
- U. Isci, M. Beyreis, N. Tortik, S. Z. Topal, M. Glueck, V. Ahsen, F. Dumoulin, T. Kiesslich and K. Plaetzer, Photodiagn. Photodyn. Ther., 2016, 13, 40–47 CrossRef CAS PubMed.
- W. Kuzyniak, E. A. Ermilov, D. Atilla, A. G. Gurek, B. Nitzsche, K. Derkow, B. Hoffmann, G. Steinemann, V. Ahsen and M. Hopfner, Photodiagn. Photodyn. Ther., 2016, 13, 148–157 CrossRef CAS PubMed.
- J. Y. Liu, J. Li, X. Yuan, W. M. Wang and J. P. Xue, Photodiagn. Photodyn. Ther., 2016, 13, 341–343 CrossRef CAS PubMed.
- B. Y. Zheng, M. R. Ke, W. L. Lan, L. Hou, J. Guo, D. H. Wan, L. Z. Cheong and J. D. Huang, Eur. J. Med. Chem., 2016, 114, 380–389 CrossRef CAS PubMed.
- K. Sakamoto, T. Kato, E. Ohno-Okumura, M. Watanabe and M. J. Cook, Dyes Pigm., 2005, 64, 63–71 CrossRef CAS.
- F. Mitzel, S. FitzGerald, A. Beeby and R. Faust, Chem.–Eur. J., 2003, 9, 1233–1241 CrossRef CAS PubMed.
- Z. Musil, P. Zimcik, M. Miletin, K. Kopecky and J. Lenco, Eur. J. Org. Chem., 2007, 4535–4542, DOI:10.1002/ejoc.200700275.
- A. Kozyrev, M. Ethirajan, P. Chen, K. Ohkubo, B. C. Robinson, K. M. Barkigia, S. Fukuzumi, K. M. Kadish and R. K. Pandey, J. Org. Chem., 2012, 77, 10260–10271 CrossRef CAS PubMed.
- L. Luan, L. Ding, W. Zhang, J. Shi, X. Yu and W. Liu, Bioorg. Med. Chem. Lett., 2013, 23, 3775–3779 CrossRef CAS PubMed.
- P. Zimcik, M. Miletin, H. Radilova, V. Novakova, K. Kopecky, J. Svec and E. Rudolf, Photochem. Photobiol., 2010, 86, 168–175 CrossRef CAS PubMed.
- P. Shao and M. F. Bai, Chem. Commun., 2012, 48, 9498–9500 RSC.
- C. Kohl, T. Weil, J. Qu and K. Mullen, Chemistry, 2004, 10, 5297–5310 CrossRef CAS PubMed.
- W. D. Jang, Y. Nakagishi, N. Nishiyama, S. Kawauchi, Y. Morimoto, M. Kikuchi and K. Kataoka, J. Controlled Release, 2006, 113, 73–79 CrossRef CAS PubMed.
- D. K. Muli, B. L. Carpenter, M. Mayukh, R. A. Ghiladi and D. V. McGrath, Tetrahedron Lett., 2015, 56, 3541–3545 CrossRef CAS.
- P. Shao and M. Bai, Chem. Commun., 2012, 48, 9498–9500 RSC.
- P. Shao, N. Jia, S. Zhang and M. Bai, Chem. Commun., 2014, 50, 5648–5651 RSC.
- A. Saraste, Herz, 1999, 24, 189–195 CrossRef CAS PubMed.
- P. Golstein and G. Kroemer, Trends Biochem. Sci., 2007, 32, 37–43 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06348b |
| This journal is © The Royal Society of Chemistry 2017 |